
Extending the π-conjugation system of covalent organic frameworks for more efficient photocatalytic H2O2 production†
Maojun
Deng
a,
Jiamin
Sun
a,
Andreas
Laemont
a,
Chunhui
Liu
 a,
Linyang
Wang
a,
Laurens
Bourda
ab,
Jeet
Chakraborty
a,
Kristof
Van Hecke
b,
Rino
Morent
c,
Nathalie
De Geyter
c,
Karen
Leus
ac,
Hui
Chen
*a and
Pascal
Van Der Voort
*a
a,
Linyang
Wang
a,
Laurens
Bourda
ab,
Jeet
Chakraborty
a,
Kristof
Van Hecke
b,
Rino
Morent
c,
Nathalie
De Geyter
c,
Karen
Leus
ac,
Hui
Chen
*a and
Pascal
Van Der Voort
*a
aCOMOC-Center for Ordered Materials, Organometallics and Catalysis, Department of Chemistry, Ghent University, Krijgslaan 281-S3, 9000 Ghent, Belgium. E-mail: huichen@xauat.edu.cn; Pascal.Vandervoort@ugent.be
bXStruct, Department of Chemistry, Ghent University, Krijgslaan 281-S3, 9000 Ghent, Belgium
cRUPT-Research Unit Plasma Technology, Department of Applied Physics, Ghent University, Sint-Pietersnieuwstraat 41 B4, 9000 Gent, Belgium
First published on 14th March 2023
Abstract
The photocatalytic oxygen reduction reaction (ORR) towards hydrogen peroxide (H2O2) is a promising but challenging alternative to the industrial anthraquinone process. Crystalline porous covalent organic frameworks (COFs), a new generation of semiconductors, have attracted significant attention because they exhibit broad visible-light harvesting, possess tunable energy levels, and facilitate facile separation and migration of photogenerated charge pairs. The modulation of the energy levels of COFs could facilitate charge transport, resulting in better photocatalytic performance. Herein, the π-conjugation system of COFs was extended via a post-sulfurization process, leading to a considerably enhanced photocatalytic activity. During the sulfurization process, imine-linked COFs were converted into thiazole-linked COFs to extend the conjugation structure of COFs in the x and y directions, resulting in higher transport of electrons. The thiazole-linked 4PE-N-S presents the best photocatalytic H2O2 generation performance with a rate of 1574 μmol g−1 h−1, which is about 5.8 and 3.7 times higher than that of imine-linked 4PE-N and 4PE-TT, respectively. Our findings provide new prospects for the design and synthesis of highly active organic photocatalysts for solar-to-chemical energy conversion.
Introduction
Hydrogen peroxide (H2O2), a green and cheap chemical, is commonly utilized in industrial manufacturing, sterilization, disinfection, and chemical synthesis due to its strong oxidative nature.1–3 Traditionally, hydrogen peroxide is produced almost exclusively via the anthraquinone oxidation (AO) process. This process involves the sequential hydrogenation and oxidation of an alkyl anthraquinone precursor dissolved in a mixture of organic solvents, followed by liquid–liquid extraction to recover H2O2.4 In this process, the multistep manipulation requires significant energy input and generates tremendous waste, which is contrary to sustainable development. For this reason, a more sustainable green alternative strategy for hydrogen peroxide generation is attractive but challenging.In the last few decades, society has put more effort on the development of clean and sustainable production processes. Amongst the numerous candidates, photocatalysis is one of the most promising ones, which allows an environmentally friendly and sustainable strategy to perform catalytic reactions under very mild conditions.5 Several inorganic semiconductors (TiO2, ZnO, BiOCl and nitrogen-doped carbon materials (C3N4)) have been reported as photocatalysts for H2O2 production via the oxygen reduction reaction (ORR).2,6–9 While great progress is being made, the development of tunable and metal-free crystalline photocatalysts working in the visible range would be a major step forward. In 2005, a prominent discovery was made by Omar Yaghi10 who showed that it is possible to assemble organic building units towards crystalline porous materials since then referred to as Covalent Organic Frameworks (COFs). The creation of crystalline nanoporous frameworks based merely on organic (metal-free) monomers was a new concept. COFs are materials made completely by covalent linkage of molecular groups consisting of light elements (C, H, N), using the concept of dynamic covalent chemistry, i.e. the bonding is reversible allowing error corrections to improve crystallinity. In general, COFs are (i) highly chemically and thermally stable, (ii) excellent photo-absorbers and semiconductors, (iii) (photo)catalytically active, (iv) crystalline, (v) with tunable band positions, (vi) with chemically tunable functionalities and facile post-synthesis modification, and (vii) are highly porous for efficient diffusion.11–16 COFs are typically prepared using two organic precursors that either form infinite layers that are stacked in the z-direction by π–π interactions or form 3D structures. The beauty of the COF synthesis approach lies in the fact that these (typically aromatic) linkers can be functionalized to change the electronic properties of the final material. This allows to make photoactive COFs with no additional (metal) catalysts required.
In photocatalytic processes, there are in general three essential steps to convert light energy into chemical energy, namely the light-harvesting process, the generation and migration of photogenerated electron–hole pairs, and surface redox reactions.17 COFs are good semiconductors to ensure sufficient mobility for the separation and migration of electron–hole pairs.18 The advantage of COFs compared to other heterogeneous photocatalysts is their capacity to ensure efficient separation of electron–hole pairs through columnar π arrays, which also significantly narrows their bandgaps.17 Additionally, the π–π stacking interaction, the long-range order, and the regularly integrated building blocks enable the transport and transformation of photogenerated charges.19 Recently, our group reported for the first time the use of two imine-linked COFs for the photocatalytic metal-free production of H2O2 through the ORR.20 Inspired by Wurster-type systems, two imine-linked COFs, which consist of (diarylamino)benzene units, were synthesized. This report triggered interest in the synthesis of other imine-linked COFs with a variety of linkers for photocatalytic H2O2 production.20–27 However, there is still room to improve π-delocalization, because the strong polarization of the nitrogen atoms partly impedes the charge transfer.28,29 Lotsch's group reported on the utilization of a post-synthetic sulfur-assisted chemical strategy to convert an imine-linked COF into a thiazole-linked COF.30 This strategy provides an extraordinary platform that not only improves the stability of COFs but also their conjugation. Herein, we hypothesize that extending the π-conjugation system of COFs could significantly improve the photocatalytic H2O2 production efficiency. As a proof of concept, we present our results using a post-sulfurization process, so the imine-linked COFs were converted into thiazole-linked COFs, to generate highly conjugated and ultrastable photocatalysts for H2O2 production. Firstly, 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetraaniline (4PE) and naphthalene-2,6-dicarbaldehyde (N) were used to synthesize an imine-linked COF (named 4PE-N). After that, a simple post-sulfurization process was applied to obtain a thiazole-linked COF (named 4PE-N-S). Also, an imine-linked COF that incorporates a thieno[3,2-b]thiophene linker (4PE-TT) was synthesized for comparison (Fig. 1 and Fig. S1, S2†).
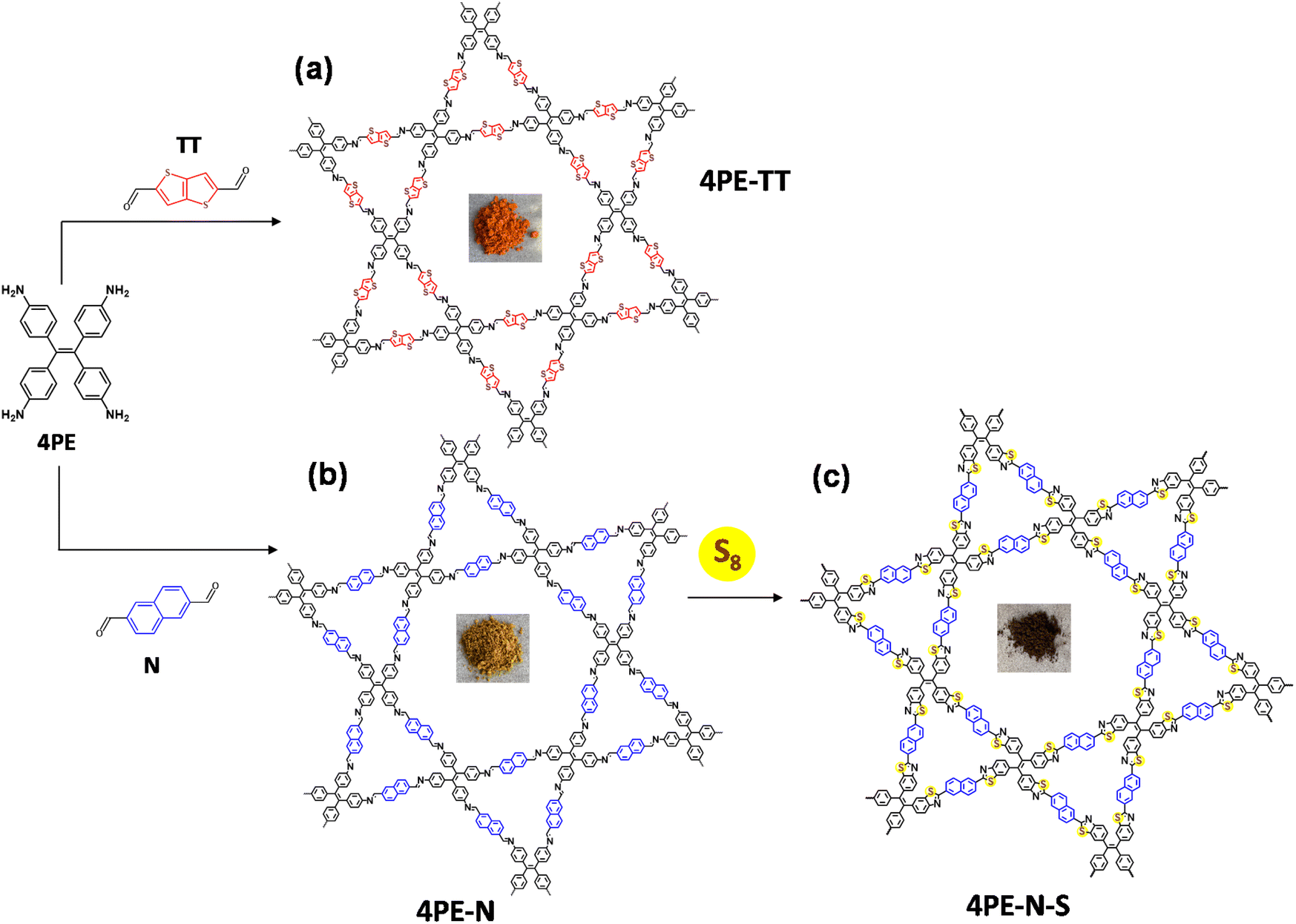 | ||
| Fig. 1 Schematic for the synthesis of (a) 4PE-TT, (b) 4PE-N, and (c) 4PE-N-S COFs. | ||
Results and discussion
The crystalline structures of 4PE-N, 4PE-N-S, and 4PE-TT were determined by means of powder X-ray diffraction (PXRD) analysis. As shown in Fig. 2a and b, 4PE-N and 4PE-N-S possess a similar structure. More specifically, the experimental PXRD diffraction patterns of the 4PE-N and 4PE-N-S COFs revealed intense reflection peaks in the low-angle region at 2θ = 2.43°, 4.10°, 4.84°, 6.43°, 7.24°, 8.7°, and 9.7° which corresponds to the (100), (110), (200), (210), (300), (310) and (400) planes, respectively, whereas the presence of a broad diffraction peak and intense bulge at a higher 2θ value (∼19.73°) of the 4PE-N-S COF can be identified as characteristic of the amorphous phase. The experimental PXRD diffraction patterns of 4PE-TT exhibited strong diffraction peaks at 2θ = 2.49°, 4.28°, 4.92°, 6.63°, 7.47°, 8.96°, 9.94°, and 19.94°, which were assigned to the (100), (110), (200), (210), (300), (310) (400), and (001) planes, respectively (Fig. 2c). All the diffraction peaks follow the P6 space group that represents a kagome 2D layered network. In addition, the structural models for the three COFs were constructed, showing a good match with the eclipsed AA stacking mode over staggered AB stacking. Pawley refinements of the full experimental PXRD profiles were carried out and the refinement results yield unit cell parameters of a = b = 42.31 Å, c = 5.30 Å, and α = β = 90°, γ = 120° with Rwp = 5.97% and Rp = 3.15% for 4PE-N; a = b = 40.85 Å, c = 5.09 Å, α = β = 90°, γ = 120° with Rwp = 4.68% and Rp = 3.34% for 4PE-N-S; a = b = 41.50 Å, c = 4.52 Å, α = β = 90°, and γ = 120° with Rwp = 6.4% and Rp = 4.78% for 4PE-TT. More detailed elemental parameters and atomic positions are presented in Tables S1 and S2.†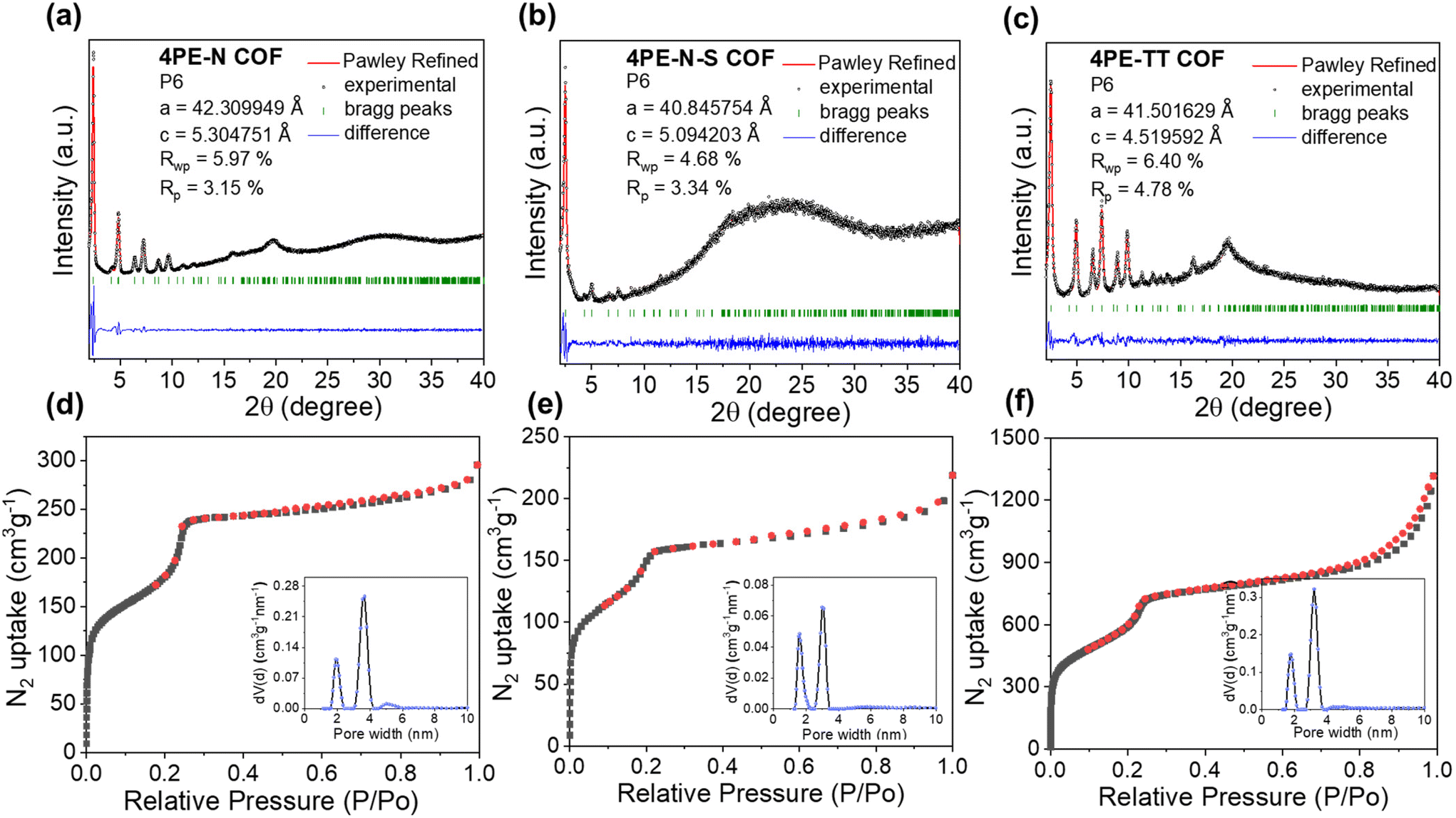 | ||
| Fig. 2 The PXRD patterns and Pawley refinements of (a) 4PE-N-COF, (b) 4PE-N-S-COF and (c) 4PE-TT-COF. N2 adsorption/desorption isotherms and pore size distributions (inset) of (d) 4PE-N-COF, (e) 4PE-N-S-COF and (f) 4PE-TT-COF. | ||
The chemical structures of the 4PE-N, 4PE-N-S, and 4PE-TT materials were analyzed by Fourier-transform infrared (FT-IR) spectroscopy and X-ray photoelectron spectroscopy (XPS). In the FT-IR spectra of 4PE-N and 4PE-TT (Fig. 3a and Fig. S3†), a typical imine bond (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) vibration at 1620 cm−1 was observed, which confirms the successful condensation of the amino and aldehyde building blocks. After post-sulfurization of 4PE-N, the imine bond vibration in the newly generated 4PE-N-S disappeared, while four new peaks at 1595, 1308, 945, and 795 cm−1 were observed, which were attributed to the thiazole linkage.30–34 This observation indicates the successful conversion of the imine-linkage in the 4PE-N material into the thiazole-linkage in 4PE-N-S. The X-ray photoelectron spectroscopy (XPS) analysis further confirmed the existence of C 1s, N 1s and S 2p in the 4PE-N-S and 4PE-TT COF framework structures (Fig. 3b and Fig. S4†). For 4PE-N-S, the peaks at ∼165.64 eV and ∼164.44 eV observed in the S 2p spectra are attributed to S 2p1/2 and S 2p3/2 of the C–S moiety, respectively (Fig. 3c). The binding energy exhibits a positive shift compared to S8 (164.0 eV), which is due to the formation of a “C–S” bond with the imine carbon atom.33 As shown in Fig. S4,† the N 1s spectrum of 4PE-N, 4PE-N-S and 4PE-TT could be deconvoluted into two peaks, centered at ∼400.10 and ∼398.80 eV, which are assigned to N–H (amine group) and NC (imine linkage) bonds, respectively. The peak at ∼400.86 eV in the spectrum of the 4PE-N-S COF is ascribed to the NC–S bond of the thiazole linkage. In addition, three distinct binding energy peaks centered at ∼288.30 eV, ∼285.70 eV and ∼284.90 eV are observed in the C 1s spectra, proving the existence of “π–π*”, “C–N” and “CC/C–C” bonds in the three COFs. Moreover, the peak at ∼284.23 eV in the 4PE-TT and 4PE-N-S COFs can be attributed to the “C–S” bond. Additionally, elemental analysis indicates the presence of sulfur in the 4PE-N-S COF and the experimental sulfur content (15.07 wt%) is consistent with the theoretically predicted value (15.77 wt%) (Table S3†).
N–) vibration at 1620 cm−1 was observed, which confirms the successful condensation of the amino and aldehyde building blocks. After post-sulfurization of 4PE-N, the imine bond vibration in the newly generated 4PE-N-S disappeared, while four new peaks at 1595, 1308, 945, and 795 cm−1 were observed, which were attributed to the thiazole linkage.30–34 This observation indicates the successful conversion of the imine-linkage in the 4PE-N material into the thiazole-linkage in 4PE-N-S. The X-ray photoelectron spectroscopy (XPS) analysis further confirmed the existence of C 1s, N 1s and S 2p in the 4PE-N-S and 4PE-TT COF framework structures (Fig. 3b and Fig. S4†). For 4PE-N-S, the peaks at ∼165.64 eV and ∼164.44 eV observed in the S 2p spectra are attributed to S 2p1/2 and S 2p3/2 of the C–S moiety, respectively (Fig. 3c). The binding energy exhibits a positive shift compared to S8 (164.0 eV), which is due to the formation of a “C–S” bond with the imine carbon atom.33 As shown in Fig. S4,† the N 1s spectrum of 4PE-N, 4PE-N-S and 4PE-TT could be deconvoluted into two peaks, centered at ∼400.10 and ∼398.80 eV, which are assigned to N–H (amine group) and NC (imine linkage) bonds, respectively. The peak at ∼400.86 eV in the spectrum of the 4PE-N-S COF is ascribed to the NC–S bond of the thiazole linkage. In addition, three distinct binding energy peaks centered at ∼288.30 eV, ∼285.70 eV and ∼284.90 eV are observed in the C 1s spectra, proving the existence of “π–π*”, “C–N” and “CC/C–C” bonds in the three COFs. Moreover, the peak at ∼284.23 eV in the 4PE-TT and 4PE-N-S COFs can be attributed to the “C–S” bond. Additionally, elemental analysis indicates the presence of sulfur in the 4PE-N-S COF and the experimental sulfur content (15.07 wt%) is consistent with the theoretically predicted value (15.77 wt%) (Table S3†).
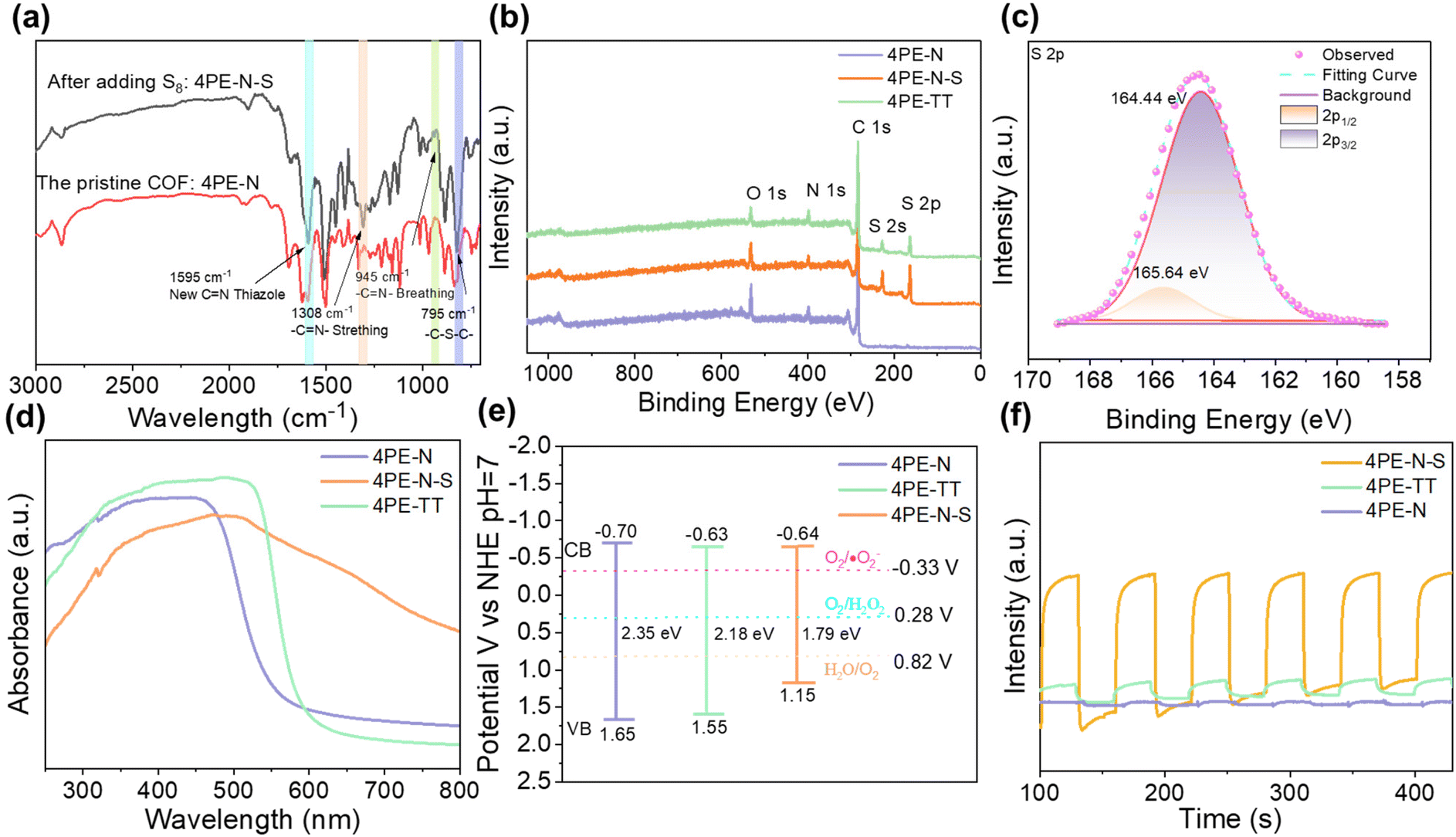 | ||
| Fig. 3 (a) FT-IR spectrum of 4PE-N and 4PE-N-S COFs, (b) survey XPS spectrum of all samples in the region of C 1s, N 1s, O 1s, and S 2p, (c) XPS spectra of 4PE-N-S in the region of S 2p, (d) UV-vis spectra, (e) band alignment and (f) photocurrent responses of 4PE-N, 4PE-N-S and 4PE-TT COFs. | ||
Nitrogen sorption measurements at 77 K were performed to determine the permanent porosity of the COFs. As shown in Fig. 2d–f, the isotherms exhibit a steep uptake in the low-pressure region (P/P0 < 0.01), indicating the presence of micropores. Additionally, a second rapid increase in gas uptake is observed around P/P0 = 0.22, which implies the presence of mesopores. In other words, the overall isotherms display both microporous and mesoporous characteristics, indicating that these COFs possess typical kagome-type structures. The Brunauer–Emmett–Teller (BET) surface areas of 4PE-N, 4PE-N-S, and 4PE-TT were 819 m2 g−1, 539 m2 g−1 and 2129 m2 g−1, respectively, while the total pore volumes (at P/P0 = 0.99) were calculated to be 0.49 cm3 g−1, 0.33 cm3 g−1, and 2.03 cm3 g−1, respectively. The experimental pore size distribution was analyzed by means of Quenched-Solid Density Functional Theory (QSDFT) carbon kernels for cylindrical-type pores. As shown in Fig. 2d–f and Fig. S5,† the average pore sizes of 4PE-N, 4PE-N-S, and 4PE-TT are 1.9/3.8 nm, 1.7/3.2 nm, and 1.8/3.3 nm. These values match well with the theoretical calculation. Furthermore, the morphologies of 4PE-N, 4PE-N-S and 4PE-TT were examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. S6,† no obvious changes in the shape were detected after post-modification with S8. The TEM images revealed a spherical hollow structure with domain sizes of ∼300 nm (Fig. S7 and S8†), and energy-dispersive X-ray spectroscopy (EDS) mapping analysis clearly showed the uniform distribution of all the elements within the COFs.
The chemical stability of 4PE-N, 4PE-N-S, and 4PE-TT COFs was assessed by immersing them into common organic solvents (N,N-dimethylformamide, toluene, methanol, 1,4-dioxane), strong acid (12 M HCl) and strong base (14 M NaOH) for 3 days. After the treatment, the COFs were collected and measured by PXRD. No drastic changes in their crystalline features were observed in the PXRD pattern of the three COFs after the treatment in the common organic solvents (Fig. S9†). Despite the initial loss in crystallinity as a consequence of the modification process (from 81% to 56% calculated crystallinity), the stability of the 4PE-N-S COF significantly improved as can be observed from the %crystallinity presented in Table S4.† Interestingly, it can be noted that upon immersion in strongly acidic media for 3 days, the structure of 4PE-N and 4PE-TT COFs was destroyed. It is worth mentioning that the S-stabilized 4PE-N-S COF retained most of its crystallinity (from 56% to 44%) in strong acid and basic media after 3 days of treatment (Table S4†). The higher chemical stability of the 4PE-N-S COF is due to the fact that chemically labile imine moieties have been converted to very strong benzothiazole moieties. In addition to chemical stability, thermal stability was also examined. The thermogravimetric analysis (TGA) results in Fig. S10† show that the three COFs exhibit remarkable thermal stability up to 440 °C under a N2 atmosphere.
The optical properties of 4PE-N, 4PE-N-S, and 4PE-TT COFs were assessed to verify the usability of these COFs as photocatalysts to produce H2O2 under visible light irradiation. Solid-state ultraviolet/visible (UV–vis) absorption experiments were carried out at room temperature. As shown in Fig. 3d, the UV-vis spectra indicate that 4PE-N and 4PE-TT absorb light in the UV and visible light regions. Interestingly, 4PE-N-S exhibits a significantly broader absorption, and it can be activated in the UV, visible, and even near-infrared regions. The broader absorption of the latter framework can be attributed to the extended π-conjugation due to the conversion of the imine linkage into the thiazole linkage. Based on the UV-vis absorption results, the optical band gaps of the COFs were calculated using the Tauc-plot (Fig. S11†). The 4PE-N and 4PE-TT materials exhibit similar band gaps (2.35 eV vs. 2.18 eV). In contrast, the band gap of 4PE-N-S dramatically narrowed and it was analyzed to be 1.79 eV. This can be explained by the introduction of a conjugated thiazole linkage, which ensures an extended conjugation in the x and y directions of the COF structure, resulting in higher conductivity for the transmission of electrons.35 To gain more insights into the electronic properties of all three COFs, impedance spectroscopy was performed at AC frequencies of 1000, 2000 and 3000 Hz to acquire Mott–Schottky (M–S) plots. Previously, Mott–Schottky analysis was confirmed to be performed at a pH corresponding to the isoelectric point of the material.36,37 The zeta potential shows that the isoelectric points of the 4PE-N, 4PE-N-S and 4PE-TT COFs are found at approximately pH = 5 (Fig. S12†). As illustrated in Fig. S13,† the positive slopes of the Mott–Schottky plots suggest an n-type semiconductor behaviour for these COFs. The conduction bands (CBs) of 4PE-N, 4PE-TT, and 4PE-N-S were estimated to be −0.70 V, −0.63 V and −0.64 V vs. NHE, respectively. The CB values of these COFs are more negative than the redox potential of O2/˙O2− (−0.33 V vs. NHE, pH = 7), which means that it is theoretically feasible to photocatalytically produce H2O2via an oxygen reduction reaction (ORR) pathway with all three COFs. It is important to note that due to the extended π-conjugation system in 4PE-N-S, the valence band (VB) position of 4PE-N-S is +1.15 V vs. NHE, which is smaller compared to that of 4PE-N and 4PE-TT, (1.65 and 1.55 V vs. NHE). The more positive VB position of 4PE-N-S results in stronger oxidation capacity.
Furthermore, to investigate the charge separation and migration behaviour of the synthesized COFs, transient photocurrent intensity, electrochemical impedance spectroscopy (EIS), and photoluminescence (PL) experiments were performed. As shown in Fig. 3f, the synthesized COFs exhibit a clear photocurrent response, indicating that electron–hole pairs are produced under visible light irradiation. The photocurrent intensity of 4PE-N-S (∼4.33 μA cm−2) is remarkably higher than those of 4PE-N (∼0.09 μA cm−2) and 4PE-TT (∼0.50 μA cm−2), which can be ascribed to the higher π-conjugation, evidencing a more efficient charge separation for 4PE-N-S. Moreover, EIS was employed to investigate the interfacial properties between the electrode and the electrolyte. As shown in Fig. S14,† the capacitance arc of 4PE-N-S is smaller than that of 4PE-N and 4PE-TT, implying that the electron mobility was improved after extending the π-conjugation system, and thus the electron–hole recombination rate was effectively prohibited.27 To provide additional evidence for this point of view, the photoluminescence (PL) spectra of the COFs were recorded. Compared to 4PE-N and 4PE-TT, 4PE-N-S exhibits the strongest fluorescence quenching, which suggests that the highly π-conjugated thiazole linkage facilitates the separation of the photogenerated electron–hole pairs and inhibits their recombination (Fig. S15†). Based on all these results, it is clear that the extended π-conjugated system can effectively separate photogenerated charges and prevent their recombination, which is beneficial for photocatalysis.
So far, we have shown that 4PE-N, 4PE-TT, and 4PE-N-S COFs exhibit high crystallinity, permanent porosity, outstanding stability, and promising photoredox properties. Therefore, these COFs were employed as metal-free and green photocatalysts for the production of H2O2. The photocatalytic H2O2 generation experiment was carried out both with and without an electron sacrificial agent under visible light (420 < λ < 700 nm) irradiation. As shown in Fig. 4a, even in the absence of an additional electron sacrificial agent, the 4PE-N and 4PE-TT COFs display remarkable photocatalytic H2O2 generation performance after 6 h of irradiation. The H2O2 generation rates of the 4PE-N and 4PE-TT COFs are 270 μmol g−1 h−1 and 421 μmol g−1 h−1, respectively. Compared to 4PE-N and 4PE-TT, the H2O2 generation rate of 4PE-N-S significantly increased. Under the same experimental conditions, the 4PE-N-S material presents the best photocatalytic H2O2 generation performance with a rate of 1574 μmol g−1 h−1, which is about 5.8 and 3.7 times higher compared to 4PE-N and 4PE-TT respectively. This is one of the best photocatalytic H2O2 generation performances compared to those of similar materials (Table S5†). Moreover, more free electrons participate in photocatalytic H2O2 generation when ethanol is added as a sacrificial agent into the reaction mixtures (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol = 9:1). Ethanol acts as an electron and proton donor during the reaction, to effectively trap holes and promote the separation of electrons and holes.20 As shown in Fig. 4b, the photocatalytic H2O2 generation rates are 546, 624, and 2237 μmol g−1 h−1 for 4PE-N, 4PE-TT, and 4PE-N-S, respectively, in the presence of ethanol. It is important to note that the H2O2 generation rate of 4PE-N-S is higher compared to most of the previously reported COF materials, such as the EBA-COF (1820 μmol g−1 h−1), the TF50-COF (1739 μmol g−1 h−1) and the CoPc-BTM-COF (2096 μmol g−1 h−1).22,27,38 In addition, in order to verify that the excellent H2O2 production performance of the 4PE-N-S COF is due to the formation of thiazole bonds rather than the heat treatment during synthesis, a comparative experiment was carried out. In this experiment, the 4PE-N COF was subjected to the same heating procedure as in the 4PE-N-S COF but without the addition of S8. The obtained material did not show a change in the H2O2 production rate compared to the pristine, non-heated 4PE-N COF.
:ethanol = 9:1). Ethanol acts as an electron and proton donor during the reaction, to effectively trap holes and promote the separation of electrons and holes.20 As shown in Fig. 4b, the photocatalytic H2O2 generation rates are 546, 624, and 2237 μmol g−1 h−1 for 4PE-N, 4PE-TT, and 4PE-N-S, respectively, in the presence of ethanol. It is important to note that the H2O2 generation rate of 4PE-N-S is higher compared to most of the previously reported COF materials, such as the EBA-COF (1820 μmol g−1 h−1), the TF50-COF (1739 μmol g−1 h−1) and the CoPc-BTM-COF (2096 μmol g−1 h−1).22,27,38 In addition, in order to verify that the excellent H2O2 production performance of the 4PE-N-S COF is due to the formation of thiazole bonds rather than the heat treatment during synthesis, a comparative experiment was carried out. In this experiment, the 4PE-N COF was subjected to the same heating procedure as in the 4PE-N-S COF but without the addition of S8. The obtained material did not show a change in the H2O2 production rate compared to the pristine, non-heated 4PE-N COF.
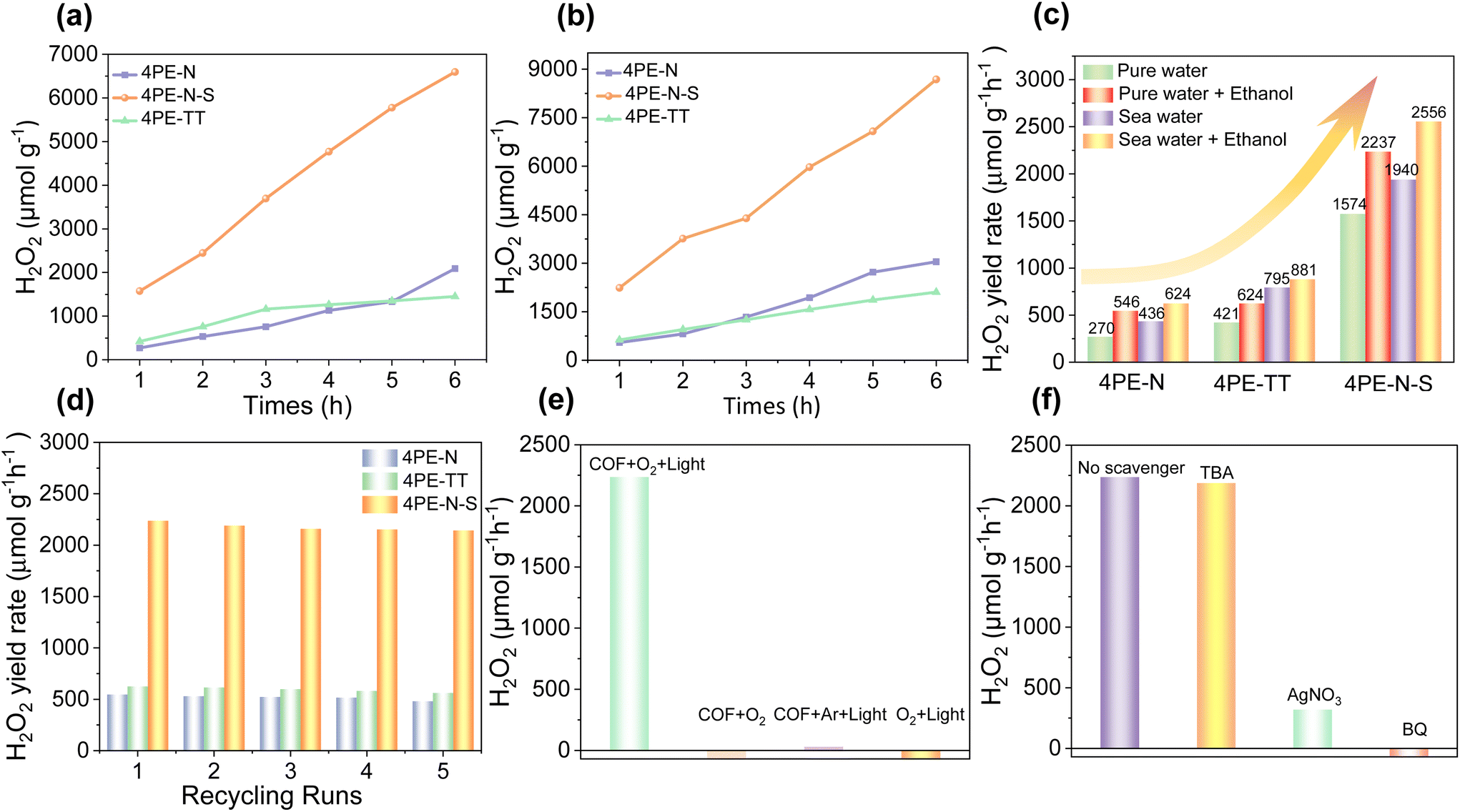 | ||
| Fig. 4 Photocatalytic production of H2O2 for 4PE-N, 4PE-N-S, and 4PE-TT COFs under the illumination of a 300 W Xe lamp: (a) pure water, (b) water:ethanol (9:1) system, (c) comparison of H2O2 production among the photocatalysts with pure water and real seawater with and without ethanol. The photocatalytic performance of three COFs with one-hour of irradiation, (d) H2O2 production for five consecutive cycles using three COFs, (e) under different reaction conditions, (f) effects of scavengers (2 mmol L−1), 5 mg of photocatalyst, water:ethanol (9:1). | ||
Recyclability is an important aspect of heterogeneous catalysts for their practical implementation. To demonstrate their reusability, recycling experiments were performed. As can be seen from Fig. 4d, these photocatalysts could be recovered and reused for at least five cycles without significant loss of catalytic activity. No apparent changes in the PXRD patterns of all three spent COFs were observed (Fig. S16†), indicating that the crystalline structures of these COFs can be retained upon five cycles of catalysis. In addition, negligible changes in the FT-IR spectrum and N2 adsorption isotherm prove that the chemical structure of the COFs was preserved (Fig. S17 and S18†). Besides, a long-term photocatalytic production of H2O2 by the 4PE-N-S COF in pure water was implemented. As shown in Fig. S19,† it can be observed that 4PE-N-S can generate 10203 μmol g−1 and 11551 μmol g−1 H2O2 after 15 and 24 hours of irradiation, respectively. Another important aspect of their practical implementation is adaptability. Natural water consists of complex ingredients, including various kinds of inorganic ions, dissolved organic matter, and microorganisms, which may affect the photocatalytic process. Therefore, seawater instead of distilled water was used for photocatalytic H2O2 generation. Surprisingly, the H2O2 generation rates of the 4PE-N, 4PE-TT, and 4PE-N-S materials were up to 624, 881, and 2556 μmol g−1 h−1, respectively (Fig. 4c). This can be explained by the presence of metal ions in seawater, such as Na+, K+, Ca2+, etc. which can ionize the catalyst surface, attract electrons, and promote the separation of electrons and holes, thereby enhancing the generation of H2O2.39–41
In order to elucidate the photocatalytic reaction mechanism, a series of radical capture and control experiments were carried out using 4PE-N-S as a photocatalyst. As shown in Fig. 4e, in the absence of 4PE-N-S or light, no detectable amount of H2O2 was observed, which suggests that COFs and light are essential for the photocatalytic H2O2 generation reaction. In addition, when using Ar instead of O2, no obvious H2O2 was detected, implying that the photosynthesized H2O2 comes from O2. It is known that H2O2 generation via the ORR mechanism can occur via two possible pathways, namely direct 2e− (eq. (1)) and indirect 2e− pathways (eq. (2) and (3)).
| O2 + 2H+ + 2e− → H2O2 (E = +0.28 V vs. NHE) | (1) |
| O2 + e− → ˙O−2 (E = −0.33 V vs. NHE) | (2) |
| ˙O2− + 2H+ + e− → H2O2 (E = +0.91 V vs. NHE) | (3) |
To confirm the ORR pathway during the photocatalytic H2O2 generation process, a series of radical capture experiments was performed. As shown in Fig. 4f, a huge decrease in the photocatalytic activity was observed when silver nitrate (AgNO3, e− scavenger) or benzoquinone (BQ, ˙O2− scavenger) was added, indicating that the photogenerated electrons (e−) and superoxide radicals (˙O2−) play a crucial role in the photocatalytic H2O2 generation reaction. In sharp contrast, the addition of tertbutyl alcohol (TBA, ˙OH scavenger) to the reaction system resulted in a negligible decrease in the H2O2 generation rate, meaning that the hydroxyl radical is not involved in the process of H2O2 formation. The radical trapping and control experiment results are consistent with the indirect 2e− ORR pathway (eq. (2) and (3)). Micro-GC measurements confirm the production of O2 (in water, without the addition of alcohol as a hole scavenger) and NMR measurements confirm the presence of benzaldehyde (with the addition of benzyl alcohol, Fig. S20 and S21†). We use benzyl alcohol in this experiment since acetaldehyde has a very low boiling point, and is therefore hard to detect. Based on these results, a plausible mechanism in Fig. S22† is proposed. Initially, upon absorption of visible light, the COF-based photocatalyst is excited, and the photogenerated electron–hole pairs are separated. The accumulated electrons in the CB transfer to the surface of the photocatalyst where they combine with the adsorbed O2 to generate ˙O2− radicals. In the following step, the ˙O2− radicals combine with protons to generate H2O2. Accordingly, ethanol is oxidized to aldehyde by the photogenerated holes (h+) from the VB, which in turn releases protons for H2O2 formation. In the pure water system (without the addition of sacrificial ethanol), the most likely anodic reaction is the oxidation of H2O towards O2 and H+ (E = +0.82 V vs. NHE).
Conclusions
In summary, three highly crystalline and porous COFs with promising photoredox properties have been prepared. These COFs exhibit excellent light absorption capacity and enhanced photo-induced charge separation and transport efficiency, endowing the as-prepared COFs with a remarkable photocatalytic activity towards O2-to-H2O2 conversion under visible-light irradiation. In particular, the relationship between the structure and photocatalytic activity was revealed. Imine-linked COFs can be easily converted into thiazole-linked COFs via a simple post-sulfurization, which results in improved conjugation, leading to a prominent increase in the photocatalytic performance. Our findings should benefit the subsequent design and fabrication of high-performance photocatalysts.Author contributions
Maojun Deng: writing – original draft. Jiamin Sun: data analysis, writing and editing the draft. Andreas Laemont and Dr. Jeet Chakraborty: validation, investigation and resources. Chunhui Liu conducted the fluorescence emission, solid-state UV-Vis absorption and elemental analysis experiments. Linyang Wang conducted the electrochemistry measurements. Prof. Kristof Van Hecke and Laurens Bourda performed the structural modeling and refinement. Prof. Rino Morent, Prof. Nathalie De Geyter and Dr. Karen Leus conducted the X-ray photoelectron spectroscopy (XPS) experiments and analysis. Dr Karen Leus revised the manuscript. Dr. Hui Chen and Prof. Pascal Van Der Voort: funding acquisition, data analysis, and revision of the manuscript. All authors contributed to the revision and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dirk Poelman (UGent) for helping with the Solid-state UV-vis measurements. M. J. D. (202107565003), J. M. S. and H. C. acknowledge the financial support from the China Scholarship Council (CSC). J. M. S. acknowledges the financial support from Ghent University (01SC0619) and we also gratefully acknowledge the financial support from the FWO (project I002418N).References
- Z. Xu, Y. Li, Y. Cao, R. Du, Z. Bao, S. Zhang, F. Shao, W. Ji, J. Yang, G. Zhuang, S. Deng, Z. Wei, Z. Yao, X. Zhong and J. Wang, J. Energy Chem., 2022, 64, 47–54 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS PubMed.
- L. F. de L. e. Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- C. Yang, S. Wan, B. Zhu, J. Yu and S. Cao, Angew. Chem., 2022, 134, e202208438 Search PubMed.
- Y. Zhang, Y. Xia, L. Wang, B. Cheng and J. Yu, Nanotechnology, 2021, 32, 415402 CrossRef CAS PubMed.
- R. Qu, W. Zhang, N. Liu, Q. Zhang, Y. Liu, X. Li, Y. Wei and L. Feng, ACS Sustainable Chem. Eng., 2018, 6, 8019–8028 CrossRef CAS.
- Z. Jiang, Y. Zhang, L. Zhang, B. Cheng and L. Wang, Chin. J. Catal., 2022, 43, 226–233 CrossRef CAS.
- B. Zhu, B. Cheng, J. Fan, W. Ho and J. Yu, Small Struct., 2021, 2, 2100086 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- M. Najafi, S. Abednatanzi, P. G. Derakhshandeh, F. Mollarasouli, S. Bahrani, E. S. Behbahani, P. Van Der Voort and M. Ghaedi, Coord. Chem. Rev., 2022, 454, 214332 CrossRef CAS.
- S. Abednatanzi, M. Najafi, P. G. Derakhshandeh and P. Van Der Voort, Coord. Chem. Rev., 2022, 451, 214259 CrossRef CAS.
- S. Ma, Z. Li, J. Jia, Z. Zhang, H. Xia, H. Li, X. Chen, Y. Xu and X. Liu, Chin. J. Catal., 2021, 42, 2010–2019 CrossRef CAS.
- Z.-B. Zhou, P.-J. Tian, J. Yao, Y. Lu, Q.-Y. Qi and X. Zhao, Nat. Commun., 2022, 13, 2180 CrossRef CAS PubMed.
- S. Yang, H. Lv, H. Zhong, D. Yuan, X. Wang and R. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115655 CAS.
- X. Zhao, H. Pang, D. Huang, G. Liu, J. Hu and Y. Xiang, Angew. Chem., Int. Ed., 2022, 61, e202208833 CAS.
- H. Chen, H. S. Jena, X. Feng, K. Leus and P. Van Der Voort, Angew. Chem., 2022, 134, 61 Search PubMed.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- S. Fu, E. Jin, H. Hanayama, W. Zheng, H. Zhang, L. Di Virgilio, M. A. Addicoat, M. Mezger, A. Narita, M. Bonn, K. Müllen and H. I. Wang, J. Am. Chem. Soc., 2022, 144, 7489–7496 CrossRef CAS PubMed.
- C. Krishnaraj, H. S. Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed.
- Y. Zhang, J. Qiu, B. Zhu, M. V. Fedin, B. Cheng, J. Yu and L. Zhang, Chem. Eng. J., 2022, 444, 136584 CrossRef CAS.
- H. Wang, C. Yang, F. Chen, G. Zheng and Q. Han, Angew. Chem., Int. Ed., 2022, 61, 134 Search PubMed.
- C. Wu, Z. Teng, C. Yang, F. Chen, H. B. Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 2110266 CrossRef CAS PubMed.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- L. Li, L. Xu, Z. Hu and J. C. Yu, Adv. Funct. Mater., 2021, 31, 2106120 CrossRef CAS.
- F. Tan, Y. Zheng, Z. Zhou, H. Wang, X. Dong, J. Yang, Z. Ou, H. Qi, W. Liu, Z. Zheng and X. Chen, CCS Chem., 2022, 4, 3751–3761 CrossRef CAS.
- L. Zhai, Z. Xie, C.-X. Cui, X. Yang, Q. Xu, X. Ke, M. Liu, L.-B. Qu, X. Chen and L. Mi, Chem. Mater., 2022, 34, 5232–5240 CrossRef CAS.
- Q. Yang, M. Luo, K. Liu, H. Cao and H. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- R. Paul, S. Chandra Shit, H. Mandal, J. Rabeah, S. S. Kashyap, Y. Nailwal, D. B. Shinde, Z. Lai and J. Mondal, ACS Appl. Nano Mater., 2021, 4, 11732–11742 CrossRef CAS.
- W.-R. Cui, C.-R. Zhang, R.-P. Liang and J.-D. Qiu, J. Mater. Chem. A, 2021, 9, 25611–25620 RSC.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 2003735 CrossRef CAS.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., 2020, 132, 16730–16736 Search PubMed.
- M. Alves Fávaro, D. Ditz, J. Yang, S. Bergwinkl, A. C. Ghosh, M. Stammler, C. Lorentz, J. Roeser, E. A. Quadrelli, A. Thomas, R. Palkovits, J. Canivet and F. M. Wisser, ACS Appl. Mater. Interfaces, 2022, 14, 14182–14192 CrossRef PubMed.
- R. Beranek, Adv. Phys. Org. Chem., 2011, 2011, 786759 Search PubMed.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao and Z. Kang, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed.
- A. Gopakumar, P. Ren, J. Chen, B. V. Manzolli Rodrigues, H. Y. Vincent Ching, A. Jaworski, S. V. Doorslaer, A. Rokicińska, P. Kuśtrowski, G. Barcaro, S. Monti, A. Slabon and S. Das, J. Am. Chem. Soc., 2022, 144, 2603–2613 CrossRef CAS PubMed.
- W.-K. Han, H.-S. Lu, J.-X. Fu, X. Liu, X. Zhu, X. Yan, J. Zhang, Y. Jiang, H. Dong and Z.-G. Gu, Chem. Eng. J., 2022, 449, 137802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04459e |
| This journal is © The Royal Society of Chemistry 2023 |