
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable cell-laden silk acid hydrogel†
Ziyang
Sun
a,
Haoran
Liu
a,
Dandan
Dai
a,
Hao
Lyu
a,
Ruochuan
Huang
a,
Wenzhao
Wang
a and
Chengchen
Guo
 *abc
*abc
aSchool of Engineering, Westlake University, Hangzhou, Zhejiang 310023, China. E-mail: guochengchen@westlake.edu.cn
bResearch Center for Industries of the Future, Westlake University, Hangzhou, Zhejiang 310030, China
cWestlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang 310024, China
First published on 29th November 2023
Abstract
This study presents an injectable cell-laden hydrogel system based on silk acid, a carboxylated derivative of natural silk fibroin, which exhibits promising applications in biomedicine. The hydrogel is produced under physiological conditions (37 °C and pH 7.4) via physical crosslinking. Notably, the hydrogel demonstrates remarkable cytocompatibility, enabling efficient cell encapsulation, and exhibits good injectability. These promising results strongly indicate the potential of silk acid hydrogel for transformative applications, including 3D cell culture, targeted cell delivery, and tissue engineering.
Cell-laden hydrogels hold tremendous promise in tissue engineering and regenerative medicine. Their primary use is to mimic the natural extracellular matrix environment in which cells reside in the body, offering a way to culture and control cells in a three-dimensional (3D) format.1–3 Cell-laden hydrogels have been used in various applications, from cell delivery and disease modeling to the development of engineered tissues for implantation in patients. Ideal cell-laden hydrogels should satisfy several criteria, including biocompatibility to ensure no adverse immune reactions, mechanical strength tailored to the specific tissue they are designed to replace, efficient mass transport capabilities for the adequate diffusion of nutrients and waste products, and suitable degradability to allow new tissue to supersede the hydrogel structure gradually.4–7
Silk fibroin derived from silkworm cocoons is a natural protein recognized for its excellent mechanical properties, biocompatibility, and degradability.8–10 Silk fibroin hydrogels can be made via self-assembly of silk fibroin under specific chemical or physical treatments.11–14 Nevertheless, prior studies on silk hydrogels have indicated that the formation of hydrogels typically requires non-physiological conditions, such as high temperatures, low pH, and the addition of organic solvents and compounds, or necessitates harsh treatments like vortexing and ultrasonication.15–19 The poor cytocompatibility resulting from these conditions restricts the utility of silk fibroin hydrogels as cell-laden hydrogels.
In this study, we produced silk acid (SA) with a carboxylation degree of 9.5% by an efficient chemical modification on the regenerated silk fibroin (RSF) (Fig. 1a and Fig. S1, ESI†).20 Subsequently, by dissolving the SA in a HEPES solution (pH 7.4), the same buffer used in the cell culture medium, and incubating at 37 °C, the spontaneous formation of the SA hydrogel was observed (Fig. 1b). In contrast, unmodified silk fibroin did not exhibit hydrogel-forming properties under the same conditions. The SA hydrogel was formed under physiological conditions without cytotoxic chemicals or harsh treatments. Consequently, it holds tremendous potential for cell encapsulation in tissue engineering, cell therapies, and 3D cell culture applications. Herein, we have thoroughly investigated the conditions that influence the self-assembly process and the structural change of proteins during gelation, elucidating the self-assembly mechanism of the SA hydrogel. We have demonstrated the injectability of the SA hydrogel and loaded various types of cells into the injectable SA hydrogel to show its potential applications in cell encapsulation, cell delivery, and 3D cell culture.
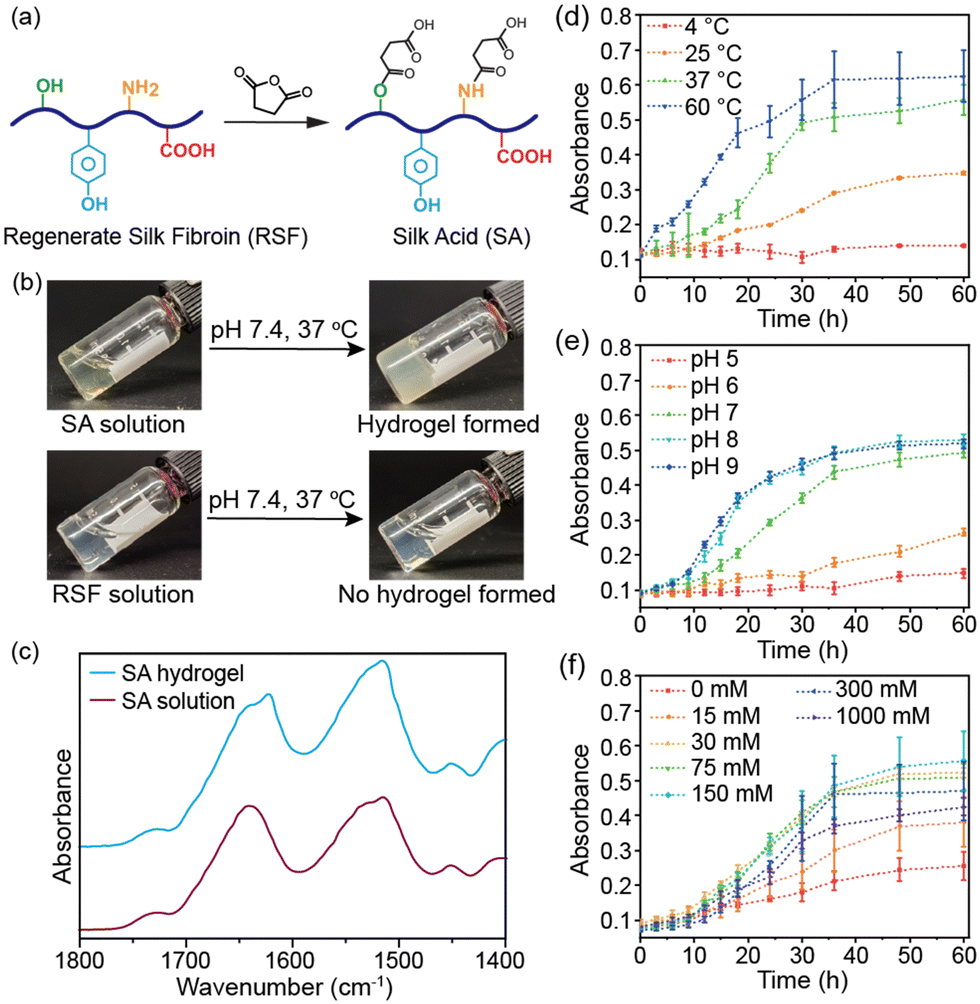 | ||
| Fig. 1 (a) Silk acid (SA) is prepared by carboxylation of regenerated silk fibroin (RSF). (b) Silk acid hydrogel is formed under physiological conditions (37 °C and pH 7.4), while no hydrogel was formed for RSF. (c) The FTIR spectra of the SA solution and the SA hydrogel after freeze-drying. The absorbance changes of the SA solutions with time at different temperatures (d), different pH values (e), and different NaCl concentrations (f). Data are presented as mean ± s.d., n = 4. | ||
The SA hydrogel turned opaque upon gelation, similar to the silk fibroin hydrogel (Fig. 1b).21 The Fourier-transform infrared spectroscopy (FT-IR) spectra revealed a shift in the peaks of the amide I from 1645 to 1620 cm−1 after self-assembly, indicating the formation of β-sheet structures (Fig. 1c). The β-sheet structures further transform into a semi-crystalline structure, leading to reduced light transmittance of the SA hydrogel. The self-assembly process of SA was monitored by measuring the absorbance of the hydrogel system. During incubation at 37 °C, the absorbance of the SA solution increased gradually in the initial 12 hours, indicating that it remained in a solution state during this early stage (Fig. 1d). However, after 18 hours, the absorbance significantly increased, reaching a plateau after 36 hours of incubation. This result suggests that the self-assembly of SA to form a hydrogel required approximately 36 hours at 37 °C. Conversely, under the same conditions, the absorbance of silk fibroin solution remained low, indicating negligible self-assembly (Fig. S2a, ESI†). The self-assembly of SA is energy-dependent, with an accelerated rate at 60 °C within 24 hours and no occurrence at 4 °C within three days (Fig. 1d). Additionally, a higher concentration of SA led to faster self-assembly while a 1 wt% SA solution did not form a hydrogel (Fig. S2b, ESI†).
Subsequently, we investigated the influence of the buffer solution. SA showed a similar self-assembly process in HEPES, PBS, and Tris–HCl buffer solutions (Fig. S2c, ESI†). However, the pH value had a significant effect on its self-assembly. In weak acid solution (pH ∼5 or 6), SA failed to self-assemble into a hydrogel within three days (Fig. 1e). In contrast, increasing the pH value accelerated the self-assembly process, with gelation occurring in approximately 24 hours in weak basic solution (pH ∼8 or 9). Moreover, ionic strength was also an influencing factor. Self-assembly of SA was slow when dissolved in buffer solution without adding NaCl, indicating that sufficient ionic strength was necessary for the gelation of SA (Fig. 1f). However, increasing the NaCl concentration over 300 mM slowed down the self-assembly process. The optimal NaCl concentration was 75–300 mM, which aligns with the physiological level.
Circular dichroism (CD) spectroscopy and FT-IR spectroscopy were further used to characterize the secondary structural changes of SA during the sol–gel transition. SA presented a random coil structure with a prominent band at 195 nm in the CD spectrum at the beginning and after 12 hours of incubation. However, as the self-assembly progressed, the band at 219 nm appeared and was gradually enhanced, indicating the secondary structural transition into β-sheet structures (Fig. 2a). The FT-IR results of SA with increased incubation time confirmed this structural change. The deconvolution analysis of the amide I band demonstrated a progressive increase in β-sheet content during the self-assembly, with the β-sheet content reaching approximately 14% after 48 hours (Fig. 2b and Fig. S3, ESI†). Based on the above characterization, the self-assembly of SA can be attributed to a structural transformation from a random coil structure to a β-sheet structure. The β-sheet structure then further aggregates into protofibril and fibril structures.22,23
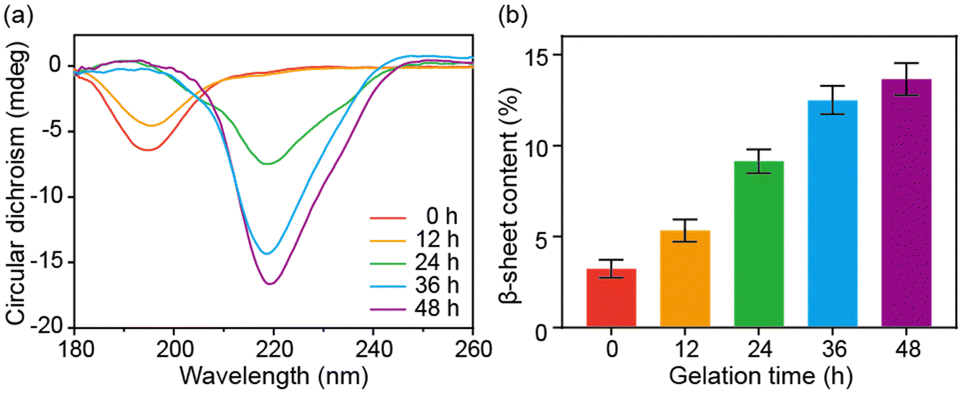 | ||
| Fig. 2 (a) The CD spectra of the SA solutions with different incubation times. (b) The β-sheet content of the SA in solutions with different incubation times. The β-sheet content was determined with the deconvolution of the amide I band in the FTIR spectra. Data are presented as mean ± s.d., n = 4. | ||
Fluorescent dyes were used to probe the structural transformation of SA during the self-assembly.24 After 12 hours of incubation, the enhancement of the fluorescence intensity of Nile red was observed, indicating the formation of hydrophobic domains within the SA solution (Fig. 3a).25 Meanwhile, the fluorescence intensity of thioflavin T (ThT) and 8-anilinonaphthalene-1-sulfonic acid (ANS) showed no significant increase during the initial 12 hours. Subsequently, after 24 hours of incubation, the fluorescence intensity of ThT and ANS showed a pronounced increase, confirming the presence of the fibril structures within SA hydrogel (Fig. 3a).26,27 Similar to SF, the highly repetitive amino acid motifs in SA were able to fold into β-sheet structures, which further arranged into fibril structures.28 Therefore, the formation of β-sheet structures and β-sheet-enriched fibril structures after 24 hours of incubation was consistent with the CD and FT-IR results. Confocal microscopy was used to observe the formation of fibril structures in SA hydrogel (Fig. 3b). After incubating for 12 hours, certain regions of SA exhibited fluorescence enhancement, indicating the presence of β-sheet-enriched fibril structures in these regions. As the incubation time increased, the area of fluorescence enhancement expanded and eventually formed a network. Furthermore, nanofibril structures were observed within the fluorescence-enhanced regions.
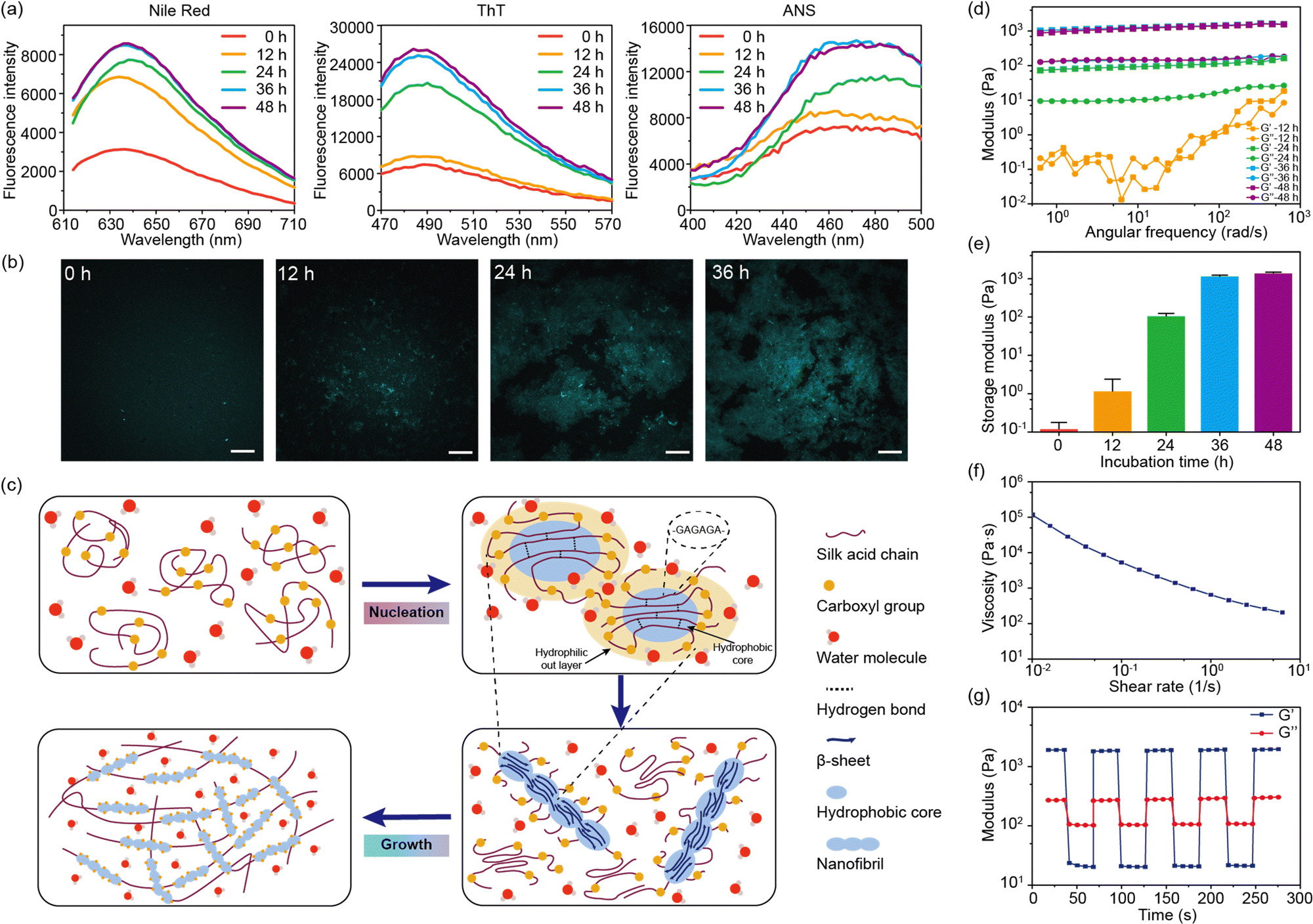 | ||
| Fig. 3 (a) The fluorescence spectra of the SA solutions loaded with Nile red, ThT, and ANS at different incubation times. (b) Confocal images of the SA solution loaded with ThT at different incubation times. The scale bar is 10 μm. (c) Self-assembly mechanism of SA during the sol–gel transition. (d) Rheological study on the SA hydrogels with different incubation times. (e) Storage modulus of the SA hydrogel with different incubation times. (f) Flow curve of the SA hydrogel after 36 h of incubation. (g) Low-high strain cycles on the SA hydrogel after 36 h of incubation. Data were presented as mean ± s.d., n = 3. | ||
Based on these findings, the self-assembly of SA undergoes two stages: nucleation and growth (Fig. 3c). Initially, SA is in the random coil conformation after dissolution in water. Due to the electrostatic repulsion between carboxyl groups, the hydrophobic domains of SA, mainly composed of GAGA sequences, are confined in the core of the SA molecules. The formation of the hydrophobic cores is confirmed by the enhanced fluorescence of Nile red after 12 hours of incubation. Driven by the hydrophobic interaction, the hydrophobic cores of SA undergo dehydration, forming intra- or inter-molecular hydrogen bonds that further lead to the formation of β-sheet structures.21 This process is referred to as nucleation. While unmodified silk fibroin takes several days for nucleation, SA accomplishes this within a day under physiological conditions. This acceleration may be owing to the altered charge distribution by the carboxyl groups in SA. This is also correlated with the pH-dependent self-assembly. Under basic conditions (pH = 8–9), most carboxyl groups are deprotonated, leading to a stronger electrostatic interaction that promotes the formation of hydrophobic cores. Meanwhile, under acidic conditions (pH = 5–6), the nucleation process is significantly suppressed. Moreover, the nucleation process is also suppressed by high NaCl concentrations (>300 mM) due to the strong electrostatic shielding effect of electrolytes.
During the growth stage, the hydrophobic domains aggregate with the accelerated formation of β-sheet structures, forming nanofibril structures (Fig. 3c). This is reflected by the enhancement of the β-sheet structures in the CD and FT-IR spectra of SA during incubation for 12–36 hours. The nanofibrils further aggregate and intertwine to form the SA hydrogel network. However, this process is influenced by the ionic strength of the solution. When no NaCl is added to the solution, the strong electrostatic repulsion between carboxyl groups may inhibit the aggregation of hydrophobic domains and the formation of nanofibril structures, leading to a slow self-assembly. This result implies that an optimal concentration of NaCl is critical to the sol–gel transition of SA.
We also characterized the rheological properties of SA during the sol–gel transition (Fig. 3d). In the initial 12 hours, SA remains in a solution state with a low modulus below 1 Pa. After incubation for 24 hours at 37 °C, the modulus increased to about 100 Pa, indicating the occurrence of a gel state (G′ > G′′). After incubation for 48 hours, the storage modulus would reach a plateau around 1000 Pa (Fig. 3e). Remarkably, the modulus of the SA hydrogel was much lower than that of the silk fibroin hydrogel, which was tens to hundreds of kPa.17,29 This is due to the low content of β-sheet structures in the SA hydrogel (14% from FT-IR results). Due to electrostatic repulsion between carboxyl groups, the growth of the fibril structure and the size of the crystalline structure are significantly inhibited. Moreover, the repulsive effect would also loosen the fibril network, leading to a low modulus and viscosity. However, the loosened network offers shear-thinning properties and injectability of the SA hydrogel. As the shear rate increases, the viscosity of the SA hydrogel drops dramatically (Fig. 3f). Under high strain (100%), the SA hydrogel behaves like a low-modulus solution but quickly reverts to a gel state under minimal strain (2%) (Fig. 3g). Such property makes the SA hydrogel easily extruded through a 21-gauge needle.
The SA hydrogel offers excellent potential for 3D cell culture. Herein, three different cell lines, including fibroblasts (L929), an endothelial cell line (HUVEC), and bone marrow-derived mesenchymal stem cells (rBMSCs), were seeded inside the SA hydrogel. After 24 hours and 72 hours of incubation, all three cell lines survived within the SA hydrogel (Fig. 4a and Fig. S5, ESI†). Among them, rBMSCs showed the best survival rate of about 95% (Fig. 4b). Moreover, the SA hydrogel can support cell volume expansion, cell growth, and cell proliferation (Fig. S6 and S7, ESI†). We further investigated the cell viability after injection. Further tests on cell viability post-injection revealed that after encapsulating cells inside the SA hydrogel and injecting them through a 21-gauge needle, all cell lines maintained a survival rate of over 90% after 24 hours. This suggests that the shear-thinning property of the SA hydrogel minimizes the shear force during injection, protecting cells from rupture (Fig. 4c and d). Therefore, the SA hydrogel emerges as a promising bio-ink for cell-laden bioprinting. In addition, the SA hydrogel showed enzymatic degradability (Fig. S8, ESI†), similar to the reported SA films.20
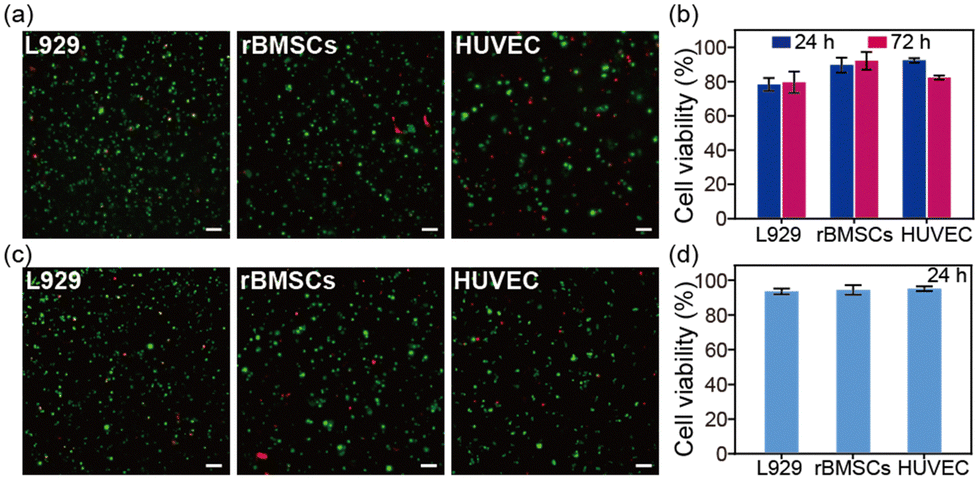 | ||
| Fig. 4 (a) L929, rBMSCs, and HUVEC were seeded and cultured inside the SA hydrogels for 72 hours. Cells were stained with calcein AM and PI for live/dead staining. Scale bar: 100 μm. (b) Cell viability was calculated from staining images with ImageJ. (c) The SA hydrogel was encapsulated with L929, rBMSCs, and HUVECs and loaded into the syringes. After extruding through a 21G needle and incubating for 24 hours, the cells were stained with calcein AM and PI for live/dead staining. The scale bar is 100 μm. (d) Cell viability after injection was calculated from staining images with ImageJ. Data are presented as mean ± s.d., n = 3. | ||
In conclusion, a chemically modified silk fibroin hydrogel system was developed based on silk acid. The silk acid hydrogel can be produced under physiological conditions (pH 7.4, 150 mM NaCl, 37 °C) with a modulus close to the extracellular matrix (1–10 kPa), supporting 3D cell culture and cell delivery through injection.30 We believe that the silk acid hydrogel will emerge as a promising biomaterial in cell delivery, tissue regeneration, and organoid development.
This work was supported by the Key R&D Program of Zhejiang Province (No. 2023SDXHDX0004), the National Natural Science Foundation of China (No. 52103129), the HRHI program (No. 202209004) of Westlake Laboratory of Life Sciences and Biomedicine, and the Foundation of Westlake University. The authors thank Min Wang, the Instrumentation and Service Center for Physical Sciences and the Instrumentation and Service Center for Molecular Science at Westlake University for the technical support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Unagolla and A. C. Jayasuriya, Appl. Mater. Today, 2020, 18, 100479 CrossRef PubMed.
- J. Z. Yang, Y. S. Zhang, K. Yue and A. Khademhosseini, Acta Biomater., 2017, 57, 1–25 CrossRef CAS PubMed.
- J. Malda, J. Visser, F. P. Melchels, T. Jungst, W. E. Hennink, W. J. A. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- A. Skardal, M. Devarasetty, H. W. Kang, I. Mead, C. Bishop, T. Shupe, S. J. Lee, J. Jackson, J. Yoo, S. Soker and A. Atala, Acta Biomater., 2015, 25, 24–34 CrossRef CAS PubMed.
- P. Rastogi and B. Kandasubramanian, Biofabrication, 2019, 11, 042001 CrossRef CAS PubMed.
- Y. Zhang, Y. Xu and J. Gao, Biomater. Sci., 2023, 11, 3784–3799 RSC.
- C. Guo, C. Li, X. Mu and D. L. Kaplan, Appl. Phys. Rev., 2020, 7, 011313 CAS.
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS PubMed.
- W. Huang, S. Ling, C. Li, F. G. Omenetto and D. L. Kaplan, Chem. Soc. Rev., 2018, 47, 6486–6504 RSC.
- U. J. Kim, J. Y. Park, C. M. Li, H. J. Jin, R. Valluzzi and D. L. Kaplan, Biomacromolecules, 2004, 5, 786–792 CrossRef CAS PubMed.
- S. Nagarkar, T. Nicolai, C. Chassenieux and A. Lele, Phys. Chem. Chem. Phys., 2010, 12, 3834–3844 RSC.
- H. Zheng and B. Zuo, J. Mater. Chem. B, 2021, 9, 1238–1258 RSC.
- Y. Lyu, Y. Liu, H. He and H. Wang, Gels, 2023, 9, 431 CrossRef CAS PubMed.
- X. L. Wu, J. Hou, M. Z. Li, J. N. Wang, D. L. Kaplan and S. Z. Lu, Acta Biomater., 2012, 8, 2185–2192 CrossRef CAS PubMed.
- X. Q. Wang, J. A. Kluge, G. G. Leisk and D. L. Kaplan, Biomaterials, 2008, 29, 1054–1064 CrossRef CAS PubMed.
- T. Yucel, P. Cebe and D. L. Kaplan, Biophys. J., 2009, 97, 2044–2050 CrossRef CAS PubMed.
- Z. Zhu, S. Ling, J. Yeo, S. Zhao, L. Tozzi, M. J. Buehler, F. Omenetto, C. Li and D. L. Kaplan, Adv. Funct. Mater., 2018, 28, 1704757 CrossRef PubMed.
- B. Maity, S. Samanta, S. Sarkar, S. Alam and T. Govindaraju, ACS Appl. Bio Mater., 2020, 3, 3544–3552 CrossRef CAS PubMed.
- Z. Sun, R. Huang, H. Lyu, X. Yu, W. Wang, J. Li, X. Lu and C. Guo, Adv. Healthcare Mater., 2023, 2301439 CrossRef CAS PubMed.
- A. Matsumoto, J. Chen, A. L. Collette, U. J. Kim, G. H. Altman, P. Cebe and D. L. Kaplan, J. Phys. Chem. B, 2006, 110, 21630–21638 CrossRef CAS PubMed.
- J. A. J. Housmans, G. Q. Wu, J. Schymkowitz and F. Rousseau, FEBS J., 2023, 290, 554–583 CrossRef CAS PubMed.
- A. Morriss-Andrews and J. E. Shea, Annu. Rev. Phys. Chem., 2015, 66, 643–666 CrossRef CAS PubMed.
- M. F. Pignataro, M. G. Herrera and V. I. Dodero, Molecules, 2020, 25, 4854 CrossRef CAS PubMed.
- D. L. Leiske, I. C. Shieh and M. L. Tse, Langmuir, 2016, 32, 9930–9937 CrossRef CAS PubMed.
- R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, R. K. Grover, R. Roy and S. Singh, J. Struct. Biol., 2005, 151, 229–238 CrossRef CAS PubMed.
- A. Ahmad, V. N. Uversky, D. Hong and A. L. Fink, J. Biol. Chem., 2005, 280, 42669–42675 CrossRef CAS PubMed.
- Z. Gong, L. Huang, Y. Yang, X. Chen and Z. Shao, Chem. Commun., 2009, 7506–7508 RSC.
- J. W. Liu, H. W. Sun, Y. W. Peng, L. G. Chen, W. Xu and R. Shao, Molecules, 2022, 27, 3418 CrossRef CAS PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04280d |
| This journal is © The Royal Society of Chemistry 2024 |