
RuS2@CN-x with exposed (200) facet as a high-performance photocatalyst for selective C–C bond cleavage of biomass coupling with H–O bond cleavage of water to co-produce chemicals and H2†
Xinze
Li
a,
Jiliang
Ma
 *ab,
Hongquan
Fu
c,
Zhendong
Liu
a,
Junqiang
Zhang
a,
Rui
Cui
a,
Yanzhu
Guo
a,
Shuangquan
Yao
b and
Runcang
Sun
*a
*ab,
Hongquan
Fu
c,
Zhendong
Liu
a,
Junqiang
Zhang
a,
Rui
Cui
a,
Yanzhu
Guo
a,
Shuangquan
Yao
b and
Runcang
Sun
*a
aLiaoning Key Lab of Lignocellulose Chemistry and BioMaterials, Liaoning Collaborative Innovation Center for Lignocellulosic Biorefinery, College of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian 116034, China. E-mail: jlma@dlpu.edu.cn; rcsun3@dlpu.edu.cn; Tel: +86-0411-86323652; Fax: +86-0411-86323652
bGuangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control. College of Light Industrial and Food Engineering, Guangxi University, Nanning 530004, China
cCollege of Chemistry and Chemical Engineering, Chemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, China West Normal University, Nanchong, Sichuan 637000, China
First published on 23rd March 2023
Abstract
The selective photocatalytic C–C bond cleavage of biomass coupling with H–O bond cleavage of water for co-producing chemicals and H2 is of great significance, but limited successes have been reported. In this study, a RuS2@CN-x photocatalyst with only exposed crystal face (200) was prepared by anchoring RuS2 nanoparticles on carbon nitride (CN) nanosheets and successfully used in photocatalytic C–C bond cleavage of biomass-based sugars coupling with water splitting to co-generate lactic acid and hydrogen. Interestingly, the RuS2 (200) crystal face combined with the interfacial synergism between CN and RuS2 in RuS2@CN-x effectively improved visible light absorption and enhanced the transfer/separation efficiency of photo-generated carriers. The RuS2@CN-2 exhibited superior photocatalytic activity towards synchronous water splitting and biomass conversion to co-produce hydrogen (8698.3 μmol g−1 h−1) and lactic acid (91.5%). Furthermore, the experimental and DFT calculation results indicated that the exposed crystal face (200) of RuS2 significantly enhanced the photocatalytic activity of RuS2@CN-2. Moreover, this reaction system not only has excellent recyclability but also displays excellent universality for small molecule biomass-derived monosaccharides and macromolecular xylan. This work provides a novel route to efficiently co-produce hydrogen and fine chemicals from photocatalytic C–C bond cleavage of biomass coupling with H–O bond cleavage of water.
Introduction
Following the rapid growth of global industry and economy, energy shortage and environmental pollution have become two major problems facing the world today. Therefore, countries worldwide have begun to look for environment-friendly and sustainable energy. Hydrogen is a readily available and clean new energy.1–5 As hydrogen is one of the best clean energies to replace fossil fuels, where many methods are used to produce hydrogen, such as hydrogen production from fossil fuels,6,7 electrolytic water8,9 and photocatalytic water splitting, it has a high energy density (143 MJ kg−1) and no carbon emissions during combustion.10,11 Hydrogen production from fossil fuels is an inexpensive and mature technology. Nevertheless, the excessive use of fossil fuels leads to a series of irreversible problems, such as energy shortage and the greenhouse effect. On the other hand, hydrogen production from electrolytic water is highly efficient and less polluting. Meanwhile, the high price and limited precious metal resources increase the cost of this technology and hinder its large-scale application and promotion. Therefore, many scholars have gradually turned their attention to the photocatalytic hydrogen evolution technology,12–15 which is a low-energy consumption and environmentally friendly method. In addition, the technology is easier to build and expand. Recently, some sacrificial agents (e.g., Na2SO3, methanol, triethanolamine) were always added to the photocatalytic system to improve the efficiency of photocatalytic hydrogen evolution.16–19 However, the utilization of the sacrificial agents has not been studied in detail. Yang et al.20 prepared a MoS2/O-ZnIn2S4 photocatalyst for hydrogen evolution using Na2SO3 and Na2S as sacrificial agents, but the end products of sacrificial agents have not been further studied.Biomass is a renewable organic resource with wide distribution and easy access on the earth, which can be used to prepare a series of fine chemicals via selective oxidation.21–25 Lactic acid is one of the three largest organic acids in the world, which can be obtained from the selective oxidation of biomass. Currently, lactic acid has been widely used in the food, chemical industry, medicine, and other fields due to its unique advantages,26 resulting in many methods for synthesizing it. Among them for converting biomass into lactic acid, photocatalytic technology has the benefits of non-pollution and low cost. However, contrary to splitting water into hydrogen, biomass conversion only utilizes the oxidation half-reaction, which restricts the economic benefits. Therefore, it is necessary to couple photocatalytic water splitting and biomass conversion to co-produce hydrogen and lactic acid by using redox reactions in one system to improve economic benefits. Furthermore, to accelerate the redox reaction, Pt is often added to capture electrons (e−) in the conduction band to effectively separate e− and holes (h+).27–29 For example, Liu et al.30 developed a PACN photocatalyst using a phytic acid hydrothermal method, and Yang et al.31 prepared a K/O@CN-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N photocatalyst via a simple strategy. These two photocatalysts realized the coupling of photocatalytic water splitting and biomass conversion to co-produce hydrogen and lactic acid. Unfortunately, an adequate amount of chloroplatinic acid was added to the system, dramatically increasing the application cost and polluting the environment. Consequently, developing an economical and environment-friendly photocatalyst is necessary to efficiently co-produce lactic acid and hydrogen without a co-catalyst.
N photocatalyst via a simple strategy. These two photocatalysts realized the coupling of photocatalytic water splitting and biomass conversion to co-produce hydrogen and lactic acid. Unfortunately, an adequate amount of chloroplatinic acid was added to the system, dramatically increasing the application cost and polluting the environment. Consequently, developing an economical and environment-friendly photocatalyst is necessary to efficiently co-produce lactic acid and hydrogen without a co-catalyst.
Carbon nitride (CN) has gained extensive attention due to its simple preparation process and low cost. Nevertheless, CN suffers from the drawbacks of a high electron–hole recombination rate and low visible light utilization, limiting its photocatalytic activity.32–35 Recently, several studies have reported that CN combined with metal sulfides can effectively enhance the transfer/separation efficiency of photo-generated carriers, thereby indirectly improving its photocatalytic activity. For example, combining CN microtubules with ZnIn2S4 nanosheets36 and decorating CN nanosheets with Bi2S3 quantum dots.37 In addition to the above metal sulfides, RuS2 is a semiconductor with a narrow band gap, high conductivity, and hydrogen adsorption free energy similar to Pt.38–41 Furthermore, RuS2 has been reported to form photocatalysts with TiO2, SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 and other materials.43 For instance, Maheu et al. compounded RuS2 and TiO2 to obtain RuS2/TiO2 and its photocatalytic activity gradually enhanced with increasing RuS2 content.44 The obtained photocatalyst combining RuS2 and CN has broad application prospects in realizing water splitting and biorefinery.
42 and other materials.43 For instance, Maheu et al. compounded RuS2 and TiO2 to obtain RuS2/TiO2 and its photocatalytic activity gradually enhanced with increasing RuS2 content.44 The obtained photocatalyst combining RuS2 and CN has broad application prospects in realizing water splitting and biorefinery.
Herein, a hydrothermal-calcination strategy was adopted to anchor RuS2 nanoparticles onto CN to obtain RuS2@CN. The strong interface interaction between CN and RuS2 significantly enhanced the transfer and separation efficiency of photo-generated carriers, improving photocatalytic activity. Furthermore, RuS2@CN showed excellent performance in the co-production of hydrogen and lactic acid under a synergetic effect of the RuS2 co-catalyst. As a result, the optimal RuS2@CN-2 sample exhibited the best photocatalytic performance. The influences of ˙OH and h+ on the co-production of hydrogen and lactic acid were also investigated. In addition, under optimal conditions, the photocatalytic hydrogen release reached 8698.3 μmol g−1 h−1, and the yield of lactic acid reached 91.5%.
Results and discussion
As shown in Fig. 1A, RuS2@CN-x was successfully prepared using a novel hydrothermal-calcination method to anchor RuS2 nanoparticles onto CN nanosheets. The scanning electron microscopy (SEM) images of CN and RuS2 exhibit the structure of 2D nanosheets and 3D nanoparticles (Fig. S1†), respectively. The SEM image of RuS2@CN-2 shows that many nanoparticles are loaded on the surface of CN nanosheets (Fig. 1B). From Fig. 1C, D, and S2,† the transmission electron microscopy (TEM) images of RuS2@CN-2 reveal that the lattice spacing of 0.28 nm corresponds to the (200) crystal plane of RuS2.45,46 Furthermore, the elemental mapping images indicate that C, N, Ru, and S are uniformly spread over the surface of RuS2@CN-2 (Fig. 1E–H). The N2 adsorption–desorption isotherms of CN and RuS2@CN-2 exhibit the type IV isotherm patterns, indicating the microporosity of CN and RuS2@CN-2 (Fig. 1I). Meanwhile, the BET surface area of RuS2@CN-2 (15.45 m2 g−1) is slightly higher than that of CN (13.07 m2 g−1). The result indicates that RuS2@CN-2 can provide more photocatalytic active sites in the reaction system. As shown in Fig. 1J, the pore width of CN and RuS2@CN-2 are mainly centred at 5 nm.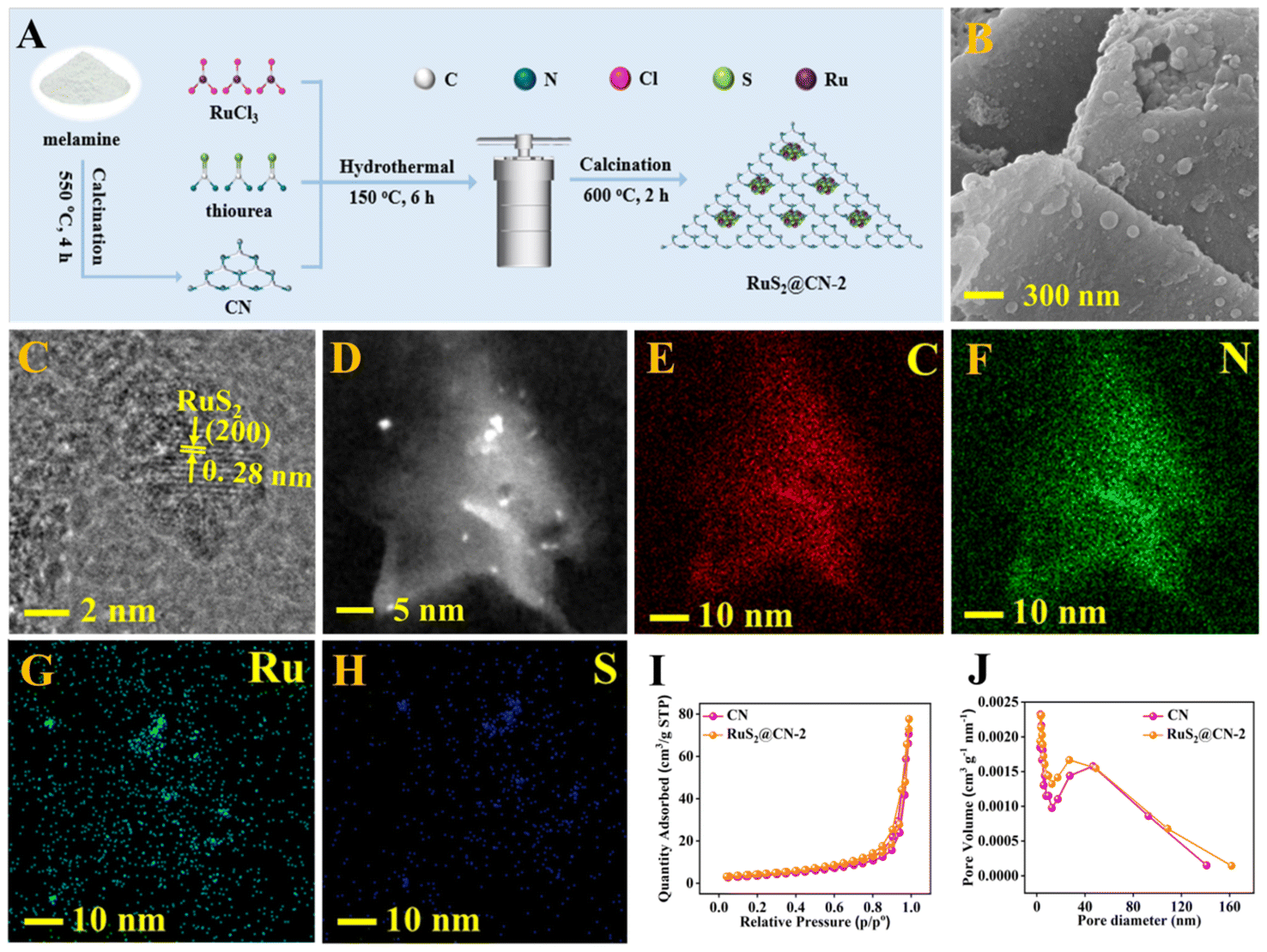 | ||
| Fig. 1 Preparation scheme of RuS2@CN-x (A). SEM image (B) and TEM images (C and D) of RuS2@CN-2. Elemental mapping images of RuS2@CN-2: C (E), N (F), Ru (G), S (H). N2 adsorption–desorption isotherms of CN and RuS2@CN-2 (I). Pore width distribution of CN and RuS2@CN-2 (J). | ||
X-ray diffraction (XRD) analysis was employed to explore crystal phase components of CN, RuS2@CN-2, and RuS2. It can be seen that the XRD pattern of RuS2@CN-2 is similar to that of CN (Fig. 2A). CN peaks show two positions, 12.7° and 27.6°, corresponding to the lattice planes of (100) and (002), respectively. As compared with CN, the diffraction peak (100) in the XRD pattern of RuS2@CN-2 disappeared, and the diffraction peak (002) was relatively weaker, where it was seen or suggestive of the in-plane structure of CN being changed or destroyed. Moreover, a new diffraction peak appeared at 31.8°, corresponding to the (200) lattice plane of RuS2. In addition, the XRD pattern of pure RuS2 exhibited diffraction peaks centred at 31.8°, 27.5°, 45.7°, and 54.2° corresponding to (111), (200), (220), and (311) lattice planes of standard RuS2 (JCPDS No. 73-1677), respectively.47 The above results indicate that RuS2 nanoparticles were successfully anchored on CN, which agrees with the TEM results. Furthermore, the optical absorption features of RuS2@CN-0.5, RuS2@CN-1, RuS2@CN-2, and RuS2@CN-3 from the Fourier transform infrared spectra did not change as compared with CN (Fig. 2B). The characteristic peaks at 805, 1050–1700, and 3050–3600 cm−1 can be attributed to the tri-s-triazine units, heterocycles, and vibration absorptions of NH/OH groups in CN, respectively.48 The results show that the CN framework was undamaged after anchoring RuS2 nanoparticles on CN. To study the chemical composition of CN and RuS2@CN-2, X-ray photoelectron spectroscopy (XPS) measurements were conducted. Fig. S3† illustrates that the survey spectrum verifies the presence of C, N, S, O, and Ru in RuS2@CN-2. Furthermore, the N 1s spectra of CN and RuS2@CN-2 are displayed in Fig. 2C, where three peaks appeared at 401.0 eV, 399.9 eV, and 398.3 eV representing the C–N–H, (C)3–N, and C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, respectively. For the C 1s spectra of CN and RuS2@CN-2 (Fig. 2D), they were associated with three carbon signal peaks, corresponding to N–CN (288.2 eV), C–N (286.3 eV), and C–C (284.7 eV) band, respectively. Fig. 2E shows the Ru 3p spectra of RuS2@CN-2. The distinct peaks centred at 461.5 and 484.2 eV are assigned to Ru 3p3/2 and Ru 3p1/2 states of Ru–S bonds.49 Meanwhile, the S 2p spectra are presented in Fig. 2F, and two peaks can be observed at approximately 163.3 and 162.1 eV, which are ascribed to the S 2p1/2 and S 2p3/2, respectively. They are also related to Ru–S bonds.50,51 The discovery of Ru–S bonds further proves the successful preparation of RuS2@CN-2.41
C, respectively. For the C 1s spectra of CN and RuS2@CN-2 (Fig. 2D), they were associated with three carbon signal peaks, corresponding to N–CN (288.2 eV), C–N (286.3 eV), and C–C (284.7 eV) band, respectively. Fig. 2E shows the Ru 3p spectra of RuS2@CN-2. The distinct peaks centred at 461.5 and 484.2 eV are assigned to Ru 3p3/2 and Ru 3p1/2 states of Ru–S bonds.49 Meanwhile, the S 2p spectra are presented in Fig. 2F, and two peaks can be observed at approximately 163.3 and 162.1 eV, which are ascribed to the S 2p1/2 and S 2p3/2, respectively. They are also related to Ru–S bonds.50,51 The discovery of Ru–S bonds further proves the successful preparation of RuS2@CN-2.41
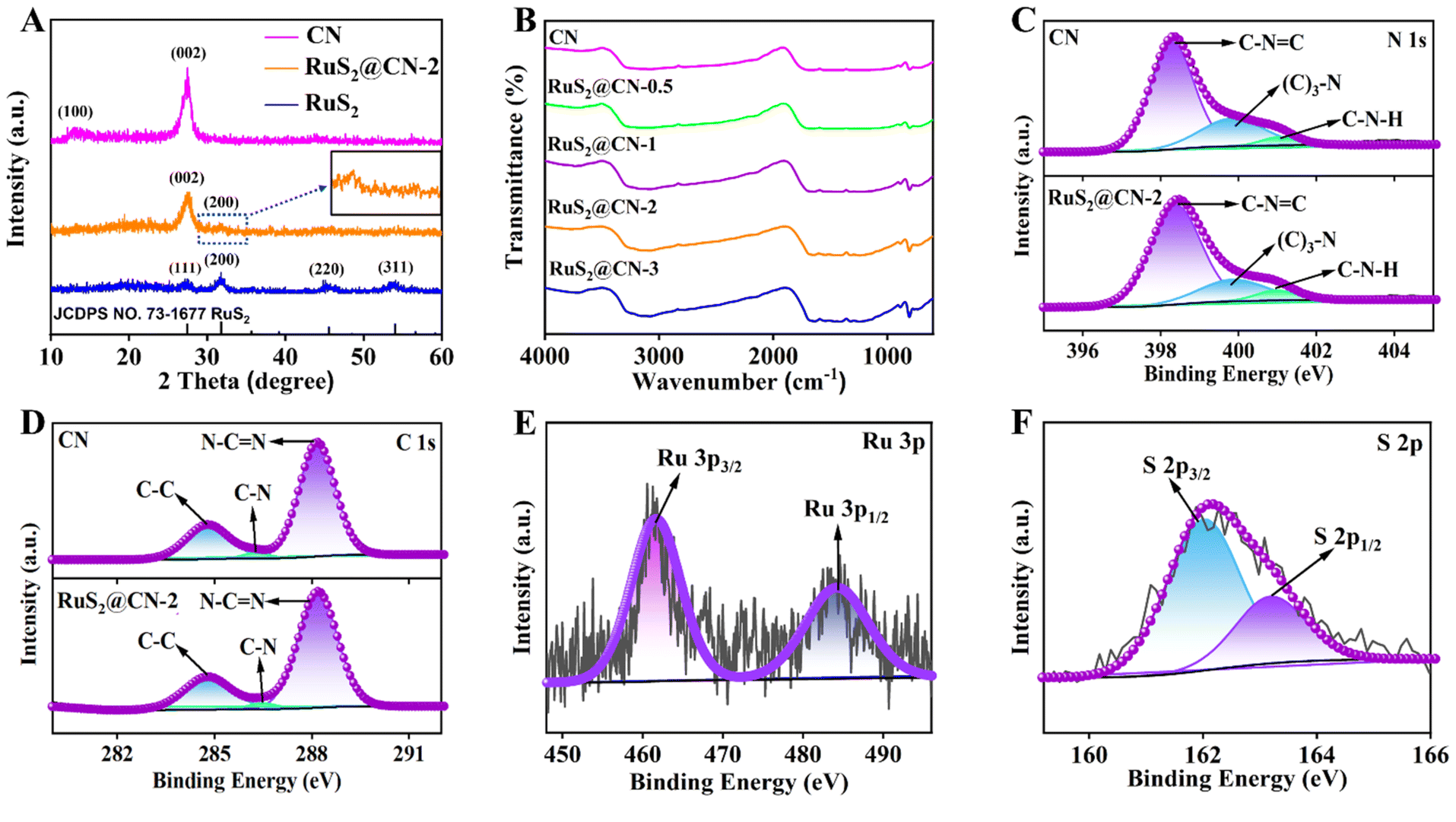 | ||
| Fig. 2 XRD patterns of CN, RuS2@CN-2, and RuS2 (A). FT-IR spectra of as-prepared samples (B). XPS high-resolution spectra of CN and RuS2@CN-2: N 1s (C), C 1s (D), Ru 3p (E) and S 2p (F). | ||
To further study the optical properties of CN and RuS2@CN-2, ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) was investigated. As compared with CN, the RuS2@CN-2 showed a red-shift in the visible light absorption range, indicating that RuS2@CN-2 has stronger light capture capability. As RuCl3 content increases, the visible light absorption range is further red-shifted (Fig. 3A and S4†). The band gaps of CN and RuS2@CN-2 were tested to be 2.33 and 1.85 V, respectively. Mott–Schottky (M–S) and ultraviolet photoelectron spectroscopy (UPS) were used to study semiconductor-based photocatalyst types and electronic band structures. Fig. 3B exhibits that the M–S slopes of CN and RuS2@CN-2 are positive, proving CN and RuS2@CN-2 are both n-type semiconductors. In addition, the flat band potential of CN (−1.20 V) and RuS2@CN-2 (−0.74 V) vs. Ag/AgCl were calculated using the horizontal intercept of the maximum slope. Generally, the flat band potential was 0.2 V less than the conduction band (CB) position. Therefore, CBs of CN and RuS2@CN-2 were −1.40 and −0.94 V vs. Ag/AgCl, respectively. After calculating the reference CB electrode as a normal hydrogen electrode (NHE, pH = 7), CBs of CN and RuS2@CN-2 were converted to −1.20 and −0.74 V vs. NHE, respectively. For the UPS analysis of CN and RuS2@CN-2, the work function (Φ) is first calculated. Fig. 3C shows the work functions (Φ) of CN and RuS2@CN-2 are 2.90 eV and 3.72 eV, respectively. Afterwards, the valence band (VB) positions can be calculated using this formula: EVB (vs. vacuum) = ESECO + φ, in which the ESECO is the secondary electron cut-off energy. As a result, the EVB (vs. vacuum) were 5.57 eV (CN) and 5.55 eV (RuS2@CN-2), respectively. Moreover, the EVB (vs. vacuum) should be converted to EVB (vs. NHE.) using this equation: EVB (vs. NHE) = EVB (vs. vacuum) − 4.44. After calculation, the EVB of CN and RuS2@CN-2 were 1.13 V and 1.11 V vs. NHE., respectively. Based on the above analysis, the relative band structure scheme of CN and RuS2@CN-2 was conducted (Fig. S5†). Moreover, to investigate the charge recombination rate of CN and RuS2@CN-2, photoluminescence (PL) spectroscopy was conducted. Fig. 3D shows that the PL intensity of CN is greater than that of RuS2@CN-2, suggesting charge recombination difficulty in RuS2@CN-2. This indicates that the unique structure obtained after anchoring RuS2 on CN inhibited the recombination of charges. Fig. 3E shows the photocurrent response of CN, RuS2, RuS2@CN-0.5, RuS2@CN-1, RuS2@CN-2, and RuS2@CN-3. As compared with individual CN and RuS2, RuS2@CN-x displays higher photocurrent intensity, with RuS2@CN-2 having the highest photocurrent intensity, indicating it has the strongest photo-generated charge separation capacity. The electrochemical impedance spectroscopy (EIS) Nyquist plots of CN, RuS2, RuS2@CN-0.5, RuS2@CN-1, RuS2@CN-2, and RuS2@CN-3 are displayed in Fig. 3F. Consistent with the results of the photocurrent response, the Nyquist curve radius of RuS2@CN-2 is the smallest as compared with the other samples, suggesting the lowest electron transfer resistance of RuS2@CN-2. The results demonstrate that anchoring of RuS2 nanoparticles on CN effectively improved photo-generated carrier transfer and separation efficiency.
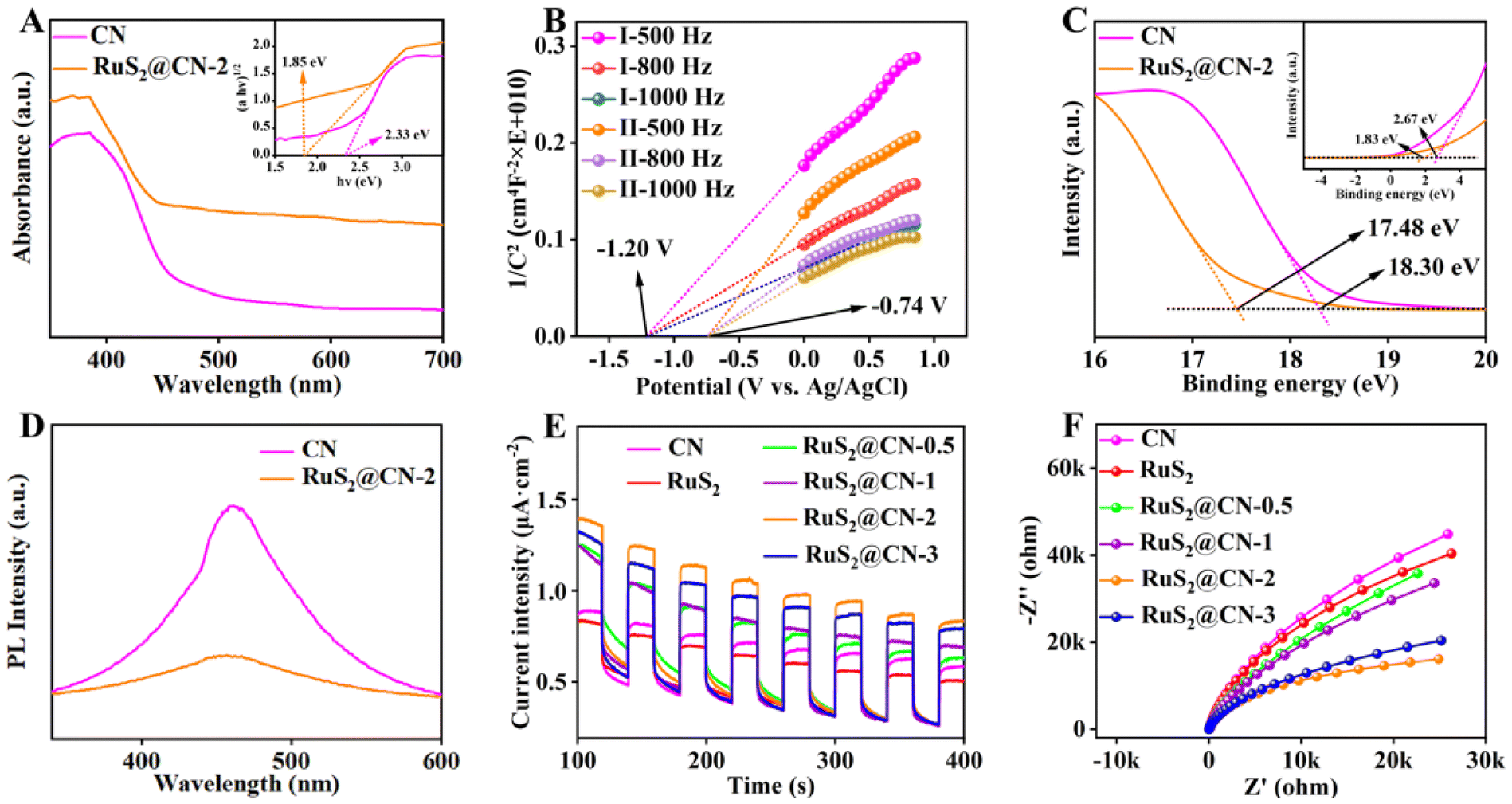 | ||
| Fig. 3 UV-vis DRS of CN and RuS2@CN-2 (A). Mott–Schottky plots of CN (I) and RuS2@CN-2 (II) at frequencies of 500, 800, 1000 Hz in 0.5 M Na2SO4 (B). UPS of CN and RuS2@CN-2 (C). PL of CN and RuS2@CN-2 (D). Periodic ON/OFF photocurrent responses of as-prepared samples in 0.5 M Na2SO4 electrolyte under visible light irradiation (λ > 420 nm) at 0.5 V vs. Ag/AgCl electrode (E). EIS of as-prepared samples (F). | ||
To investigate the performance of these photocatalysts, the following experiments were conducted with 10 W visible light illumination. First, the influences of various photocatalysts on hydrogen production and the yield of lactic acid were studied. Fig. 4A displays that the hydrogen production and the yield of lactic acid grew with the RuCl3 content. As the volume of RuCl3 solution (20 mg mL−1) reached 2 mL, the photocatalytic hydrogen production rate (462.7 μmol g−1 h−1) and lactic acid yield (69.3%) of RuS2@CN-2 was about 7.0 times and 1.3 times than those of CN, respectively. Yet, the hydrogen production rate decreased when the volume of the RuCl3 solution was further increased, which could probably be caused by the aggregation of RuS2 nanoparticles.52 The results agree with the characterization of EIS and transient photocurrent response. It validates the synergistic co-catalysis of RuS2 nanoparticles towards CN, enhancing the photocatalytic activity. Furthermore, xylose conversion and the yield of by-products were increased with the RuCl3 content (Fig. S6A†). Thereafter, the influence of RuS2@CN-2 dosages on their photocatalytic performances was investigated. Fig. 4B shows that the hydrogen evolution and lactic acid yield first grew and decreased with increasing catalyst dosage. The hydrogen release rate reached a peak of 502.7 μmol g−1 h−1 at 50 mg. Possibly caused by the impeded light transmission and absorbance due to the excess amount of RuS2@CN-2, the hydrogen release decreased when the amount of RuS2@CN-2 further increased.53–55 The trend of xylose conversion and by-product yields were similar to that of hydrogen release, which reached a maximum at 50 mg (Fig. S6B†). Moreover, pH plays an essential role in this system. Further work was conducted in KOH solutions of different concentrations, as numerous investigations found that the acidic and neutral environments were not conducive to water splitting and xylose conversion.56Fig. 4C displays that the hydrogen production and lactic acid yield were continuously increased with the concentration of KOH solution. The lactic acid yield and the hydrogen evolution rate reached the maximum of 88.4% and 4106.4 μmol g−1 h−1 with 5 M KOH concentration, respectively. Meanwhile, the by-product yields and xylose conversion grew with the KOH concentration. The xylose conversion reached 94.6% with 5 M of KOH concentration (Fig. S6C†). Furthermore, the influence of different irradiation times on the photocatalytic co-production of hydrogen and lactic acid was studied. The hydrogen release rate enhanced from 1176.8 μmol g−1 h−1 to 8698.3 μmol g−1 h−1 with the irradiation time ranging from 1 h to 9 h. At the same time, the lactic acid yield reached 90.3% after 9 h of reaction. The by-product yields and xylose conversion grew gradually with the increasing irradiation time (Fig. S6D†).
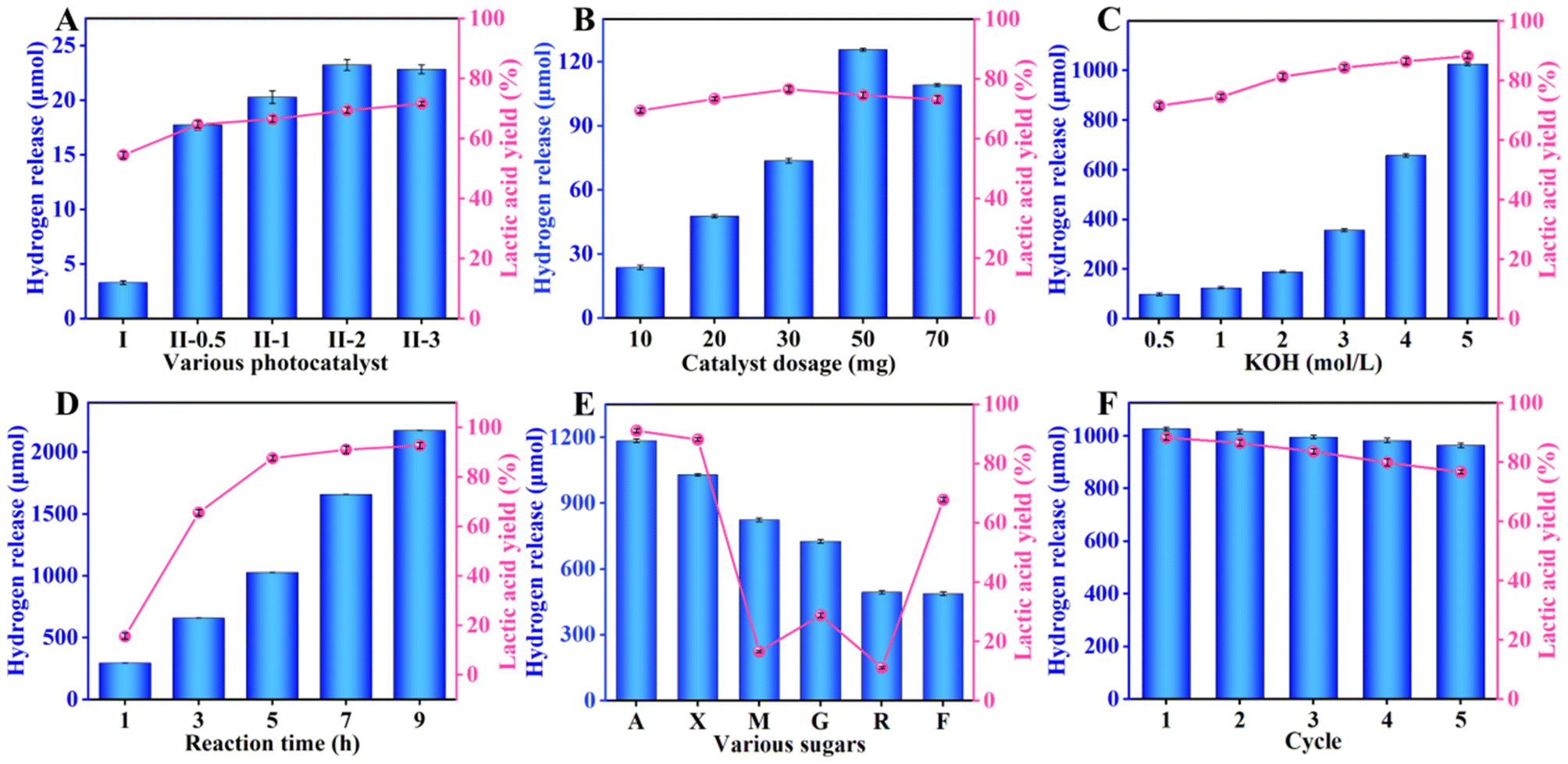 | ||
| Fig. 4 The effects of various conditions on co-production of hydrogen and lactic acid, (A) different photocatalysts (I: CN, II: RuS2@CN), (B) catalyst dosages (5.0 h, 30 °C), (C) KOH concentrations (10.0 mg RuS2@CN-2, 5.0 h), (D) irradiation time (10 mg RuS2@CN-2, 5.0 M KOH). (E) The selective oxidation of different biomass-derived sugars to hydrogen and lactic acid photocatalyzed by RuS2@CN-2. (F) Recycling test of RuS2@CN-2. | ||
To investigate the universality of RuS2@CN-2, xylose in the system was replaced by other biomass-based monosaccharides. A mixture of different monosaccharides (arabinose, xylose, mannose, glucose, rhamnose, and fructose; 100 mg), RuS2@CN-2 (50 mg), and KOH solution (5 M; 30 mL) was irradiated for 5 h. As Fig. 4E shows, the hydrogen evolution rates of different monosaccharides were 4741.2 μmol g−1 h−1 (arabinose), 4106.4 μmol g−1 h−1 (xylose), 3303.3 μmol g−1 h−1 (mannose), 2910.9 μmol g−1 h−1 (glucose), 1978.9 μmol g−1 h−1 (rhamnose), and 1956.6 μmol g−1 h−1 (fructose). The lactic acid yields of these monosaccharides were 91.5% (arabinose), 88.4% (xylose), 16.5% (mannose), 29.0% (glucose), 11.2% (rhamnose), and 67.9% (fructose). The photocatalytic activity of hexoses was worse than pentoses. The above performance studies were based on small-molecule monosaccharides. Hence, the photocatalytic activity of RuS2@CN-2 was explored with macromolecular sugar as a sacrificial agent. The study was conducted under the optimal condition of 5 h. As displayed in Fig. S7,† the hydrogen production rate is 772.3 μmol g−1 h−1. The lactic acid yield, formic acid yield, and acetic acid yield were 0.532 g gxylan−1, 0.059 g gxylan−1, and 0.022 g gxylan−1, respectively. These results indicated that RuS2@CN-2 could be applied to macromolecular and small molecular biomass reaction systems. Furthermore, to explore the repeatability and stability of RuS2@CN-2, recycling experiments were conducted for 5 h under optimal conditions (50 mg RuS2@CN-2, and 5.0 M KOH), thereby proving that RuS2@CN-2 has excellent repeatability and stability in this system, as depicted in Fig. 4F. Additionally, the lactic acid yield and hydrogen evolution rate, after five cycles, were 76.9% and 3859.3 μmol g−1 h−1, respectively, being 87.0% and 93.9% of that of the first cycle. Finally, the influence of monosaccharides on hydrogen evolution was studied. As anticipated, the hydrogen production rate in the optimal reaction system without monosaccharide was 387.0 μmol g−1 h−1, which was far less than that of the reaction system with xylose (8698.3 μmol g−1 h−1). This is because monosaccharide as a sacrificial reagent can rapidly remove the photo-induced holes to transform lactic acid, thus improving the hydrogen evolution rate.
To further illustrate the superiority of this reaction system, a series of previous reports were carefully investigated (Fig. 5). For the traditional water splitting to hydrogen, a certain number of sacrificial agents were added to the corresponding reaction systems, which promoted the hydrogen release. However, the conversions of those sacrificial agents have not been investigated, resulting in poor atomic economy. Meanwhile, the corresponding hydrogen releases (3.46, 1.29, 0.171, 3.086, 1.01, 1.686, 0.35, 0.792, 2.267 and 7.301 mmol g−1 h−1 for 15%ReS2@CN, CuInS2/g-C3N4, NV-g-C3N4, g-C3N4/OLC, PC-CN, g-C3N4/UFR-NC, Co/P/CN-sc, GD/g-C3N4, Cd/Mo(0.38)-VC and CdS/FePc, respectively) in those reaction systems were lower that of this work (Table S1†).57–66 As compared with the 300 W or 350 W Xe lamp, the light source used in this work is 10 W visible light, leading to low energy consumption (Table S1,† entries 1, 2, 4–10).57,58,60–66 Furthermore, the conversion of arabinose (sacrificial agent) was also studied, and a 91.5% yield of lactic acid was obtained. Recently, the conversions of sacrificial agents were studied in some works. As compared with the reported works, this work has excellent hydrogen release (8.698 mmol g−1 h−1), which is higher than those reported (Table S2†).67–71 Overall, this work shows excellent performance in coupling water splitting to hydrogen and biomass conversion, resulting in good environmental and economic benefits.
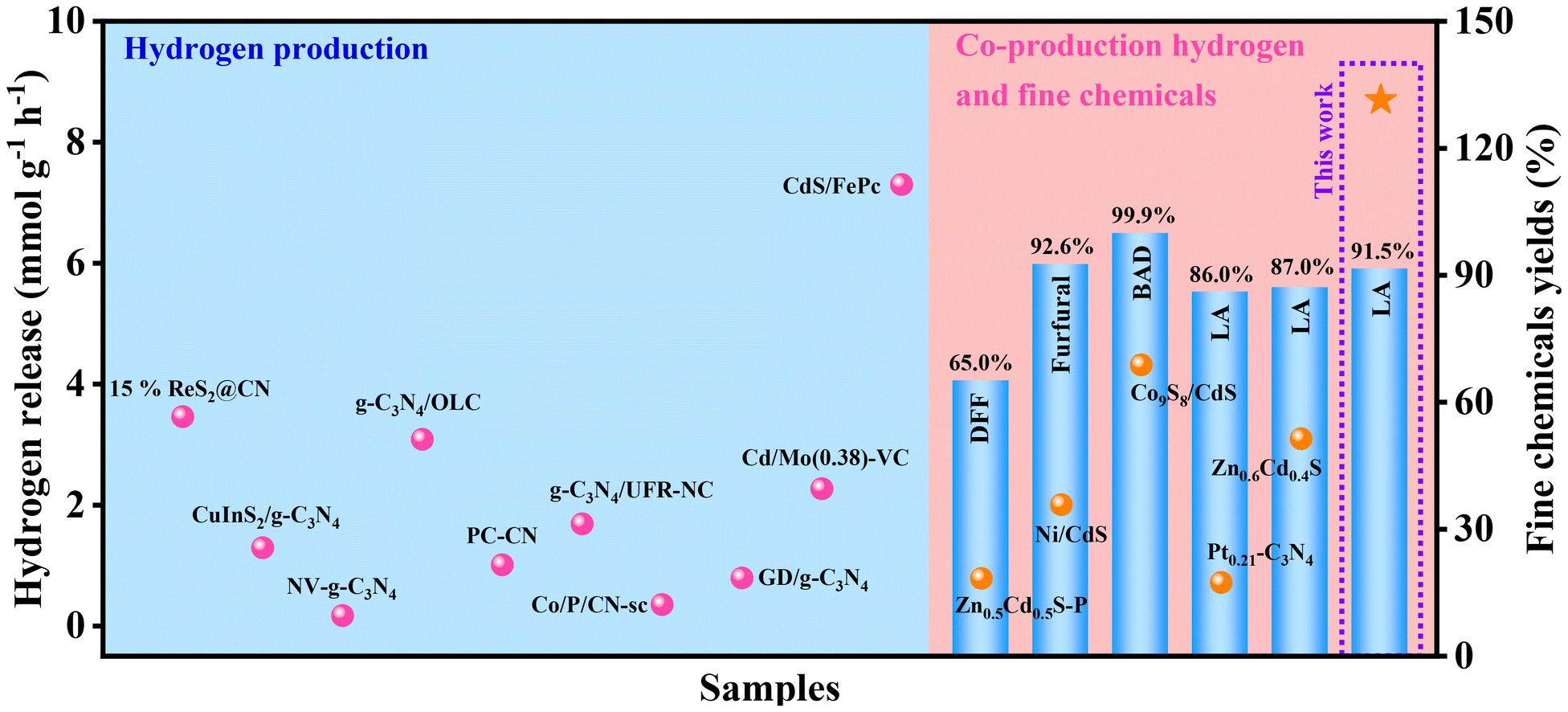 | ||
| Fig. 5 Performance map for the systems of co-production of hydrogen and fine chemicals or hydrogen production (DFF: 2,5-diformylfuran, BAD: benzaldehyde, LA: lactic acid). | ||
To study the influence of various active oxidation species on water splitting and biomass conversion, electron spin resonance (ESR) characterization was carried out. The e−, h+, and ˙OH in the reaction system cannot be detected directly. Thus, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 5,5-dimethyl-1-pyrrole N-oxide (DMPO) were used as trapping agents to detect these active oxidation species. Firstly, TEMPO was applied to detect e− and h+. For e−, the intensity of TEMPO was the highest under the control conditions (Fig. 6A). It then decreased with the increased irradiation time in the RuS2@CN-2 system. The results indicate that the e− was generated and its contents increased gradually with the irradiation time. Meanwhile, the TEMPO ESR spin-labelling of h+ was analogous to the e− (Fig. 6B) due to the consistent pairing of e− and h+. Furthermore, it is known that ˙OH can form adducts of DMPO/˙OH with DMPO. The DMPO/˙OH can be captured. As shown in Fig. 6C, no obvious peak can be seen under the control condition. The results proved that DMPO/˙OH was not formed. The peak intensities of DMPO/˙OH increased and displayed signal peaks with a 1:2:2:1 intensity when the irradiation time reached 5 min. After irradiation in 10 min, the peak intensity continued to increase. Finally, poisoning experiments were performed to study the effect of different active oxidation species on the lactic acid yield and hydrogen release under optimal conditions. Different from the above experiments, 0.05 M of KI and IPA were added to the system, respectively. As depicted in Fig. 6D, the hydrogen release and lactic acid yield decreased after adding a sacrificial agent into the corresponding system, in which the IPA addition system had the greatest effect on lactic acid yield, indicating that the ˙OH played a significant role in this system.
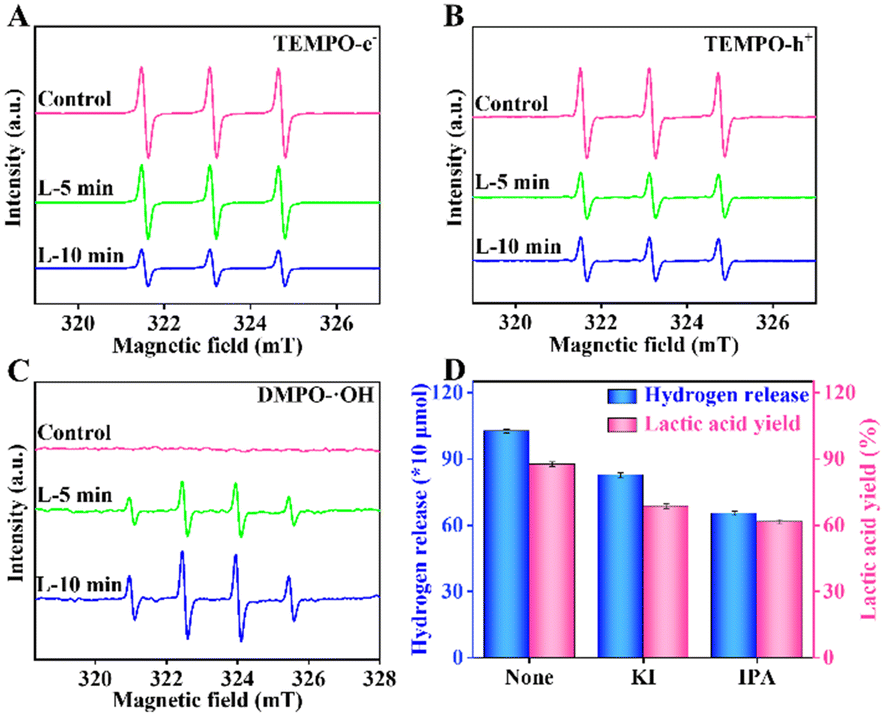 | ||
| Fig. 6 TEMPO ESR spin-labelling for e− (A) and h+ (B) of RuS2@CN-2. DMPO ESR spin-labelling for ˙OH of RuS2@CN-2 (C). The effects of different oxidation active species on the photocatalytic performances (D). | ||
The DFT calculations were used to clarify the hydrogen evolution reaction mechanism on RuS2@C3N4. The surface of RuS2@C3N4 can adsorb relevant species (H, 2H, and H2) in solution. As shown in Fig. 7, obviously, it was very easy for H formation in RuS2@C3N4-H with an exothermic process of −3.60 eV. However, the second H from H2O also can adsorb on RuS2@C3N4-H (−2.4 eV) to form RuS2@C3N4-2H intermediate with energy barriers of 1.2 eV. Moreover, it was very easy for H2 formation on surfaces of RuS2@C3N4 with an energy barrier of about −0.9 eV. The stable product of RuS2@C3N4-H2 is the most stable in thermodynamics because they are at the lowest point of the deep well.72–75
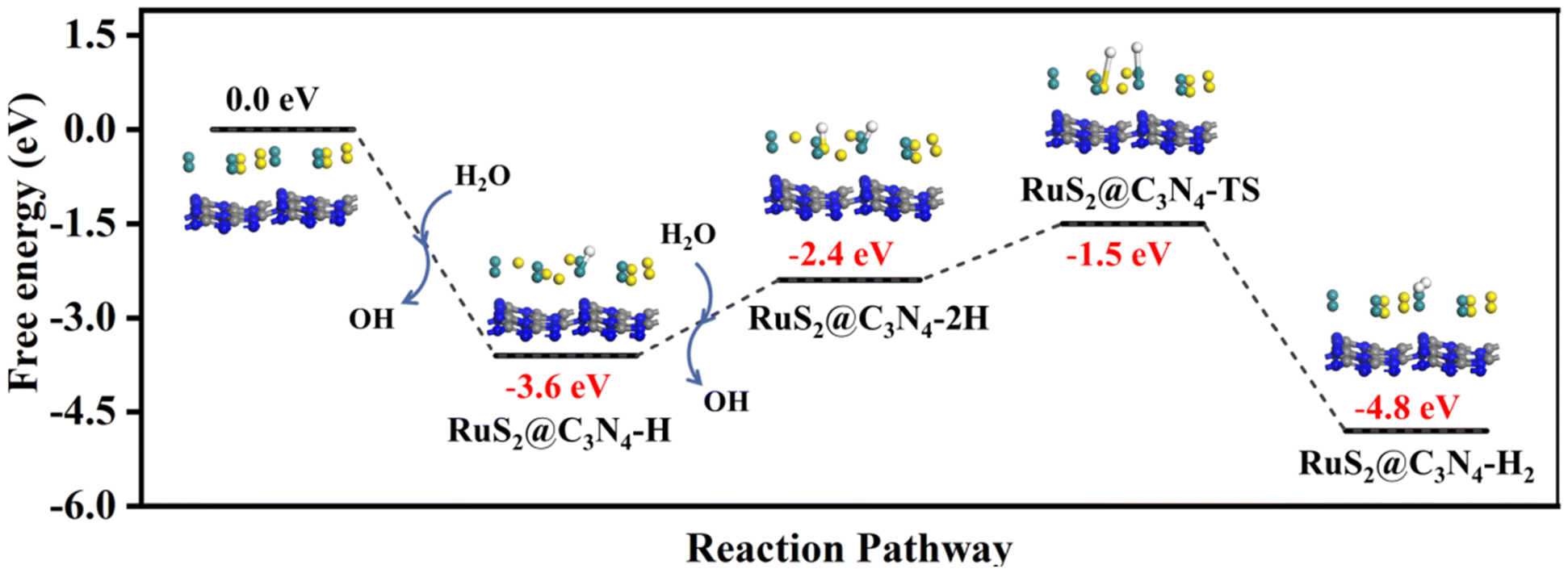 | ||
| Fig. 7 The hydrogen generation pathway calculated by DFT. | ||
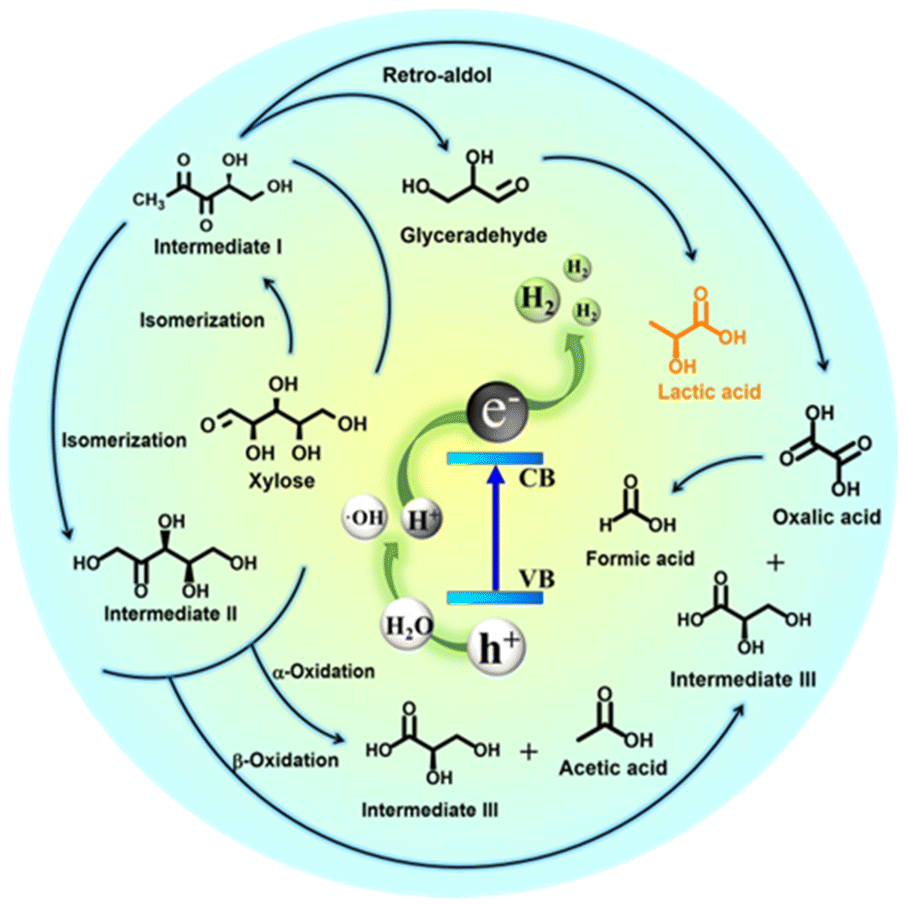
According to the above results and previous investigations,76–79 we proposed a possible mechanism for photocatalytic xylose conversion coupled with water splitting via RuS2@CN-2. These results of ESR and poisoning tests provided a basis for this mechanism. When RuS2@CN-2 was illuminated by visible light, RuS2@CN-2 absorbed photons with sufficient energy, and e−–h+ pairs were generated (Fig. 8). The obtained e− jumped to CB, and the h+ left on VB. Afterwards, the h+ reacted with H2O to produce ˙OH and protons.80 Meanwhile, with electrons reacting with protons to generate hydrogen, resulting in a reduction half-reaction, RuS2 nanoparticles anchored on the CN surface to receive protons as reduction sites.
 | ||
| Fig. 8 Possible reaction mechanism of the photocatalytic co-production of hydrogen and lactic acid via RuS2@CN-2. | ||
In the oxidation half-reaction, xylose was first transformed into intermediate I by isomerization. Then, intermediate II was generated via isomerization from intermediate I. Subsequently, xylose and the above products were converted to form intermediate III, acetic acid, and oxalic acid by α-oxidation and β-oxidation. The formic acid was produced after the C–C bond of oxalic acid was broken. Moreover, intermediate I synthesized oxalic acid and glyceraldehyde via the Retro-aldol mechanism, and glyceraldehyde was further converted to lactic acid.30
Conclusions
In summary, a novel RuS2@CN photocatalyst was prepared through a hydrothermal-calcination strategy, where RuS2 nanospheres were anchored on CN. The hydrogen production and lactic acid yield of RuS2@CN-2 were 14.18 times and 1.27 times than that of CN under the same conditions, respectively. The results further indicate that adding RuS2 promoted the transfer of photo-generated electrons, thus enhancing the photocatalytic activity. Moreover, providing more active sites for redox reactions, the interfacial interaction between RuS2 and CN evidently improved the transfer and separation efficiency of photo-generated charge carriers. In the optimal reaction system, which can be applied to macromolecular xylan and different biomass-derived monosaccharides, the highest hydrogen production rate of RuS2@CN-2 reached 8698.3 μmol g−1 h−1 and the highest lactic acid yield was 91.5% (monosaccharide: arabinose, catalyst dosage: 50 mg, KOH concentration: 5 M, irradiation time: 5 h). Poisoning experiment results show that ˙OH plays a significant major role in the oxidation half-reaction. The RuS2@CN-2 also demonstrated excellent reusability and stability. Following the above results, this study provides a new idea for designing and constructing excellent photocatalysts to realize the photocatalytic co-production of hydrogen and lactic acid.Experimental section
Preparation of CN photocatalysts
The pristine carbon nitride (CN) was prepared using a typical calcination strategy, in which 6.0 g of melamine was first added to a porcelain boat. Then, the porcelain boat containing melamine was transferred to a tubular furnace and then calcinated at 550 °C for 4 h with a ramping rate of 5 °C min−1. Amongst, the calcination process was carried out in an N2 atmosphere (99.99%, 30 mL min−1).Preparation of RuS2@CN-x photocatalysts
The RuS2@CN was prepared using a hydrothermal-calcination method. Initially, 1.0 g of CN, X mL of RuCl3 solution (20.0 mg mL−1), and a certain amount of thiourea were added to 25 mL of deionized water (notes: the weight ratio of RuCl3 and thiourea is 1:2), and the solution mixture was ultrasonicated for 20.0 min. Then, the obtained mixture was sealed in a Teflon-lined autoclave (50.0 mL) and kept at 150 °C for 6 h. The obtained grey solid precursors were washed with deionized water several times and dried overnight at 60 °C. The dried solid powders were calcinated at 500 °C with a heating rate of 5 °C min−1 for 2 h under the N2 atmosphere to give photocatalysts named RuS2@CN-x. Furthermore, pure RuS2 was prepared using the same method except for the introduction of CN.
Photocatalytic biomass conversion coupled with water splitting
The photocatalytic experiments were performed at 30 °C using Perfectlight PCX 50C multichannel photochemical reaction system with 10 W visible light irradiation. Generally, a certain amount of RuS2@CN-x and 100 mg of xylose were homogeneously dispersed in various concentrations of KOH solution (30 mL). Afterwards, the solution was sonicated for 10 min. The obtained systems were irradiated by visible light (10 W, 380 nm < λ < 760 nm) different times. Prior to irradiation, the system was degassed with 101 kPa of Ar gas 5 times. Finally, the hydrogen was detected by gas chromatography (GC) and the liquid products were detected by high-performance liquid chromatography (HPLC),81 respectively.Universality test
Typically, 100.0 mg of various biomass-derived monosaccharides (glucose, rhamnose, xylose, mannose, arabinose, and fructose) and 50.0 mg of RuS2@CN-2 were added to 5.0 M of KOH solution (30 mL). The reaction systems were sonicated for 10 min and degassed with 101 kPa Ar gas 5 times. After 5 h irradiation, the yields of hydrogen and lactic acid were detected by GC and HPLC.Recycling test
50.0 mg of RuS2@CN-2 and 100.0 mg of xylose were dispersed in 5.0 M of KOH solution (30 mL). The mixture was degassed 5 times with Ar (101 kPa) after 10 min of sonication. Afterwards, the reactor was sealed and then illuminated for 5 h by visible light at 30 °C. After the reaction, RuS2@CN-2 was filtered, washed, and dried at 60 °C overnight for the next cycle.Poisoning text
RuS2@CN-2 (50.0 mg) and xylose (100 mg) were added to 5.0 M of KOH solution (30 mL). Then, 0.01 M of KI, Trp, BQ, or IPA were inserted into the above reaction system, respectively. The obtained reactors were sonicated for 10 min and then degassed with Ar 5 times. Finally, they were conducted for 5 h at 30 °C with 10 W visible light irradiation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22008018), China Association for Science and Technology “Youth Talent Promotion Project” (No. YESS20210216), Dalian Outstanding Young Scientific and Technological Talents (2022RY24), the Foundation (No. 2021KF08) of Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, College of Light Industry and Food Engineering, Guangxi University. We also thank the Shiyanjia lab (https://www.shiyanjia.com) for the XPS analysis.References
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. F. Dong, R. Sougrat, G. T. Harrison, Y. L. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. D. Xu, L. Y. Zhao, A. Basu, S. D. Wolf, T. D. Anthopoulos, J. R. Durrant and L. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- Y. Q. Wu, Q. Wu, Q. Q. Zhang, Z. Z. Lou, K. F. Liu, Y. D. Ma, Z. Y. Wang, Z. K. Zheng, H. F. Cheng, Y. Y. Liu, Y. Dai, B. B. Huang and P. Wang, Energy Environ. Sci., 2021, 15, 1271–1281 RSC.
- L. Han, F. Jing, J. Zhang, X. Z. Luo, Y. L. Zhong, K. Wang, S. H. Zhang, D. H. Teng, Y. Liu, J. Chen, C. Yang and Y. T. Zhou, Appl. Catal., B, 2021, 282, 119602 CrossRef CAS.
- B. Yan, Y. He and G. W. Yang, J. Mater. Chem. A, 2022, 10, 1899–1908 RSC.
- D. N. Tritton, F. K. Tang, G. B. Bodedla, F. W. Li, C. S. Kwan, K. C. F. Leung, X. J. Zhu and W. Y. Wong, Coord. Chem. Rev., 2022, 459, 214390 CrossRef CAS.
- W. J. Li, Y. Y. Wang, L. Xu, Y. L. Tang, X. Y. Wu and J. H. Liu, Energy Convers. Manage., 2022, 256, 115344 CrossRef CAS.
- S. Salahshoor and S. Afzal, Int. J. Hydrogen Energy, 2022 DOI:10.1016/j.ijhydene.2022.08.202.
- J. A. Bau, A. H. Emwas, P. Nikolaienko, A. A. Aljarb, V. Tung and M. Rueping, Nat. Catal., 2022, 5, 397–404 CrossRef CAS.
- D. B. Adam, M. C. Tsai, Y. A. Awoke, W. H. Huang, C. H. Lin, T. Alamirew, A. A. Ayele, Y. W. Yang, C. W. Pao, W. N. Su and B. J. Hwang, Appl. Catal., B, 2022, 316, 121608 CrossRef CAS.
- Z. X. Zeng, F. Ye, S. H. Deng, D. X. Fang, X. J. Wang, Y. F. Bai and H. Xiao, Chem. Eng. J., 2021, 444, 136561 CrossRef.
- W. Li, Q. Ma, X. Wang, S. A. He, M. Li and L. F. Ren, Nano Converg., 2019, 494, 275–284 CrossRef CAS.
- B. Q. Xia, B. He, J. J. Zhang, L. Q. Li, Y. Z. Zhang, J. G. Yu, J. R. Run and S. Z. Qiao, Adv. Energy Mater., 2022, 22, 2201449 CrossRef.
- H. P. Peng, T. Yang, H. P. Lin, Y. Xu, Z. H. Wang, Q. H. Zhang, S. H. Liu, H. B. Geng, L. Gu, C. Wang, X. Fan, W. X. Chen and X. Q. Huang, Adv. Energy Mater., 2022, 22, 01688 Search PubMed.
- C. Z. Han, S. H. Xiang, M. T. Ge, P. X. Xie, C. Zhang and J. X. Jiang, Small, 2022, 14, 2202072 CrossRef PubMed.
- X. Wu, M. C. Zhang, Y. S. Xia, C. L. Ru, P. Y. Chen, H. Zhao, L. Zhou, C. L. Gong, J. C. Wu and X. B. Pan, J. Mater. Chem. A, 2020, 63, 1325–1335 Search PubMed.
- Y. Y. Zou, C. Y. Guo, X. L. Cao, T. X. Chen, Y. L. Kou, L. G. Zhang, T. Wang, N. Akram and J. D. Wang, Int. J. Hydrogen Energy, 2022, 47, 84 Search PubMed.
- S. W. Cao, B. J. Shen, Q. Huang and Z. Chen, Appl. Surf. Sci., 2018, 442, 361–367 CrossRef CAS.
- L. Wang, X. L. Geng, L. Zhang, Z. H. Liu, H. Wang and Z. Y. Bian, Chemosphere, 2022, 286, 131558 CrossRef CAS PubMed.
- W. C. Lin, J. Jayakumar, C. L. Chang, L. Y. Ting, M. H. Elsayed, M. Abdellah, K. Zheng, A. M. Elewa, Y. T. Lin, J. J. Liu, W. S. Wang, C. Y. Lu and H. H. Chou, Appl. Catal., B, 2021, 298, 120577 CrossRef CAS.
- Y. H. Peng, X. L. Guo, S. F. Xu, Y. N. Guo, D. S. Zhang, M. J. Wang, G. S. Wei, X. L. Yang, Z. Li, Y. Zhang and F. H. Tian, J. Energy Chem., 2021, 75, 276–284 CrossRef.
- J. L. Ma, D. N. Jin, Y. C. Li, D. Q. Xiao, G. J. Jiao, Q. Liu, Y. Z. Guo, L. P. Xiao, X. H. Chen, X. Z. Li, J. H. Zhou and R. C. Sun, Appl. Catal., B, 2021, 283, 119520 CrossRef CAS.
- Y. H. Zhang, S. C. Yang, Z. Wang, H. J. Qin, G. J. Lyu, J. C. Chen and G. H. Yang, J. Catal., 2015, 413, 843–857 CrossRef.
- J. L. Ma, Y. C. Li, D. N. Jin, Z. Ali, G. J. Jiao, J. Q. Zhang, S. Wang and L. R. C. Sun, Green Chem., 2020, 22, 6384–6392 RSC.
- Z. P. Huang, N. C. Luo, C. F. Zhang and F. Wang, Small, 2022, 6, 197–214 Search PubMed.
- K. A. Davis, S. Yoo, E. W. Shuler, B. D. Sherman, S. Lee and G. Leem, Nano Converg., 2021, 8, 7658 Search PubMed.
- X. P. Yang, K. N. Liu, J. L. Ma and R. C. Sun, Green Chem., 2021, 24, 5894–5903 RSC.
- Y. Q. Zhu, T. Wang, T. Xu, Y. X. Li and C. Y. Wang, Appl. Surf. Sci., 2019, 464, 36–42 CrossRef CAS.
- B. Lin, H. An, X. Q. Yan, T. X. Zhang, J. J. Wei and G. D. Yang, Appl. Catal., B, 2017, 210, 173–183 CrossRef CAS.
- X. B. Zhou, Y. F. Li, Y. Xing, J. S. Li and X. Jiang, Dalton Trans., 2019, 48, 15068–15073 RSC.
- K. N. Liu, J. L. Ma, X. P. Yang, Z. D. Liu, X. Z. Li, J. Q. Zhang, R. Cui and R. C. Sun, Chem. Eng. J., 2022, 437, 135232 CrossRef CAS.
- X. P. Yang, J. L. Ma, S. F. Sun, Z. D. Liu and R. C. Sun, Green Chem., 2022, 24, 2104–2113 RSC.
- L. Chen, X. Liang, H. X. Wang, Q. Q. Xiao and X. Q. Qiu, Chem. Eng. J., 2022, 442, 136115 CrossRef CAS.
- Z. P. Yu, Y. F. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. F. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- L. Wang, R. Lian, Y. Zhang, X. L. Ma, J. W. Huang, H. She, C. L. Liu and Q. Z. Wang, Appl. Catal., B, 2022, 315, 121568 CrossRef CAS.
- X. Ma and H. F. Cheng, Appl. Catal., B, 2022, 314, 121497 CrossRef CAS.
- K. H. Chen, X. W. Wang, Q. Y. Li, Y. N. Feng, F. F. Chen and Y. Yu, Chem. Eng. J., 2021, 418, 129476 CrossRef CAS.
- R. T. Guo, X. Y. Liu, H. Qin, Z. Y. Wang, X. Shi, W. G. Pan, Z. G. Fu, Z. Y. Tang, P. Y. Tang, P. Y. Jia, Y. F. Miao and J. W. Gu, Appl. Surf. Sci., 2020, 500, 144059 CrossRef CAS.
- J. X. Wu, X. X. Xi, W. Zhu, Z. Yang, P. An, Y. j. Wang, Y. M. Li, Y. F. Zhu, W. Q. Yao and G. Y. Jiang, Chem. Eng. J., 2010, 442, 136334 CrossRef.
- X. Y. Lu, A. Tong, D. Luo, F. Jiang, J. D. Wei, Y. C. Huang, Z. Jiang, Z. Lu and Y. H. Ni, J. Mater. Chem. A, 2022, 10, 4594–4600 RSC.
- Q. H. Shen, C. C. Du, Q. Q. Chen, J. Tang, B. Wang, X. H. Zhang and J. H. Chen, Mater. Today Phys., 2022, 23, 100625 CrossRef CAS.
- N. Su, D. Q. Zhu, P. F. Zhang, Y. H. Fang, Y. X. Chen, Z. Fang, X. T. Zhou, C. M. Li and H. J. Dong, Inorg. Chem., 2022, 61, 15600–15606 CrossRef CAS PubMed.
- T. V. Nguyen, S. S. Kim and O. B. Yang, Catal. Today, 2004, 5, 59–62 CAS.
- A. P. Gaikwad, D. Tyagi, C. A. Betty and R. Sasikala, Appl. Catal., A, 2016, 517, 91–99 CrossRef CAS.
- C. Maheu, E. Puzenat, P. Afanasiev, L. Cardenas and C. Geantet, Catal. Today, 2021, 377, 166–175 CrossRef CAS.
- X. Y. Gao, B. Li, X. Z. Sun, B. F. Wu, Y. P. Hu, Z. C. Ning, J. Li and N. Wang, Chin. Chem. Lett., 2021, 32, 3591–3595 CrossRef CAS.
- Z. Zhang, C. Jiang, P. Li, K. G. Yao, Z. L. Zhao, J. T. Fan, H. Li and H. J. Wang, Small, 2021, 17, 2007333 CrossRef CAS PubMed.
- Y. J. Xia, W. Q. Wu, H. Wang, S. L. Rao, F. Y. Zhang and G. F. Zou, Nanotechnology, 2020, 31, 145401 CrossRef CAS PubMed.
- S. J. Lv, Y. H. Ng, R. X. Zhu, S. Li, C. X. Wu, Y. X. Liu, Y. F. Zhang, L. Jing, J. G. Deng and H. X. Dai, Appl. Catal., B, 2021, 297, 120438 CrossRef CAS.
- J. Yu, Y. N. Guo, S. S. Miao, M. Ni, W. Zhou and Z. P. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 34098–34107 CrossRef CAS PubMed.
- K. Krishnamoorthy, P. Pazhamalai and S. J. Kim, Electrochim. Acta, 2017, 227, 85–94 CrossRef CAS.
- Y. L. Zhu, H. A. Tahini, Y. Wang, Q. Lin, Y. Liang, C. M. Doherty, Y. Liu, X. Y. Li, J. Lu, S. C. Smith, C. Selomulya, X. W. Zhang, Z. P. Shao and H. T. Wang, J. Mater. Chem. A, 2019, 7, 14222 RSC.
- S. W. Cao, X. F. Liu, Y. P. Yuan, Z. Y. Zhang, Y. S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef CAS.
- M. I. Badawy, M. Y. Ghaly and M. E. M. Ali, Desalination, 2011, 267, 250–255 CrossRef CAS.
- M. A. Raja and V. Preethi, Int. J. Hydrogen Energy, 2020, 45, 7574–7583 CrossRef.
- D. X. Peng, Y. T. Wang, H. F. Shi, W. Jiang, T. Jin, Z. H. Jin and Z. Chen, J. Colloid Interface Sci., 2022, 613, 194–206 CrossRef CAS PubMed.
- K. N. Liu, J. L. Ma, X. P. Yang, D. N. Jin, Y. C. Li, G. J. Jiao, S. Q. Yao, S. L. Sun and R. C. Sun, J. Alloys Compd., 2022, 906, 164276 CrossRef CAS.
- T. Y. Yang, Y. Y. Shao, J. D. Hu, J. F. Qu, X. G. Yang, F. Y. Yang and C. M. Li, Chem. Eng. J., 2022, 448, 137613 CrossRef CAS.
- X. X. Li, K. Y. Xie, L. Song, M. J. Zhao and Z. P. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 24577–24583 CrossRef CAS PubMed.
- Y. W. Liao, J. Yang, G. H. Wang, J. Wang, K. Wang and S. D. Yan, Rare Met., 2022, 41, 396–405 CrossRef CAS.
- Y. X. Shi, Q. Zhao, J. Y. Li, G. Y. Gao and J. F. Zhi, Appl. Catal., B, 2022, 308, 121216 CrossRef CAS.
- P. Wen, K. F. Zhao, H. Li, J. S. Li, J. Li, Q. Ma, S. M. Geyer, L. Jiang and Y. J. Qiu, J. Mater. Chem. A, 2020, 8, 2995–3004 RSC.
- H. N. Che, G. B. Che, P. J. Zhou, C. B. Liu, H. J. Dong, C. X. Li, N. Song and C. M. Li, Chem. Eng. J., 2020, 382, 122870 CrossRef CAS.
- W. X. Li, W. Li, Z. W. Guo, Y. Song, S. R. Tang, Y. N. Ma, X. Q. Xing and Q. Wang, ACS Appl. Mater. Interfaces, 2022, 13, 52560–52570 CrossRef PubMed.
- Q. L. Xu, B. C. Zhu, B. Cheng, J. G. Yu, M. H. Zhou and W. K. Ho, Appl. Catal., B, 2019, 255, 117770 CrossRef CAS.
- Y. G. Lei, K. H. Ng, Y. C. Zhu, Y. Z. Zhang, Z. X. Li, S. Xu, J. Y. Huang, J. Hu, Z. Chen, W. L. Cai and Y. K. Lai, Appl. Catal., B, 2023, 452, 139325 CAS.
- W. J. Zhang, X. Z. Zhou, J. Z. Huang, S. W. Zhang and X. J. Xu, J. Mater. Sci. Technol., 2022, 115, 199–207 CrossRef.
- H. F. Ye, R. Shi, X. Yang, W. F. Fu and Y. Chen, Appl. Catal., B, 2018, 233, 70–79 CrossRef CAS.
- G. Q. Han, Y. H. Jin, R. A. Burgess, N. E. Dickenson, X. M. Cao and Y. Z. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- C. Xia, C. Y. Xue, W. X. Bian, J. Liu, J. J. Wang, Y. J. Wei and J. B. Zhang, ACS Appl. Nano Mater., 2021, 4, 2743–2751 CrossRef CAS.
- E. Y. Wang, A. Mahmood, S. G. Chen, W. H. Sun, T. Muhmood, X. F. Yang and Z. P. Chen, ACS Catal., 2022, 12, 11206–11215 CrossRef CAS.
- H. Zhao, C. F. Li, X. Yong, P. Kumar, B. Palma, Z. Y. Hu, G. V. Tendeloo, S. Siahrostami and S. Larter, iScience, 2021, 24, 102109 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 77, 3865–3868 CAS.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- S. K. Cushing, F. Meng, J. Zhang, B. Ding, C. Chen, C. Chen, R. S. Liu, A. D. Bristow, J. Bright, P. Zheng and N. Wu, ACS Catal., 2017, 7, 1742–1748 CrossRef CAS.
- M. S. Holm, S. Saravamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- L. Yang, J. Su, S. Carl, J. G. Lynam, X. K. Yang and H. F. Lin, Appl. Catal., B, 2015, 162, 149–157 CrossRef CAS.
- F. Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef PubMed.
- M. Noor, F. Sharmin, M. A. Mamun, S. Hasan, M. A. Hakim and M. A. Basith, J. Alloys Compd., 2022, 895, 162639 CrossRef CAS.
- Z. D. Liu, K. N. Liu, R. C. Sun and J. L. Ma, Chem. Eng. J., 2022, 446, 137128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00367a |
| This journal is © The Royal Society of Chemistry 2023 |