
Speciation of potential-dependent fouling on copper foil electrodes during electrochemical hydrogenation and hydrogenolysis of furfural in strong acid†
Andrew S.
May
 and
Elizabeth J.
Biddinger
*
and
Elizabeth J.
Biddinger
*
Department of Chemical Engineering, The City College of New York, CUNY, New York, New York 10031, USA. E-mail: ebiddinger@ccny.cuny.edu
First published on 19th September 2023
Abstract
The electrochemical transformation of sustainable feedstocks such as biomass derived species can help to electrify the chemical industry and reduce reliance on petroleum. Furfural (FF), one such biomass derived species, can be electrochemically reduced to form furfuryl alcohol (FA) and 2-methylfuran (MF) which are used to produce thermally stable molds and resins, and a fuel candidate or additive, respectively. Using Cu electrodes allows for selective production of FA or MF, dependent on the pH of solution, with MF requiring a low pH. The highly acidic conditions to produce MF also drive homogeneous side reactions and foul the Cu electrode with time. To better understand the nature of the build up on the electrodes, accelerated fouling conditions of high initial concentrations of FF and very low pH were investigated. At concentrations of 200 mM FF in 0.5 M H2SO4, we observed fouling of the Cu electrode during electrochemical hydrogenation and hydrogenolysis (ECH) of FF. The fouling of the electrode was also shown to be potential dependent, with poly(furfuryl alcohol) (pFA) forming at −560 mV RHE while an amorphous carbon resembling soot was observed more significantly at −700 mV RHE. The fouling of the Cu electrodes was shown to increase the polarization resistance during FF reduction, hindering the rates of reactions to FA and MF. Through control studies, it was found that the fouling is electrochemically-driven and requires the starting reactant FF to be present. By investigating and understanding the fouling of Cu electrodes during the electrochemical hydrogenation and hydrogenolysis of FF, catalysts can be better designed to inhibit fouling and be used for longer durations or for more cycles before requiring a treatment to regain activity or replacement. Additionally, while this work shows the detriment of carbon fouling on the electrode, pFA is a desirable product. Process intensification through development of a FF to pFA electrochemical step could be achieved through electroreduction with high concentration of FF in 0.5 M H2SO4.
Introduction
Limiting the production of greenhouse gases coming from the chemical industry is a challenge currently being faced which can be addressed through the design and implementation of sustainable processes. Electrochemical processes have benefits in terms of sustainability compared to their non-electrochemical counterparts,1–4 with greater benefit reached through biomass upgrading.5 Electrochemical processes can be considered greener due to being able to avoid upstream CO2 generation production of reactant hydrogen, eliminate toxic reagents, and operate at ambient conditions.5,6 Additionally, electrochemical processes can have a modular small-scale design that is more flexible to intermittent energy usage.5,6 In electrochemical reduction reactions the protons for hydrogenation can be supplied from the electrolyte instead of externally supplied hydrogen gas. Not requiring hydrogen gas reduces the overall CO2 emissions, as the hydrogen gas is conventionally produced via steam reforming followed by water gas shift reactions, which emits a large amount of CO2. Renewable biomass is also replenishable on a much shorter time scale compared to fossil fuels and lessens the concerns regarding long term availability of feedstocks.7 Biomass upgrading depots have recently been proposed as one example using biomass as a feedstock on a distributed scale.8,9 Electrochemical upgrading of the bio-oil can allow for transportation of the oil or use.While many biomass derived species exist, furfural (FF) has been identified as a top 30 building block molecule10 with a production estimated to be approximately 420![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year in 2024.11 FF is used to produce crucial commodity chemicals such as furfuryl alcohol (FA), 2-methylfuran (MF), furan, and tetrahydrofuran.12,13 While traditional thermocatalytic methods dominate FF reactions currently, electrochemical methods have been of great interest recently. For example, the electrochemical hydrogenation and hydrogenolysis (ECH) of FF is used to produce FA and MF, respectively. Nilges and Schröder showed that Cu electrodes can produce both FA and MF in 0.5 M H2SO4 with a high selectivity of 80% to MF, whereas Ni and Pt metal electrodes would be more selective to FA than MF, and electrodes of C, Fe, Pb, and Al would largely produce the dimer species hydrofuroin.14 Over a Cu catalyst, the pH has been shown to be crucial in achieving a high selectivity to MF, with the selectivity shifting towards FA as the pH increases.15–19 While the acidic conditions have been shown to greatly increase the selectivity to the MF species, acidic conditions have also been shown to be responsible for homogeneous side reactions occurring in the electrolyte consuming FA and MF products.15,17,19,20 We have shown that the sparging of nitrogen gas will allow for higher MF yields in more acidic conditions despite the side reactions, due to the facile evaporation of MF into a solvent trap that does not contain acid.19,20 The applied potential controls the current (reaction rate) of the process. We have previously shown that by running at higher current density, the mass balance in FF ECH can be improved as the duration of experiment can be shortened, limiting side reactions.15 An increase to the magnitude of the reducing potential typically increases the conversion of FF reduction, but comes at the cost of lowering the faradaic efficiency to FA and MF.20 A higher selectivity to MF can be achieved by increasing the applied potential from −500 mV RHE to −800 mV RHE, but will decrease with further increase in potential to −900 mV RHE.20
000 tons per year in 2024.11 FF is used to produce crucial commodity chemicals such as furfuryl alcohol (FA), 2-methylfuran (MF), furan, and tetrahydrofuran.12,13 While traditional thermocatalytic methods dominate FF reactions currently, electrochemical methods have been of great interest recently. For example, the electrochemical hydrogenation and hydrogenolysis (ECH) of FF is used to produce FA and MF, respectively. Nilges and Schröder showed that Cu electrodes can produce both FA and MF in 0.5 M H2SO4 with a high selectivity of 80% to MF, whereas Ni and Pt metal electrodes would be more selective to FA than MF, and electrodes of C, Fe, Pb, and Al would largely produce the dimer species hydrofuroin.14 Over a Cu catalyst, the pH has been shown to be crucial in achieving a high selectivity to MF, with the selectivity shifting towards FA as the pH increases.15–19 While the acidic conditions have been shown to greatly increase the selectivity to the MF species, acidic conditions have also been shown to be responsible for homogeneous side reactions occurring in the electrolyte consuming FA and MF products.15,17,19,20 We have shown that the sparging of nitrogen gas will allow for higher MF yields in more acidic conditions despite the side reactions, due to the facile evaporation of MF into a solvent trap that does not contain acid.19,20 The applied potential controls the current (reaction rate) of the process. We have previously shown that by running at higher current density, the mass balance in FF ECH can be improved as the duration of experiment can be shortened, limiting side reactions.15 An increase to the magnitude of the reducing potential typically increases the conversion of FF reduction, but comes at the cost of lowering the faradaic efficiency to FA and MF.20 A higher selectivity to MF can be achieved by increasing the applied potential from −500 mV RHE to −800 mV RHE, but will decrease with further increase in potential to −900 mV RHE.20
We have previously shown that with repeated use in FF ECH, Cu electrodes became covered in carbonaceous material that inhibited the production rate to FA and MF.21 Deng showed that coking and polymerization occurs in the electrochemical upgrading of bio-oils and how to tune the coke structures using current density and reaction time to produce coke for desired applications.22,23 Systems containing bio-oils or furanics have shown electropolymerization reactions or coking at oxidative potentials24 or at highly negative reductive potentials (−5400 mV RHE).25 While fouling is not desired in the selective production to FA and MF, electrochemical polymerization can be a desired reaction.25
In this work we show the impact of the applied potential on the fouling of Cu electrodes during FF ECH in conditions designed to accelerate fouling. FF ECH was done with 200 mM concentrations of FF in 0.5 M H2SO4 with a Cu electrode. The electrode was used three times to further foul the electrode. In the ECH of FF to produce FA and MF, we show that a polymer or amorphous carbon resembling soot would form in the selected conditions. The carbon deposition on the electrode led to deactivation and decreased rates (partial current densities) to FA and MF. FTIR characterization showed that poly(furfuryl alcohol) (pFA) was formed at a moderately negative applied potential. Amorphous carbon was formed at both potentials, however it was more prevalent at a more negative applied potential. We also identified that the fouling is electrochemically driven and requires the presence of the starting material, FF. Understanding the electrode fouling in FF ECH is important so that more robust systems can be implemented to extend catalyst lifetime for ECH of FF to FA and MF.
Additionally, an understanding of catalyst performance will allow for more detailed models regarding production of FA and MF. This work showed that pFA could be produced via electrochemical reduction of FF in acidic conditions with 200 mM FF. There is benefit in targeting the pFA and collecting it, as a common use for FA is the acidic polymerization to form pFA.
Experimental
Materials
To construct Cu electrodes, Cu wire (99.999%, 0.5 mm diameter from Alfa Aesar) was attached to a 1.5 by 2.0 cm Cu foil (99.999%, 0.1 mm thick from Alfa Aesar). The Pt gauze counter electrode was made by welding a Pt wire (99.95%, 0.5 mm diameter from Alfa Aesar) onto a 2.5 by 2.5 cm Pt gauze (99.9%, 52 mesh, 0.1 mm thick from Alfa Aesar). The reference electrode was a 3.0 M sodium chloride Ag/AgCl electrode (MR-5275, from BASi). DI water for solutions was 18.2 MΩ cm produced in lab via Milli-Q Direct 8 water purification system. Acetone (optima grade from Fisher) and hydrochloric acid (37%, from Fisher) diluted to 10 vol% in water were used for Cu electrode preparation. Chloroform (ethanol as stabilizer, HPLC grade from Beantown Chemical), sodium chloride from Acros, and sodium sulfate anhydrous from Fisher were used in sample workup. Acetonitrile (optima grade from Fisher), furfural (99% from Sigma-Aldrich), furfuryl alcohol (98% from Acros), 2-methylfuran (99% with BHT as stabilizer from Sigma), and sulfuric acid (93–98% trace metal grade from Fisher) were used in preparation of calibration samples and reactor electrolytes. Furfural was vacuum distilled in lab to remove color and increase purity at a pressure of 5 mbar and a temperature increasing from ambient to approximately 40 °C. Nitrogen gas (5.0 ultra high purity from Praxair) was used to sparge the catholyte.ECH cell setup
A two compartment H-cell separated by a Nafion 117 membrane was used, as depicted in our previous work.17 A solvent trap was connected to a cathode compartment port, and nitrogen gas was sparged at 60 mL min−1 into the catholyte to promote evaporation of volatile species into the solvent trap for collection. The solvent trap consisted of 60 mL of acetonitrile chilled to −15 °C. The anolyte was 30 mL of 0.5 M sulfuric acid. The catholyte was 35 mL of 0.5 M sulfuric acid in a 20:80 v:v acetonitrile:water cosolvent with 200 mM of FF. The H-cell was kept in a water bath at 25 °C. Stir bars were placed into the anode and cathode chambers and stirred at 600 rpm via magnetic stirrer. The Cu electrode was sonicated in acetone for 10 minutes, rinsed with DI water, submerged into 10 vol% HCl for 1 minute, and then rinsed prior to the first electrolysis experiment.
A Gamry Interface 1000 potentiostat was used for electrochemical experiments. Electrochemical impedance spectroscopy (EIS) was run potentiostatically at −560 mV vs. RHE in all cases. An AC voltage amplitude of 10 mV was applied in a frequency range of 100 kHz to 0.1 Hz.
Chronoamperometry was run for 3 hours at an applied potential of −560 mV RHE or −700 mV RHE and iR corrected manually post experiment. Electrochemical experiments were repeated at least 3 times. Electrolytes were sparged with nitrogen gas at 60 mL min−1 for 30 minutes prior to use, and during electrochemical experiment.
FA ECH experiments were done with 15 mM of FA in lieu of FF at −560 mV and −700 mV RHE.
A Cu submerged in electrolyte containing 30 mM of FA and 100 mM FF for 3 hours experiment was done with no applied potential.
Analysis
0.5 mL samples were taken via syringe from the catholyte, anolyte, and solvent trap. The solvent trap sample was diluted by adding 1 mL of acetonitrile. Catholyte and anolyte samples underwent a workup to remove the aqueous and acidic components of the solution before GCMS analysis. For the workup, the sample was first diluted to 5 mL with chloroform, then 200 mg of NaCl was added and the sample was shaken. After settling the sample would phase separate into an aqueous and organic phase. The organic phase was removed by syringe and further dried with 100 mg of Na2SO4. The dried samples were then removed by syringe and syringe filtered into a vial. Samples from the anolyte did not show any furanics from membrane crossover.Calibration curves were produced to relate the concentration of furanic (FF, FA, MF) to the corresponding peak area of GC-MS spectra.
Worked up samples were run through a gas chromatograph mass spectrometer (GC-MS) from Shimadzu (Model QP 2010Ultra), equipped with a TR-WAXMS column (30 m long, 0.25 mm diameter, 0.25 micron thick) from Thermo Scientific. The method file used ran a temperature ramp of 5 °C min−1 from 40 °C to 160 °C and had an injection temperature of 250 °C.
X-ray photoelectron spectroscopy (XPS) was used to investigate the chemical composition and functional groups present in the fouled Cu surfaces. Fouled Cu electrodes were dried at 40 °C overnight under vacuum to be suitable for XPS analysis. A PHI 5000 VersaProbe II spectrometer from Physical Electronics, equipped with a monochromatic Al Kα radiation source, and operated at 10−8 Torr pressure was used.
A FEI Helios NanoLab 660 from Thermo Fisher was used for SEM and EDS imaging. Due to low conductivity, fouled Cu samples were coated with 20 nm of Au using a Leica EM ACE600 coater. Scans were taken using 1.6 nA at 10 kV.
A confocal Raman Microscope alpha300R from WITec equipped with a Zeiss EC Epiplan-Neofluar Dic 100×/0.9 objective lens was used to collect Raman spectra of fouled Cu samples. Scans were taken with a 532 nm laser. The spectrometer was calibrated with a Si sample using 50 co-added scans at 0.1 s scan time with 20 mW power laser. Fouled Cu electrodes were sampled with a 5 mW power laser, 50 co-added scans with 0.1 s scan time.
A Nicolet is50 FTIR bench with a built-in ATR (Diamond) from Thermo Fisher Scientific was used equipped with a DTGS detector. Spectra consisted of 32 co-added scans with a 4 cm−1 resolution.
Results and discussion
To investigate the fouling of Cu electrodes during the ECH of FF, conditions were selected to intentionally promote fouling. These conditions included consecutive use of the Cu electrode in bulk electrolysis with high concentrations of FF (200 mM), acidic electrolyte of 0.5 M H2SO4, and long experimental run time of three hours. Experiments were performed in a 2-compartment H-cell attached to a solvent trap to collect volatile species. The electrodes were used for three consecutive experiments: ECH1, ECH2, and ECH3. The electrolytes and solvent trap were sampled during all sets of ECH, and the electrodes had ex situ characterization after ECH. A depiction of this experimental procedure is shown in Fig. 1a. During ECH, the Cu electrodes underwent fouling due to formation of carbonaceous material on the surface of the Cu electrode. When bare Cu flag electrodes were used under accelerated fouling conditions, the activity of the electrode was negatively impacted at both −560 and −700 mV RHE. With each use of the electrode, the final conversion decreased, a trend observed at both −560 and −700 mV RHE. For the more negative potential of −700 mV RHE, the conversion was 43.3% after ECH1 and 23.4% after ECH3. At −560 mV RHE, the conversion was 60.1% after ECH1 and 20.7% after ECH3. With a more negative potential, it is expected that the greater driving force will increase the rate of FF ECH to FA and MF, and hence reach a higher conversion.18,20,21 Surprisingly, the conversion at the less negative potential was found to be higher than the conversion at a more negative potential for the first and second trials of ECH, shown in Fig. 1b–d. After ECH3, the conversion reached were very similar. Reaction data is also tabulated in Tables S1 and S2.† In previous work, we have shown that conversion generally increases with more negative potentials,17,20,21 however, the accelerated fouling conditions likely have influenced the conversion results.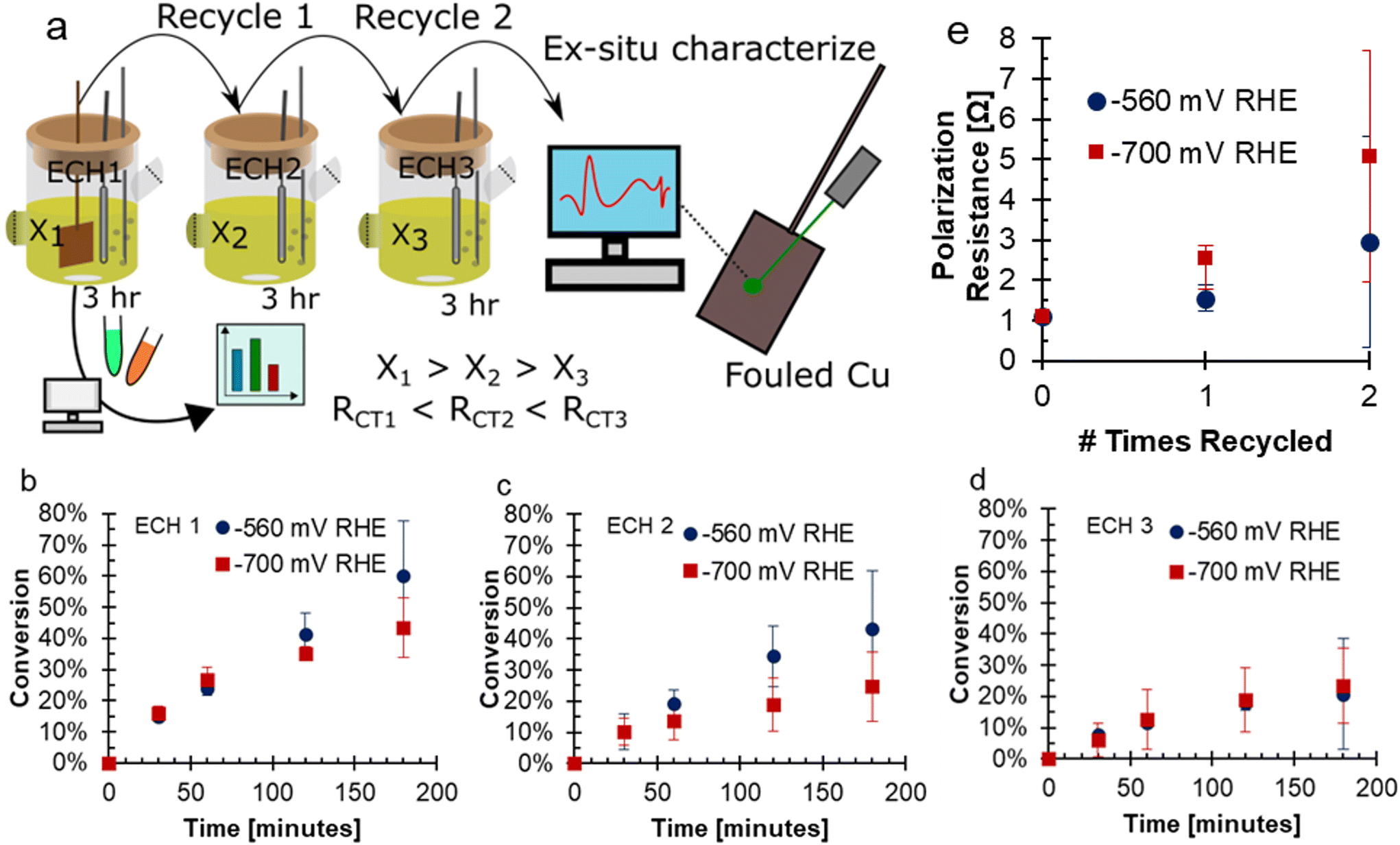 | ||
| Fig. 1 (a) A schematic showing the experimental procedure of this work, noting that the ECH cell depicts only the cathodic chamber while a full cell was used as depicted in ref. 17. Conversion of FF during 3 hours ECH with recycle showing (b) ECH1, (c) ECH2 (d) ECH3. Conditions: 200 mM FF, 0.5 M H2SO4, 80:20 vol% water:acetonitrile, 60 mL N2 sparging, 25 °C, Cu electrode. (e) Polarization resistance before ECH determined using EIS. X-Axis shows the number of uses prior to the EIS. EIS conditions: −560 mV RHE RMS, 10 mV amplitude, 0.1 Hz to 100 kHz. | ||
In the FF ECH system on Cu in acid, the conversion of FF can produce both FA and MF through parallel reactions.20 Additionally, due to the acidic electrolyte, homogeneous side reactions occur which consume FA and MF and negatively impact the mass balance closure.15 The mass balance closure is a measure of the quantified furanics (FF, FA, and MF), which will decrease when products such as humins, that are difficult to quantify, are produced.15 The mass balance, as well as the selectivity to FA and MF over consecutive ECH at either −560 or −700 mV RHE are shown in Fig. 2. The mass balance is the accounting of quantified products FF, FA, and MF, and did not reach 100% due to homogeneous acidic side reactions or fouling.15,17,19,20Fig. 2a–c shows that the mass balance closure at −560 mV RHE decreased with time throughout ECH, but the decrease was less severe with subsequent ECH. The higher mass balance with subsequent ECH was due to a lower amount of converted FF when the electrode was recycled and hence less FA and MF going through side reactions. This was because the side reactions were concentration dependent.19,20 At the end of ECH, the mass balance was found to be 65.9%, 73.7%, and 82.9% when run at −560 mV RHE with consecutive use. A similar trend was shown when ECH was run at −700 mV RHE (Fig. 2d–f), where the mass balance reached was 72.1%, 79.9%, and 81.6% with consecutive use at −700 mV RHE.
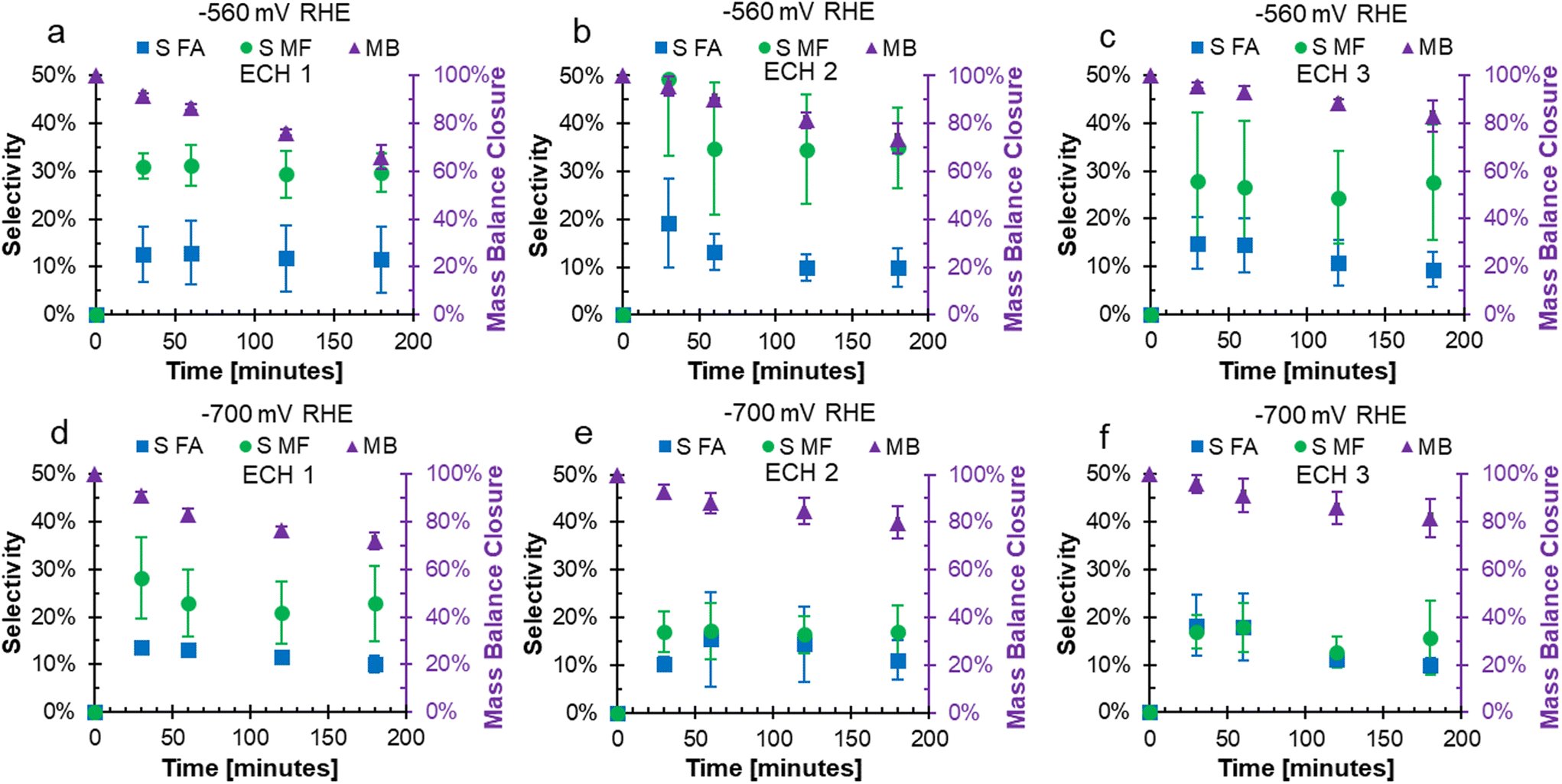 | ||
| Fig. 2 Mass balance and selectivity to FA and MF from consecutive ECH of FF with recycled Cu foil electrode. At −560 mV RHE: (a) ECH1, (b) ECH2, (c) ECH3. At −700 mV RHE: (d) ECH1, (e) ECH2, (f) ECH3. Conditions: 200 mM FF, 0.5 M H2SO4, 80:20 vol% water:acetonitrile, 3 hours duration ECH, 60 mL N2 sparging, Cu electrode. | ||
At −560 mV RHE, the selectivity to MF was stable around 30% throughout ECH1 to ECH 3 while the selectivity to FA decreased over the duration of ECH. For example, the selectivity to FA dropped by 1.2%, 9.4%, and 5.5% during ECH1, ECH2, and ECH3 between the first and final samples. This decrease in selectivity was due to the rate of homogeneous side reactions exceeding the rate of FA production.19 At −700 mV RHE, the selectivity towards MF dropped early on during ECH1 but then remained constant. The selectivity of MF after 30 minutes on ECH1 was 28.2% and dropped to 22.8% at 3 hours. The selectivity to MF then remained fairly stable around 18% during ECH2 and ECH3. The selectivity towards FA had a small decrease during the ECH experiments at −700 mV RHE, again due to the rate of side reactions exceeding that of the electrochemical FA production. The resulting selectivity was lower than our previous work at 100 mM FF where fouling was not observed,18 which suggests that additional furanics were lost due to fouling. One possibility is that carbon on the surface from fouling acted catalytically, as carbon shows a lower selectivity to MF production compared to Cu.14 It could also be the case that an intermediate to MF was remaining on the catalyst and was the cause of the fouling. Similar trends were found for the faradaic efficiencies to FA and MF, shown in Fig. S1.† The mass of the electrode was measured prior to use and after the final use of ECH to determine the mass change due to fouling. The average mass on the electrode after continued ECH was 0.00228 g at −560 mV RHE and 0.00306 g at −700 mV RHE. While the impacts of the fouling was severe, the mass accounted for only 1.27 and 1.98% of the unaccounted for mass from ECH at −560 mV and −700 mV RHE, respectively.
The decrease in current was due to an increase in the polarization resistance at the electrode surface. The polarization resistance prior to ECH at both potentials is shown in Fig. 1e. The polarization resistance was found using EIS, with representative samples shown in Fig. S2.† At −560 mV RHE the polarization resistance increased with consecutive use from 1.10 Ω to 1.56 Ω, and then 2.97 Ω. A larger increase in the polarization resistance was seen with use at −700 mV RHE, from 1.11 Ω, to 2.55 Ω, and then 5.09 Ω. The increase in polarization resistance with consecutive use was an indicator that the fouling had caused the decreased conversion with electrode recycling. The charge transfer resistance of the reaction increased more significantly between consecutive ECH at −700 mV RHE compared to −560 mV RHE. The greater charge transfer resistance of the −700 mV RHE fouled electrode supports the more significant deactivation of the electrode.
Once used, the electrodes had material buildup which could be seen by eye as a darkening of the electrode surface which was likely causing the increasing polarization resistance and decrease to conversion. To visualize and determine the chemical composition of the material found on the recycled electrodes, SEM/EDX was used. SEM/EDX showed high coverage of carbonaceous material on electrodes used three times at either −560 mV RHE or −700 mV RHE. The carbonaceous fouling had different features and chemical composition between the −560 mV RHE and −700 mV RHE used electrodes. The high coverage of carbonaceous material on the surface of the −560 mV RHE fouled Cu electrode and −700 mV RHE electrodes can be seen in the EDX scans presented in Fig. 3a and b. On the −560 mV RHE fouled Cu electrode, the surface had small features located throughout but otherwise appeared smooth, and no Cu was detected with EDX. In comparison, the −700 mV RHE fouled electrode (Fig. 3b) showed a rough surface with small, dispersed Cu sites throughout the electrode. For comparison, SEM/EDX images of a bare Cu electrode are shown in Fig. 3c. The bare Cu electrode is smooth and had significantly less C and O contribution, with a higher Cu contribution than the fouled electrodes. The C and O contributions on the bare Cu were attributed to time spent in atmosphere during sample transfer to XPS. The composition of the surfaces in atomic percentage can be found in Table 1. The bare Cu had a significantly higher Cu at% with C and O at% due to exposure to atmosphere. The Cu fouled at −560 mV RHE had the highest at% to C and had an undetectable Cu at%. The Cu electrode fouled at −700 mV RHE had a higher O and Cu at% compared to the −560 mV RHE fouled electrode.
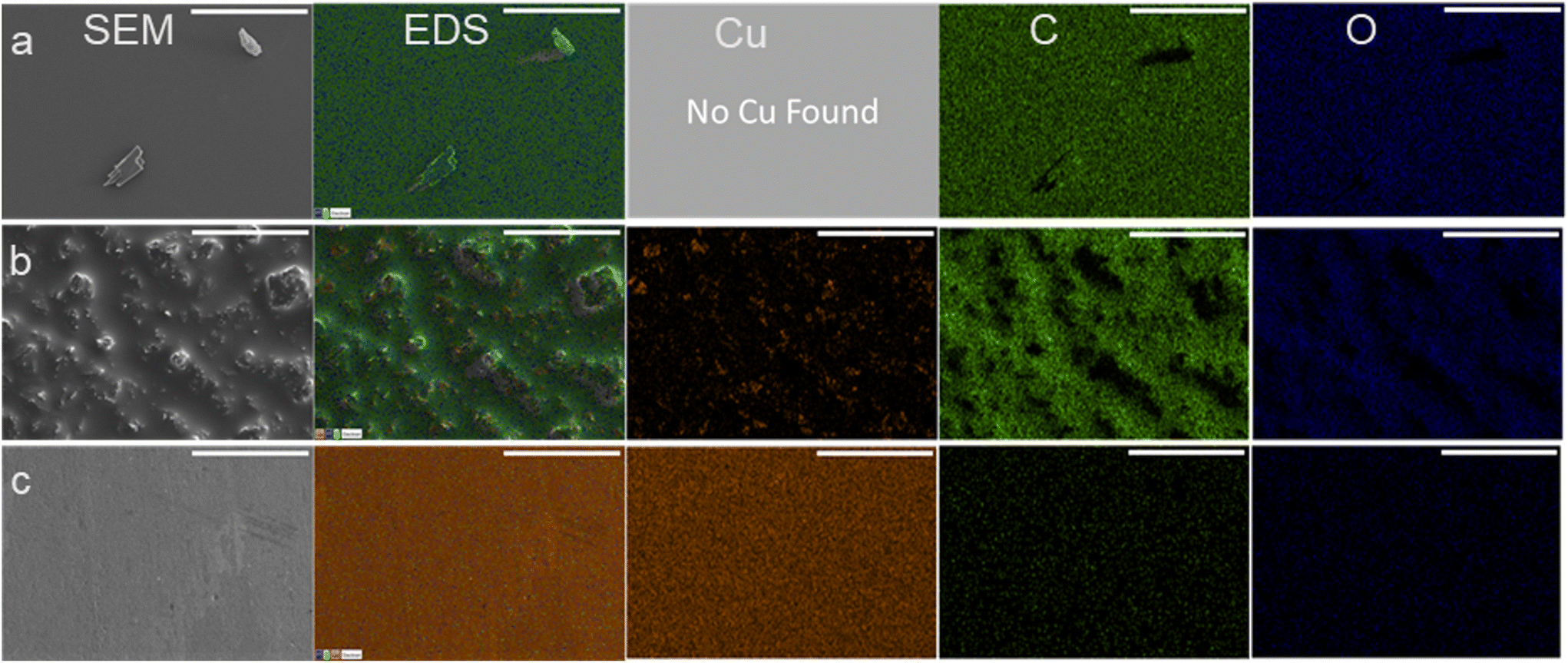 | ||
| Fig. 3 SEM/EDX images at 500× magnification for (a) −560 mV RHE fouled Cu (used three times), (b) −700 mV RHE fouled Cu (used three times), (c) bare Cu. Scale bar shown is 100 μm for all images. Columns from left to right are labeled on top row. | ||
| Sample | At% Cu | At% C | At% O |
|---|---|---|---|
| −560 mV RHE fouled | 0 | 85.7 | 14.3 |
| −700 mV RHE fouled | 4.1 | 78.7 | 17.3 |
| Bare Cu | 87.7 | 12.0 | 0.3 |
While a high coverage of C was on the electrodes after use, determined using EDX, further analysis into what functional groups were found in the carbonaceous fouling was done using FTIR, Raman, and XPS. The electrode fouled at −560 mV RHE through consecutive ECH showed bands on the FTIR spectra suggestive of a polymer material. The spectra of the −700 mV RHE electrode had some similar bands as the −560 mV RHE fouled electrode, however they were not as intense and required magnification to see. The −700 mV RHE electrode did show an increasing absorbance as the wavenumber decreased, which can be seen by carbon blackbody absorbance.26
Due to the polymer like bands seen on the spectra of the −560 mV RHE electrode, a polymerization could have occurred at −560 mV RHE. One possible polymer material that could form in the conditions of our electrochemical experiment is poly(furfuryl alcohol), which is commonly produced through reacting FA in acidic media. To investigate this possibility, commercial pFA was also studied using FTIR, and showed almost complete overlap with the −560 mV RHE fouled electrode. FTIR spectra of the −560 mV RHE and −700 mV RHE fouled Cu electrode surfaces and commercial pFA can be seen in Fig. 4, with pFA bands of interest seen in literature27–29 labelled. The primary bands of pFA are also summarized in Table S3.† While most of the bands overlapped between commercial pFA and the −560 mV RHE fouled Cu electrode, the –CH2 stretch showed a larger shift of nearly 30 wavenumber. Shifts in the –CH2 stretch have also been seen in samples of pFA produced via thermocatalytic process due to the pyrolysis temperature.27 Acetonitrile has been shown to react electrochemically with FF in polymerization reactions on Pt electrodes at highly negative potential (−5.4 V RHE).25 If acetonitrile was polymerizing in our sample, we would expect to see a C triple bond N band at 2250 cm−1 or the a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch between 1690–1640 cm−1 which are both likely to be found in acetonitrile containing polymers.25 In our current study, we found that acetonitrile did not polymerize with FF as no band was seen in the commercial pFA or the −560 mV RHE fouled Cu polymer at 2250 cm−1 or between 1690–1640 cm−1.
N stretch between 1690–1640 cm−1 which are both likely to be found in acetonitrile containing polymers.25 In our current study, we found that acetonitrile did not polymerize with FF as no band was seen in the commercial pFA or the −560 mV RHE fouled Cu polymer at 2250 cm−1 or between 1690–1640 cm−1.
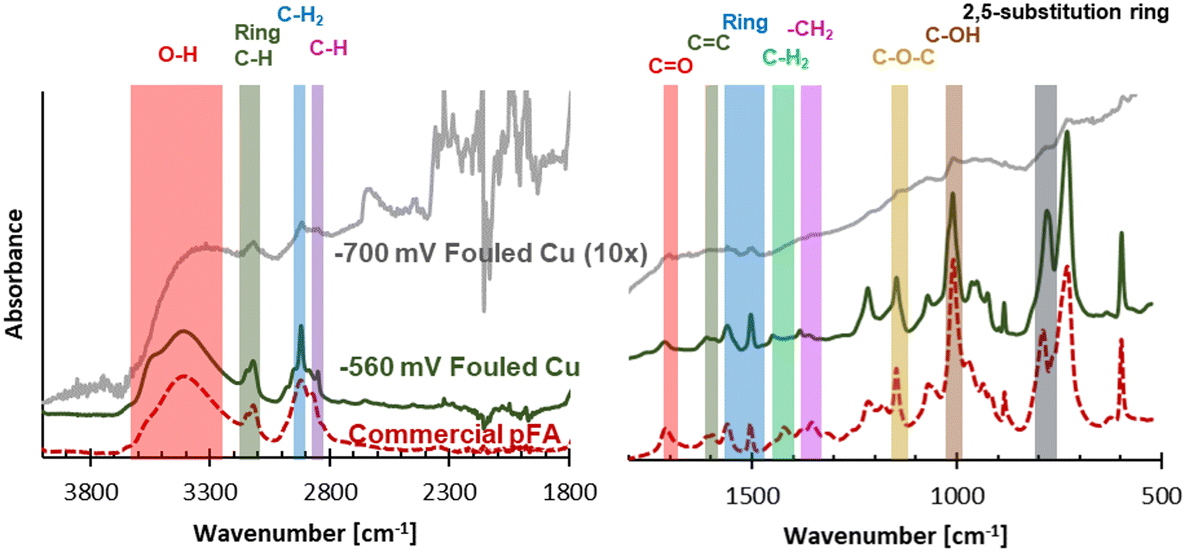 | ||
| Fig. 4 FTIR spectra of −560 mV RHE fouled Cu polymer, commercial pFA, and 10× magnified −700 mV RHE fouled Cu. Shaded areas indicate key pFA bands summarized in Table S3.† | ||
Due to the similar structures of the furanic species investigated in this work (FF, FA, MF), spectra for fouled Cu samples were compared to spectra for FF, FA, and MF. The spectra comparison of Cu samples and furanics, including pFA, can be seen in Fig. S3.† All spectras compared showed similarities due to all being furanic, however the similarities between the polymer formed and pFA are nearly identical while the −560 mV RHE fouled Cu electrode only shared some overlap with other furanics. A comparison was also made between bare Cu and fouled Cu electrodes, which is shown in Fig. S4.† The Bare Cu showed very little absorption from FTIR, while the −700 mV RHE fouled Cu showed a broad absorption growing with decreasing wavenumber without strong features present in the −560 mV RHE fouled Cu. The results of the FTIR analysis showed strong evidence that pFA was formed on the Cu surface during ECH of FF at −560 mV RHE. When the Cu was used for consecutive experiments at a more negative potential (−700 mV RHE), it was found that a polymer was not formed on the surface, however the surface was instead covered in another carbonaceous material. Although FTIR analysis showed small intensity features of the pFA it was within an increasing baseline which suggested a non-polymeric carbon material due to the broad absorption of carbon material.26
Poly(furfuryl alcohol) can be produced through the reaction of FA in acid, and so the conditions of ECH conducted in this work could promote the production of pFA from product FA.28,30 The formation reaction of pFA from FA has been suggested to proceed via two routes,28 shown in Scheme 1.
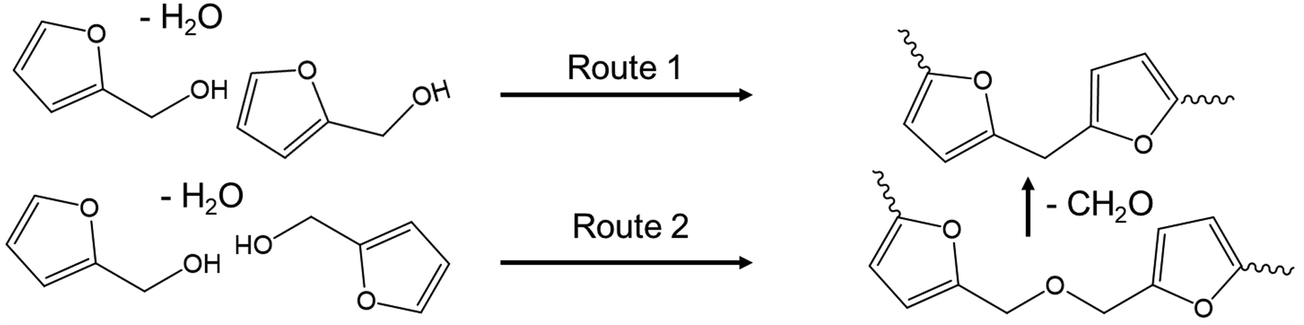 | ||
| Scheme 1 Reaction pathway for pFA formation from FA in acid. | ||
In our system, we saw the pFA was formed during FF ECH to FA and MF, however this could be due to either an electrochemical or non-electrochemical process. To decipher whether the pFA formed on the Cu electrode at −560 mV RHE was due to an electrochemical bias or homogeneously, experiments without electrochemistry were completed. In the non-electrochemical experiments, a Cu electrode was submerged for 3 hours into solution containing 100 mM FF and 30 mM FA to imitate the concentrations of FF and FA found in solution during FF ECH. If pFA was formed during the experiment without electrochemistry, it could be the case that FA produced during ECH would react homogeneously and then collide with the electrode. SEM/EDX images at 5000× magnification were taken (Fig. 5) which showed that while carbon did accumulate onto the Cu foil submerged into electrolyte (no electrochemistry), the Cu was not covered as completely as samples under a potential bias. The submerged Cu sample had 45.3 at% Cu, 52.8 at% C, and 1.9 at% O. FTIR spectra for the Cu submerged in electrolyte did not show overlap with commercial pFA or fouled Cu samples, spectra can be seen in Fig. S3 and S4.† This suggested that the applied potential played a key role in the polymerization reaction leading to pFA formation on the −560 mV RHE fouled Cu.
 | ||
| Fig. 5 SEM/EDX images at 5000× magnification for a Cu foil electrode submerged into catholyte solution containing 100 mM of FF and 30 mM of FA for 3 hours. Scale bar shown is 10 μm. | ||
The FTIR spectra of the submerged Cu (Fig. S3 and S4†) closely resembled the spectra of humins samples seen in other works.31–33 Patil and Lund studied humins formed from 5-hydroxymethylfurfural (HMF) and found that FTIR spectra of humins in the 600–1800 cm−1 region did not have sharp bands such as 5-hydroxymethylfurfural but rather an envelope of several overlapping bands.31 Additionally they found that humins produced though either hydrothermal and acid catalysis routes resulted in similar FTIR bands, however the intensity differed within the 1550–1750 and 1150–1350 cm−1 regions.31 It was shown that while some bands seen in HMF were also found the in humins samples due to the presence of furan groups and hydroxyl groups, there were bands that occurred within the humins sample not seen in the HMF samples.31 Notably, a carbonyl band was not found in the humins sample, and instead multiple bands were found 50 to 90 cm−1 higher and 45 to 65 cm−1 lower than the band at 1665 cm−1 expected for carbonyl of HMF.31 For the Cu submerged sample in our work the FTIR spectra had the same characteristics as the humins produced from HMF, due to the many similarities between HMF and FF. The FTIR spectra of the Cu submerged solution (Fig. S3 and S4†) had an envelope of bands in the 700–1800 cm−1 region as well as bands at higher and lower wavenumbers compared to the carbonyl band at 1671 cm−1.
Furthermore, Tsilomelekis et al., reported that the spectra for humins derived from different molecules, namely 5-hydroxymethylfurfural, glucose, and fructose showed very similar spectra features, with major bands at the same wavenumbers.32 For all three species, the corresponding humins had 2 or 3 bands above and below the carbonyl band, as well as a band at 758 cm−1.32 The same features were found in the Cu submerged sample in our work, suggesting the humins from furfural or furfuryl alcohol have similar structure to those derived from the species mentioned by Tsilomelekis et al.32 and Patil and Lund.31
The previous FTIR results for the −700 mV RHE fouled Cu showed a broad absorption increasing at lower wavenumber, suggestive of amorphous carbon or coking.26 Raman spectroscopy has commonly been used to study different carbon structures through the presence and characteristic of the D and G bands. Hence, Raman spectroscopy was used to further analyze the materials on the fouled Cu electrodes. The Raman spectra of the −700 mV RHE fouled Cu electrode showed evidence of carbonization and the spectra was deconvoluted to show D and G bands, and a broad 2D band (Fig. 6). The D band was centered at 1361 cm−1 and the G band was centered around 1585 cm−1. The ratio of the intensity of the deconvoluted D and G bands, ID/IG, was found to be 0.652. For disordered carbon structures, the full width at half max (FWHM) has been used to investigate the carbon structure.34,35 For the −700 mV RHE fouled Cu, the D band FWHM was found to be 216 cm−1 and the G band FWHM is found to be 108 cm−1. These values are found to match closely with disordered carbon such as soot or activated carbon.34,35 The FWHM of the 2D band centered at 2966 cm−1 has a FWHM of 559, again similar to disordered carbon.34 The electrode fouled at −560 mV RHE showed different spectra depending on the area scanned, labeled as polymerized and carbonized in Fig. 6. The polymerized area was where the polymeric material was located, while the carbonized area were regions where polymer was not visible by eye. The polymerized area did not show evidence of amorphous carbon as no D and G band were seen. The carbonized area of the electrode showed similar D and G bands to the −700 mV RHE fouled sample, suggesting that the soot formation could also occur at −560 mV RHE.
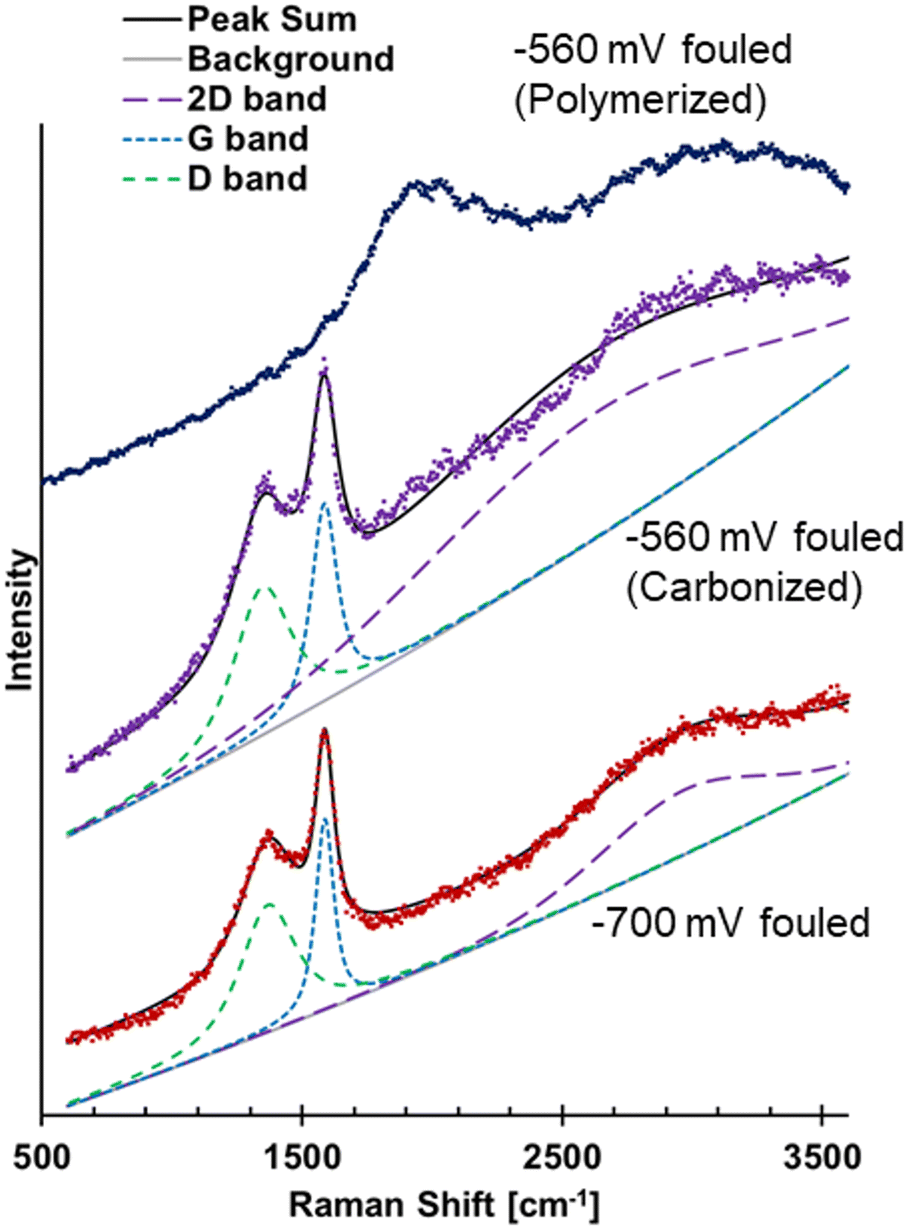 | ||
| Fig. 6 Raman spectra of −700 mV RHE fouled Cu showing D, G, and 2D bands, spectra of −560 mV RHE fouled Cu showing spectra from a carbonized area of electrode and a polymerized area of the electrode. | ||
The form of carbon and oxygen at the surface of the fouled electrodes was probed using XPS analysis and compared to commercial pFA. C 1s and O 1s spectra for the fouled samples and pFA on Cu are shown in Fig. 7. Due to the fouling, the surfaces of the samples were non-metallic. For non-metallic samples, mixed Gaussian and Lorentzian line shapes that are more Gaussian typically provide strong fits to experimental data.36 A 10% Lorentzian line shape was selected for deconvolution and peak fitting of the fouled and pFA on Cu XPS samples. The FWHM of samples were selected to remain within roughly 10% of each other between peaks of the deconvolutions. Spectra were adjusted to aliphatic carbon at 284.8 eV. The C 1s spectra were deconvoluted into 4 peaks at 284.8 eV (C–C), 286.3 eV (C–O, C–O–C),37–40 287.7 eV (CO),37–40 and 288.8 eV (OC–O)38,40 as shown in Fig. 7a. Similar C 1s and O 1s envelopes were observed for pFA on Cu and −560 mV RHE fouled Cu samples. The −700 mV RHE fouled Cu sample had a different shape, where the peak at 284.8 eV (assigned to C–C) was much larger in comparison to the peak at 286.3 eV assigned to C–O. The large peak for the C–O group on −560 mV RHE fouled Cu and pFA samples was likely due to the presence of C–O–C group in the furan ring and the C–OH group of the pFA molecule. While C–O was present in the −700 mV RHE, it was overshadowed by the large C–C band which was likely more intense due to the presence of amorphous carbon. CO and O–CO were seen in all samples, which could be due to residues of unreacted FF or presence of oxygen during transfer and drying of sample leading to oxidation. O 1s spectra were deconvoluted into 3 peaks at 532.2 eV (CO),37,40,41 533.2 eV (C–O–C),37,40,41 and 534.2 eV (O–CO)40,41 shown in Fig. 7b. The pFA on Cu and −560 mV RHE fouled Cu had similar ratios of the three peaks, while the −700 mV RHE sample had a higher proportion of the CO group. Overall, the −700 mV RHE fouled Cu had a higher proportion of aliphatic C compared to the polymer containing sample, with a higher preference for O to be double bonded with C compared to polymer containing samples. Cu signal was not observed on fouled electrodes or pFA on Cu foil through XPS, at% are included in Table S4.†
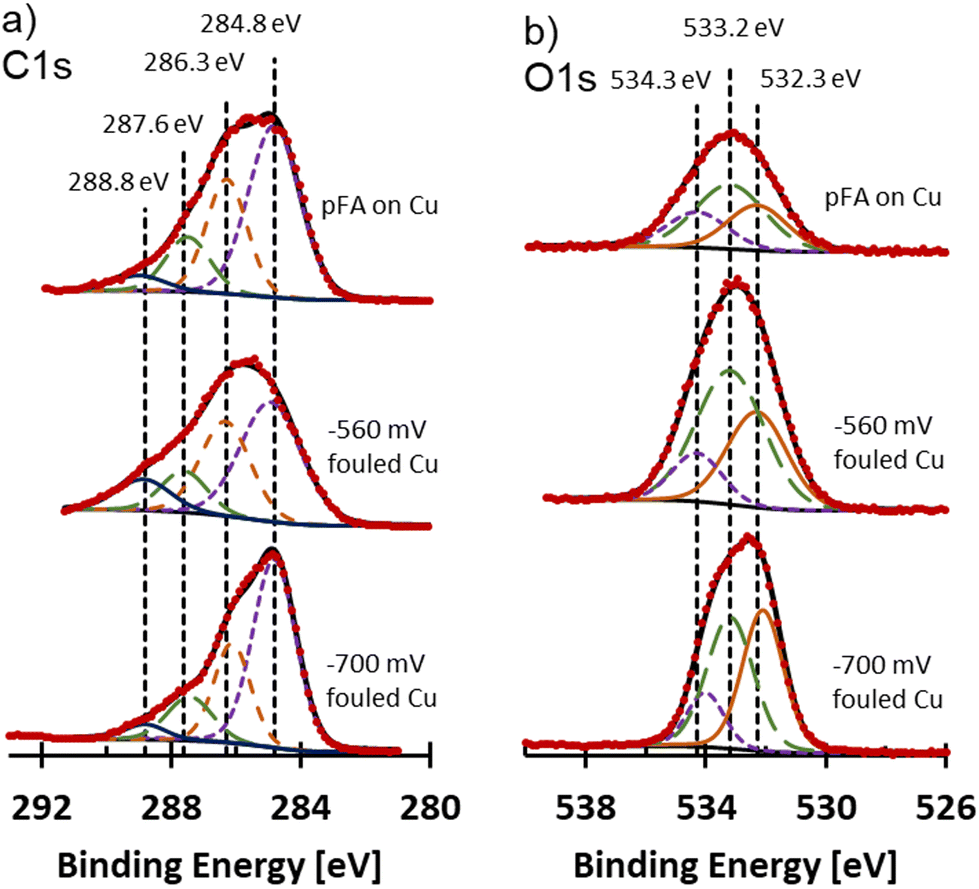 | ||
| Fig. 7 (a) C 1s and (b) O 1s XPS spectra for pFA on Cu, −560 mV RHE fouled Cu, and −700 mV RHE fouled Cu. Vertical dotted lines mark the assignments for C 1s and O 1s spectra for deconvolution. | ||
FA ECH was performed to investigate whether the applied potential would promote the pFA polymerization or coking reactions through adsorption of FA molecules produced during FF ECH. In this experiment, 15 mM of FA was added to the electrolyte in lieu of 200 mM FF to mimic the concentration of FA found in the ECH reactions. While MF can be found in the catholyte, it is at low concentrations near 1 mM when gas sparging is present due to the facile evaporation.19 For this reason, MF was not added to the solution. FA ECH was done over three experiments of 3 hours at −560 mV, however at −700 mV the system would overload after the first experiment and was hence stopped after the first 3-hour experiment. In both cases, no polymer, coke, or other furanic material was found on the surface. During the ECH of FA experiment at −560 mV RHE, the conversion of FA reached an average of (40.9 ± 2.59)%. MF was observed at an amount near the lower detection limit of GCMS (<0.57% selectivity) (Fig. 8). It has also been shown previously that the electrochemical route of FA to MF largely does not occur.16,20 While FA ECH did not produce MF, the concentration of FA decreased during the experiment. FA is known to undergo side reactions homogeneously due to the acidic conditions, and using the homogeneous kinetics from our previous work,19 the expected conversion in FA was found to be 39.4%. The unreacted FA and the estimated consumption due to side reactions accounted for 98.7% of the initial FA, showing that it was unlikely that mass was lost due to electrochemical reactions of FA. The composition of the electrolyte before and after FA ECH, along with the conversion and mass balance can be seen in Fig. 8. Reaction data can be found in Table S5.† The lack of FA ECH reactivity has also previously been reported by our group and others.16,20 FTIR spectra for the FA ECH Cu foils (Fig. S5†) did not show any bands, but showed a change in absorbance towards the lower wavenumber. The Raman spectra of the electrodes did not show evidence of any coke at either potential (Fig. S6†). The Raman spectra of a cleaned Cu foil is included in Fig. S6,† and has a band near 1500 cm−1 corresponding to the Cu. No bands for oxides of Cu were observed in the spectra from the electrodes used during FA ECH, nor was any D, G, or 2D bands.
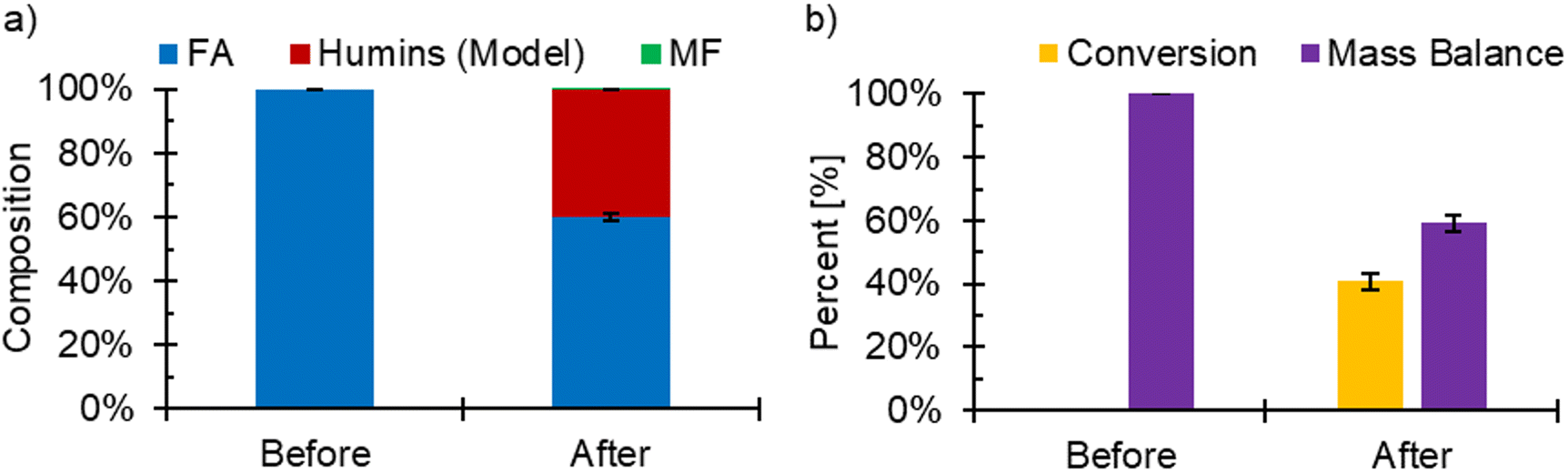 | ||
| Fig. 8 (a) The composition of the FA ECH electrolyte before and after 3 h ECH. (b) The conversion and mass balance from FA ECH experiments. Conditions: −560 mV RHE, 80:20 vol% acetonitrile:water, 0.5 M H2SO4, 15 mM FA, N2 sparging 60 mL min−1, Cu electrode. | ||
The non-electrochemical submersion experiments and FA ECH experiments did not show a pFA material. Since neither experiment resulted in pFA or coke formation, we can conclude that the FF ECH electrodes were fouled through an electrochemical reaction with FF as the reactant, as opposed to a polymerization in solution followed by collision onto the electrode. While the homogeneous side reactions did occur in the FA ECH experiments, the electrochemical HER reaction also occurred. The HER reaction likely prevented the accumulation of humins on the electrode, as humins were found when no electrochemical bias was present.
Conclusions
Experimental conditions (200 mM FF, 0.5 M H2SO4, 3 h duration, recycled 3 times) were selected to intentionally promote fouling of Cu electrodes during FF ECH to better understand the fouling. The development of carbonaceous layers occurred during FF ECH and was shown to depend on the applied potential, resulting in a polymeric layer at moderately negative potentials (−560 mV RHE) and an amorphous carbon layer at more negative potentials (−700 mV RHE). While the fouling showed severe impacts onto the reaction conversion, it was not the main cause of mass balance losses as it accounted for 1.27% and 1.98% of the missing mass from −560 mV RHE and −700 mV RHE electrolysis, respectively. The polymer material formed was found to be pFA, which is a desirable product to industry and warrants investigation as a primary product of FF electroreduction in the future. The amorphous carbon found at more negative potentials was similar to soot. We have shown that the applied potential plays a key role in the production of pFA on the electrode, as experiments with no applied potential did not lead to production of pFA. Furthermore, the production of pFA was found to be due to an electrochemical reaction consuming FF and not FA, as only when starting with FF was pFA (or coke) formed on the Cu electrode. To promote the production of FA and MF from FF ECH, it is undesired to produce the pFA polymer or unorganized carbon layers as both were found to lower the conversion towards ECH of FF to FA and MF substantially. The unorganized carbon layer produced at −700 mV RHE caused more intense deactivation of FF ECH to FA and MF, and care should be taken to avoid the formation of the amorphous carbon fouling. Avoiding the amorphous carbon layer can be done by monitoring the applied potential, and not going to very negative applied potentials beyond −560 mV RHE. Fouling can also be decreased by reducing the concentration of FF below 200 mM, as it is not observed when starting at lower concentrations.19,20 Techniques to limit these fouling phenomena are required so that the production rates towards FA and MF can remain high. Catalysts can be designed to inhibit fouling and have longer lifespans for use in reactors for FF ECH to FA and MF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the financial support of the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science program, under award DE-SC0019134. The authors acknowledge the use of the Surface Science and Imaging facilities at CUNY Advanced Science Research Center.References
- J. L. Barton, Electrification of the chemical industry, Science, 2020, 368(6496), 1181–1182 CrossRef CAS PubMed.
- Z. J. Schiffer and K. Manthiram, Electrification and decarbonization of the chemical industry, Joule, 2017, 1(1), 10–14 CrossRef.
- L. Du, Y. Shao, J. Sun, G. Yin, C. Du and Y. Wang, Electrocatalytic valorisation of biomass derived chemicals, Catal. Sci. Technol., 2018, 8(13), 3216–3232 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364(6438), eaav3506 CrossRef CAS PubMed.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, Electrochemical routes for the valorization of biomass-derived feedstocks: From chemistry to application, ACS Energy Lett., 2021, 6(4), 1205–1270 CrossRef CAS.
- E. J. Biddinger and M. A. Modestino, Electro-organic syntheses for green chemical manufacturing, Electrochem. Soc. Interface, 2020, 29(3), 43–47 CrossRef CAS.
- R. Kajaste, Chemicals from biomass – managing greenhouse gas emissions in biorefinery production chains – a review, J. Cleaner Prod., 2014, 75, 1–10 CrossRef CAS.
- P. L. Eranki, B. D. Bals and B. E. Dale, Advanced regional biomass processing depots: a key to the logistical challenges of the cellulosic biofuel industry, Biofuels, Bioprod. Biorefin., 2011, 5(6), 621–630 CrossRef CAS.
- C. H. Lam, S. Das, N. C. Erickson, C. D. Hyzer, M. Garedew, J. E. Anderson, T. J. Wallington, M. A. Tamor, J. E. Jackson and C. M. Saffron, Towards sustainable hydrocarbon fuels with biomass fast pyrolysis oil and electrocatalytic upgrading, Sustainable Energy Fuels, 2017, 1(2), 258–266 RSC.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: I. Results of screening for potential candidates from sugars and synthesis gas, United States, 2004 Search PubMed.
- Furfural market by raw material (sugarcane bagasse, corncob, rice husk and others), application (derivatives (furfural alcohol and other derivatives), solvent) and region (Asia-Pacific, Americas, Europe, Middle East and Africa) – Global forecast to 2024. Dublin, Ireland; 2019. Contract no.: CH 7382.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba and M. López Granados, Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels, Energy Environ. Sci., 2016, 9(4), 1144–1189 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, How catalysts and experimental conditions determine the selective hydroconversion of furfural and 5-hydroxymethylfurfural, Chem. Rev., 2018, 118(22), 11023–11117 CrossRef CAS PubMed.
- P. Nilges and U. Schröder, Electrochemistry for biofuel generation: production of furans by electrocatalytic hydrogenation of furfural, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- S. Jung and E. J. Biddinger, Electrocatalytic hydrogenation and hydrogenolysis of furfural and the impact of homogeneous side reactions of furanic compounds in acidic electrolytes, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, Mechanisms of furfural reduction on metal electrodes: Distinguishing pathways for selective hydrogenation of bioderived oxygenates, J. Am. Chem. Soc., 2017, 139(40), 14120–14128 CrossRef CAS PubMed.
- A. S. May and E. J. Biddinger, Strategies to control electrochemical hydrogenation and hydrogenolysis of furfural and minimize undesired side reactions, ACS Catal., 2020, 10(5), 3212–3221 CrossRef CAS.
- A. S. May, S. M. Watt and E. J. Biddinger, Kinetics of furfural electrochemical hydrogenation and hydrogenolysis in acidic media on copper, React. Chem. Eng., 2021, 6(11), 2075–2086 RSC.
- A. S. May and E. J. Biddinger, Modeling competing kinetics between electrochemical reduction of furfural on copper and homogeneous side reactions in acid, Energy Fuels, 2022, 36(18), 11001–11011 CrossRef CAS.
- S. Jung and E. J. Biddinger, Controlling competitive side reactions in the electrochemical upgrading of furfural to biofuel, Energy Technol., 2018, 6, 1370–1379 CrossRef CAS.
- S. Jung, A. N. Karaiskakis and E. J. Biddinger, Enhanced activity for electrochemical hydrogenation and hydrogenolysis of furfural to biofuel using electrodeposited Cu catalysts, Catal. Today, 2019, 323, 26–34 CrossRef CAS.
- W. Deng, X. Wang, C. H. Lam, Z. Xiong, H. Han, J. Xu, L. Jiang, S. Su, S. Hu, Y. Wang and J. Xiang, Evolution of coke structures during electrochemical upgrading of bio-oil, Fuel Process. Technol., 2022, 225, 107036 CrossRef CAS.
- W. Deng, S. S. A. Syed-Hassan, C. H. Lam, X. Hu, X. Wang, Z. Xiong, H. Han, J. Xu, L. Jiang, S. Su, S. Hu, Y. Wang and J. Xiang, Polymerization during low-temperature electrochemical upgrading of bio-oil: Multi-technique characterization of bio-oil evolution, Energy Convers. Manage., 2022, 253, 115165 CrossRef CAS.
- J. L. J. Hallal, A. M. S. Lucho and R. S. Gonçalves, Electrochemical polymerization of furfural on a platinum electrode in aqueous solutions of potassium biphthalate, Mater. Res., 2005, 8, 23–29 CrossRef CAS.
- A. M. S. Lucho and R. S. Gonçalves, Polyfurfural film synthesized by electrochemical reduction of furfural on bright platinum electrode, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42(6), 791–799 CrossRef.
- M. Bradley, Carbon black analysis comparison with diamond ATR and germanium crystals, Thermo Electron Corporation, Madison, WI, USA, 2005 Search PubMed.
- C. L. Burket, R. Rajagopalan, A. P. Marencic, K. Dronvajjala and H. C. Foley, Genesis of porosity in polyfurfuryl alcohol derived nanoporous carbon, Carbon, 2006, 44(14), 2957–2963 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Acid-catalyzed polycondensation of furfuryl alcohol: mechanisms of chromophore formation and cross-linking, Macromolecules, 1996, 29(11), 3839–3850 CrossRef CAS.
- G. Nanni, J. A. Heredia-Guerrero, U. C. Paul, S. Dante, G. Caputo, C. Canale, A. Athanassiou, D. Fragouli and I. S. Bayer, Poly(furfuryl alcohol)-polycaprolactone blends, Polymers, 2019, 11(6), 1069 CrossRef PubMed.
- A. P. Dunlop and F. N. Peters, The nature of furfuryl alcohol, Ind. Eng. Chem., 1942, 34(7), 814–817 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Formation and growth of humins via aldol addition and condensation during acid-catalyzed conversion of 5-hydroxymethylfurfural, Energy Fuels, 2011, 25(10), 4745–4755 CrossRef CAS.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Molecular structure, morphology and growth mechanisms and rates of 5-hydroxymethyl furfural (HMF) derived humins, Green Chem., 2016, 18(7), 1983–1993 RSC.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, Formation, molecular structure, and morphology of humins in biomass conversion: influence of feedstock and processing conditions, ChemSusChem, 2013, 6(9), 1745–1758 CrossRef CAS PubMed.
- R. Escribano, J. J. Sloan, N. Siddique, N. Sze and T. Dudev, Raman spectroscopy of carbon-containing particles, Vib. Spectrosc., 2001, 26(2), 179–186 CrossRef CAS.
- A. Merlen, J. G. Buijnsters and C. Pardanaud, A guide to and review of the use of multiwavelength raman spectroscopy for characterizing defective aromatic carbon solids: from graphene to amorphous carbons, Coatings, 2017, 7(10), 153 CrossRef.
- G. H. Major, N. Fairley, P. M. A. Sherwood, M. R. Linford, J. Terry, V. Fernandez and K. Artyushkova, Practical guide for curve fitting in X-ray photoelectron spectroscopy, J. Vac. Sci. Technol., A, 2020, 38(6), 061203 CrossRef CAS.
- R. Janus, A. Wach, P. Kuśtrowski, B. Dudek, M. Drozdek, A. M. Silvestre-Albero, F. Rodríguez-Reinoso and P. Cool, Investigation on the low-temperature transformations of poly(furfuryl alcohol) deposited on MCM-41, Langmuir, 2013, 29(9), 3045–3053 CrossRef CAS PubMed.
- X. H. Men, Z. Z. Zhang, H. J. Song, K. Wang and W. Jiang, Functionalization of carbon nanotubes to improve the tribological properties of poly(furfuryl alcohol) composite coatings, Compos. Sci. Technol., 2008, 68(3), 1042–1049 CrossRef CAS.
- P. Janus, R. Janus, P. Kuśtrowski, S. Jarczewski, A. Wach, A. M. Silvestre-Albero and F. Rodríguez-Reinoso, Chemically activated poly(furfuryl alcohol)-derived CMK-3 carbon catalysts for the oxidative dehydrogenation of ethylbenzene, Catal. Today, 2014, 235, 201–209 CrossRef CAS.
- E. R. Edwards, S. S. Oishi and E. C. Botelho, Analysis of chemical polymerization between functionalized MWCNT and poly(furfuryl alcohol) composite, Polímeros, 2018, 28, 15–22 CrossRef.
- M. P. Woods, E. J. Biddinger, P. H. Matter, B. Mirkelamoglu and U. S. Ozkan, Correlation between oxygen reduction reaction and oxidative dehydrogenation activities over nanostructured carbon catalysts, Catal. Lett., 2010, 136, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated data, assignment of primary pFA bands, faradaic efficiency, representative EIS plots, FTIR spectra for FF ECH fouled electrodes and furanics, FTIR and Raman spectra after FA ECH. See DOI: https://doi.org/10.1039/d3gc02222f |
| This journal is © The Royal Society of Chemistry 2023 |