
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdditive-free red phosphorus/Ti3C2Tx MXene nanocomposite anodes for metal–ion batteries†
Chandrasekar M
Subramaniyam
 a,
Min-A
Kang
a,
Jian
Li
a,
Armin
VahidMohammadi
b and
Mahiar Max
Hamedi
*a
a,
Min-A
Kang
a,
Jian
Li
a,
Armin
VahidMohammadi
b and
Mahiar Max
Hamedi
*a
aDepartment of Fibre and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, 100 44 Stockholm, Sweden. E-mail: mahiar@kth.se
bDepartment of Materials Science and Engineering, A.J. Drexel Nanomaterials Institute, Drexel University, Philadelphia, Pennsylvania, USA
First published on 24th October 2022
Abstract
Herein, we report on scalable, environmentally benign, and additive-free, high-performance anodes for alkali–metal–ion batteries (MIBs, where M = Li+, Na+, K+). The intercalators in these anodes are the red phosphorus (RP) nanoparticles of uniform size (∼40 nm), which are dispersible and blend with water-dispersed Ti3C2Tx MXene, forming a highly viscous aqueous slurry to fabricate additive-free nanocomposite electrodes. We further enhanced their performance using a very low weight percentage of various carbonaceous nanomaterials. Our RP-MWCNT/MXene nanocomposite anodes exhibited enhanced ion transport and low charge transfer resistance, delivering specific capacities of 1293.7 mA h g−1 at 500 mA g−1 and 263.3 mA h g−1 at 2600 mA g−1 for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in Li+ cells, 371.6 mA h g−1 at 500 mA g−1 in Na+ cells, and 732.8 mA h g−1 at 50 mA g−1 in K+ cells. Our work shows a path towards fabricating nanoarchitectured electrodes using sustainable materials to eliminate inert polymer binders, toxic processing solvents, and rare earth elements from the battery fabrication process for next-generation alkali–metal–ion batteries.
000 cycles in Li+ cells, 371.6 mA h g−1 at 500 mA g−1 in Na+ cells, and 732.8 mA h g−1 at 50 mA g−1 in K+ cells. Our work shows a path towards fabricating nanoarchitectured electrodes using sustainable materials to eliminate inert polymer binders, toxic processing solvents, and rare earth elements from the battery fabrication process for next-generation alkali–metal–ion batteries.
1. Introduction
There is an urgent need to develop high energy density storage devices, ideally batteries, for (1) stationary grid storage1 to harness and store intermittent renewable energy resources such as sun and wind and (2) develop plug-in electric vehicles (EVs) as the transportation system is being electrified. These factors have created a global demand for ∼30 GW h battery-based energy storage each year by 2040.2 The lithium–ion battery (LIB) is the best available battery technology, but the electrodes contain rare earth metals, notably lithium and cobalt, which are neither the constituents of biomass nor cost effective. Hence, it is high time to use earth-abundant and cheap elements in the LIB electrode architecture and move beyond Li technologies.1,3Recent advances in nanotechnology have offered new opportunities for Na- and K-based batteries,4,5 herein referred to as alkali–metal–ion-batteries (MIBs). Besides these, phosphorous (P), which is also abundant and mined in large quantities for fertilizers, presents opportunities to be used as an anode in different battery chemistries.3,4,6–12 Pure phosphorus exists in 3 allotropes: white (most unstable), red (amorphous, room temperature stable), and black (layered crystalline and energy consuming to synthesize).5,9 Among these, red phosphorus (RP) has attracted interest because of its high theoretical Li, Na, and K storage capacities (e.g. 2596 mA h g−1 (P + 3M+ ↔ M3P + 3e−) where M = Li+ and Na+ and 865 mA h g−1vs. K).6 However, RP's low conductivity and high volume change upon M+ intake have kept RP from reaching and retaining its theoretical capacities. Conventional additives widely used in the fabrication of electrodes for LIBs (i.e. binders such as carboxymethyl cellulose or polyvinylidene fluoride and conductive carbon) have failed to mitigate these shortcomings during battery cycling. Therefore, new electrode architectures are still needed to realize high capacity practically in long-cycling RP anodes.
Learning from the success of silicon anodes,13–16 one tactic is to reduce the RP particle size to the nanometer range, which provides shorter and faster ion transport, enhancing their rate capability, but the problems associated with conductivity and volume change still remain unsolved. To overcome these shortcomings, RP nanoparticles were covalently bonded onto high-surface–area carbon materials (carbon nanotubes, graphene, etc.). Though promising, there are still issues that limit the electrochemical performance of these composites, mainly due to the random distribution of uneven particle sizes over the carbonaceous nanomaterials.17
In this study, we addressed both these challenges by fabricating stable dispersions of RP nanoparticles in ethanol, and further exploited the water dispersions of Ti3C2Tx MXene as conductive binders to form nanocomposite anodes for MIBs. We also explored the addition of 2 other low-dimensional materials, multi-walled carbon nanotubes (MWCNTs) and reduced-graphene-oxide (rGO), to the RP/MXene composite to further enhance its cycle stability.
The wettability and mechanical property of aqueous Ti3C2Tx nanosheets enabled the fabrication of a robust and continuous nanoscopic conductive network, wherein the high-capacity RP nanoparticles are held in contact with the current collector during cycling. Using various electrochemical analytical techniques, we found that the RP-MWCNT/MXene anode composite showed the best long cycling stabilities of 1293.7 mA h g−1 at 500 mA g−1 and 263.3 mA h g−1 at 2600 mA g−1 for 10000 cycles in lithium–ion cells and a better rate of 371.6 mA h g−1 at 500 mA g−1 in sodium–ion cells. The production and use of nanoparticulate RP in water enables, for the first time, nanocomposite anodes fabricated from viscous MXene slurries for high performance anodes in alkali metal–ion batteries (MIBs).
2. Results and discussion
2.1 Synthesis of RP nanoparticles and conventional additive-free composites
Large-scale synthesis of stably dispersed, uniformly sized RP nanoparticles in ethanol can be used for the self-assembly of various nanocomposites with other nanomaterials. Here, we developed a route, shown schematically in Fig. 1a, which starts with bulk RP containing rigid and uneven ∼3–5 μm sized particles (Fig. S1a and b, ESI†) using a scalable ball-mill technique. The ball-milled RP contained particles of drastically reduced sizes, between ∼150–300 nm, congregated into porous particles (Fig. S1c and d, ESI†). These particles were subsequently dispersed in ethanol using high-energy probe sonication to obtain uniformly sized RP nanoparticles, as confirmed by morphology and particle-size distribution using field emission scanning electron microscopy (FESEM) (Fig. 1b and Fig. S1e, ESI†) and transmission electron microscopy (TEM) images (Fig. S1f, ESI†). The RP nanoparticles had an average particle size of ∼40 nm, with a Gaussian distribution of standard deviation 0.23 nm (Fig. 1c). They retained their amorphous phase, as shown by the selected area electron diffraction (SAED) pattern (Fig. S1f insert, ESI†). Ball milling also induced defects and active sites, leading to an enhanced surface area of 25.49 m2 g−1 compared to 1.47 m2 g−1 for bulk RP.9 This 17-fold enhancement of the surface area could help promote electrolyte percolation and electron transport.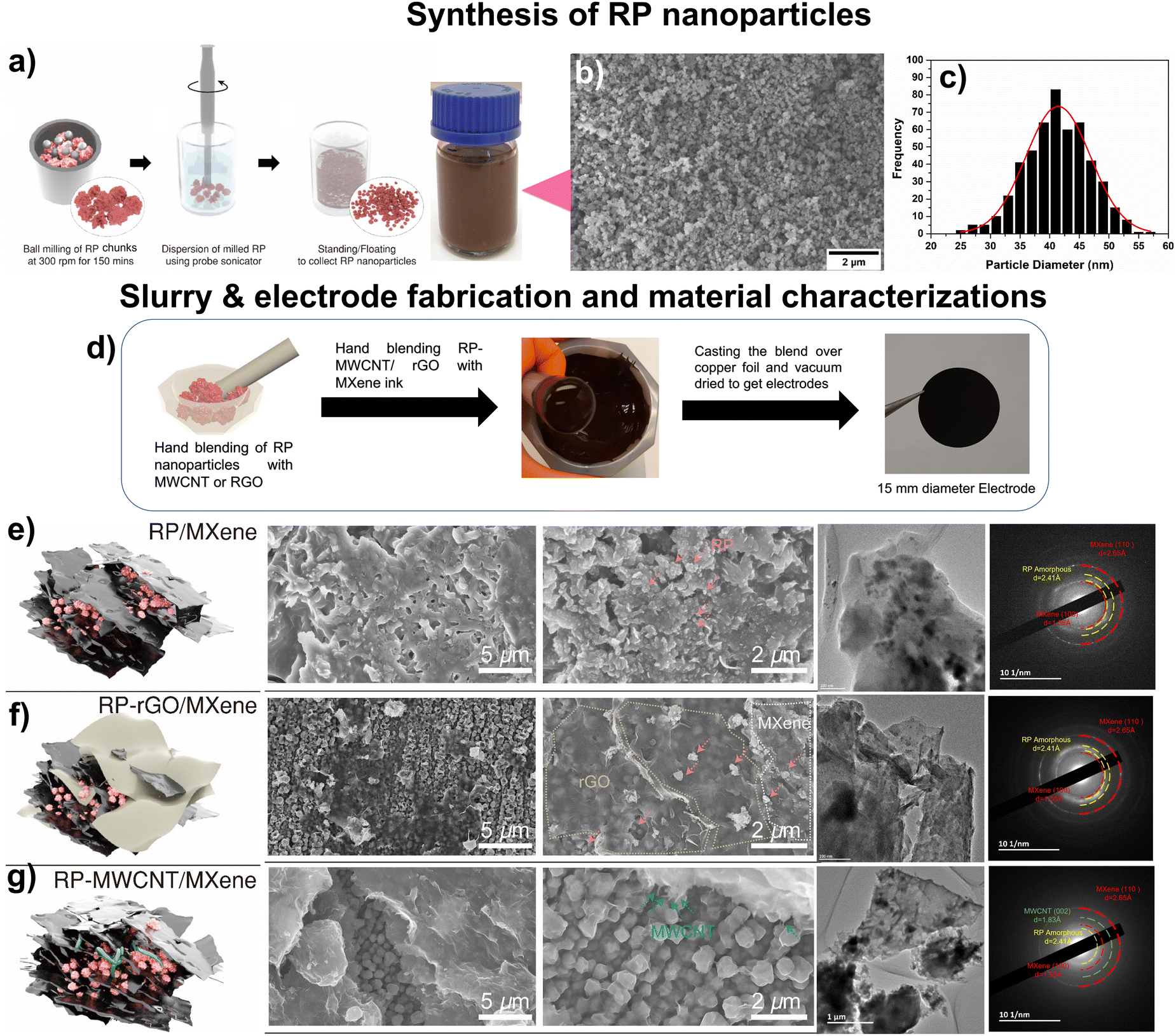 | ||
| Fig. 1 (a) Schematic of red phosphorus (RP) nanoparticle synthesis using ball-milling technique. (b) FESEM depicting the uniform distribution of RP nanoparticles and (c) corresponding histogram showing the particle-size distribution. (d) Scheme showing the electrode preparation using mortar and pestle. The morphology, particle distribution and phase purity were obtained using FESEM, TEM and SAED pattern, respectively. (e) FESEM images of RP/MXene nanocomposite wherein the RP nanoparticles are embedded between the MXene sheets, quite similar to (f) RP-rGO/MXene, while in (g) RP-MWCNT/MXene nanocomposites, the RP nanoparticles are engulfed completely by the MWCNTs and then covered with MXene sheets, resulting in robust electrodes. These morphological observations were well substantiated by the corresponding TEM images and SAED patterns. | ||
To fabricate the nanocomposites, we used MXenes, a class of 2-dimensional (2D) nanosheets with the general formula Mn+1XnTx where M = early transition elements, X = C and/or N, Tx = surface functionalities such –O, –OH, etc., and n = 1, 2, 3, or 4.20 We used aqueous MXene ink as both the conductive additive and the binder to form self-assembled nanocomposites of MXenes and our RP nanoparticles. We also studied the effects of 1D (MWCNT) and 2D (rGO) carbonaceous materials added to these MXene inks to assemble a continuous conductive network wherein the high-capacity RP nanoparticles are held in contact with these low-dimensional materials (Fig. 1d).
Atomic force microscopy (AFM) images confirmed that the MXenes consisted of individual nanosheets with a thickness and lateral dimension of 2 nm and 200 nm, respectively (Fig. S2, ESI†). The hydrophilic nature of MXene ink exhibits a high negative surface charge that is essential for the surface interaction and dispersion of RP nanoparticles over the respective low-dimensional carbonaceous materials whose morphologies are shown in Fig. 1e–g. From the FESEM and TEM images, we confirmed that the RP nanoparticles were randomly self-assembled and stacked into layers to form the RP/MXene nanocomposites (Fig. 1e). For the RP-rGO/MXene nanocomposites, however, we observed that the RP nanoparticles were dispersed over the rGO sheets (Fig. S3a–c, ESI†) and subsequently stacked between the MXene nanosheets (Fig. 1f). The same stacking trend could be seen in the RP-MWCNT/MXene nanocomposites; however, here the MWCNT completely engulfed the RP nanoparticles (Fig. S4a–d, ESI†), providing enhanced particle-to-particle interconnectivity when blended with MXene ink, as observed from the FESEM and TEM images (Fig. 1g). These observations agreed well with their respective EDS compositional analyses (Fig. S3d and S4e, ESI†).
Using these three differently morphological, self-assembled nanocomposites, we analyzed the phase purity, surface composition, and structure of all the electrode nanocomposites using XRD (Fig. 2a and b), Raman spectroscopy (Fig. 2c) and X-ray photoelectron spectroscopy (XPS, Fig. 2d and e).
 | ||
| Fig. 2 (a) XRD with (b) enlarged portions between 5–25° of MXene, RP, RP/MXene, rGO, RP-rGO/MXene, MWCNT and RP-MWCNT/MXene nanocomposites. The encircled section in (b) depicts the peak shifts from 7.46° in pure MXene to 6.44° in RP-MWCNT/MXene composites attributed to the enhanced blending of RP and MWCNT with MXene ink. These observations, supported by the Raman spectra in Fig. 2c, demonstrate their structural and phase purities. For MXene, the region 230–470 cm−1 represents in-plane (Eg) vibrations of surface groups attached to the Ti atoms which help in the surface interactions and self-assembly to make the composites while the region 580 cm−1 (marked as *1) is related to the carbon vibrations used to identify the surface atoms in electrochemical studies. The region 300–500 cm−1 representing B1, A1 and E1 corresponds to red phosphorus. The slight shifts of peaks corresponding to the MXene Eg and the MWCNT D and G bands show surface interactions as depicted by XRD. (c) XPS survey spectra of MXene and its composites. (d) High resolution spectra of RP-MWCNT/MXene composites for C 1s, P 2p, Ti 2p, and O 1s. | ||
Fig. 2a and b depict the XRD patterns of MXene, the RP nanoparticles and their 3 nanocomposites in addition to the MWCNT and rGO components. The characteristic (0002) peak at 2Θ = 7.46° signifies the successful synthesis of high purity MXene nanosheets while those at 2Θ = 15.41° and 2Θ = 33.12° demonstrate the amorphous phase and pure phase of RP nanoparticles, leaving aside the substrate peaks. The RP/MXene, RP-rGO/MXene and RP-MWCNT/MXene exhibited similar XRD peaks with slight shifts in peak positions toward lower angles. For instance, in the RP-MWCNT/MXene nanocomposite, the characteristic (0002) peak of MXene is positioned at 2Θ = 6.44° as compared to pure MXene nanosheets. However, the MXene (0002) peak is weaker in the composites, as it is present in smaller amounts and the flakes have a higher degree of misorientation due to the blending with RP nanoparticles and highly porous multi-components like rGO and MWCNT. The XRD analysis is well supported and corroborated by Raman spectroscopy (Fig. 2c) and X-ray photoelectron spectroscopy (Fig. 2d and e). The sharp peak of MXene at 580 cm−1 depicts highly ordered C atoms in the few-layered Ti3C2Tx MXenes after delamination.21 The three characteristic peaks of RP nanoparticles at 352, 393 and 458 cm−1 correspond to high purity monoclinic RP. The rGO spectrum exhibited broad D and G peaks at 1346.6 cm−1 and 1593.8 cm−1, respectively, while for MWCNT, the broad G peak is positioned at 1580 cm−1. The peak intensity and position in the composites remained the same between 200–350 cm−1 while we observed a remarkable red-shift of the rGO G-band from 1593.76 to 1584.84 cm−1 which was assigned to the π–π interaction of electron transfer from P to C in the graphite plane.9 The amplitude of the RP Raman peak vanished in RP-MWCNT/MXene, probably due to the layered RP nanoparticles stacking with MXene nanosheets as manifested in the FESEM, TEM images and XRD patterns (Fig. 2a and b).
We used X-ray photoelectron spectroscopy (XPS) to study the chemical interactions and electronic states of the various elements (mainly C, O, P, Ti) present on the surface of the composites. Fig. 2d and e show the complete XPS survey spectra and high-resolution (or medium-resolution detail) spectra of the C 1s, O 1s, Ti 2p and P 2p signals for MXene and various composites. The pristine MXene sample contains mainly the four elements C, Ti, O and F while the survey spectra of composites also exhibited the presence of the element phosphorus (P). The high-resolution C 1s signals have several peaks in the region from 285 eV to 289 eV, corresponding to different carbon functional groups, like C–C, C–H and/or carbon–oxygen. The peak at ca. 282 eV (281–283 eV) corresponds to carbides, particularly to the Ti–C bond in Ti3C2Tx (MXene) at 281.6 eV.12,22 The Ti 2p peak at about 455.5 eV is most likely from the added Ti3C2Tx MXene.
All the Ti 2p spectra for each composite are similar and have the Ti 2p3/2 peak position at 455.5 eV.22,23 The P 2p signal is shown as two peaks: the minor peak at about 130 eV corresponds to unoxidized P, such as in red phosphorous, and the major peak at the higher binding energy of about 133–135 eV corresponds to oxidized P. For the O 1s signal, the major part of the total oxygen signal most likely corresponds to inorganic oxides, particularly Ti–O–P at 456.4 eV.22,24–26 From Table S1 (ESI†), it can been seen that the intensity of the P–O peak is more pronounced in RP-MWCNT/MXene (90%) than in the other composites, substantiating that P–O bonds are formed due to the formation of Ti–O–P bonds while compositing with MXene. Also, it is observed that P–O binds at a higher energy (133–135 eV) than P–P (130 eV). The P–O bond formation is vital for long cycling stability. It is anticipated that the P–P bonds are formed during the charge/discharge process, which results in huge volume change leading to loss of contact with the carbonaceous material and the current collector and results in capacity fade. Meanwhile, the P–O (in Ti–O–P) bonds with higher energies (335–585 kJ mol−1) enable robust particle–particle binding with the electronic conducting Ti–O matrix which helps in long cycling stability.12 Therefore, the Ti–O–P bonds formed in the composites are vital in integrating the particle–particle network in the metallic conducting Ti–O matrix, enabling a robust electrode with better electrochemical performances. More detailed XPS peak calculations are provided in ESI.†
2.2 Electrochemical characterizations
We employed these RP-MXene composites as negative electrodes for various MIBs (where M = Li+, Na+, K+) half cells in which RP imitates a ‘conversion reaction’ involving LIB (Li+ + 3P ↔ Li3P + 3e−),9,11,12 SIB (Na+ + 3P ↔ Na3P + 3e−),1,4,11 or KIB (K+ + P ↔ KP + e−)6,7 based on their respective electrochemical potentials. We systematically studied the electrochemical storage capabilities of the RP/MXene, RP-rGO/MXene and RP-MWCNT/MXene nanocomposites against alkali metals and elucidated their electrochemical properties such as galvanostatic charge–discharge (CV, rate test), their ion diffusivity in each system using the galvanostatic intermittent titration technique (GITT), and their charge transfer resistance using electrochemical impedance spectroscopy (EIS). Based on these electrochemical performances, we optimized the best possible material design using low-dimensional carbonaceous materials for enhanced, long-term electrochemical activity. To avoid ambiguity, we report the specific capacity in terms of weight percentage (i.e. 70%) in all these composites, taking into account the 2D and 1D carbonaceous content as active material in each of their composites and neglecting the specific capacity contributed by MXene, which is comparatively low (Fig. S5, ESI†).Lithium–ion battery (LIB). We tested the electrochemical activity of these three RP-MXene nanocomposites in a half-cell coin cell configuration against lithium metal between 0.002–3 V. Fig. S6a and b (ESI†) show various electrochemical characterization plots such as cyclic voltammograms (CVs), charge–discharge curves and rate tests (Fig. 3d and g). In the CVs (Fig. S6a, ESI†), the RP/MXene exhibited three cathodic peaks while there were two cathodic peaks for the RP-MWCNT/MXene (Fig. 3b) and RP-rGO/MXene nanocomposites. RP/MXene exhibited a discharge peak at 1.28 V corresponding to the reduction of TiO2 in the MXene, while the same was not observed in other composites, indicating an excellent blending leading to surface interaction between the low-dimensional carbonaceous materials and MXene nanosheets. The onset of the conversion reaction involving a multiple e− transfer of P with Li+ begins at 1 V. Further discharge to 0.002 V involves the conversion of the P-rich phase to a Li-rich phase with LixP corresponding to first discharge capacities of 1836.2, 2784 and 2219.8 mA h g−1, respectively, for the RP/MXene, RP-rGO/MXene and RP-MWCNT/MXene nanocomposites (Fig. 3c and Fig. S6b, ESI†). There was a subsequent formation of a resistive solid–electrolyte interface (SEI) at the electrode–electrolyte interface upon reaching 0.002 V due to electrolyte decomposition. During charging, there were two distinct peaks in all composites at 1.2 V and 1.28 V corresponding to the delithiation of the Li-rich phase (LixP) to a P-rich phase (Li(x−δ)P) upon reversing the polarity that correspond to the first charge capacities of 1300.6, 1673.1 and 1562.1 mA h g−1, respectively, for RP/MXene, RP-rGO/MXene and RP-MWCNT/MXene. The CV curves were identical in the subsequent cycles, representing reversibility upon achieving stable state. We cycled the cells at various current densities from 50–2600 mA g−1 for 100 cycles. The RP-MWCNT/MXene composite exhibited enhanced fast charge–discharge capability, reaching its highest value of 1293.7 mA h g−1 at 260 mA g−1 (Fig. 3d). For long cycling, RP-MWCNT/MXene delivered an excellent high capacity of 1077.6 mA h g−1 at 260 mA g−1 (for 500 cycles) and 263.3 mA h g−1 at 2600 mA h g−1 (1C) even after 10
000 cycles (Fig. 3a). To the best of our knowledge, these results are among the best for additive-free RP electrodes at rate of 1C for 10000 cycles (Table S2, ESI†).
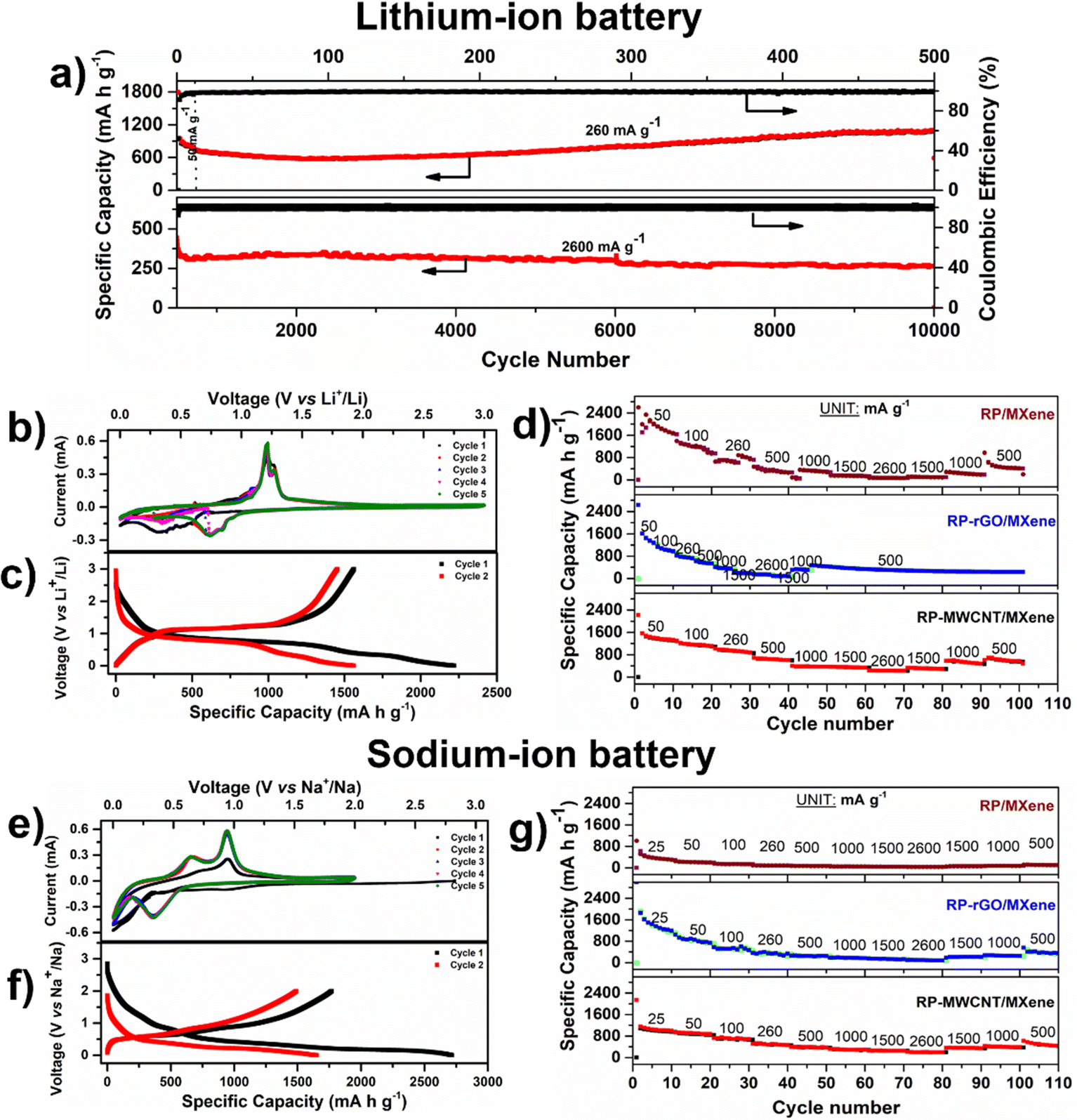 | ||
| Fig. 3 Electrochemical performance of RP composites as anodes for LIB cycled between 0.002–3 V using 1 M LiPF6 in 1:1 (v/v) EC:DEC electrolyte and for SIB from 0.002–2 V using 1 M NaPF6 in 1:1 (v/v) EC:DEC + 10 vol% FEC electrolyte. (a) Long cycle stability plot of RP-MWCNT/MXene composites at 50, 260 and 2600 mA g−1 for 10000 cycles of lithium battery. (b and e) Cyclic voltammetry of RP-MWCNT/MXene composites for 5 cycles obtained at 0.1 mV s−1 and corresponding (c and f) charge–discharge of 1st and 2nd cycles at 50 mA g−1. (d and g) Rate capability plots of the nanocomposites at various current densities (mA g−1). The test procedure for RP-rGO/MXene used the same current densities (CDs) with 5 cycles each and shows extended cyclability at 500 mA g−1 up to 100 cycles. | ||
Sodium–ion battery (SIB). Only a few reports exist on RP-MXene-based materials as anodes for SIB,11,22,25–28 as shown in Table S2 (ESI†). Analogous to the LIBs, we tested these composites as anodes for SIBs cycled between 0.002–2 V against sodium. According to the CV plots in Fig. S6c (ESI†), all composites exhibited one broad cathodic peak at 0.35 V, starting from 0.55 V and extending to 0.15 V. This peak is attributed to the conversion plateau wherein the sodium-rich phase was formed in accordance with the conversion reaction Na+ + x P ↔ NaxP + xe− which corresponds to first specific discharge capacities of 1350.5, 2075.3 and 2716.2 mA h g−1, respectively, for RP/MXene, RP-rGO/MXene (Fig. S6d, ESI†) and RP-MWCNT/MXene composites (Fig. 3f). There were two anodic peaks upon charging at 0.64 V and 0.95 V which correspond to the desodiation of the Na-rich phase (NaxP) to a P-rich phase (Na(x−δ)P) upon reversing the polarity that correspond to first specific charge capacities of 667.2, 1078.9 and 1767.8 mA h g−1, respectively. The irreversible trend disappeared in the subsequent cycles, demonstrating reversibility upon achieving a stable state. With the enhanced Na+ diffusivity, the composites exhibit similar trends in the rate capability test when cycled at different current densities, as shown in Fig. 3g. We cycled the cells at various current densities from 25–2600 mA g−1 for 100 cycles. The RP-MWCNT/MXene nanocomposites exhibited enhanced charge–discharge capacities of 371.6 mA h g−1 even after 100 cycles at 500 mA g−1 compared to other composites. Our work shows the highest initial discharge (2716.2 mA h g−1) and charge capacity (1767.8 mA h g−1) compared to previous reports (Table S2, ESI†).
Potassium–ion battery (KIB). Owing to the low electrochemical potential of K metal (μo = −2.92 V) and its large atomic radius, KIB exhibits lower specific capacity compared to SIB and LIB. These three nanocomposites were tested as anodes for KIBs cycled between 0.002–2.5 V. The CVs (Fig. S6e, ESI†) showed a significantly broad cathodic peak between 0.5–1.0 V pertaining to the conversion of the P-phase to a potassium-rich phase followed by SEI formation at lower voltage, which corresponds to first specific discharge capacities of 1540.5, 1629, and 2341.6 mA h g−1, respectively, for RP/MXene, RP-rGO/MXene and RP-MWCNT/MXene (Fig. S6f, ESI†) and their respective first specific charge capacities were 475.8, 400.1, and 732.8 mA h g−1. We report the highest discharge (2341.6 mA h g−1) and charge capacities (732 mA h g−1) for a RP-MXene based composite as anode for KIB compared to other reported RP-carbon-based electrodes (Table S2, ESI†).
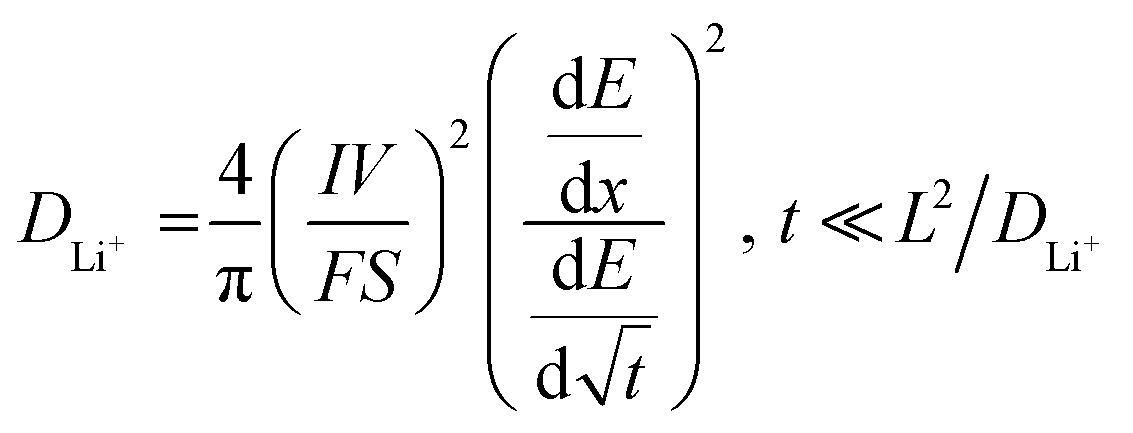 | (1) |
485 C mol−1), I is the applied current (A), S is the surface area of the electrode (cm2) and L is the diffusion length (cm).
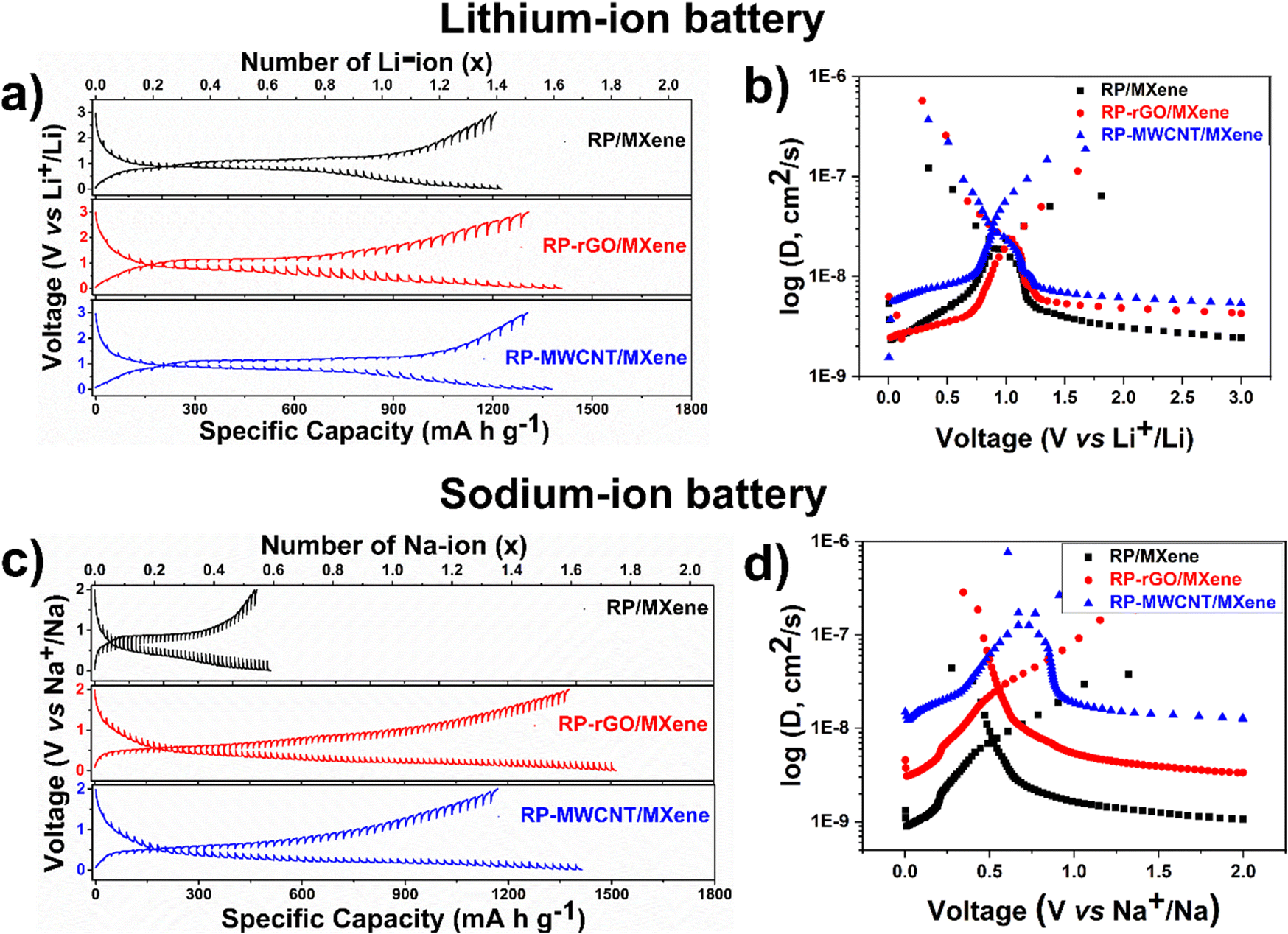 | ||
| Fig. 4 GITT charge–discharge plots and corresponding M+ chemical diffusion coefficients in the composites at various voltages: (a and b) LIB, (c and d) SIB. GITT was measured after second charge–discharge cycle to evade the effects of solid–electrolyte interface formed during first cycle. | ||
Using this equation, we estimated the Li–ion diffusivities against each potential, as shown in Fig. 4a and b. The Li+ diffusion in the active RP nanoparticles varied between 10−9 to 10−6 cm2 s−1 with each curve spiralling downwards. During discharge, the Li+ diffusivity was enhanced at voltages below 1 V pertaining to the conversion reaction (lithiation) in the descending order of RP-MWCNT/MXene > RP-rGO/MXene > RP/MXene, while the RP-rGO/MXene composite showed slightly enhanced diffusivity during the charging process. Accordingly, the Li+ diffusivities (cm2 s−1) were 3.19 × 10−8 for RP-MWCNT/MXene, 2.35 × 10−8 for RP-rGO/MXene, and 1.92 × 10−8 for RP/MXene. The lithium diffusivity is greatly enhanced in these composites compared to those previously reported.9,12
We observed similar trends for Na+ and K+ diffusivities in SIBs and KIBs. Using eqn (1), we also calculated Na–ion diffusivities in the composites and plotted them against various potentials, as shown in Fig. 4c and d. During discharge, the Na+ diffusivity was enhanced at voltages below 0.75 V pertaining to the conversion reaction (sodiation) in the ascending order of RP/MXene < RP-rGO/MXene < RP-MWCNT/MXene and showed the same trend during the charging process. Accordingly, the Na+ diffusivities (cm2 s−1) were calculated to be 7.53 × 10−9 (RP/MXene) < 2.70 × 10−8 (RP-rGO/MXene) < 1.57 × 10−7 (RP-MWCNT/MXene). For the KIBs, the corresponding GITT and K+ diffusivities at various potentials during discharge are plotted in Fig. S7a and b (ESI†). During discharge, the K+ diffusivity was enhanced at lower voltages pertaining to the conversion reaction in the descending order of RP-MWCNT/MXene > RP-rGO/MXene > RP/MXene with values varying between 5 × 10−9–6 × 10−7 cm2 s−1.
These observations show that the metal–ion diffusivities are enhanced by using non-conventional highly viscous, water-dispersed MXene ink as an additive along with highly porous carbonaceous materials (rGO and MWCNT) providing a continuous network for electron transfer and the shortest path for metal–ion transport during cycling.
2.3 Designing of Electrode for long cycle stability
In electrochemical performance, in terms of cycle stability, ion diffusivity and ohmic overpotential (from GITT plot) and charge transfer resistance, the RP(0D)-MWCNT(1D)/MXene (2D) composites outperformed the others. As represented in Fig. 5, the 1D MWCNT serves the following purposes: (1) it encapsulates the inert and non-conducting RP nanoparticles and thereby improves the conductivity for electron transport; (2) it helps to relieve the strain induced due to volume change during the cycling process while keeping the RP nanoparticles intact; (3) in the composite with MXene, MWCNT helps prevent stacking of the MXene sheets during cycles, thereby maintaining the conductive medium throughout the electrode and providing less charge transfer resistance for faster diffusion pathways for the electrolyte and ions, as observed in the GITT and EIS data. This is more evident from the post-cycling morphological analysis using FESEM, shown in Fig. S9a and b (ESI†). Even after 10000 cycles, the RP particles are still intact and observable beneath the MXene sheets whose elemental compositions are identified using EDS (Fig. S10 and S15, ESI†), forming a connectivity network throughout the electrode. In the RP-rGO/MXene and RP/MXene nanocomposites, the RP particle active material loses interfacial contact with the conducting networks rGO and MXene, probably due to the stacking of nanosheets, as seen in the FESEM of electrodes after 100 cycles (Fig. S9c–f, ESI†) and the corresponding compositional analysis (Fig. S11 and S12, ESI†). This affects the overall, long-term cycling performance of these electrodes, as we also observed for SIBs (Fig. S13–S15, ESI†).
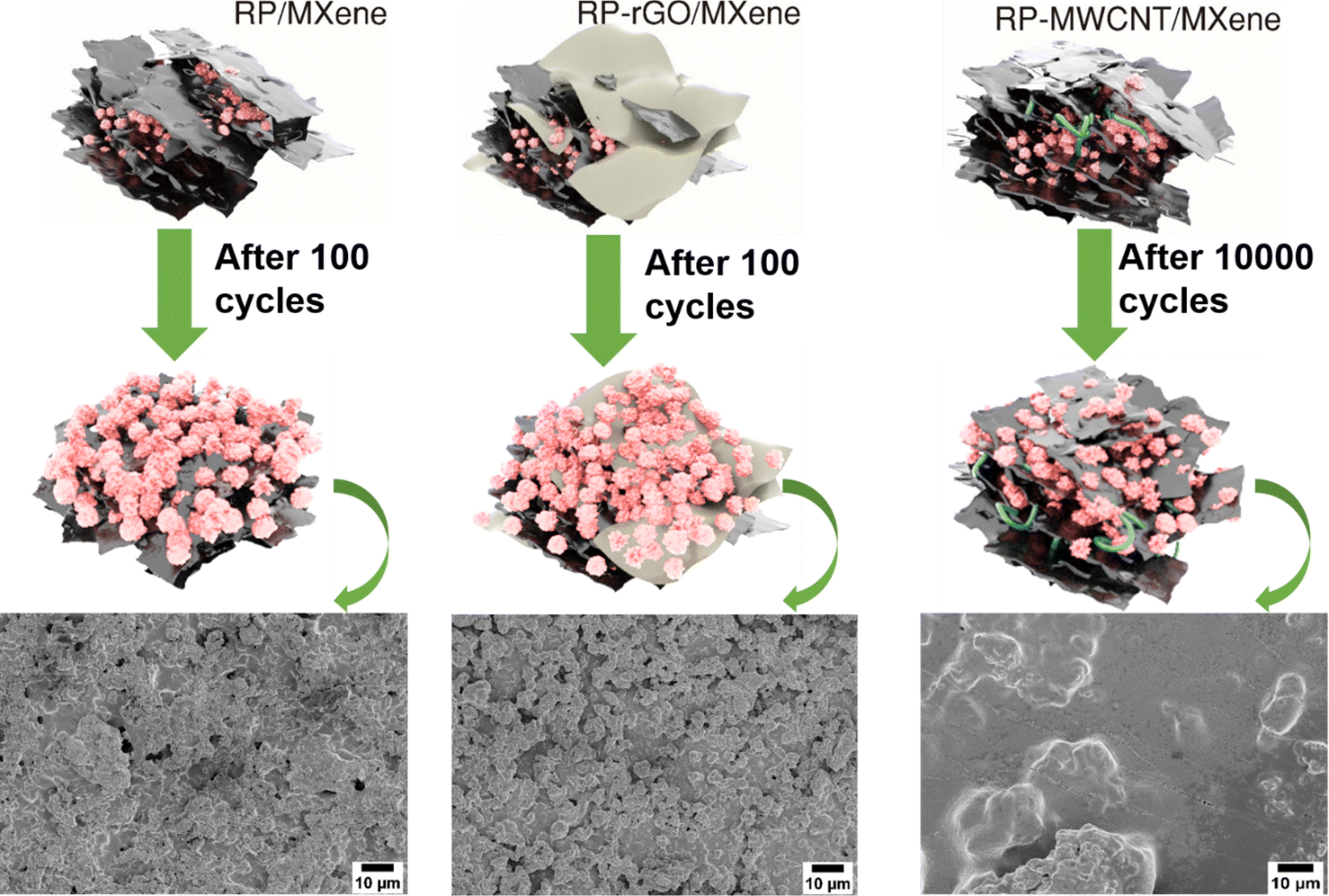 | ||
| Fig. 5 Schematic representations of morphological changes after cycling and their corresponding ex situ SEM images. | ||
3. Conclusions
We successfully produced RP nanoparticles by a facile, scalable ball-milling technique and uniformly dispersed them in eco-friendly polar solvent. The uniformity of the nano-sized (∼40 nm), stably dispersed RP particles with high surface area (25.49 m2 g−1) allowed us to fabricate conventional additive-free composite battery anodes by mixing them with Ti3C2Tx MXene, also dispersed in water. We added small amounts of other low-dimensional carbon nanomaterials to enhance the life cycles of our anodes. The highly viscous water-dispersed MXene ink serves dual functionalities: it binds the RP nanoparticles and highly porous carbon (rGO/MWCNT) to provide a continuous network for the fastest electron transfer and the shortest path for ion transport. Among these electrodes, RP-MWCNT/MXene nanocomposites exhibited excellent long-cycle stability due to the enhanced ionic transport and low electronic resistance stemming from the synergetic effect of MWCNT engulfing the RP nanoparticles and acting as a bridge to prevent the stacking of MXene nanosheets. We achieved LIBs performing at a 1C rate for 10000 cycles and the highest discharge–charge capacities for SIBs and KIBs compared to other reported RP-MXene anodes (Table S2, ESI†). Though these initial specific capacities for SIB and KIB are promising, further research is needed on their electrode compositions to enhance their lifetimes.
Our work shows a path toward the rational design and fabrication of advanced self-assembled nanocomposite electrodes29 from water, using an earth abundant element, RP in this case, as the intercalating agent and MXene as a binder and electronic conductor. Future work can optimize the geometry and composition of new MXenes to decrease their concentration (we think that they could be as low as 1–3 wt% based on preliminary results) and to decrease the stability of the electrodes. MXene can also be used as the cathode30 with water-dispersed intercalators which we think can be fabricated using similar strategies as the ones presented here. These strategies combined can eliminate toxic solvents, binders and rare earth materials from the battery fabrication process and enable a next generation of green energy storage devices.
4. Experimental details
All chemicals were purchased from Sigma Aldrich and used without any further purification unless otherwise mentioned.4.1 Synthesis of red phosphorus nanoparticles (RP)
RP chunks/bulk particles were ball milled under an inert argon atmosphere with a tungsten-carbide ball to a powder weight ratio of 20:1 at 300 rpm for 150 minutes. The powders were collected, dispersed in ethanol using high-energy probe sonication, and centrifuged at 1500 rpm for 30 minutes. The uniformly dispersed stable RP nanoparticles were collected from the supernatant.
4.2 Synthesis of reduced graphene oxide (rGO)
First, graphene oxide (GO) was synthesised by Tour's improved method18 wherein KMnO4 (18 g) was slowly added in 6 equal portions to a 9:1 acid mixture containing 360 mL H2SO4 and 40 mL H3PO4, followed by 3 g of graphite flakes (Asbury Carbons, USA, Grade: 3805), and the reaction mixture was heated at 50 °C with continuous stirring for 12 h. The room temperature cooled mixture was then poured onto ice (400 mL) containing 3 mL of 30% H2O2 and washed several times at 6000 rpm with Millipore water. The final product was freeze dried to obtain GO. The as-prepared GO was dispersed in Millipore water at 2 g L−1 under vigorous stirring. The mild reducing agent ascorbic acid of equal weight ratio or equal quantity to GO was added and the stirring was maintained at 50 °C for 10 h. The resultant reduced solid sediments were collected and washed several times with Millipore water and ethanol, then vacuum dried at 50 °C overnight to obtain reduced graphene oxide (rGO).
4.3 Synthesis of RP nanocomposites
The low-dimensional carbonaceous materials 1D MWCNT (Sigma Aldrich) and 2D reduced graphene oxide (rGO) were used to make nanocomposites with the RP nanoparticles. The RP nanocomposites contain fixed 7:3 weight ratios of RP nanoparticles to MWCNT and rGO. The nanocomposites were prepared by hand grinding, using a mortar and pestle of appropriate quantity, RP nanoparticles and low dimensional carbonaceous materials (1D MWCNT, 2D rGO) for ∼40 minutes. The samples were marked as RP-MWCNT and RP-rGO, accordingly.
4.4 Preparation of MXene ink
In a typical delamination of the MAX phase (Ti3AlC2),19 2 g of LiF was completely dissolved in 40 mL of 6 M HCl solution under constant stirring for 15 mins followed by the slow addition of 2 g of MAX phase powder at a rate of 200 mg min−1 in an ice bath to avoid excessive heat generation during the addition. The etchant solution was then maintained at 50 °C for 24 h at 550 rpm under constant stirring. The room temperature cooled etchant was washed several times with Millipore water until its pH ≈ 6. The obtained MXene powders were dispersed in deaerated Millipore water and probe sonicated at 35% amplitude for 1 h in an ice bath. The dispersed solution was centrifuged at 3500 rpm for 1 h and the final supernatant was collected and labelled as MXene. To obtain MXene ink, the obtained supernatant MXene dispersion was centrifuged at 5000 rpm and re-dispersed in 10 mL Millipore water by hand shaking vigorously for 15 minutes. Its concentration was determined from the net weight of the MXene film obtained by the vacuum filtration of a known volume of MXene ink using a Celgard membrane.4.5 Electrochemical studies
CR2032 half-cell configured coin cells were used to test the electrochemical performances of the as-prepared nanocomposites against lithium, sodium and potassium for LIB, SIB and KIB, respectively, in an argon-filled glove box (MBraun, Germany) maintained at <0.5 ppm H2O and <0.5 ppm O2. The RP nanocomposites (RP nanoparticles, RP-MWCNT and RP-rGO) were blended with MXene ink at a weight ratio of 7:3 respectively using a mortar and pestle, tape cast over a copper current collector using a doctor's blade, and vacuum dried at 80 °C overnight. The dried electrodes were cut into 15 mm diameter circular discs each weighing about ∼0.8 mg cm−2 (net wt). Half-cell type coin cells were fabricated using these electrodes as the working electrode while lithium, sodium and potassium metals were used in the LIB, SIB and KIB, respectively, as counter and reference electrodes. The electrodes were impregnated with 1 M LiPF6 in 1:1 (v/v) EC:DEC for LIB, 1 M NaPF6 in 1:1 (v/v) EC:DEC + 10 vol% FEC for SIB and 0.8 M KTFMSi in DME for KIB. The galvanostatic cycling of the assembled cells was tested using a Landt multichannel battery tester (Landt, China CT2001A) discharging from OCV to 0.002 V and charging to 3 V for LIB while SIB and KIB were cycled, respectively, between 0.002–2 V and 0.002–2.5 V at a specific current density (mA g−1). The cyclic voltammetry (CV) was performed at a scan rate of 0.1 mV s−1 and potentiostatic electrochemical impedance spectroscopy (PEIS) was performed in the frequency range of 0.1 MHz to 10 mHz against lithium, sodium and potassium using a BioLogic electrochemical work station (VMP3, France).
4.6 Material characterizations
The phase purities of the RP nanoparticles, MXene, and RP-MWCNT and RP-rGO nanocomposites were identified using a PANalytical X’Pert PRO powder X-ray diffractometer with Cu Kα irradiation scanning from 10–90° with a 5° min−1 scan rate and 0.02° step size. Raman spectroscopy (Jobin Yvon, HR800 UV with 514 nm laser wavelength) was employed to determine the structures and compositions of composites. Morphology and elemental composition were obtained using field emission scanning electron microscope (FESEM, Hitachi SEM4800, Hitachi Corp., Japan) coupled with energy dispersive spectroscopy (EDS). Particle size and distributions were analysed using a transmission electron microscope (TEM, JOEL JEM 2010F, Japan) operated at 200 kV with 10 Å resolution and were processed with Gatan Micrograph software. The MXene nanosheet's depth profile was obtained using atomic force microscope (AFM) with a NanoSCope V controller (Bruker Corp., USA). Surface area and pore-size distribution were studied using the Brunauer–Emmett–Teller method (BET, Micromeritics ASAP 2020). X-ray photoelectron spectra (XPS) were recorded using a Kratos AXIS UltraDLD X-ray photoelectron spectrometer (Kratos Analytical, Manchester, UK). The samples were analysed using a monochromatic Al X-ray source. The analysis area was below 1 mm2, with most of the signal coming from an area of about 700 μm × 300 μm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Chandrasekar M Subramaniyam and Mahiar Max Hamedi acknowledge the Knut and Alice Wallenberg Foundation for funding to carry out the research. RISE Research Institutes of Sweden, Department of Material and Surface Design, is thanked for performing the XPS analyses, Mikael Sundin for help with the XPS runs and Dr Marie Ernstsson for help evaluating the results and interpretation of data.References
- E. Quartarone, T. Eisenmann, M. Kuenzel, C. Tealdi, A. G. Marrani, S. Brutti, D. Callegari and S. Passerini, J. Electrochem. Soc., 2020, 167, 080509 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- R. Rajagopalan, Y. Tang, X. Ji, C. Jia and H. Wang, Adv. Funct. Mater., 2020, 30, 1909486 CrossRef CAS.
- G.-L. Xu, R. Amine, A. Abouimrane, H. Che, M. Dahbi, Z.-F. Ma, I. Saadoune, J. Alami, W. L. Mattis, F. Pan, Z. Chen and K. Amine, Adv. Energy Mater., 2018, 8, 1702403 CrossRef.
- L. Li, Y. Zheng, S. Zhang, J. Yang, Z. Shao and Z. Guo, Energy Environ. Sci., 2018, 11, 2310–2340 RSC.
- W.-C. Chang, J.-H. Wu, K.-T. Chen and H.-Y. Tuan, Adv. Sci., 2019, 6, 1801354 CrossRef PubMed.
- C. Isaac, A. Jack, L. Hyeon Jeong, X. Albert, I. Johannes and P. Mauro, A Red Phosphorus-Graphite Anode for K-ion Batteries, Mater. Today Energy, 2021, 21, 100840 CrossRef.
- D. Su, J. Dai, M. Yang, J. Wen, J. Yang, W. Liu, H. Hu, L. Liu and Y. Feng, Nanoscale, 2021, 13, 6635–6643 RSC.
- C. M. Subramaniyam, Z. Tai, N. Mahmood, D. Zhang, H. K. Liu, J. B. Goodenough and S. X. Dou, J. Mater. Chem. A, 2017, 5, 1925–1929 RSC.
- Y. Wu, S. Hu, R. Xu, J. Wang, Z. Peng, Q. Zhang and Y. Yu, Nano Lett., 2019, 19, 1351–1358 CrossRef CAS PubMed.
- S. Zhang, X.-Y. Li, W. Yang, H. Tian, Z. Han, H. Ying, G. Wang and W.-Q. Han, ACS Appl. Mater. Interfaces, 2019, 11, 42086–42093 CrossRef CAS PubMed.
- S. Zhang, H. Liu, B. Cao, Q. Zhu, P. Zhang, X. Zhang, R. Chen, F. Wu and B. Xu, J. Mater. Chem. A, 2019, 7, 21766–21773 RSC.
- A. Casimir, H. Zhang, O. Ogoke, J. C. Amine, J. Lu and G. Wu, Nano Energy, 2016, 27, 359–376 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- P. Xiong, P. Bai, S. Tu, M. Cheng, J. Zhang, J. Sun and Y. Xu, Small, 2018, 14, 1802140 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- W. Tian, A. VahidMohammadi, Z. Wang, L. Ouyang, M. Beidaghi and M. M. Hamedi, Nat. Commun., 2019, 10, 2558 CrossRef PubMed.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu, B. Chen, Y. Mi and J. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- S. A. Ansari and M. H. Cho, Sci. Rep., 2016, 6, 25405 CrossRef CAS PubMed.
- S. Zhang, H. Ying, R. Guo, W. Yang and W.-Q. Han, J. Phys. Chem. Lett., 2019, 10, 6446–6454 CrossRef CAS PubMed.
- T. Zhang, X. Jiang, G. Li, Q. Yao and J. Y. Lee, ChemNanoMat, 2018, 4, 56–60 CrossRef CAS.
- R. Zhao, Z. Qian, Z. Liu, D. Zhao, X. Hui, G. Jiang, C. Wang and L. Yin, Nano Energy, 2019, 65, 104037 CrossRef CAS.
- X. Guo, W. Zhang, J. Zhang, D. Zhou, X. Tang, X. Xu, B. Li, H. Liu and G. Wang, ACS Nano, 2020, 14, 3651–3659 CrossRef CAS PubMed.
- H. Li, A. Liu, X. Ren, Y. Yang, L. Gao, M. Fan and T. Ma, Nanoscale, 2019, 11, 19862–19869 RSC.
- F. B. Ekaterina Pomerantseva, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, 969 Search PubMed.
- M. Zhang, J. Zhou, J. Yu, L. Shi, M. Ji, H. Liu, D. Li, C. Zhu and J. Xu, Chem. Eng. J., 2020, 387, 123170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00168c |
| This journal is © The Royal Society of Chemistry 2022 |