
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyacrylonitrile-containing amphiphilic block copolymers: self-assembly and porous membrane formation†
Lea
Gemmer
a,
Bart-Jan
Niebuur
 b,
Christian
Dietz
c,
Daniel
Rauber
d,
Martina
Plank
e,
Florian V.
Frieß
a,
Volker
Presser
dfg,
Robert W.
Stark
c,
Tobias
Kraus
bh and
Markus
Gallei
*ag
b,
Christian
Dietz
c,
Daniel
Rauber
d,
Martina
Plank
e,
Florian V.
Frieß
a,
Volker
Presser
dfg,
Robert W.
Stark
c,
Tobias
Kraus
bh and
Markus
Gallei
*ag
aPolymer Chemistry, Universität des Saarlandes, Campus C4 2, 66123 Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bINM – Leibniz-Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
cPhysics of Surfaces, Institute of Materials Science, Technische Universität Darmstadt, Peter-Grünberg-Straße 2, 64287 Darmstadt, Germany
dDepartment of Materials Science and Engineering, Saarland University, 66123 Saarbrücken, Germany
eErnst-Berl Institute of Technical and Macromolecular Chemistry, Technische Universität Darmstadt, Peter-Grünberg-Straße 4, 64287 Darmstadt, Germany
fINM - Leibniz-Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany
gSaarene, Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
hColloid and Interface Chemistry, Universität des Saarlandes, Campus D2 2, 66123 Saarbrücken, Germany
First published on 5th October 2023
Abstract
The development of hierarchically porous block copolymer (BCP) membranes via the application of the self-assembly and non-solvent induced phase separation (SNIPS) process is one important achievement in BCP science in the last decades. In this work, we present the synthesis of polyacrylonitrile-containing amphiphilic BCPs and their unique microphase separation capability, as well as their applicability for the SNIPS process leading to isoporous integral asymmetric membranes. Poly(styrene-co-acrylonitrile)-b-poly(2-hydroxyethyl methacrylate)s (PSAN-b-PHEMA) are synthesized via a two-step atom transfer radical polymerization (ATRP) procedure rendering PSAN copolymers and BCPs with overall molar masses of up to 82 kDa while maintaining low dispersity index values in the range of Đ = 1.13–1.25. The polymers are characterized using size-exclusion chromatography (SEC) and NMR spectroscopy. Self-assembly capabilities in the bulk state are examined using transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS) measurements. The fabrication of isoporous integral asymmetric membranes is investigated, and membranes are examined by scanning electron microscopy (SEM). The introduction of acrylonitrile moieties within the membrane matrix could improve the membranes’ mechanical properties, which was confirmed by nanomechanical analysis using atomic force microscopy (AFM).
Introduction
Nanostructured polymer membranes composed of block copolymers (BCP) have been subject to intense research due to their promising applications in for instance waste-water purification, selective separations, as well as mining and harvesting of components.1,2 Water purification addresses one of the world's most serious environmental issues.3 BCP membranes have gained particular interest for use in microfiltration, ultrafiltration, reverse osmosis, and nanofiltration.4–6 The mechanical stability of these membranes is a key to their performance, and introducing mechanically more stable building blocks to the BCPs used in membrane fabrication could enhance their long-term stability.4Polyacrylonitrile (PAN) is a rigid and solvent-resistant material because of the small size of the acrylonitrile moiety in combination with its highly polar nature.7,8 Acrylonitrile is widely used as a comonomer to increase the mechanical stability and solvent resistance in commercial plastics. Polyacrylonitrile-nonwovens are often used as membrane support material,9,10 and phase inversion membranes based on PAN and Poly(styrene-co-acrylonitrile) (PSAN) have been subject of research for a while due to their outstanding solvent resistance, mechanical robustness and thermal properties.7 The research often focuses on polymer blends,11,12 composite or hybrid membranes,13,14 as well as their fabrication based on the non-solvent induced phase separation (NIPS) process9,10,14,15 or electrospinning methods.16,17 While isoporous asymmetric membranes can be produced via the self-assembly and non-solvent induced phase separation (SNIPS) process, the use of P(S)AN-based BCPs has not been reported in this context. This would enable a new platform of BCP-based robust membranes.
The SNIPS process is a procedure to fabricate isoporous integral asymmetric membranes from amphiphilic BCPs, wherein the self-assembly is the underlying key mechanism. As has been shown for some well-studied polymers, this membrane formation process is predictable and controllable.2,18,19 In the first reporting of a BCP-membrane prepared via SNIPS by Peinemann and Abetz in 2007,20 PS-b-poly(4-vinylpyridine) (4VP) was cast from a solvent-mixture of N,N-dimethylformamide (DMF) and tetrahydrofurane (THF), and since then PS-b-P4VP has been widely used for ultrafiltration membrane production. Many more BCPs have been used to fabricate SNIPS membranes based on the deepened knowledge in the following years.5,19,21 For example, the introduction of PHEMA as hydrophilic block bearing accessible hydroxy-moieties by Schöttner et al.22 paved the way to multiple post-modification possibilites.23–26
The formation of a membrane via the SNIPS process includes three essential steps: preparing a semi-concentrated solution of the BCP in a solvent mixture, casting the membrane solution by doctor blading and, after an appropriate evaporation time, freezing the membrane in its non-equilibrium state by immersion in a precipitation bath. The process itself is complex and many parameters have to be considered, whilst being strongly dependent on each other, when introducing a new BCP.27,28 For example, it depends on the molecular parameters of the BCP, the solvents, the composition of the casting solution, the additives, the film thickness, and the evaporation time.18 Within a recent work, Blagojevic and Müller identified the most important influencing parameters using simulations.29
Although valuable work has been published in this field,4,19,21 there is still need for improvement regarding the membranes’ mechanical stability. Amongst others,30 our group has approached this issue by incorporating UV-crosslinkable moieties into a methacrylate-based amphiphilic BCP prepared by ATRP,31 leading to improved solvent resistance and mechanical stability. Nonetheless, regarding mechanical and chemical robustness, BCPs containing PAN-segments or copolymers thereof could be a benefit without the necessity of post-treatments.
Morphologies in the bulk-state can only be observed above a certain molar mass of each BCP-segment, and a narrow molar mass distribution is necessary to obtain ordered morphologies. In phase equilibrium, these morphologies can be spheres, cylinders, gyroid or bicontinuous morphologies, and lamellae.32–34 In general, the gyroid or bicontinuous morphologies are challenging to obtain, as precise tuning of distinct parameters is required.34–37 When the system is out of equilibrium or on the border of equilibrium-state morphologies, also mixed or other complex morphologies can be found, for example the so-called worm-like cylinder morphology.38–40 Morphologies of polyacrylonitrile-containing BCPs in the bulk state are scarcely investigated, even though they are of academic interest and technological relevance because of their functionality and post-treatment possibilities.41–46
Acrylonitrile has been introduced to a variety of controlled polymerization methods.44,47–55 A few examples of well-defined amphiphilic BCPs containing PSAN can be found, which are shortly presented here. Besides some other reports,44–47 Quémener et al. produced an amphiphilic triblock copolymer with an overall molar mass of 130 kDa containing polyethylene oxide (PEO) as a middle block via nitroxide-mediated polymerization (NMP) method, observing micelle formation and self-assembly phenomena.46 Using the reversible addition–fragmentation chain-transfer (RAFT) polymerization method, Fan et al. synthesized a variety of amphiphilic BCPs starting the PSAN polymerization with hydrophilic macro-chain-transfer-agents (macro-CTAs).56 Among other BCPs, they synthesized poly(hydroxyethyl methacrylate)-b-PSAN (PHEMA-b-PSAN) with a molar mass of 19 kDa and a dispersity index (Đ) of 1.3.56 This is, to the best of our knowledge, the only BCP of this kind published so far.
Within the last two decades, ATRP has become a popular method for the synthesis of functional BCPs due to its compatibility with a huge variety of functional monomers for polymerization.54,55,57 However, especially acrylonitrile and styrene have caused issues for classical ATRP synthesis as side reactions with the copper catalyst occur.53,58 This resulted in limitations in achievable molecular weights of PAN,53,59 PS58,60 and PSAN61,62 and the re-initiation capability with a second monomer. This issue was overcome by the introduction of the activators regenerated by electron transfer (ARGET) ATRP,63,64 where extremely low copper concentrations of 30–50 ppm are generally used. Following this method, high molecular weight PAN (Mn, GPC = 161 kDa, Đ = 1.18 and Mn, GPC = 216 kDa, Đ = 1.45),65 PS (Mn, GPC = 72 kDa, Đ = 1.18 and Mn, GPC = 185 kDa, Đ = 1.35)66 and PSAN (Mn, GPC = 99 kDa, Đ = 1.22 and Mn, GPC = 188 kDa, Đ = 1.25)67 could then be synthesized.
However, long-chain BCPs obtained from ATRP are still scarcely reported. The highest molar mass reported here is PEO5000-b-PSAN with 101 kDa (SEC vs. PS standards) and a dispersity index value of 1.19 prepared by Pietrasik et al.,67 followed by poly(butyl acrylate)-b-PAN with up to 59 kDa (SEC vs. PS standards) and a dispersity index value as low as 1.15, prepared by Tang et al. by means of the classical ATRP method.68 From a viewpoint of application in the field of membranes, a certain chain length of the BCP segments as well as the controllability of the hydrophilic block content is of utmost importance in order to tailor the membrane formation process, and to tailor the pores’ sizes and properties.21,69 For that reason, the protocols developed by Pietrasik and co-workers are adjusted and used for parts of this work.
In this work we introduce a two-step ATRP procedure as a synthetic route to obtain well-defined high-molar mass PSAN-based amphiphilic BCPs. The PSAN-block is investigated with respect to its local mechanical stability by nanomechanical characterization using atomic force microscopy (AFM) measurements. Subsequently the BCPs are investigated regarding their microphase separation capabilities in the bulk state and in solvent mixtures, as well as their membrane formation by means of the SNIPS process. Both the ATRP as well as the SNIPS process are scalable methods.70–73
Experimental section
Materials
Solvents and reagents were purchased from Alfa Aesar, Sigma-Aldrich (Merck), Fisher Scientific, VWR or TCI and used as received unless otherwise stated. For ARGET-ATRP the following purification procedures were carried out: the monomers 2-(trimethylsilyloxy) ethyl methacrylate (HEMA-TMS), acrylonitrile and styrene were passed over a column filled with basic aluminum oxide. HEMA-TMS was stored at 4 °C in Ar atmosphere until used. Styrene was passed over a column at least three times, styrene and acrylonitrile were subsequently dried with calcium hydride, distilled under reduced pressure and stored in the glovebox (GB) at −16 °C in nitrogen atmosphere until used. The ligands N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA) and tris(2-dimethylaminoethyl)amin (Me6TREN) as well as the copper salt CuIICl2 were degassed and stored in the GB until used. CuICl was washed with glacial acetic acid (degassed) and ethanol (abs., degassed), filtered, dried and heated in vacuum before stored in the GB until used. The preparation of the catalyst solutions was conducted in the GB. For this purpose, a copper salt, degassed anisole and the ligand required were mixed in a glass vial and stirred overnight. The solution of the reducing agent was prepared in an analogous manner before use. Regarding the synthesis, all steps outside of the glovebox were carried out under Schlenk conditions. The subscript used with PSAN refers to the calculated repeating units. The subscripts used in PSAN-b-PHEMA BCPs refer to the calculated volume fractions of each BCP segment, the superscripts refer to the calculated overall molar mass of the BCP (in kDa, calculations see Table 1).| Polymer |
M
n, GPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/kDa a/kDa |
M
n, GPCb/kDa (PHEMA-Bz) |
Đb |
M
n, PHEMAc/kDa (calc) |
M
n, ges, cald/kDa |
x
PHEMAe/mol% |
Φ
PHEMAf/vol% |
|---|---|---|---|---|---|---|---|
| a Molar mass Mn in kg mol−1 determined via SEC-MALLS in THF. b Determined via SEC in THF, in case of BCP the PHEMA moieties were benzoyl-protected (–Bz). c Calculated from 1H-NMR spectra. d Calculated from Mn, PSAN from (a) and Mn, PHEMA from (c). e Calculated from the corresponding 1H-NMR spectra. f Calculated from xPHEMA using the molar mass of HEMA and a polymer density of 1.42 g cm−3.78 | |||||||
| PSAN 738 | 62.6 | 1.17 | — | — | — | — | |
| PSAN87-b-PHEMA1376 | 86.5 | 1.25 | 13.8 | 76.4 | 11.1 | 12.8 | |
| PSAN83-b-PHEMA1782 | 90.2 | 1.24 | 19.2 | 81.8 | 14.8 | 17.0 | |
| PSAN 403 | 34.2 | 1.13 | — | — | |||
| PSAN88-b-PHEMA1241 | 51.7 | 1.18 | 7.1 | 41.3 | 10.7 | 12.4 | |
| PSAN84-b-PHEMA1645 | 51.0 | 1.19 | 11.0 | 45.2 | 14.0 | 16.1 | |
| PSAN80-b-PHEMA2047 | 56.0 | 1.21 | 12.5 | 46.7 | 17.4 | 19.9 | |
| PSAN76-b-PHEMA2450 | 62.2 | 1.18 | 15.4 | 49.6 | 20.7 | 23.6 | |
Methods
Standard size-exclusion chromatography (SEC) with THF (HPLC grade) as the solvent was performed with a 1260 Infinity II (Agilent Technologies) system with a flow rate of 1 mL min−1 on an SDV column set from polymer standard service (PSS, SDV 103 Å, SDV 105 Å, SDV 106 Å, 5 μm) with a PSS SECurity2 RI/UV detector. Calibration was carried out using polystyrene (PS) standards from PSS (Polymer Standard Service, Mainz, Germany). For multi-angle laser light scattering (MALLS) a PSS SLD 7000 detector was used. SEC with DMF (HPLC grade) as the solvent was performed with a Waters (Milford, MA, USA) system composed of a 515 HPLC Pump, a 2487 UV-detector at 260 nm and a 2410 RI-detector at 40 °C, with DMF (1 g L−1 LiBr) as the mobile phase (flow rate 1 mL min−1) on a GRAM column set (GRAM 30, GRAM 1000, GRAM 1000) from PSS at 60 °C. Calibration was carried out using PMMA standards. The software PSS WinGPC UniChrom V 8.31 was used for data acquisition and evaluation of the measurements.Differential scanning calorimetry (DSC) was carried out using a Netzsch DSC 214 Polyma in aluminum crucibles with a heating rate of 10 K min−1 and nitrogen as both protective and purge gas in flow rates of 60 mL min−1 and 40 mL min−1, respectively. Thermogravimetric analysis (TGA) was conducted on a NETZSCH TGA Libra in a crucible made of aluminum oxide with nitrogen flow of 20 mL min−1 and heating rate of 10 K min−1. Both TGA and DSC data were evaluated using NETZSCH Proteus Thermal Analysis 8.0.1. In the DSC thermograms used the second heating cycles are displayed with exo up.
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II 400 spectrometer with a 9.4 T Ultrashield Plus Magnet, a BBFO probe, and referenced by using the solvent signals.74 For processing and evaluation of the spectra MestReNova 14.2.0 was used.
Instrumentation details for diffusion NMR investigations can be found in the ESI.†
For transmission electron microscopy (TEM)-measurements, samples were cut by ultramicrotomy from surface to surface at room temperature (Reichert Ultracut by Leica Microsystems, Wetzlar, Germany). Ultra-thin slices with a thickness of ∼40–60 nm were prepared using a diamond trim knife cryotrim 45 and a diamond cutting knife cryo 45° by Diatome (for wet cutting, water was used as the liquid), and samples were placed on a copper grid. Staining was conducted by vapour staining with a diluted solution of OsO4 in water. TEM images were obtained using a JEOL JEM-2100 LaB6 electron microscope (JEOL Ltd Tokyo, Japan) with 200 kV acceleration voltage, 0.14 nm line resolution and a Gatan Orius SC1000 camera (Gatan Inc. Pleasanton, CA, USA) in the brightfield mode.
Scanning electron microscopy (SEM) was carried out on a Zeiss Sigma VP device (GeminiSEM 500) using the software SmartSEM Version 6.07. The samples were mounted on an aluminum stud using adhesive copper tape and sputter-coated with approximately 6 nm platinum using an Automatic Turbo Coater PLASMATOOL 125 SIN 2020_131 from Ingenieurbüro Peter Liebscher. High-resolution micrographs were acquired using an in-lens detector with acceleration voltages between 1 and 3 kV in high-current mode and a 20 μm aperture.
Small-Angle X-ray Scattering (SAXS) experiments were performed on a Xeuss 2.0 system (Xenocs SAS, Grenoble, France). The incident X-ray beam from a Copper Kα source (wavelength λ = 0.154 nm) was collimated and focused on the sample with a spot size of 0.25 mm2. 2D scattering intensity patterns were collected using a Pilatus 300K detector (pixel size 172 × 172 μm2), located at a sample-detector distance of approximately 2500 mm, determined using a silver behenate standard. For each measurement, the acquisition time was 3600 s. As no signs of anisotropic scattering were observed, the scattering patterns were azimuthally averaged to obtain the scattered intensity I in dependence on momentum transfer q = 4π × sin(θ/2)/λ, with θ being the scattering angle. The free-standing films were placed directly into the beam, without the need of a sample container.
Nanomechanical characterization was performed in the PeakForce tapping mode using a Dimension Icon atomic force microscope (AFM) from Bruker AXS (Santa Barbara, CA). We used an NSC-35 cantilever (Type C) from Mikromasch (Wetzlar, Germany) and removed the two other cantilevers mechanically from the chip prior to the measurement. This type of cantilever has a typical tip radius of 8 nm, a spring constant of 5.4 N m−1 and a resonant frequency of 150 kHz (as provided by the datasheet of the manufacturer). The optical lever deflection sensitivity was calibrated by pushing the tip against a stiff sapphire surface and linearly relating the deflection (volts) with the motion of the z-piezo (nanometer). Subsequently, the exact spring constant of the cantilever was determined by the thermal noise method.75 Images (300 × 300 nm2, 256 × 256 pixel) were taken with a line rate of 1 Hz, setpoint force of 10 nN and a cantilever drive frequency of 1 kHz. Topography images were first-order flattened to remove sample tilt and to correct for drift during image acquisition.
Block copolymer synthesis
SEC PSAN (DMF vs. PMMA):
M
n = 43200 g mol−1; Mw = 54200 g mol−1; Đ = 1.25.
SEC PSAN (THF vs. PS):
M
n = 44000 g mol−1; Mw = 49500 g mol−1; Đ = 1.11.
SEC-MALLS:
M
w = 37200 g mol−1.
1 H-NMR (300 MHz, 300 K, CDCl3) δ = 8.01 (1H, DMF residue), 7.26 (CHCl3), 7.13 (w, PS aromatic), 6.75 (w, PS aromatic), 2.95 (3H, CH3, DMF residue), 2.88 (3H, CH3, DMF residue), 2.6–1.2 (w, multiple H, PAN, PS aliphatic, bb) ppm.
:2:1, stirred for 3 h and precipitated in water to obtain the de-protected polymer PSAN-b-PHEMA. The polymer was washed with water thoroughly to get rid of any excess of acid and then dried at 40 °C in vacuum. This synthesis yielded 467.9 mg (54.5%) polymer. The largest amount of BCP synthesized was 1.1 g. For characterization the PSAN-b-PHEMA was protected with benzoic acid. Therefore, 25 mg of the polymer and 950 mg of benzoic acid were dissolved in 2 mL of pyridine and stirred for 3 days. The polymer PSAN-b-PHEMA(-Bz) was precipitated in cold methanol and dried in vacuum.
SEC PSAN-b-PHEMA (DMF vs. PMMA):
M
n = 58300 g mol−1; Mw = 73800 g mol−1; Đ = 1.27.
SEC PSAN-b-PHEMA(-Bz) (THF vs. PS):
M
n = 51000 g mol−1; Mw = 60600 g mol−1; Đ = 1.19.
1 H-NMR PSAN-b-PHEMA (300 MHz, 300 K, py-d5) δ = 8.73 (py), 7.58 (py), 7.5–6.6 (w, PS ar), 7.22 (py), 4.97 (w, PHEMA, OH), 4.38 (w, PHEMA, CH2), 3.66 (w, PHEMA, CH2), 3.6–1.3 (w, multiple H, AN, PS and PHEMA bb), 1.59 (H2O) ppm.
Block copolymer film preparation
Formation of membranes via SNIPS
:1:1 by weight) by stirring for several hours to obtain a clear solution of 20.3 mass% (polymer/solvent). When completely dissolved, 0.2–0.3 mL of the polymer solution was cast on a THF-conditioned polyester nonwoven support (type FLPD 85, Freudenberg Performance Materials) on a glass plate with a doctor blade using the blade gap of 100 μm. After the evaporation time of 14 s the plate was immersed into a water bath, precipitated for 20 min, then dried over night at room temperature and subsequently in vacuum at 40 °C. Membranes with a size of 10 cm × 5 cm were obtained.
Results and discussion
Synthesis and characterization of PSAN-b-PHEMA
The synthesis of poly(styrene-co-acrylonitrile)-block-poly(hydroxyethyl methacrylate) (PSAN-b-PHEMA) was conducted in a two-step ATRP procedure. First, as it is shown in Scheme 1, PSAN 4 was synthesized with an azeotropic molar composition of styrene 1 and acrylonitrile 2via ARGET-ATRP with α-bromo-tert-butyl isobutyrate 3 as the initiator, CuIICl2/Me6TREN as the catalyst system, and Sn(EH)2/Me6TREN as the reducing agent according to literature.67 In a second step, the PSAN macroinitiator 4 was converted into a BCP via ATRP with HEMA-TMS 5 with CuICl/PMDETA as the catalyst system, taking advantage of the halogen exchange reaction.57,76 The halogen exchange to chloride was necessary to ensure re-initiation because of the low reactivity of the bromide terminated poly(styryl) chain ends in the copolymer macroinitiator.61 Deprotection of the hydroxy moieties in the obtained PSAN-b-(PHEMA-TMS) by treatment with diluted hydrochloric acid lead to the desired amphiphilic PSAN-b-PHEMA 6. | ||
| Scheme 1 Two-step ATRP synthetic route leading to the PSAN-b-PHEMA 6. In the first step, styrene 1 and acrylonitrile 2 were polymerized via an ARGET-ATRP protocol with tris(2-dimethylaminoethyl)amine (Me6TREN) as the ligand and tin(II)-diethylhexanoate (Sn(EH)2) with Me6TREN as the reducing agent. In the second step, the PSAN-macroinitiator 4 and HEMA-TMS 5 were treated via an ATRP protocol with N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA) as the ligand, followed by de-protection with diluted hydrochloric acid in a mixture of DMF and THF to obtain the final product. | ||
The synthesized BCPs were analyzed with SEC, 1H-NMR spectroscopy, TGA and DSC. An exemplary set of characterization data is compiled in Fig. 1, while all other spectra, molar mass distributions and thermograms of the investigated BCPs are compiled in the ESI (Fig. S1–S5†).
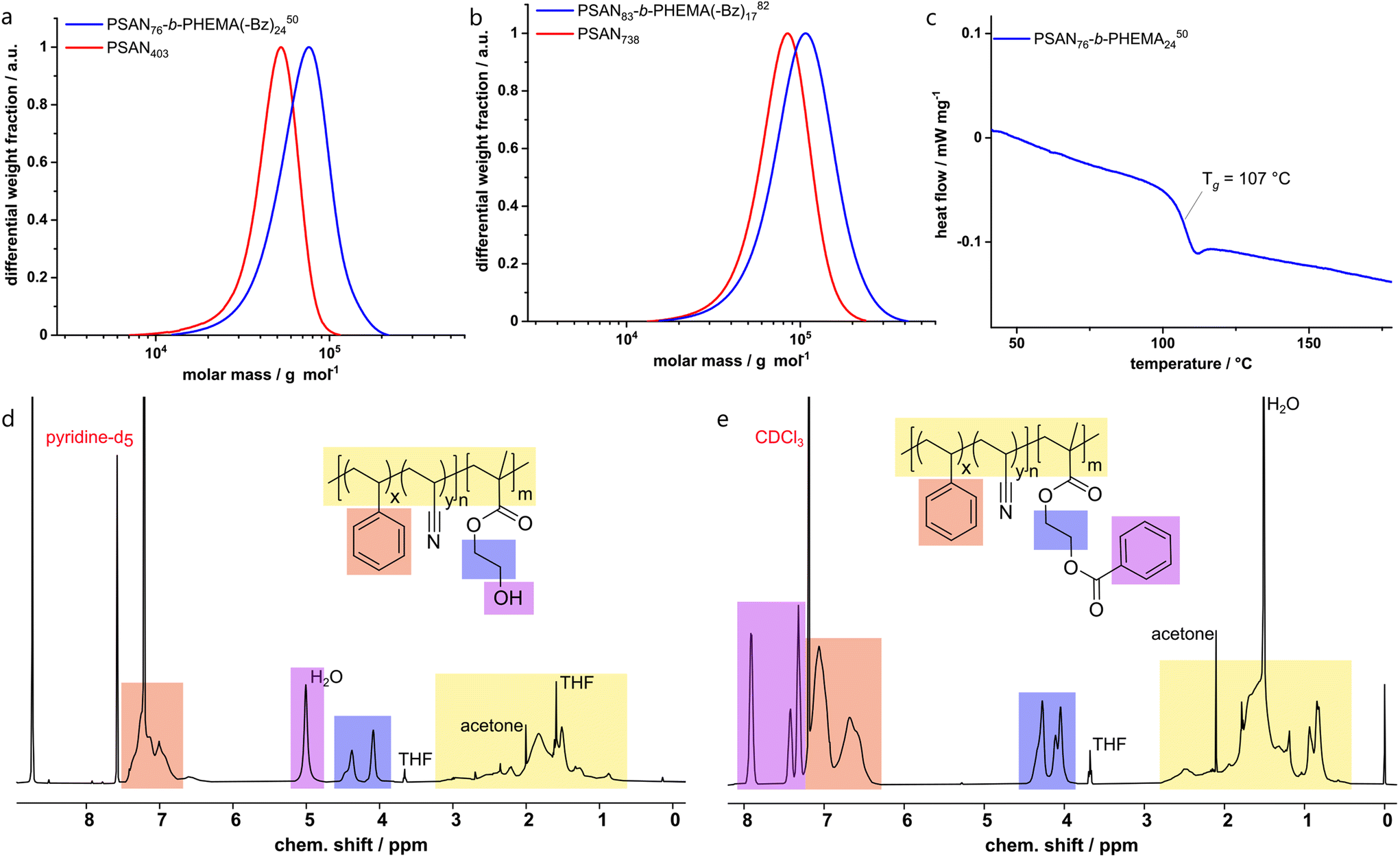 | ||
| Fig. 1 Exemplary characterization of PSAN macroinitiators and BCPs. SEC in THF of (a) PSAN403 and PSAN76-b-PHEMA(–Bz)2450 and (b) PSAN738 and PSAN83-b-PHEMA(–Bz)1782, (c) DSC thermogram of PSAN76-b-PHEMA2450 with one glass transition at 107 °C, 1H-NMR spectra of (d) PSAN76-b-PHEMA2450 in pyridine-d5 and (e) PSAN76-b-PHEMA(–Bz)2450 in CDCl3. | ||
The macroinitiators and the BCPs exhibited uniform molar mass distributions with a clear shift towards higher molar masses after conversion of the second monomer. This is a good indication for a complete and uniform re-initiation of the PSAN macroinitiator. Still, the presence of low amounts of macroinitiator cannot be excluded due to the significant overlap of the molar mass distributions. We gained further indications by conducting diffusion-ordered NMR spectroscopy (DOSY) investigations, which are presented in the ESI (Fig. S7–S9†). For this purpose, we compared the diffusion coefficients of the macroinitiator PSAN403 (Fig. S7†) with the BCP PSAN80-b-PHEMA2047 (Fig. S8†) under the same experimental conditions (solvent, concentration, temperature and setup). All integrals of the signals from the different copolymer blocks for both the macroinitiator and the BCP could be well fitted with monoexponential functions. This is a common observation for single-diffusion particles, whereas overlapping signals from species with distinct diffusion-coefficients must be fitted with biexponential functions. The applicability of the monoexponential fitting indicates narrow and uniform molecular weight distributions. For the macroinitiator, we found diffusion coefficients that were similar for the two different repeating units (D(PSAN403) ≈ 4.22 m2 s−1). For the BCP PSAN80-b-PHEMA2047 we found significantly lower diffusion-coefficients (D(PSAN80-b-PHEMA2047) ≈ 3.25 m2 s−1), clearly showing the increase in molecular weight as to be expected from the Stokes–Einstein relation (D = kBT × (6πηRH)−1; with kB the Boltzmann constant, T the temperature, η the viscosity of the solution and RH the hydrodynamic radius, related to the molecular mass of macromolecules). From the applicability of the monoexponential fit for the BCP PSAN80-b-PHEMA2047 a low to negligible amount of unreacted macroinitiator can be assumed. The diffusion behavior of the two macromolecules in solution is also depicted by the comparison of the two DOSY-plots (chemical shift vs. D), given in Fig. S9.†
In order to obtain absolute molar masses, SEC of the macroinitiators was conducted using a multi-angle laser-light scattering (MALLS) detector with THF as the eluent. For a direct comparison of the molar mass distributions of the BCPs in SEC with THF as the eluent the hydrophilic PHEMA moieties were protected with benzoyl groups (–Bz) using benzoic acid to ensure solubility and to avoid interactions with the column setup. 1H-NMR spectra of the deprotected PSAN76-b-PHEMA2450 and the corresponding benzoyl-protected PSAN76-b-PHEMA(–Bz)2450 are also shown in Fig. 1. The overall molar masses as compiled in Table 1 were calculated using the molar masses of the macroinitiators obtained by SEC-MALLS detection and molar compositions determined from the corresponding 1H-NMR spectra of the BCPs. The DSC thermogram of the BCP PSAN76-b-PHEMA2450 revealed a glass-transition temperature at 107 °C, which is assigned to the PSAN-block. The glass transition temperature of the PHEMA segment could not be detected due to the small amounts of water present in the sample.22,77
All polymers presented in this work feature narrow molar mass distributions and high molar masses. The length of the second block segment increased with increasing reaction time (Fig. S6†), which allows for targeted BCP synthesis by using the procedure presented here. Within our work at hand, this is the first preparation of an amphiphilic BCP based on PSAN featuring molar masses exceeding 20 kDa with controllable chain length of the hydrophilic block using an ATRP procedure. BCPs with molar masses between 51 and 90 kDa featuring dispersity indices between 1.18 and 1.25 were obtained. This is a basic prerequisite paving the way for the development of functional PSAN-based materials. Nanomechanical characterization is presented in the following to analyze the improvement of the materials’ properties of PSAN compared to the typical PS block segments.
AFM-investigation of PSAN
Before investigating the self-assembly capability of the obtained BCPs in the bulk state and its membrane formation, the morphology of the first block segment PSAN was investigated using AFM. The incorporation of acrylonitrile into a polymeric material can lead to mechanical improvements. To evaluate the change of mechanical properties at the nanometer length scale, a film of PSAN was investigated by AFM and compared to a film of PS, which is commonly used as a matrix material in SNIPS membranes.To this end, nanomechanical characterization on both films was performed in the PeakForce tapping mode. In this mode, the cantilever is sinusoidally driven, but far below its resonant frequency in contrast to standard tapping mode. Consequently, the cantilever deflection can be directly related to the instantaneous tip–sample force via the spring constant when scanning across the surface. The resulting force-versus-distance curves were fitted to the Hertz contact mechanics model allowing for the extraction of the elastic modulus in each pixel (Fig. 2b and d). The average value of the elastic modulus measured on the PS film was EPS = 6.0 GPa (Fig. 2b) whereas EPSAN = 28.3 GPa on the PSAN film (Fig. 2d). The surface roughness of the PSAN film was considerably larger, what could lead to potential errors in determining the elastic modulus owing to variations of the tip–sample contact area during the scan. However, when the highest elastic modulus values from the obtained image (see violet mask, Fig. 2d) were excluded, the corrected average value of the elastic modulus EPSAN,corr = 9.1 GPa found on the PSAN sample was still reasonably larger than that of PS. This demonstrates the increase in mechanical strength of the PSAN-domain in contrast to PS on a polymeric film. Hence, replacing functional PS-BCP based materials with PSAN-based ones can bring substantial improvements in mechanical stability. As described in the introduction, in BCP based membranes PS is the most used nonpolar matrix block segment, which could be outperformed upon AN incorporation.
 | ||
| Fig. 2 Nanomechanical characterization of PS (a and b) and PSAN (c and d) films. Topography images (a) and (c) were obtained by the PeakForce tapping mode where the feedback was set on a constant tip–sample force Fpeak = 10 nN. Elastic modulus maps (b) and (d) were deduced from fitting a Hertz contact mechanics model to each force-versus-distance curve. | ||
Microphase separation of PSAN-b-PHEMA in the bulk state
PHEMA is a building block that has been scarcely investigated regarding its microphase separation capability in the bulk state.22,79,80 The BCP investigated in this work carries functional moieties in both BCP segments, resulting in a highly interesting material whose overall properties are yet to be discovered. Aside from that, the capability of a BCP to form isoporous membranes by SNIPS depends strongly on their phase separation capabilities, as χ·N (where χ is the Flory-Huggins interaction parameter) is one of the most important variables herein. To investigate the self-assembly capability of PSAN-b-PHEMA, BCP bulk films with PHEMA volume fractions ranging from 12 to 24 vol% and varying overall molar masses were characterized with TEM imaging and SAXS measurements, which also probes the long-range order. For this purpose, BCP films were solvent-cast from DMF and subsequently treated with a solvent-annealing protocol (cf. Experimental section). In Table 2, the morphologies determined by the combined methods are listed, as well as the interdomain distances, if determinable. For the latter, Bragg-peak positions from SAXS measurements are considered (repeat distance DSAXS = 2π/q0), as well as a graphical evaluation of TEM-images (repeat distance DTEM). The TEM images and SAXS data are presented in Fig. 3. Both methods depict the phase separation capability of PSAN-b-PHEMA, detailed in the following.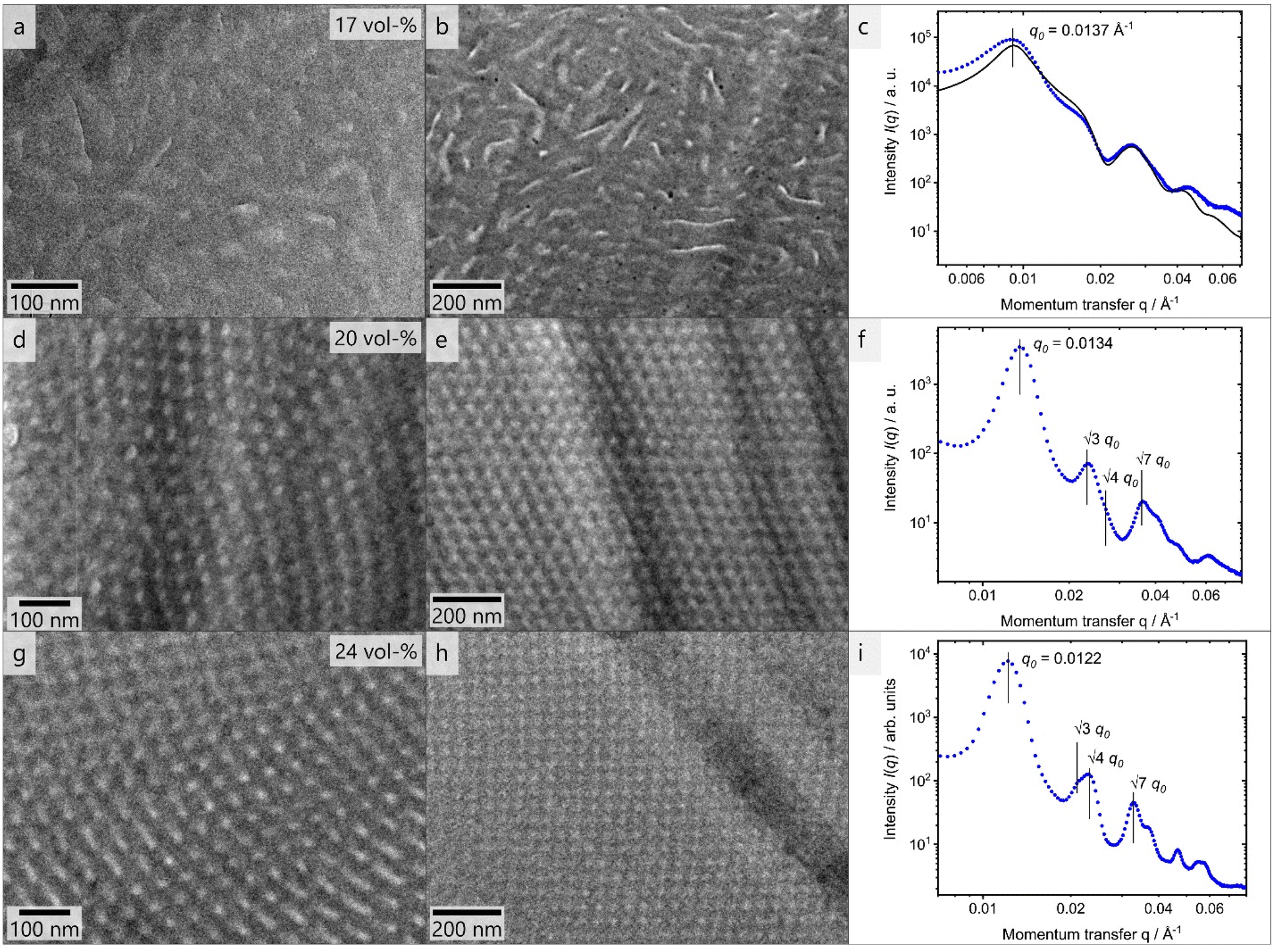 | ||
| Fig. 3 Bulk morphology investigations via TEM of thin slices of BCPs PSAN83-b-PHEMA1782 (a and b) PSAN80-b-PHEMA2047 (d and e) PSAN76-b-PHEMA2450 (g and h) in two magnifications. The corresponding SAXS curves (c, f and i) are presented with blue dots while the black line represents a model plot given within Fig. S3, ESI.† In the first image of each row the volume fractions of PHEMA are imprinted. | ||
| Polymer |
Φ
PHEMAa/vol% |
Morphologyb observed |
D
SAXSc/nm |
D
TEMd/nm |
|---|---|---|---|---|
| a See Table 1. b rcp: randomly close packed spheres; hex+: hexagonally arranged cylinders plus secondary structure. c Interdomain distance, calculated from q0 using d = 20πq0−1. d Measured graphically from middle-point to middle-point using imageJ at more than 90 points. | ||||
| PSAN 738 | 0 | — | ||
| PSAN87-b-PHEMA1376 | 13 | rcp | 56 | — |
| PSAN83-b-PHEMA1782 | 17 | rcp | 71 | — |
| PSAN 403 | 0 | — | ||
| PSAN88-b-PHEMA1241 | 12 | rcp | 33 | — |
| PSAN84-b-PHEMA1645 | 16 | rcp | 46 | — |
| PSAN80-b-PHEMA2047 | 20 | hex+ | 47 | 49 ± 6 |
| PSAN76-b-PHEMA2450 | 24 | hex+ | 52 | 50 ± 8 |
The TEM images shown in Fig. 3 reveal microphase separation with light PHEMA-domains in a dark PSAN-matrix. The sample PSAN83-b-PHEMA1782 (Fig. 3a–c) featuring 17 vol% PHEMA exhibited microphase-separated morphologies with no obvious order visible in the TEM images. The SAXS pattern (Fig. 3c) showed a series of peaks, and was compared with a model for randomly close packed (rcp) spheres, described in more detail in the ESI.† As can be observed in Fig. 3c, all prominent features of the scattering curve were captured by the model with R = 21 nm and σ = 2 nm, and a sphere center-to-center distance of 70 nm. Therefore, it qualitatively describes the microphase-separated structure of PSAN83-b-PHEMA1782. As deviations between model and the data still exist, a minor presence of other structures cannot be excluded. The worm-like appearance of the bright PHEMA domains (Fig. 3a and b) indicates that the volume fraction of 17% is close to the phase transition from spheres to cylinders.34 The TEM images of PSAN80-b-PHEMA2047 (Fig. 3d and e) featuring 20% PHEMA as well as PSAN76-b-PHEMA2450 (Fig. 3g and h) featuring 24% PHEMA reveal distinct domains with a long-range order. This finding is supported by the SAXS measurements, shown in Fig. 3f and i for PSAN80-b-PHEMA2047 and PSAN76-b-PHEMA2450, respectively. In both curves, a pronounced Bragg peak located at q0 is observed, with secondary peaks present at √3·q0, √4·q0 and √7·q0, which are assigned to scattering from hexagonally packed cylinders.81 Also in these cases, secondary structures may be present, as there are further peaks that cannot be clearly assigned. Further transmission electron micrographs can be found in the ESI.† Hexagonal structure elements of PSAN80-b-PHEMA2047 and PSAN76-b-PHEMA2450 are highlighted in Fig. S10a and b,† while Fig. S10c–f† indicates the presence of bicontinuous structures in some areas of both samples. Additional samples with PHEMA volume contents of 12 to 16% were investigated analogously and randomly close packed spheres were found by TEM and SAXS (see Table 2 and Fig. S11† and discussion).
Comparing these results to the research on the self-assembly of PHEMA BCPs by Plank et al.80 a similar phase behavior is found. The PS-b-PHEMA with 16% PHEMA exhibits hexagonally arranged cylinders, while cylinders along with hexagonally perforated lamellae and bicontinuous domains are observed at 24% PHEMA-content. A comparison of the repeat distances also reveals an increase of domain sizes following the order PS-b-PB, PS-b-PHEMA to PSAN-b-PHEMA at comparable molar masses.80 This phenomenon is attributed to increasing interaction parameters χ, corresponding values are presented in Table S1.†80,82–84 In this section we showed that the prepared amphiphilic BCPs PSAN-b-PHEMA were capable of microphase-separating in the bulk state into distinct domains above a PHEMA-content of 16 vol% revealing highly ordered and interesting morphologies. Further detailed investigation of the bulk morphologies will be subject of future research.
Besides the microphase separation in the bulk state, micelle formation capabilities in solvent mixtures give a good indication on the amphiphilicity of a BCP85 and hence the feasibility of successful membrane fabrication using the SNIPS process.86 Therefore, in this study, micelle solutions were prepared from BCPs with differing compositions in solvent mixtures, which were characterized with TEM measurements. Spherical and interconnected micelles were observed, shown and discussed in the ESI.† Furthermore, we found that the macroinitiator PSAN734 also formed micelles when introduced to a solvent mixture, which implies that the copolymer PSAN alone could self-assemble into microstructures. The membrane forming capability of the presented BCPs is investigated as a next step.
Block copolymer membranes of PSAN-b-PHEMA
As a focus of this work, membranes were fabricated via SNIPS process using the PAN-containing BCPs. As stated in the introduction, when applying the SNIPS procedure to a new BCP there are many variables to consider. The following parameters were kept constant: THF/DMF/DOX as the solvent mixture, LiCl as the additive for the casting solution and the polyester nonwoven as a membrane support. The temperature of the precipitation bath (TPB) and of the environment (TE) as well as relative air humidity (RH) of the environment were also set as similar as possible (TE = 20–24 °C, TPB = 18–22 °C, RH = 35–41%). The variables that are of utmost importance for the membrane preparation process have been studied. The evaporation time, the THF-content of the casting solution, the polymer concentration of the casting solution, the film thickness, as well as the overall molar masses of the BCPs were varied. In the following section preparation procedures are presented which lead to isoporous asymmetrical membranes that are potentially suitable for ultrafiltration applications.As a starting point, parameters known for SNIPS membranes prepared from PS-b-PHEMA BCPs were chosen based on procedures by Schöttner et al.22 For this purpose, the polymer PSAN83-b-PHEMA1782 was cast from a 20.5 mass% (polymer/solvent) solution. The solvent mixtures consisted of THF/DMF/DOX in weight fractions of 2:1:1 (Fig. 4a–d) and 3:1:1 (Fig. 4e and f), containing 0.1 mass% LiCl. The solutions were cast using the 200 μm (Fig. 4a and b) and 100 μm (Fig. 4c–f) blade gap of the doctor blade. In Fig. 4 the corresponding scanning electron micrographs of the membrane surfaces are compiled.
 | ||
| Fig. 4 Topography images by SEM of the membrane surfaces cast from solutions of PSAN83-b-PHEMA1782 in THF/DMF/DOX 2:1:1 (a–d) and 3:1:1 (e and f) at 20.3–20.5 mass% using 200 μm (a and b) and 100 μm (c–f) blade gap with varying evaporation times of 8 s (a, c and e) and 14 s (b, d and f), respectively. At the bottom right of each image the pore diameters measured visually (ImageJ) at more than 100 pores are displayed. | ||
These topographies reveal that the membranes did not exhibit uniform pores when casting with the 200 μm blade gap, as is supported by the large variation in the pore diameters which are shown in the corresponding scanning electron micrographs in Fig. 4. The decrease of the film thickness when using the 100 μm gap led to more uniform pores in appearance and size. When increasing the THF content of the solvent mixture from 2:1:1 (Fig. 4d) to 3:1:1 (Fig. 4f), the pore structure appeared even more homogeneous and ordered, as the pore diameter of (34 ± 6) nm reflects. Additional scanning electron micrographs on the membranes’ structure after applying the SNIPS process are provided in Fig. S14.†
The membrane shown in Fig. 4f was considered the most promising and as a next step the polymer concentration in the casting solution was varied, which is shown in Fig. 5. When increasing the polymer concentration similar effects to increasing the film thickness and reducing the THF content were observed: the number and uniformity of pores decreased and more flaws like in Fig. S14† were observed. At 24.1 mass% no porous membrane structure was obtained. These variations suggest that lower film thickness as well as lower concentration and higher THF-content favor the formation of an isoporous selective layer.
 | ||
| Fig. 5 Topography images by SEM of the membrane surfaces cast from solutions of PSAN83-b-PHEMA1782 in THF/DMF/DOX 3:1:1 using the 100 μm blade gap at 20.3 mass% (a), 22.5 mass% (b) and 24.1 mass% (c) with an evaporation time of 14 s. | ||
As reducing the film thickness and the polymer concentration and increasing the THF content affected the pore formation similarly favorably, we assume that high mobility of polymeric chains and fast evaporation increase the uniformity of pores in the membranes prepared with these polymers. Therefore, the BCP PSAN84-b-PHEMA1645 featuring similar composition and a lower overall molar mass was cast using the same conditions. Due to the lower molar mass the concentration of polymer in the solvent mixture was increased. Fig. 6 shows the membrane surface resulting from a 24 mass% polymer solution in 3:1:1 THF/DMF/DOX cast using the 100 μm blade gap. Applying 8 seconds of evaporation time did not lead to any uniform pores at the surface of the membrane. After 14 seconds of evaporation time pores of a uniform size of (24 ± 7) nm and irregular shape were visible. Within the scanning electron micrographs of the membrane topography cast with 20 seconds of evaporation time, uniform and hexagonally arranged pores exhibited a mean pore diameter of (27 ± 8) nm. Larger circles with flaws in order and pore size were present at the membrane surface, as in Fig. 6c on the right side of the image. The overall membrane surface also exhibited larger areas that resemble the surface structure of the membrane shown in Fig. 6b (14 s evaporation time), as can be seen in additional images provided in Fig. S15.† BCPs with lower molar masses require higher evaporation times to form ordered structures as χ·N decreases.87
 | ||
| Fig. 6 Topography images by SEM of the membrane surfaces cast from solutions of PSAN84-b-PHEMA1645 in THF/DMF/DOX 3:1:1 using the 100 μm blade gap at 24.0 mass% with an evaporation time of 8 s (a), 14 s (b) and 20 s (c). | ||
As a result, the polymer PSAN83-b-PHEMA1782 cast from a 20.5 mass% solution in the solvent mixture THF/DMF/DOX 3:1:1 with a doctor blade featuring 100 μm blade gap rendered the most uniform and homogeneous surface pore structure. In Fig. 7, scanning electron micrographs with varying magnifications of this membranes’ surface are shown. Fig. 7a presents a larger area where the structure of the underlying non-woven is visible. As in this polymer system a high mobility was required in the casting solution, it featured a lower viscosity and therefore penetrates the support rather than to build a separate layer on top, as it is often the case in other supported phase inversion membranes. Nonetheless a smooth and uniform surface layer is formed with the herein used composition, as can be seen in Fig. 7b. First data reveal permeances at both membranes exhibiting uniform pores. More details and comparative measurements are given in the ESI.†
 | ||
| Fig. 7 Topography images by SEM increasing magnifications from (a) to (c) of the membrane surfaces cast from solutions of PSAN83-b-PHEMA1782 in THF/DMF/DOX 3:1:1 at 20.3 mass% using the 100 μm blade gap with an evaporation time of 14 s. | ||
Conclusion
In this work we produced isoporous integral asymmetric membranes prepared by the SNIPS process using well-defined polyacrylonitrile-based amphiphilic BCPs. In a first step, PSAN was synthesized by ARGET-ATRP procedure using very low amounts of copper. In a second step, PSAN was used as a macroinitiator to polymerize HEMA-TMS taking advantage of the halogen-exchange reaction. After de-protection of the BCP, PSAN-b-PHEMA was obtained. Macroinitiators and BCPs both featured uniform molar mass distributions with dispersity indices between 1.13 and 1.25, and with high BCP molar masses between 41 and 82 kDa. The fractions of PHEMA were systematically varied between 12 and 24 mass%. This is the first reported synthesis of a PSAN-based amphiphilic BCP with varying hydrophilic block content via an ATRP route. The BCPs at hand were capable of self-assembling into ordered microphase separated structures in the bulk state, as analyzed by TEM and SAXS measurements. Different morphologies were found, making these BCPs interesting and promising candidates for a variety of applications. Nanomechanical analysis was conducted via AFM, and the data suggest improvements in the mechanical robustness of PSAN compared to PS, which is commonly used as a matrix material in BCP membranes. We obtained isoporous membranes by applying the SNIPS process on our acrylonitrile-based amphiphilic BCP, which has not been reported until now. Incorporating acrylonitrile into the polymer matrix of BCP membranes that we presented here paves the way towards the fabrication of more robust membranes.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank Blandine Boßmann for help with SEC measurements, Frank Hartmann for support and discussions in the lab, and Jens Pieschel for additional help with SEM measurements. This research was partially supported by the DFG project GA2169/7-1 in association with the DFG-funded consortium for advanced paper research (DFG-PAK 962/2) (R. W. S, M. G. and L. G.). M. G. thanks for partial financial support in the frame of the cooperation platform iCARE for engineering sciences. Instrumentation and technical assistance for this work were provided by the Service Center NMR at UdS, with financial support from Saarland University and German Research Foundation DFG (project number 4772985087).References
- S. P. Nunes and K. V. Peinemann, Membrane Technology: in the Chemical Industry, Wiley–VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed.
- V. Abetz, T. Brinkmann, M. Dijkstra, K. Ebert, D. Fritsch, K. Ohlrogge, D. Paul, K. V. Peinemann, S. Pereira-Nunes, N. Scharnagl and M. Schossig, Adv. Eng. Mater., 2006, 8, 328–358 CrossRef CAS.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef CAS PubMed.
- L. Guo, Y. Wang and M. Steinhart, Chem. Soc. Rev., 2021, 50, 6333–6348 RSC.
- M. Radjabian and V. Abetz, Prog. Polym. Sci., 2020, 102, 101219 CrossRef CAS.
- J. R. Werber, C. O. Osuji and M. Elimelech, Nat. Rev. Mater., 2016, 1, 16018 CrossRef CAS.
- M. M. Wu, Acrylonitrile and Acrylonitrile Polymers, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, John Wiley & Sons, Inc., 2003, vol. 1, pp. 139 ff Search PubMed.
- J. Bandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Sons, Inc., 4th edn, 1999 Search PubMed.
- S. Choudhury, D. Fischer, P. Formanek, F. Simon, M. Stamm and L. Ionov, Polymer, 2018, 151, 171–178 CrossRef CAS.
- X. Jin, L. Li, R. Xu, Q. Liu, L. Ding, Y. Pan, C. Wang, W. Hung, K. Lee and T. Wang, Polymers, 2018, 10, 539 CrossRef PubMed.
- H. Srivastava, H. Lade, D. Paul, G. Arthanareeswaran and J. Kweon, Appl. Sci., 2016, 6, 159 CrossRef.
- B. Fang, Q. Ling, W. Zhao, Y. Ma, P. Bai, Q. Wei, H. Li and C. Zhao, J. Membr. Sci., 2009, 329, 46–55 CrossRef CAS.
- S. Caprarescu, M. C. Corobea, V. Purcar, C. I. Spataru, R. Ianchis, G. Vasilievici and Z. Vuluga, J. Environ. Sci., 2015, 35, 27–37 CrossRef CAS PubMed.
- P. Boonying, T. Narkkun, W. Naowanon, S. Amnuaypanich, W.-S. Hung and S. Amnuaypanich, Sep. Purif. Technol., 2019, 223, 203–210 CrossRef CAS.
- M. T. Tsai, L. H. Chung, G. Y. Lin, M. C. Chang, C. Y. Lee and N. H. Tai, Sep. Purif. Technol., 2020, 241, 116683 CrossRef CAS.
- R. Sallakhniknezhad, M. Khorsi, A. S. Niknejad, S. Bazgir, A. Kargari, M. Sazegar, M. Rasouli and S. Chae, Membranes, 2021, 11, 969 CrossRef CAS PubMed.
- M. Makaremi, R. T. De Silva and P. Pasbakhsh, J. Phys. Chem. C, 2015, 119, 7949–7958 CrossRef CAS.
- M. Müller and V. Abetz, Chem. Rev., 2021, 121, 14189–14231 CrossRef PubMed.
- S. P. Nunes, Sustainable Nanoscale Engineering, 2020, pp. 297–316, DOI:10.1016/b978-0-12-814681-1.00011-4.
- K. V. Peinemann, V. Abetz and P. F. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- L. Tsaur and U. B. Wiesner, Polymers, 2023, 15, 2020 CrossRef CAS PubMed.
- S. Schöttner, H.-J. Schaffrath and M. Gallei, Macromolecules, 2016, 49, 7286–7295 CrossRef.
- S. Schöttner, R. Hossain, C. Rüttiger and M. Gallei, Polymers, 2017, 9, 491 CrossRef PubMed.
- J. Wang, M. M. Rahman, C. Abetz, S. Rangou, Z. Zhang and V. Abetz, Macromol. Rapid Commun., 2018, 39, e1800435 CrossRef PubMed.
- M. M. Rahman, Macromol. Rapid Commun., 2021, 42, e2100235 CrossRef PubMed.
- M. Plank, F. V. Frieß, C. V. Bitsch, J. Pieschel, J. Reitenbach and M. Gallei, Macromolecules, 2023, 56(4), 1674–1684 CrossRef CAS.
- B. Sutisna, G. Polymeropoulos, V. Musteata, K. V. Peinemann, A. Avgeropoulos, D. M. Smilgies, N. Hadjichristidis and S. P. Nunes, Mol. Syst. Des. Eng., 2016, 1, 278–289 RSC.
- S. Rangou, K. Buhr, V. Filiz, J. I. Clodt, B. Lademann, J. Hahn, A. Jung and V. Abetz, J. Membr. Sci., 2014, 451, 266–275 CrossRef CAS.
- N. Blagojevic and M. Müller, ACS Appl. Mater. Interfaces, 2023 DOI:10.1021/acsami.3c03126.
- S. Rangou, M. Appold, B. Lademann, K. Buhr and V. Filiz, ACS Macro Lett., 2022, 11, 1142–1147 CrossRef CAS PubMed.
- F. V. Friess, Q. Hu, J. Mayer, L. Gemmer, V. Presser, B. N. Balzer and M. Gallei, Macromol. Rapid Commun., 2022, 43, e2100632 CrossRef PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 7641–7644 CrossRef CAS.
- D. A. Hajduk, P. E. Harper, S. M. Gruner, C. C. Honeker, G. Kim, E. L. Thomas and L. J. Fetters, Macromolecules, 1994, 27, 4063–4075 CrossRef CAS.
- X. Tan, J. Li and S. Guo, Macromolecules, 2018, 51, 2099–2109 CrossRef CAS.
- H.-F. Wang, H. Marubayashi and H. Jinnai, Macromolecules, 2023, 56(4), 1503–1513 CrossRef CAS.
- I. Vukovic, G. ten Brinke and K. Loos, Macromolecules, 2012, 45, 9409–9418 CrossRef CAS.
- M. Zhong, S. Jiang, Y. Tang, E. Gottlieb, E. K. Kim, A. Star, K. Matyjaszewski and T. Kowalewski, Chem. Sci., 2014, 5, 3315–3319 RSC.
- M. Kopeć, M. Lamson, R. Yuan, C. Tang, M. Kruk, M. Zhong, K. Matyjaszewski and T. Kowalewski, Prog. Polym. Sci., 2019, 92, 89–134 CrossRef.
- S. Vowinkel, C. G. Schäfer, G. Cherkashinin, C. Fasel, F. Roth, N. Liu, C. Dietz, E. Ionescu and M. Gallei, J. Mater. Chem. C, 2016, 4, 3976–3986 RSC.
- T. Fukuda, T. Terauchi, A. Goto, Y. Tsujii, T. Miyamoto and Y. Shimizu, Macromolecules, 1996, 29, 3050–3052 CrossRef CAS.
- D. Gromadzki, J. Lokaj, P. Černoch, O. Diat, F. Nallet and P. Štěpánek, Eur. Polym. J., 2008, 44, 189–199 CrossRef CAS.
- D. Quémener, G. Bonniol, T. N. T. Phan, D. Gigmes, D. Bertin and A. Deratani, Macromolecules, 2010, 43, 5060–5065 CrossRef.
- C. Detrembleur, V. Sciannamea, C. Koulic, M. Claes, M. Hoebeke and R. Jérôme, Macromolecules, 2002, 35, 7214–7223 CrossRef CAS.
- V. Sciannamea, R. Jerome and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- Z. Zhou and G. Liu, Small, 2017, 13, 1603107 CrossRef PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- K. Matyjaszewski, S. M. Jo, H.-J. Paik and D. A. Shipp, Macromolecules, 1999, 32, 6431–6438 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, e1706441 CrossRef PubMed.
- D. Fan, J. He, J. Xu, W. Tang, Y. Liu and Y. Yang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2260–2269 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski, K. Davis, T. E. Patten and M. Wei, Tetrahedron, 1997, 53, 15321–15329 CrossRef CAS.
- K. Matyjaszewski, S. Mu Jo, H.-J. Paik and S. G. Gaynor, Macromolecules, 1997, 30, 6398–6400 CrossRef CAS.
- K. Matyjaszewski, T. E. Patten and J. Xia, J. Am. Chem. Soc., 1997, 119, 674–680 CrossRef CAS.
- N. V. Tsarevsky, T. Sarbu, B. Göbelt and K. Matyjaszewski, Macromolecules, 2002, 35, 6142–6148 CrossRef CAS.
- N. V. Tsarevsky, K. V. Bernaerts, B. Dufour, F. E. Du Prez and K. Matyjaszewski, Macromolecules, 2004, 37, 9308–9313 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2005, 39, 39–45 CrossRef.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- H. Dong, W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 2974–2977 CrossRef CAS.
- W. Jakubowski, B. Kirci-Denizli, R. R. Gil and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 32–39 CrossRef CAS.
- J. Pietrasik, H. Dong and K. Matyjaszewski, Macromolecules, 2006, 39, 6384–6390 CrossRef CAS.
- C. Tang, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2003, 36, 1465–1473 CrossRef CAS.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- S. P. Nunes, P. Z. Culfaz-Emecen, G. Z. Ramon, T. Visser, G. H. Koops, W. Jin and M. Ulbricht, J. Membr. Sci., 2020, 598, 117761 CrossRef CAS.
- T. Bucher, V. Filiz, C. Abetz and V. Abetz, Membranes, 2018, 8, 57 CrossRef PubMed.
- F. De Bon, R. G. Fonseca, F. Lorandi, A. C. Serra, A. A. Isse, K. Matyjaszewski and J. F. J. Coelho, Chem. Eng. J., 2022, 445, 136690 CrossRef CAS.
- G. J. Dunderdale, C. Urata, D. F. Miranda and A. Hozumi, ACS Appl. Mater. Interfaces, 2014, 6, 11864–11868 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- H. J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1–7 CrossRef.
- D. A. Shipp, J.-L. Wang and K. Matyjaszewski, Macromolecules, 1998, 31, 8005–8008 CrossRef CAS.
- M. Fernández-García, M. F. Torrado, G. Martínez, M. Sánchez-Chaves and E. L. Madruga, Polymer, 2000, 41, 8001–8008 CrossRef.
- R. K. Bose and K. K. S. Lau, Biomacromolecules, 2010, 11, 2116–2122 CrossRef CAS PubMed.
- C. L. Breaux, B. L. Sharp, P. J. Ludovice, C. L. Henderson, H. Li, B. Li and M. Neisser, J. Vac. Sci. Technol., B, 2019, 37, 011603-1–1-1 Search PubMed.
- M. Plank, F. Hartmann, B. Kuttich, T. Kraus and M. Gallei, Eur. Polym. J., 2020, 141, 1674–1687 CrossRef.
- I. W. Hamley and V. Castelletto, Prog. Polym. Sci., 2004, 29, 909–948 CAS.
- A. N. Semenov, Zh. Eksp. Teor. Fiz., 1985, 88, 1242–1256 CAS.
- E. Helfand and Z. R. Wasserman, Macromolecules, 2002, 9, 879–888 CrossRef.
- J. Chu, Polymer, 2000, 41, 7193–7199 CrossRef CAS.
- D. Heinz, E. Amado and J. Kressler, Polymers, 2018, 10, 960 CrossRef PubMed.
- S. P. Nunes, M. Karunakaran, N. Pradeep, A. R. Behzad, B. Hooghan, R. Sougrat, H. He and K. V. Peinemann, Langmuir, 2011, 27, 10184–10190 CrossRef CAS PubMed.
- C. Stegelmeier, A. Exner, S. Hauschild, V. Filiz, J. Perlich, S. V. Roth, V. Abetz and S. Förster, Macromolecules, 2015, 48, 1524–1530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00836c |
| This journal is © The Royal Society of Chemistry 2023 |