
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioether-based poly(2-oxazoline)s: from optimized synthesis to advanced ROS-responsive nanomaterials†
Semira
Bener
 a,
Ewa
Pavlova
b,
Hynek
Beneš
b and
Ondřej
Sedláček
*a
a,
Ewa
Pavlova
b,
Hynek
Beneš
b and
Ondřej
Sedláček
*a
aDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Prague 2 128 40, Czech Republic. E-mail: sedlacek@natur.cuni.cz
bInstitute of Macromolecular Chemistry, AS CR, Prague 6 162 06, Czech Republic
First published on 10th October 2023
Abstract
Intelligent redox-responsive polymers, such as thioether-containing macromolecules, facilitate drug delivery and triggered release in biomedical applications. Moreover, reactive oxygen species (ROS)-responsive thioether systems based on poly(2-oxazoline)s (PAOx) platforms hold great promise for the development of highly biocompatible, stimuli-responsive biomaterials. However, thioether-containing PAOx are particularly difficult to synthesize because thioethers are incompatible with the cationic ring-opening polymerization (CROP). In this study, we aim at developing an alternative route to well-defined thioether-containing PAOx by a simple post-polymerization modification of linear polyethyleneimine. First, the synthesis of ROS-responsive PAOx homopolymers was optimized. Furthermore, ROS-sensitive amphiphilic diblock copolymers poly(ethylene glycol)-block-poly(2-methylthiomethyl-2-oxazoline) were synthesized by combining CROP with 2-oxazoline side-chain interchange via a polyethyleneimine block intermediate. In an aqueous environment, the copolymers self-assembled into thioether-containing micelles. These micelles were characterized by size exclusion chromatography, nuclear magnetic resonance, matrix-assisted laser desorption/ionization-time of flight mass spectrometry, dynamic light scattering, differential scanning calorimetry, and transmission electron microscopy. In addition, treatment with diluted H2O2 destabilized the nanoparticles, thus demonstrating their oxidation-responsiveness. This approach provides key insights into the design and development of stimuli-responsive polymers for potential biomedical applications, such as drug delivery systems.
Introduction
Over the past century, several studies have analyzed supramolecular nanostructures generated by the self-assembly of amphiphilic block copolymers for their stability as drug carriers.1–4 Such nanostructures have been extensively used in bio-inspired applications, including drug delivery and nanoreactors, thanks to their broad range of topologies, including micelles, vesicles, and nanogels.5 Among these applications, self-assembled nanoparticles composed of biocompatible and biodegradable polymers have been explored for anticancer drug encapsulation for their high potential to improve the solubility of a drug in water, prolonging its blood half-life while simultaneously reducing its systemic toxicity.6–8Another key feature of these systems is their ability to trigger the controlled release of their encapsulated content. This feature has prompted extensive research on the engineering mechanisms of drug carriers and generated major advances in the design of such systems responsive to various stimuli. By harnessing intrinsic triggers, these advancements have improved excretion from the body and enhanced the therapeutic potential of targeted applications through a more effective and controlled drug release in response to pH,9 temperature,10,11 redox,12 ultrasound13 and light14,15 stimuli.
Reactive oxygen species (ROS), particularly hydrogen peroxide (H2O2), hydroxyl radicals (˙OH), the superoxide anion radical (O2˙−), and nitrites (ONOO−), play a key role in regulating fundamental physiological processes in living organisms, including cellular growth, cell signaling, modulation of protein function, and stress adaptation.16,17 While ROS are maintained at basal levels in healthy cells, elevated levels are often found in pathogenic cells, as a result of high metabolic activity or inflammation due to conditions such as cancer or neurodegenerative diseases. Many delivery systems have been recently developed to release therapeutic cargos in response to ROS for target–specific drug delivery. The oxidative response of these systems is consistently based on the oxidation of the hydrophobic moieties leading to strongly hydrophilic polymeric materials that disassemble upon oxidation, thereby releasing the incorporated cargo.18 Invariably, these hydrophilic polymers contain oxidation–sensitive moieties, such as sulfides, diselenides, thioketals, aryl boronic esters, and thioethers.19,20
Thioether-containing polymer systems have demonstrated potential in ROS-responsive applications due to the unique reactivity of sulfur to ROS.21 Polymers incorporating thioether groups can be designed to be responsive to ROS, such as glutathione, by undergoing a hydrophobic-hydrophilic phase transition from thioethers with low dipole moments to a hydrophilic sulfoxide or sulfone in response to ROS, leading to an efficient disassembly of the materials.22 Thus, their disassembly enables the targeted and controlled release of drugs or other cargos based on the hydrophobic-hydrophilic phase transition of their thioether groups.
Poly(2-oxazoline)s (PAOx) represent an emerging class of polymers for the construction of amphiphilic block copolymer nanoparticles that can be precisely controlled and adjusted for specific applications thanks to their versatility and relatively straightforward synthesis.23–27 The properties of PAOx can be fine-tuned by varying the monomer as the side-chain substituent of 2-oxazoline can be replaced widely and readily.28 Furthermore, living cationic ring-opening polymerization (CROP) provides control over the resulting copolymeric structure and access to end-functionalized polymer chains with a wide range of functional groups.29,30 Thioether-containing poly(2-oxazoline)s offer several advantages over analogous thioether-containing polymers derived from, e.g., polyacrylamides,31 even though their synthesis can be somewhat more complex. The increased flexibility of the poly(2-oxazoline) backbone,32 in contrast to acrylic polymers, could potentially enhance nanoparticle self-assembly and enable the encapsulation of drugs.33 Additionally, these more flexible polymers exhibit accelerated diffusion into cancerous tissues, leading to greater accumulation within tumors. Yet, despite their potential benefits, thioether-containing PAOx polymers are still difficult to synthesize. Polymers containing specific side chain groups, such as free amines, alcohols, thiols, and carboxylic acids cannot be prepared by CROP because of potential terminations, chain transfer, and side reactions via a nucleophilic attack on the propagating cationic oxazolinium species.34–36 In this context, 2-((methylthio)methyl)-2-oxazoline has been polymerized using methyl tosylate and methyl triflate as a more electrophilic initiator.37–39 However, this polymerization was not controlled, showing a steep initial increase in molar masses at a low monomer conversion rate, which indicates the occurrence of side reactions during the polymerization, most likely caused by the thioether bond. Moreover, the nucleophilic thioether group could induce termination and transfer reactions by attacking the oxazolinium group and forming a sulfonium ion. Nevertheless, functional groups can be incorporated into the PAOx polymeric chain by post-polymerization modifications.40–42
Post-polymerization acylation of PEI is a straightforward method for preparing well-defined, functional PAOx, as described recently.32,35,43–45 By using this approach, we can synthesize diverse poly(2-oxazoline) (PAOx) libraries using various carboxylic acids. This method also eliminates the need for functional monomer synthesis and purification, which are often challenging and time-consuming processes, and extends to the synthesis of random PAOx copolymers, especially when monomers with different reactivities yield gradient copolymer structures, such as MeOx with PhOx. Concomitantly, the reacetylation of linear polyethyleneimine (PEI) prepared via hydrolysis of poly(2-methyl-2-oxazoline) (PMeOx) with acetic anhydride for PAOx synthesis is a promising alternative to conventional CROP, particularly for monomers with functionalities that can act as terminators of CROP.46–50 Therefore, we aimed at developing an alternative route to thioether-containing PAOx by a simple post-polymerization modification of linear PEI prepared via hydrolysis of readily available PEtOx.
In this study, we report the synthesis of novel oxidation-sensitive PAOx by post-polymerization side chain interchange via a linear PEI intermediate. First, we prepared a series of three leading thioether-containing PAOx homopolymers using three different alkylthiocarboxylic acids. The polymers were well-defined and showed oxidation responsivity. In addition, we synthesized poly(ethylene glycol)-block-poly(2-methylthiomethyl-2-oxazoline) (PEG-b-PMTMeOx) amphiphilic diblock copolymers by combining CROP with the reacylation method. Once PEG-b-PEtOx was synthesized by CROP, the copolymer side chain groups were selectively removed by acidic hydrolysis, resulting in a linear PEG-b-PEI, which was further reacylated with a thioether-containing carboxylic acid. Aqueous self-assembly of the PEG-b-PMTMeOx diblock copolymers with different PAOx lengths was examined using the solvent switch method. The micelles containing thioether groups were treated with H2O2 to demonstrate the destabilization of the micellar structure. Self-assembly was then investigated by dynamic light scattering (DLS) and transmission electron microscopy (TEM).
Experimental section
Materials
Poly(ethylene glycol) monomethyl ether (PEG, 2000 g mol−1), triethylamine (TEA), p-nitrobenzenesulfonyl chloride, (benzotriazole-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyBOP), (methylthio)acetic acid, (methylthio)propionic, and (ethylthio)acetic acid were purchased from Sigma-Aldrich and used as received. 2-Ethyl-2-oxazoline (EtOx) was purchased from Sigma-Aldrich and distilled over CaH2. Linear polyethyleneimine (PEI, DP = 100) was synthesized from PEtOx, according to the literature.43Polymer characterization
Size Exclusion Chromatography (SEC) was used to determine the molar masses (Mw, mass-averaged molecular weight; Mn, number-averaged molecular weight) and dispersity (Đ = Mw/Mn) of the polyoxazoline polymers on a Watrex Streamline system equipped with a Streamline P1 Pump, a Streamline AS2 Autosampler, a Streamline CT Column Thermostat, a Streamline UV detector, and a Streamline RI detector. The separation was performed on two PLgel 5 μm mixed-D columns in a series thermostatted at 55 °C in N,N-dimethylacetamide (DMAc) containing 50 mM of LiCl at an elution rate of 0.5 mL min−1. Molar masses and dispersities were calculated against narrow dispersity poly(methyl methacrylate) standards. Linear PEI was measured using Ultimate 3000 system (Dionex) equipped with a TSKgel G5000PWXL-CP 300 × 7.8 mm SEC column. Three detectors, UV/vis, refractive index (RI) Optilab-rEX and multiangle light scattering (MALS) DAWN EOS (Wyatt Technology) were employed; with a methanol and sodium acetate buffer (0.3 M, pH 6.5) mixture (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 vol%, flow rate of 0.5 mL min−1) as mobile phase using dn/dc = 0.169.
:20 vol%, flow rate of 0.5 mL min−1) as mobile phase using dn/dc = 0.169.
Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance MSL 400 MHz NMR spectrometer at 25 °C in CD3OD or DMSO-d6. All chemical shifts are given in ppm.
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) was performed on a Microflex LT MALDI-TOF MS (Bruker Daltonics) mass spectrometer. All mass spectra were recorded at an accelerating potential of 20 kV in positive ion mode and in reflectron mode (matrix:2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB)). The samples were applied to the MALDI plate using the dried drop method. All measurements were calibrated using poly(ethylene glycol) monomethyl ether (PEG, Mn = 2000 Da).
Dynamic light scattering (DLS) measurements were used to determine the hydrodynamic diameters of the polymers in distilled water on a ZEN3600 Zetasizer Nano-ZS zeta potential analyzer (Malvern Instruments, UK). The apparent Z-averaged hydrodynamic diameter of the particles, Dh, was determined at a scattering angle of θ = 173°, and the DTS (Nano) program was used to evaluate the data.
Differential Scanning Calorimetry (DSC) was performed on a DSC Q2000 (TA Instruments, USA) with a nitrogen purge gas (50 mL min−1). Indium was used as a standard for temperature and enthalpy calibrations. Samples of about 5 mg were encapsulated into aluminum pans. DSC runs were performed with a ramp rate of 10 °C min−1 using a heating–cooling–heating cycle from −80 °C to 150 °C. The glass transition temperature (Tg) and ΔCp values were determined from the second heating run.
Transmission Electron Microscopy (TEM) observations were conducted under a Tecnai G2 Spirit Twin microscope operating at an accelerating voltage of 120 kV in bright-field imaging mode. Polymer nanoparticles (4 μL) were applied onto a copper TEM grid (300 mesh) coated with a thin, electron-transparent carbon film. To prevent oversaturation during the drying process, excess solution was removed by gently touching the bottom of the grid with filtering paper after 5 minutes of sedimentation. Subsequently, the samples were negatively stained with uranyl acetate (2 μL of 1 wt% solution applied onto the dried nanoparticles and removed after 30 seconds) and left to dry at room temperature before their observation under the TEM microscope.
Results and discussion
The synthetic approach to the preparation of thioether-containing PAOx homopolymers is illustrated in Scheme 1. This approach involved a controlled reacylation of linear polyethyleneimine (PEI). PEI was obtained from PEtOx by controlled acidic hydrolysis of its side-chain groups.51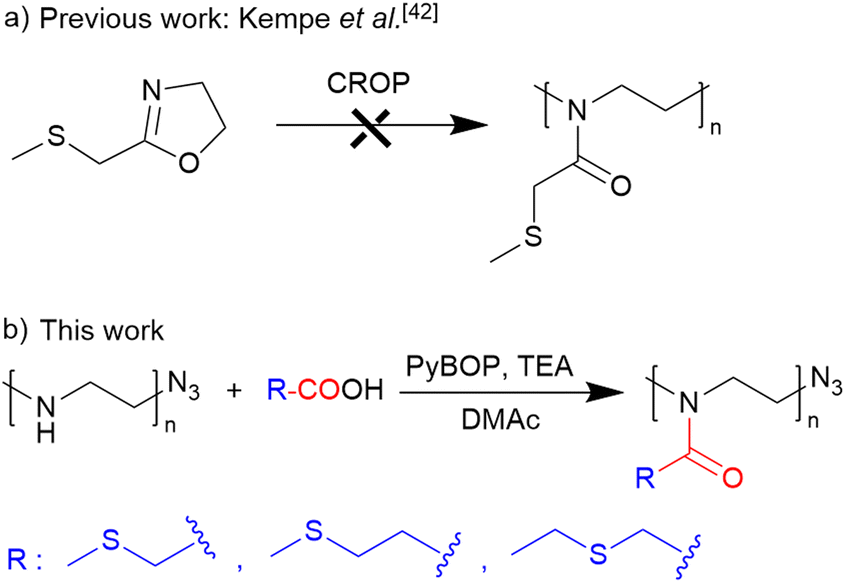 | ||
| Scheme 1 (a) Schematic representation of the unsuccessful CROP of 2-methylthiomethyl-2-oxazoline42 and (b) synthesis of thioether-containing homopolymers via acylation of well-defined PEI. | ||
The starting material, a well-defined poly(2-ethyl-2-oxazoline) (PEtOx) with a degree of polymerization (DP) of 100, was synthesized by living CROP of EtOx in acetonitrile. This polymerization was terminated with sodium azide to introduce the semitelechelic azide group for potential conjugations. Furthermore, the azide group is inert to the acylation procedure,43 unlike the frequently used hydroxy or amine groups, which can be acylated by carboxylic acids to their esters or amides. The resulting PEtOx was treated with concentrated hydrochloric acid to yield linear PEI, which was reacylated with a series of thioether-containing carboxylic acids (Table 1) to prepare a library of new PAOx.
| Polymer | R-COOHa |
M
wb (kDa) |
M
nb (kDa) |
Đ |
T
gc (°C) |
ΔCp (J g−1 °C−1) |
|---|---|---|---|---|---|---|
| a Carboxylic acid used for PEI acylation. b Determined by SEC in DMAc/LiCl. c Determined from the second heating DSC run. | ||||||
| PMTMeOx | 2-(Methylthio)acetic acid | 15.9 | 14.1 | 1.1 | 48.0 | 0.46 |
| PMTEtOx | 3-(Methylthio)propanoic acid | 20.3 | 15.2 | 1.2 | 11.5 | 0.49 |
| PETMeOx | 2-(Ethylthio)acetic acid | 16.3 | 13.3 | 1.2 | 29.4 | 0.42 |
Controlled acylation of linear PEI enables us to synthesize PAOx containing highly functional side-chain groups otherwise unattainable by CROP of the respective monomers, as previously reported.43 PEI amidation was achieved by peptide coupling using (benzotriazole-1-yloxy)-tripyrrolidinophosphonium hexafluorophosphate (PyBOP) as the coupling agent, triethylamine (TEA) as the base and DMAc as the solvent, thereby yielding the new thioether-containing PAOx homopolymers. In this study, polyethyleneimine (PEI) was reacylated with three different thioether-containing carboxylic acids (Table 1) to prepare a library of poly(2-alkylthioalkyl-2-oxazoline) (PAOx) homopolymers. The resulting thioether PAOx homopolymers were comprehensively analyzed using various techniques, including 1H NMR spectroscopy (Fig. 1 and S1†) and SEC (Fig. 2).
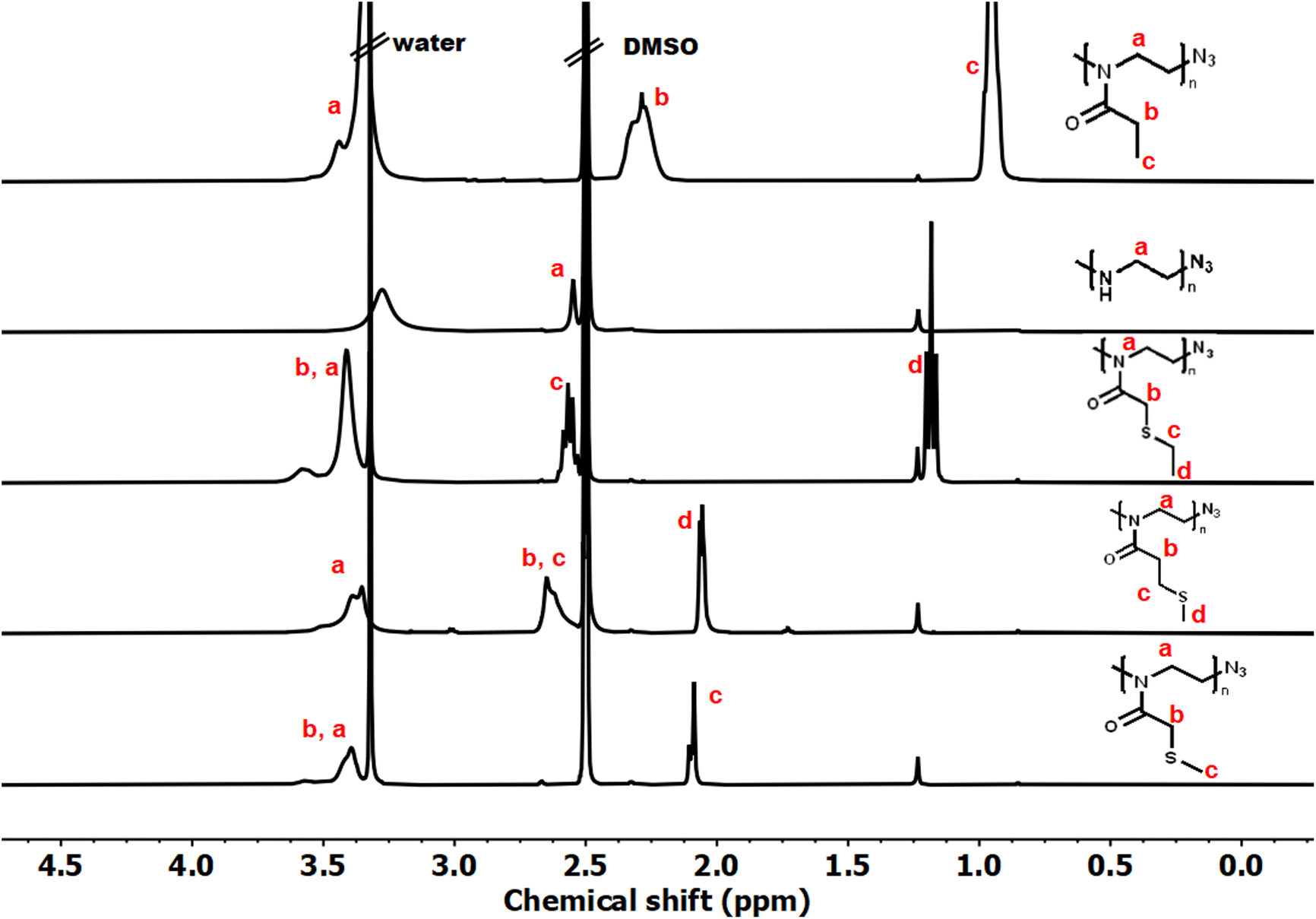 | ||
| Fig. 1 Representative 1H NMR spectra of PEtOx, PEI, PMTMeOx, PMTEtOx, and PETMeOx in DMSO-d6. | ||
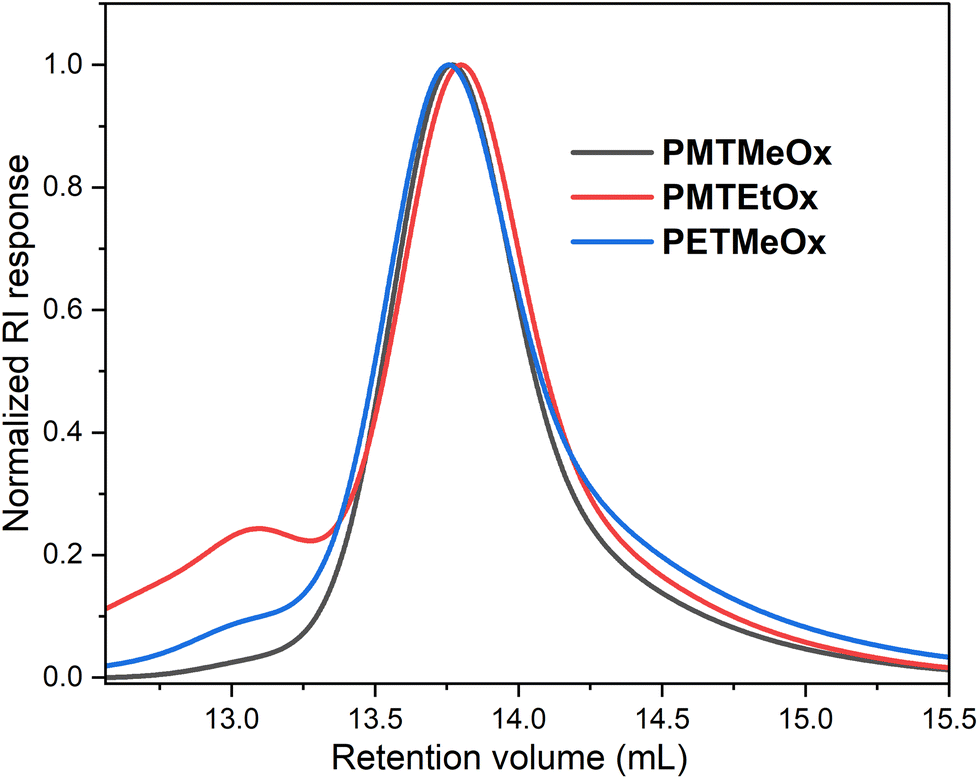 | ||
| Fig. 2 SEC traces of the homopolymers eluted with DMAc/LiCl. | ||
The structures of the homopolymers were confirmed by 1H NMR, lacking the signal at 2.8 ppm corresponding to the PEI methylene units. To rule out a potential overlap of the PEI methylene signal with the PAOx side-chain signal (PMTMeOx, PMTEtOx), the peaks were further deconvoluted by 2D 1H/13C HSQC NMR spectroscopy (Fig. S2†). Furthermore, the presence of remaining secondary amines was excluded by a negative Kaiser test with ninhydrin.43 New peaks corresponding to the PAOx side chains and the typical PAOx backbone peak at 3.4 ppm appeared in the spectra. In addition, the signals of PMTMeOx and PMTEtOx were assigned by 1H/1H COSY NMR.
Since many biological properties of polymers, such as renal clearance, biodistribution, and cellular uptake, are affected by their size, achieving a high level of uniformity in polymer size is generally desirable. All polymers prepared in this study had distinct structures, with narrow molar mass distributions (Đ ∼ 1.2). Nevertheless, the SEC chromatograms displayed minor high molar mass shoulder formations, which can be attributed to chain coupling side reactions during the acylation procedure (Fig. 2). As this high molar mass shoulder is most distinct in the chromatogram of PMTEtOx, it might arise from the base-catalyzed elimination of methanethiol followed by intramolecular chain coupling through the side chains.
The thermal characteristics of the newly synthesized thioether poly(2-oxazoline) (PAOx) homopolymers were also investigated by differential scanning calorimetry (DSC). DSC analysis revealed that all the polymers were amorphous, as indicated by the absence of melting transitions, with glass transition temperatures (Tg) ranging from 12 to 48 °C (Table 1 and Fig. 3). Amorphous polymers with low Tg have several advantageous characteristics, such as high chain flexibility and mobility. High chain flexibility accelerates dissolution. In turn, (i) the high chain mobility of low-Tg polymers facilitates both the formation of various self-assembling architectures by accelerating chain association and dissociation, promoting a reproducible equilibrium in self-assembled structures, and (ii) the development of magnetic resonance imaging (MRI) contrast agents, enabling fast transverse relaxation and enhancing MRI contrast.52 Additionally, these low-Tg polymers can serve as effective plasticizers, improving the mechanical properties of various polymer blends. All homopolymers synthesized in this study showed limited water solubility at room temperature. As such, they are highly promising candidates for stimuli-responsive systems, enabling rapid transitions from a hydrophobic to a hydrophilic state upon oxidation.
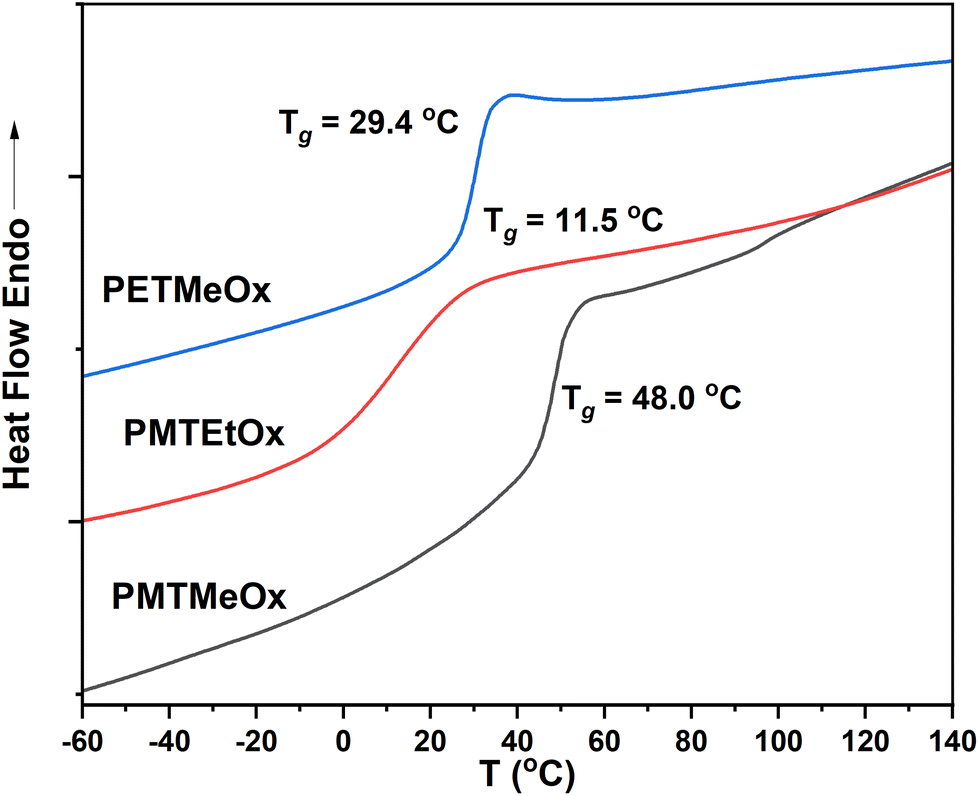 | ||
| Fig. 3 DSC curves of the homopolymers. | ||
In a subsequent procedure, we used the PMTMeOx polymer as a hydrophobic block with oxidation-responsive properties to prepare amphiphilic diblock copolymers (Scheme 2). Nosylated PEG-macroinitiator (Mn = 2 kDa) was synthesized (Fig. S3 and S4†) and chain-extended with PEtOx of varying degrees of polymerization (DP = 30, 60, and 100) through CROP in acetonitrile (Table 2). To obtain PEG-b-PMTMeOx, the PEG-b-PEtOx copolymers were subjected to controlled acidic hydrolysis in aqueous hydrochloric acid, followed by reacylation methylthioacetic acid, using the hydrolysis/reacylation protocol mentioned above (Scheme 2).
 | ||
| Scheme 2 Synthesis of thioether-containing PEG-b-PMTMeOx (*part of end groups might be acylated). | ||
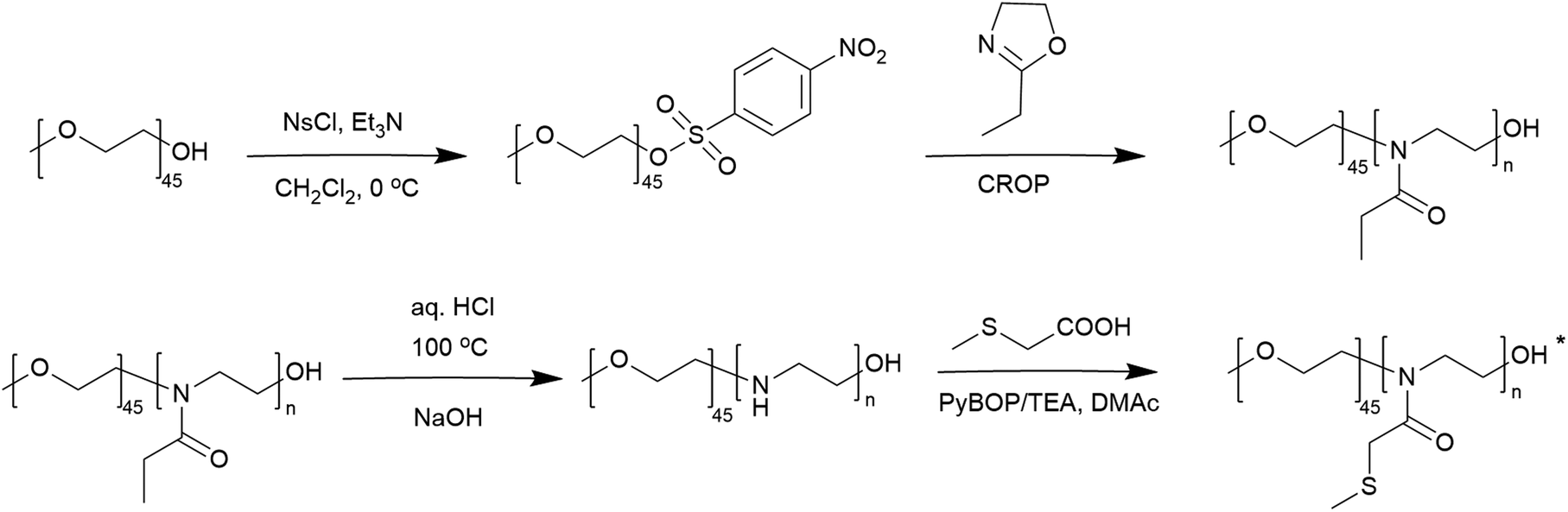
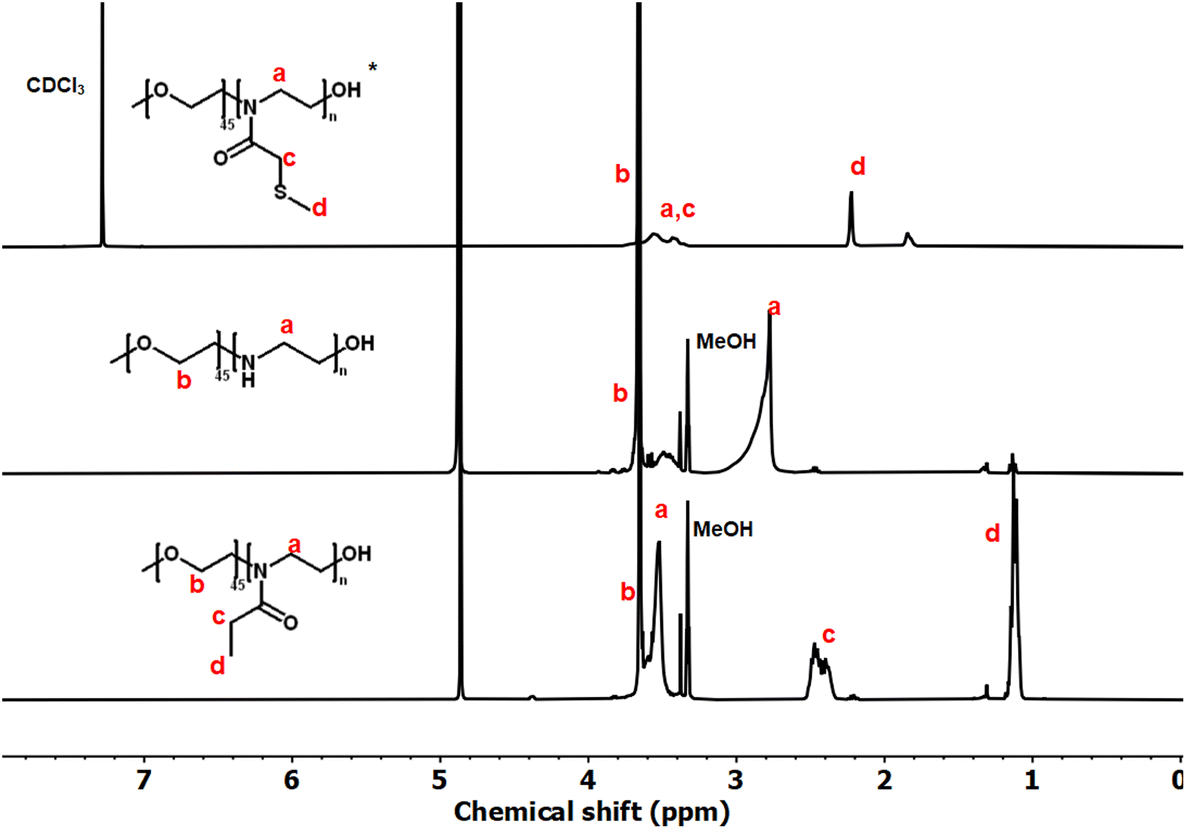
These copolymers were characterized by 1H NMR spectroscopy (Fig. 4). The spectra lacked imine signals from PEI at δ 2.77 ppm and showed new peaks assigned to PAOx side chains (δ 1.10 and 2.45 ppm). As with the homopolymers, 1H/13C HSQC NMR spectra were used for further structure confirmation to rule out potential signal overlaps (Fig. S5†). All polymers had well-defined structures with a low molar mass distribution (Đ ∼ 1.2), as confirmed by size exclusion chromatography (SEC) analysis. Moreover, SEC analysis revealed a distinct shift in molecular weight from PEG to PEG-b-PEtOx and PEG-b-PMTMeOx as each subsequent block was added, accompanied by an increase in DP (Fig. 5).
 | ||
| Fig. 4 1H NMR spectra of PEG-b-PMTMeOx in CDCl3; and PEG-b-PEI and PEG-b-PEtOx, in CD3OD (*some –OH groups were transformed into their respective esters). | ||
 | ||
| Fig. 5 SEC chromatograms of PEG, PEG-b-PEtOx, and PEG-b-PMTMeOx with different degrees of polymerizations. | ||
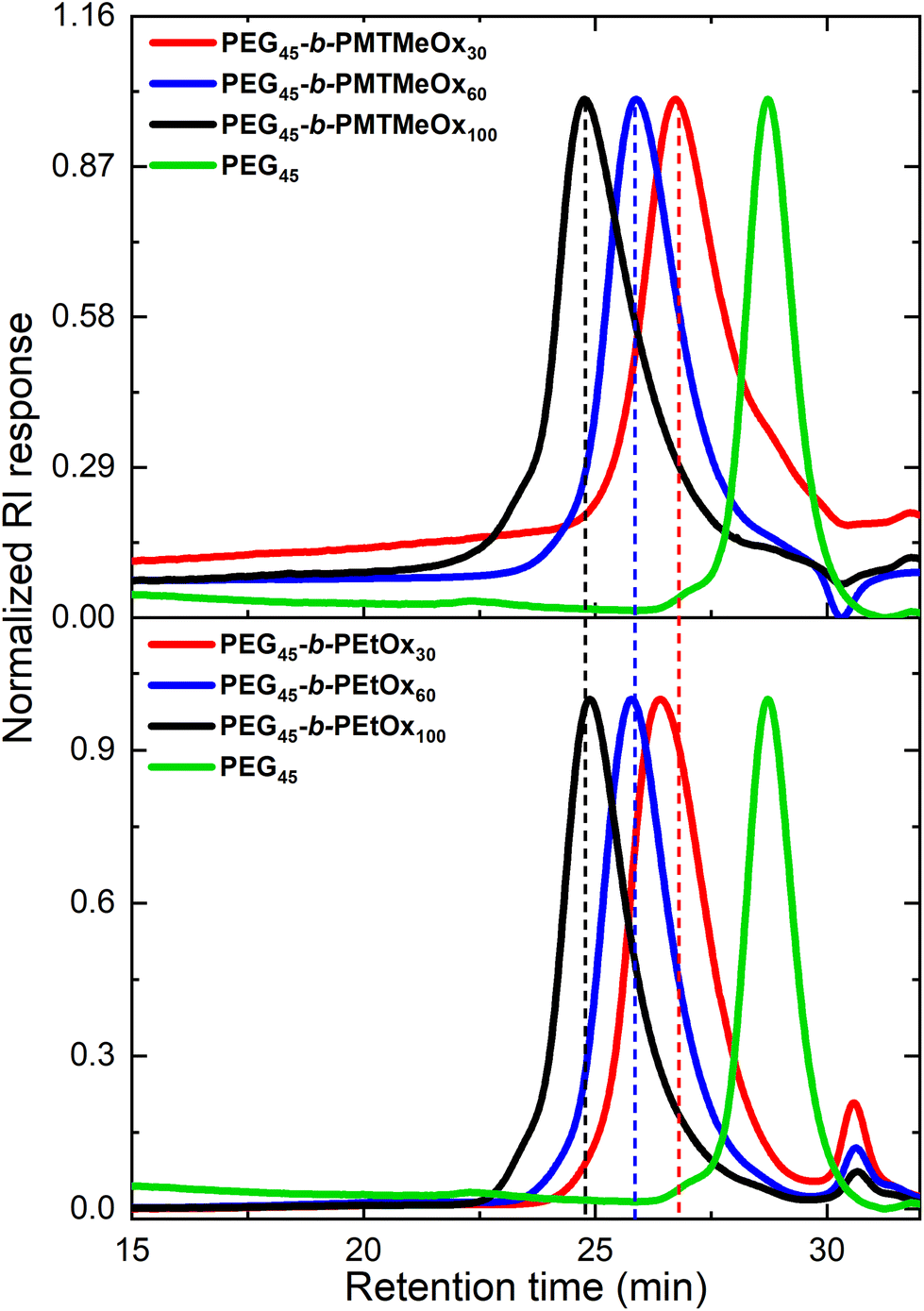
Block copolymer nanoparticles were prepared using the solvent switch method.53 The block copolymers were first dissolved in acetonitrile and then introduced to water to induce self-assembly, followed by evaporation of the organic solvent. The hydrodynamic sizes of the nanoparticles were assessed by DLS and TEM, revealing sizes ranging from 70 to 450 nm, which increased with the degree of polymerization (DP). TEM analysis of DP 30 nanoparticles indicated the presence of spherical micelles, together with aggregated clusters of approximately 60 nm, in line with our DLS measurement. Such clusters may result from hydrogen bonding interactions between the oxazoline and PEG chains (Fig. 6). However, gradual macroscopic aggregation was observed in DP 60 and 100 copolymer nanoparticles after several days of storage in an aqueous solution at room temperature. The aggregation of DP60 and DP100 copolymers might be attributed to the insufficient stabilization from the short PEG block (2 kDa). For this reason, we selected the DP 30 copolymer for further analysis of its oxidation-responsive disassembly.
 | ||
| Fig. 6 (a) Hydrodynamic size distributions of PEG45-PMTMeOxx micelles of different PMTMeOx DP measured by DLS in water (cpol = 1 mg mL−1) and (b) TEM image of PEG45-b-PMTMeOx30 nanoparticles. | ||
PEG45-PMTMeOx30 nanoparticles disassembled in the presence of H2O2, as shown by DLS (Fig. 7). The light scattering intensity assessed in DLS measurements correlated with the particle size and/or concentration in the solution. The optimal scattering intensity should range from 100 to 500 kcps. Values exceeding this range indicate either inadequate dilution or high particle concentration. In contrast, values below this range suggest that the particles are too small or excessively diluted to be detected by the laser in DLS.
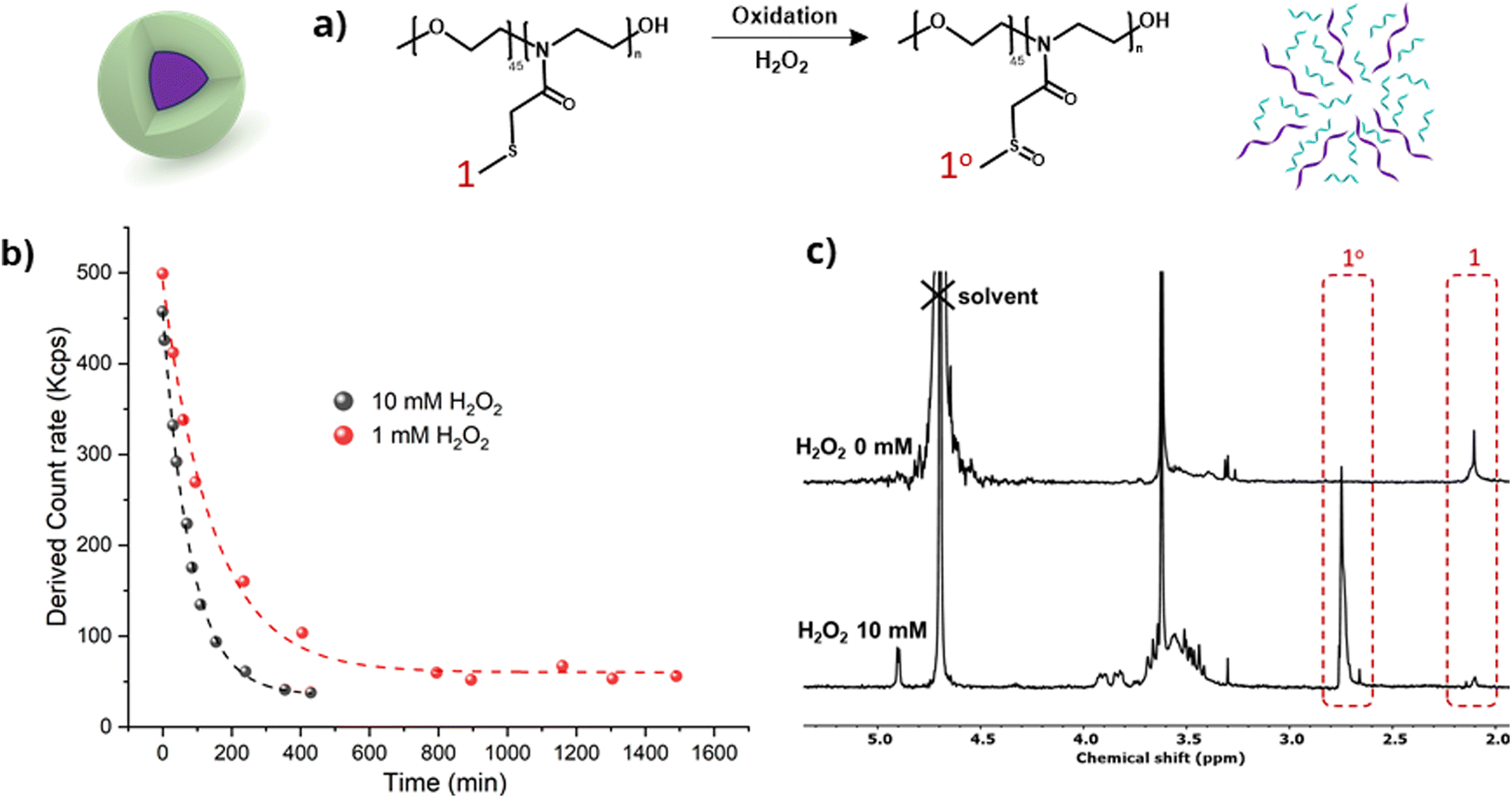 | ||
| Fig. 7 (a) Schematic illustration of the oxidative reaction of PEG-b-PMTMeOx (DP = 30) block copolymer NPs with H2O2, (b) decrease of DLS scattering light intensity of the nanoparticle solution at 1 mg mL−1 when adding 1 and 10 mM H2O2 and (c) 1H NMR spectra of PEG-b-PMTMeOx before and 24 hours after oxidation with 10 mM H2O2 in D2O. | ||
At the beginning of the experiment, the particle solutions showed a scattering intensity of 400–450 kcps, which fell within the acceptable range. However, incubation with 10 and 1 mM H2O2 exponentially decreased the light scattering intensity, which dropped below 100 kcps after 6 and 24 hours, respectively. After these intervals, only a minute fraction of particles was still detectable, thus demonstrating micelle disassembly. The dilution of polymer nanoparticles was very low (from 1 mg mL−1 to 0.83 mg mL−1), which is still clearly above the critical micelle concentration of the polymer without added peroxide (DLS measurements showed the presence of micelles at 0.5 mg mL−1).
To gain insights into the molecular response to the polymer oxidation, the oxidation process was monitored by 1H-NMR spectroscopy (Fig. 7c). The solution of oxidized nanoparticles treated with 10 mM H2O2 for 24 hours was subjected to gel filtration and lyophilization before analyzing the structures of the resulting solids by 1H NMR (D2O). The spectra clearly revealed noticeable shifts in the peaks corresponding to the methylene groups adjacent to the sulfide functionalities, confirming the oxidation of thioether to sulfoxide by H2O2. In addition, further partial oxidation of sulfoxides to sulfones cannot be entirely excluded, however, this is highly improbable due to the absence of NMR peaks at around 3 ppm.54 Such oxidation disrupted the amphiphilic balance of the block copolymers, potentially leading to the dissociation of the spherical structures. These results are in line with the findings of our previous research on thioether-based polymer systems responsive to reactive oxygen species (ROS).22,55
Conclusion
Oxidation-sensitive thioether-containing PAOx homopolymers can be easily prepared by reacylation of linear polyethyleneimine (PEI) with thioether-containing carboxylic acids. The resulting polymers show well-defined structures with low molar mass distributions, and low glass transition temperatures, making them suitable for various biomedical applications. Furthermore, these thioether-containing PAOx polymers can be used as hydrophobic blocks to prepare amphiphilic diblock copolymers with a polyethylene glycol (PEG) shell and a thioether-containing PAOx core. Treatment with hydrogen peroxide destabilizes the micellar structures, demonstrating the oxidation-responsiveness of these polymers, as confirmed by 1H NMR analysis, revealing the conversion of thioether moieties to sulfoxides. Overall, the synthetic approach developed in this study provides a valuable strategy for the synthesis of oxidation-sensitive PAOx polymers, which hold great promise in the development of drug delivery systems and other biomedical applications. Further studies should explore the biocompatibility, drug encapsulation capabilities, and stimuli-responsiveness of these polymers to expand their utility in controlled drug release applications.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank Charles University (grant PRIMUS/21/SCI/007), the Ministry of Health of the Czech Republic (NU22-08-00286), and the Czech Science Foundation (grant number 22-03102S) for funding this study and Carlos V. Melo for editing the manuscript.References
- D. Daubian, J. Gaitzsch and W. Meier, Polym. Chem., 2020, 11, 1237–1248 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef PubMed.
- D. Babuka, K. Kolouchova, L. Loukotova, O. Sedlacek, O. Groborz, A. Skarkova, A. Zhigunov, E. Pavlova, R. Hoogenboom, M. Hruby and P. Stepanek, Macromolecules, 2021, 54, 10667–10681 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- G. Wang, P. Huang, L. Wang, X. Chen, Y. Zhou, W. Huang and D. Yan, SmartMat, 2022, 3, 522–531 CrossRef CAS.
- A. Puglisi, E. Bayir, S. Timur and Y. Yagci, Biomacromolecules, 2019, 20, 4001–4007 CrossRef CAS PubMed.
- S. Bener, A. Puglisi and Y. Yagci, Macromol. Chem. Phys., 2020, 221, 2000109 CrossRef CAS.
- L. Xu, H. Wang, Z. Chu, L. Cai, H. Shi, C. Zhu, D. Pan, J. Pan, X. Fei and Y. Lei, ACS Appl. Polym. Mater., 2020, 2, 741–750 CrossRef CAS.
- Y. Kotsuchibashi, Polym. J., 2020, 52, 681–689 CrossRef CAS.
- P. Švec, O. V. Petrov, J. Lang, P. Štěpnička, O. Groborz, D. Dunlop, J. Blahut, K. Kolouchová, L. Loukotová, O. Sedláček, T. Heizer, Z. Tošner, M. Šlouf, H. Beneš, R. Hoogenboom and M. Hrubý, Macromolecules, 2022, 55, 658–671 CrossRef.
- M. Manzano and M. Vallet-Regí, Chem. Commun., 2019, 55, 2731–2740 RSC.
- M. E. Roth-Konforti, M. Comune, M. Halperin-Sternfeld, I. Grigoriants, D. Shabat and L. Adler-Abramovich, Macromol. Rapid Commun., 2018, 39, 1800588 CrossRef PubMed.
- A. Zhang, K. Jung, A. Li, J. Liu and C. Boyer, Prog. Polym. Sci., 2019, 99, 101164 CrossRef CAS.
- E. Jäger, V. Sincari, L. J. C. Albuquerque, A. Jäger, J. Humajova, J. Kucka, J. Pankrac, P. Paral, T. Heizer, O. Janouskova, R. Konefał, E. Pavlova, O. Sedlacek, F. C. Giacomelli, P. Pouckova, L. Sefc, P. Stepanek and M. Hruby, Biomacromolecules, 2020, 21, 1437–1449 CrossRef PubMed.
- M. Criado-Gonzalez and D. Mecerreyes, J. Mater. Chem. B, 2022, 10, 7206–7221 RSC.
- I. Piergentili, P. R. Bouwmans, L. Reinalda, R. W. Lewis, B. Klemm, H. Liu, R. M. De Kruijff, A. G. Denkova and R. Eelkema, Polym. Chem., 2022, 13, 2383–2390 RSC.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- Z. Zhou, Y. Wang and P. Hu, Int. J. Nanomed., 2022, 17, 2447–2457 CrossRef PubMed.
- L. Yu, M. Zhang, F.-S. Du and Z.-C. Li, Polym. Chem., 2018, 9, 3762–3773 RSC.
- F. H. Sobotta, M. T. Kuchenbrod, F. V. Gruschwitz, G. Festag, P. Bellstedt, S. Hoeppener and J. C. Brendel, Angew. Chem., Int. Ed., 2021, 60, 24716–24723 CrossRef CAS PubMed.
- J. M. Harris, M. D. Bentley, R. W. Moreadith, T. X. Viegas, Z. Fang, K. Yoon, R. Weimer, B. Dizman and L. Nordstierna, Eur. Polym. J., 2019, 120, 109241 CrossRef CAS.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- Z. M. Sahin, T. B. Kohlan, A. E. Atespare, M. Yildiz, S. Unal and B. Dizman, J. Appl. Polym. Sci., 2022, 139, e52406 CrossRef CAS.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Macromol. Rapid Commun., 2012, 33, 1648–1662 CrossRef CAS PubMed.
- O. Sedlacek and R. Hoogenboom, Adv. Thermoelectr., 2020, 3, 1900168 Search PubMed.
- G. Gil Alvaradejo, M. Glassner, R. Hoogenboom and G. Delaittre, RSC Adv., 2018, 8, 9471–9479 RSC.
- G. Vancoillie, W. L. A. Brooks, M. A. Mees, B. S. Sumerlin and R. Hoogenboom, Polym. Chem., 2016, 7, 6725–6734 RSC.
- S. Bener, G. Yilmaz and Y. Yagci, ChemPhotoChem, 2021, 5, 1089–1093 CrossRef CAS.
- H. Phan, R. Cavanagh, D. Destouches, F. Vacherot, B. Brissault, V. Taresco, J. Penelle and B. Couturaud, ACS Appl. Polym. Mater., 2022, 4, 7778–7789 CrossRef CAS.
- A. S. Gubarev, B. D. Monnery, A. A. Lezov, O. Sedlacek, N. V. Tsvetkov, R. Hoogenboom and S. K. Filippov, Polym. Chem., 2018, 9, 2232–2237 RSC.
- L. Simon, V. Lapinte, L. Lionnard, N. Marcotte, M. Morille, A. Aouacheria, K. Kissa, J. M. Devoisselle and S. Bégu, Int. J. Pharm., 2020, 579, 119126 CrossRef CAS PubMed.
- M. A. Cortez and S. M. Grayson, Macromolecules, 2010, 43, 4081–4090 CrossRef CAS.
- S. Jana and R. Hoogenboom, Polym. Int., 2022, 71, 935–949 CrossRef CAS.
- A. Krieg, C. Weber, R. Hoogenboom, C. R. Becer and U. S. Schubert, ACS Macro Lett., 2012, 1, 776–779 CrossRef CAS PubMed.
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3829–3838 CrossRef CAS.
- O. Sedlacek, B. D. Monnery and R. Hoogenboom, Polym. Chem., 2019, 10, 1286–1290 RSC.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten, B. M. Van Lankvelt and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 416–422 CrossRef CAS.
- C. Legros, M.-C. De Pauw-Gillet, K. C. Tam, S. Lecommandoux and D. Taton, Eur. Polym. J., 2015, 62, 322–330 CrossRef CAS.
- A. C. Rinkenauer, L. Tauhardt, F. Wendler, K. Kempe, M. Gottschaldt, A. Traeger and U. S. Schubert, Macromol. Biosci., 2015, 15, 414–425 CrossRef CAS PubMed.
- K. Kempe, Macromol. Chem. Phys., 2017, 218, 1700021 CrossRef.
- O. Sedlacek, O. Janouskova, B. Verbraeken and R. Hoogenboom, Biomacromolecules, 2019, 20, 222–230 CrossRef CAS PubMed.
- O. Sedlacek, D. Bera and R. Hoogenboom, Polym. Chem., 2019, 10, 4683–4689 RSC.
- W.-L. Wang, K. Kawai, H. Sigemitsu and R.-H. Jin, J. Colloid Interface Sci., 2022, 627, 28–39 CrossRef CAS PubMed.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed. Engl., 1966, 5, 875–888 CrossRef CAS PubMed.
- N. E. Göppert, M. Kleinsteuber, C. Weber and U. S. Schubert, Macromolecules, 2020, 53, 10837–10846 CrossRef.
- H. M. L. Lambermont-Thijs, F. S. Van Der Woerdt, A. Baumgaertel, L. Bonami, F. E. Du Prez, U. S. Schubert and R. Hoogenboom, Macromolecules, 2010, 43, 927–933 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Sci. Rep., 2016, 6, 33491 CrossRef PubMed.
- A. J. Van Der Vlies, J. Xu, M. Ghasemi, C. Bator, A. Bell, B. Rosoff-Verbit, B. Liu, E. D. Gomez and U. Hasegawa, Biomacromolecules, 2022, 23, 77–88 CrossRef CAS PubMed.
- G. Wang, P. Huang, M. Qi, C. Li, W. Fan, Y. Zhou, R. Zhang, W. Huang and D. Yan, ACS Omega, 2019, 4, 17600–17606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00945a |
| This journal is © The Royal Society of Chemistry 2023 |