
Ultrafast charging and ultralong cycle life in solid-state Al-ion batteries†
Xuejing
Shen
 a,
Tao
Sun
*a,
Zhanjun
Wu
*a and
Li
Tan
*b
a,
Tao
Sun
*a,
Zhanjun
Wu
*a and
Li
Tan
*b
aSchool of Aeronautics and Astronautics, State Key Laboratory of Structural Analysis for Industrial Equipment, Dalian University of Technology, Dalian 116024, China. E-mail: tsun@dlut.edu.cn; wuzhj@dlut.edu.cn
bDepartment of Mechanical & Materials Engineering, University of Nebraska, Lincoln, NE 68588, USA. E-mail: ltan4@unl.edu
First published on 28th February 2022
Abstract
Designing and fabricating solid-state batteries with high-rate capability and long cycle life remains a feat. Here, for the first time, a free-standing gel polymer electrolyte (GPE) that exhibits an ultrahigh ionic conductivity of 1.29 × 10−2 S cm−1 is used to regulate the charge transfer between the GPE and Al electrode. Full batteries with a structure of Al/GPE/3D graphene are proved to be stable under current densities from 20 to 200 A g−1, by providing a specific capacity of 122 mA h g−1, a charging rate up to 1000 A g−1 (0.24 s charging time), and a stability over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. High-flux operations are found to be essential in lowering the energy request during high-rate reactions: not only reducing the surge voltage, but also increasing the energy (237 W h kg−1@20 A g−1) and power density (469 kW kg−1@500 A g−1) in output (>17%).
000 cycles. High-flux operations are found to be essential in lowering the energy request during high-rate reactions: not only reducing the surge voltage, but also increasing the energy (237 W h kg−1@20 A g−1) and power density (469 kW kg−1@500 A g−1) in output (>17%).
Introduction
Solid-state batteries are liquid-free devices. Their robustness against external shocks, such as cutting, folding, and bending has attracted immense interest from both civil and defence sectors.1–3 Achieving high-rate operations with these robust devices is, however, often met with a severe surge in voltage and a substantial downgrade in energy density as output.4 Two main issues are found to be responsible: one is the poor mass transport inside the bulk solid-state electrolyte (SSE); another is the sluggish charge transfer at the interface between the SSE and electrodes.5,6 In the last few years, there have been efforts to tackle both issues, by increasing the ionic conductivity in the SSE (from 10−8 to 10−3 S cm−1),7–9 by reducing the energy barrier at the electrolyte/electrode interface,10–12 and by gaining more reaction sites from a porous electrode.13 The state-of-the-art solid-state batteries yet remained a long distance away from being ultrafast chargeable (no more than 30C).14–16 For example, the specific capacities of the fastest sodium-based (at 10C)15 and lithium-based (at 20C)16 batteries are only 76% and 48% of those under low-rate conditions. Even for supercapacitors that are known for their high-power capabilities, their operation mechanism built upon ion adsorption/desorption generally excludes them from providing high values of energy density.17–19 There is an unfilled gap in designing and fabricating a solid-state energy storage platform that provides both high-power and high-energy densities.Aluminum-ion batteries are considered a strong contender to fill the gap, which have compelling advantages such as rich reserves of aluminum (Al) in nature (8% vs. 2.3% for Na and 0.006% for Li), non-flammability and high anode capacity (2978 mA h g−1/8046 mA h cm−3).20–23 In 2015, Dai and co-workers revolutionized ultrafast-charging ability using an Al anode, a three-dimensional (3D) graphene foam cathode and an ionic liquid-based electrolyte.21 The low intercalation barrier of the electrolyte (AlCl4−) in the cathode is one of the main reasons behind the high-rate performances.24 The lack of a solid-electrolyte interphase (SEI) layer21 and flexibility of the electrolyte (AlCl4−, Al2Cl7−) in forming triple- (Al3Cl10−) or even polymeric complexes25,26 are another two factors that accelerate the high-rate operations up to 1000 A g−1 (104 C, 0.35 s charging time), with a cycle life of 45000 (at 40 A g−1).25 Unfortunately, such an excellent performance has never been found in solid-state platforms. In the literature, an ionic liquid-based electrolyte was usually encapsulated either within polyvinylidene fluoride (PVDF),27 polyethylene oxide (PEO),28,29 gel polymers (polyacrylamide (PAAM),30–32 polyamide (PAM),33 and poly(ethyl acrylate) (PEA)34) or inside metal–organic frameworks (MOFs).35 The highest current density to operate these batteries was 2 A g−1, by using an acrylamide-based PAAM gel polymer electrolyte (GPE).32 The corresponding specific capacity was 91 mA h g−1 (75% of that under low-rate conditions) and the cycle life was 800. While the cycling performance can be improved to 2000 by using MOFs, allowable current density was only 0.2 A g−1 (with a specific capacity of 53 mA h g−1).35 The large gaps between a liquid-electrolyte-battery and a solid-electrolyte-battery (1000 A g−1vs. 2 A g−1; 45000 cycles vs. 2000 cycles) are likely due to the poor mass-transport inside the polymers or MOFs, sluggish ion intercalation in a two-dimensional (2D) cathode, and energy-intensive charge transfer along the aluminum–SSE interface.
In this work, all three barriers above are reconfigured and lowered, respectively by designing a novel GPE with a high ionic conductivity of 1.29 × 10−2 S cm−1 (concentration of the ionic liquid is 90 wt%), by inserting an intercalation friendly 3D graphene cathode inside the GPE, and by enabling high-fractal dendrites with a large surface area as the anode. Unprecedented rate capabilities (e.g., charging current density of 800 A g−1 (8000C) with a specific capacity of 95.8 mA h g−1; 0.45 s charging time) and a long cycle life (20000 cycles at 10 A g−1) are obtained, along with a much reduced voltage surge and much improved energy and power density in output (>17%).
Results and discussion
Free-standing GPE with high ionic conductivity
Gel polymer electrolytes (GPEs) are widely used in solid-state batteries, where they generally exhibit higher ionic conductivity and better adhesion with electrodes than their ceramic and solid polymer counterparts. Here, the acrylamide (AAM) monomer was selected to fabricate the GPE. As shown in Fig. 1, the preparation process consists of the following steps: adding AAM into a cloudy suspension of aluminum chloride (AlCl3) in anhydrous dichloromethane (DCM) (step 1; Fig. 1a); adding ionic liquid (AlCl3:EMI-Cl = 1.5; EMI+ being the cation and a mixture of AlCl4− and Al2Cl7− as anions) (step 2; Fig. 1b); adding N,N-methylenebisacrylamide (MBAA) (step 3) and 2,2′-azobisisobutyronitrile (AIBN) (step 4). Subsequently, the prepared solutions were poured into casting molds and allowed to settle for 24 h. In step 1, exothermic coordination occurs between the AAM monomer and aluminum salt;30 the mixture quickly turned clear (see pictures in Fig. 1a), where DCM served as a heat sink and was later removed by evaporation (Fig. 1c).36 Thanks to the crosslinked network (steric effect),37 DCM easily evaporated and this led to the formation of a uniform GPE membrane (picture in Fig. 1c). In contrast, typical synthesis without a cross-linker30–32 (Fig. 1d) delivered a viscous GPE with a large amount of bubbles. The GPE formed in our way was free-standing (Fig. 1c), while the one from conventional synthesis was sticky and unusable for inserting a 3D graphene cathode (Fig. 1d).
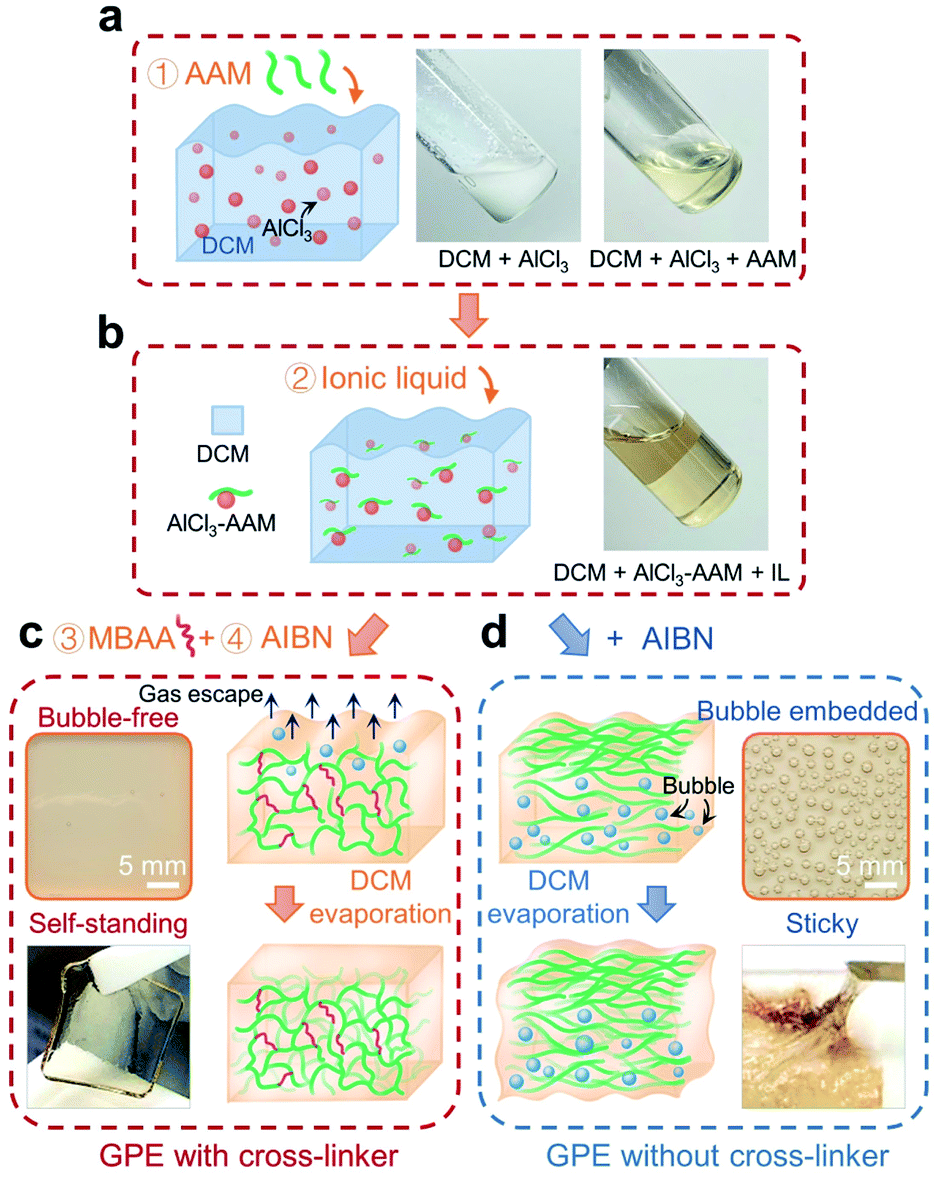 | ||
| Fig. 1 Preparation schematic of the gel polymer electrolyte (GPE). (a) The complexation of the AAM monomer with AlCl3 in DCM solvent. (b) Introducing abundant ionic liquid (IL). (c) The GPE polymerized with an MBAA cross-linker delivering a bubble-free and self-support membrane. (d) Typical fabrication showing a sticky GPE with bubbles. | ||
It should be noted that this cross-linked GPE can afford an extremely high loading of ionic liquid. In return, as shown in Fig. 2a, the ionic conductivity jumped from 0.58 × 10−2 to 1.29 × 10−2 S cm−1 when the mass loading for the electrolyte increased from 80 to 90 wt% (see Fig. S1† for details of calculation). When the amount of electrolyte was further increased to 95 wt%, an ultrahigh ionic conductivity of 1.69 × 10−2 S cm−1 was obtained. It is noteworthy to point out that a conductivity in the order of 10−2 S cm−1 has never been achieved in previous studies.27–35 Such a high conductivity, in fact, is very close to that of a pure liquid electrolyte (2.43 × 10−2 S cm−1);38 this implies that the rate of mass transport could become ideal when a full battery based on this GPE is allowed to run under fast-charging.
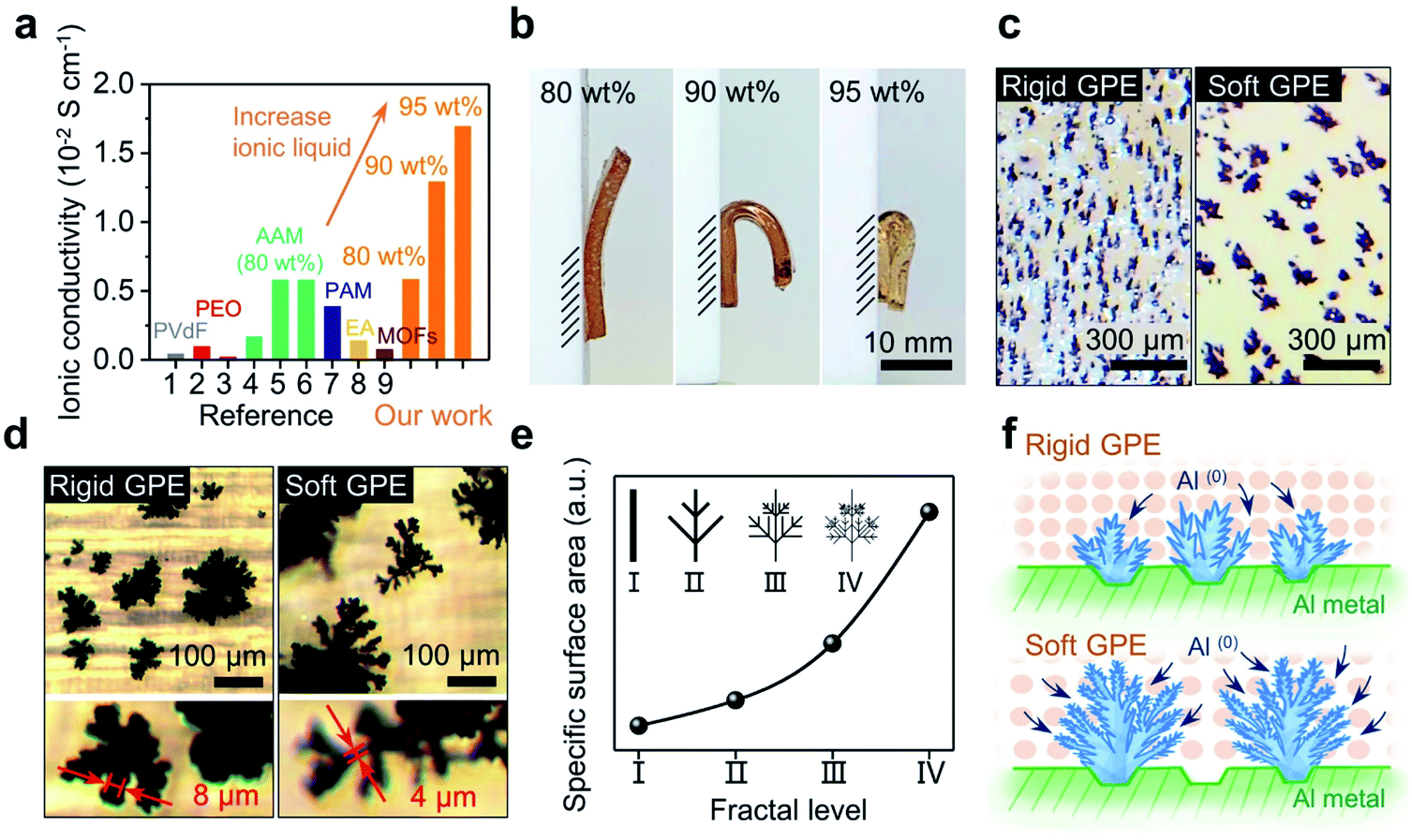 | ||
| Fig. 2 GPE membrane regulates the morphology of dendrites. (a) Ionic conductivities from state-of-the-arts and our work with different contents of ionic liquid (80–95 wt%). Reference numbers 1–9 correspond to [27]–[35], respectively. (b) GPE membranes with different contents of ionic liquid exhibited different resistance to bending under their own weights. (c) Digital microscopy image of the dendrites grown under a rigid (80 wt% ionic liquid) or a soft GPE (90 wt% ionic liquid). (d) Dendrites with a rigid GPE are smaller in size but thicker in trunk than those with a soft GPE (optical microscope (OM) images). (e) A qualitative fractal model for the dendrite reveals the contribution of fractal levels (I–IV) to the specific surface area. (f) Illustrated the dendrites grown under a rigid or a soft GPE. | ||
Effect of GPE on the deposition of aluminum
Liquid electrolyte Al-ion batteries have shown that the morphology of the Al anode is important in regulating charge transfer. Electroplated fractal dendrites on the Al anode could provide a large surface area, accommodating fast charge transfer that is otherwise sluggish.25 Will the growth of Al(0) be tuned similarly via a GPE-based electrolyte?Fig. 2b shows the side view of three free-standing GPE membranes, respectively exhibiting bendability under their own weights. When the content of ionic liquid was increased to 95 wt%, an extremely soft GPE was received (Fig. 2b-left). In contrast, decreasing the loading of ionic liquid (increasing the loading of monomers) made the GPE much more rigid (Fig. 2b-right). Then, two of these membranes (80 wt% vs. 90 wt% ionic liquid; their strength vs. strain curves are shown in Fig. S2†) were used to investigate aluminum electroplating under a high current density of 1.5 mA cm−2. After 100 s, irregular Al(0) spheres were observed; Al(0) spheres formed in the rigid GPE (80 wt% ionic liquid) are slightly smaller and denser than that in the soft GPE (90 wt% ionic liquid) (see Fig. S3†). Interestingly, as shown in Fig. 2c, these spheres became dendrites after 10 min with both GPE membranes, whereas this change in morphology was not observed in the liquid-electrolyte-battery25 (see Fig. S4† for details). Clearly, growth of Al(0) at the electrode was changed by the GPE, which perhaps localized the current injections and hence limited the nucleation sites. As a result, this induced an explosive growth of Al(0), where bulkier dendrites of Al(0) were observed with a softer GPE (Fig. 2c). Further observations showed such dendrites had branches that were thinner than those observed with a rigid GPE (Fig. 2d). This comparison indicated that the GPE membrane with low strength and high ionic liquid content is more favourable for the growth of dendrites.39 As those dendrites grew in the shape of fractal ferns, a simplified fractal model was built using the measured width of branches.40 These dendrites can be divided into four levels of fractals (I–IV), where the width of branches is halved with each increasing level. As shown in Fig. 3e, this model qualitatively gave specific surface areas of dendrites, where the value increases exponentially with fractal levels. Simply, the dendrites grown over the interface of a soft and rigid GPE (back in Fig. 2d) could be respectively assigned as level-IV and level-II. As such, one dendrite produced by a soft GPE will have the same surface area as that of five dendrites produced by a rigid GPE (see Fig. S5†). An experimental proof of this is the number of individual dendrites per unit area in the rigid GPE being almost 5 times that in the soft one (2.63 EA cm−2vs. 0.54 EA cm−2; counted from Fig. 2c). Schematically, the morphologies of dendritic Al(0) are sketched in Fig. 2f, suggesting that dendrites grown with a soft GPE will provide more nucleation sites for subsequent Al deposition. In other words, a soft GPE-based Al-ion battery may exhibit a better performance than the rigid-based one.
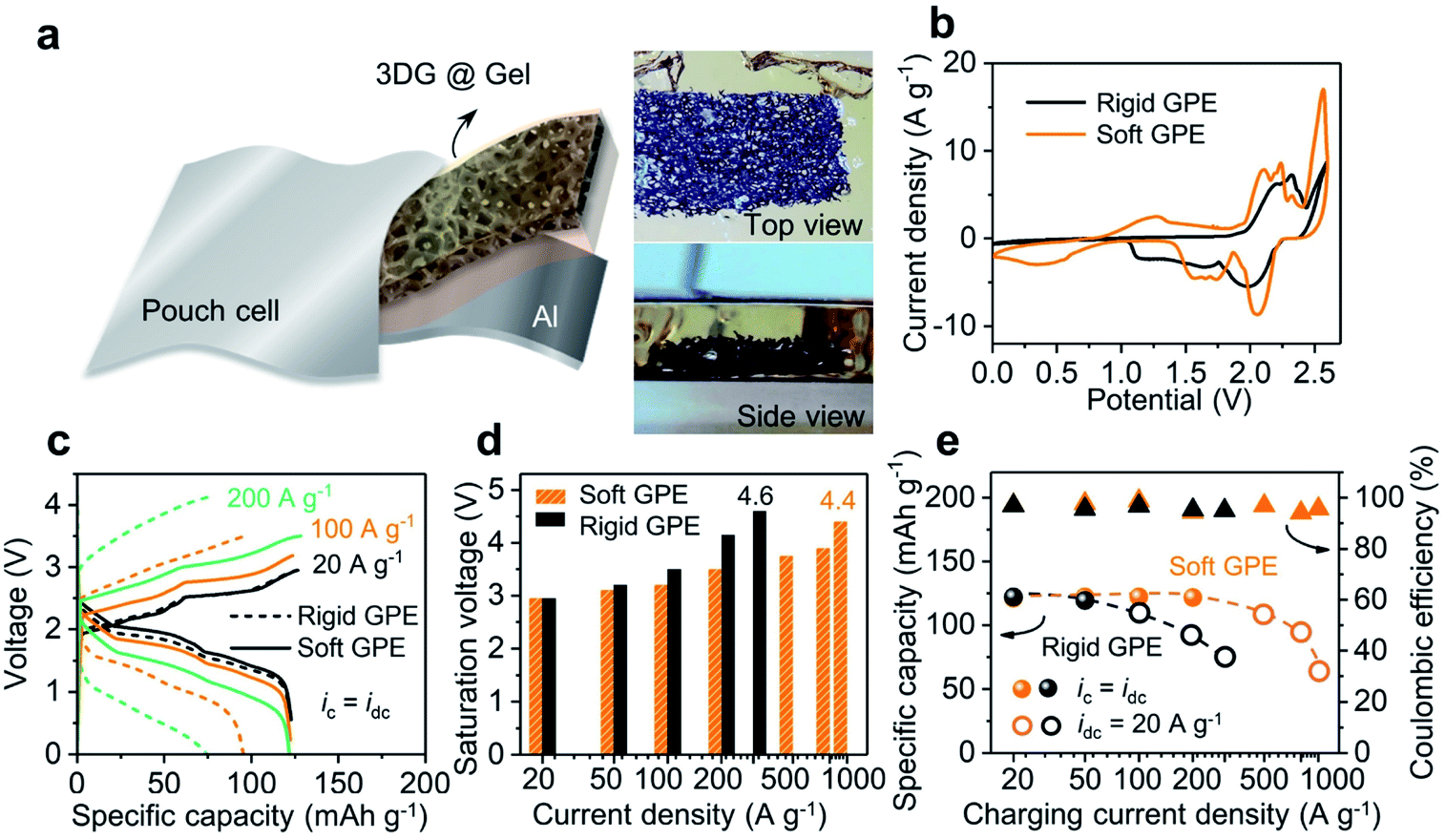 | ||
| Fig. 3 Structure and performance of the solid-state Al-ion battery. (a) Structure of the battery and top/side-view of an embedded 3D graphene cathode in the GPE membrane. (b) Redox potentials and their peak intensities varied in Al-ion batteries using a rigid GPE versus a soft one. (c) Galvanostatic charge and discharge curves at different current densities with a rigid or soft GPE membrane (ic = idc = 20–200 A g−1). (d) Comparison of saturation voltages between the device with a rigid GPE and a soft one. (e) High-rate performances of the device with a rigid or soft GPE membrane (rigid GPE: ic = idc = 20–50 A g−1; ic = 100–300 A g−1, idc = 20 A g−1; soft GPE: ic = idc = 20–200 A g−1; ic = 500–1000 A g−1, idc = 20 A g−1). | ||
Effect of GPE on the performance of solid-state Al-ion batteries
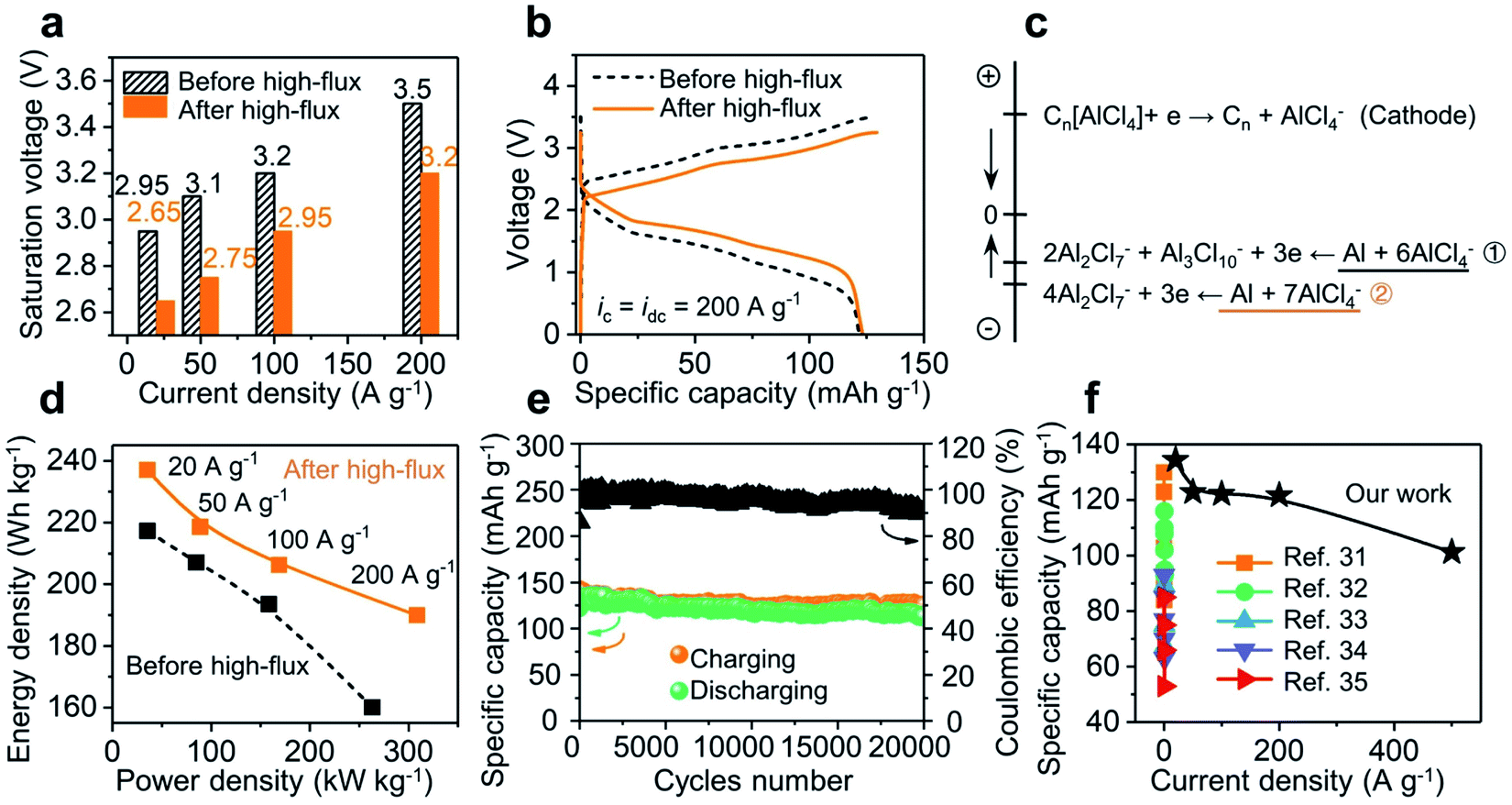
Fig. 3a illustrates the construction motif for a GPE contained Al-ion battery. In this setup, a three-dimensional (3D) network of graphene was used as the cathode, with aluminum foil being the anode. This 3D graphene was fabricated using porous nickel as the framework and later dried using supercritical CO2. The final sample has an open and continuous network structure (see Fig. S6†), which has been proved to be useful before for its capability to support high-rate and long-lasting operation of liquid-state batteries.25 As shown in Fig. 3a-left, this 3D graphene can be further embedded in GPE precursor solutions before its final settling into a free-standing membrane. The resultant pouch cell is able to power an LED bulb at different bending angles (from 180 to 90°), as well as after being rolled into a tube (radius of 1 in.; see Fig. S7†). Then, two batteries were assembled using the above rigid and soft GPE membranes (80 wt% vs. 90 wt% ionic liquid) and cyclic voltammetry (CV) was used to investigate the electrochemical reaction (shown in Fig. 3b; linear-sweep voltammetry (LSV) of the GPE in Fig. S8†). In comparison with the rigid GPE, stronger oxidation/reduction peaks, lower charging voltage plateaus and higher discharging voltage plateaus were all observed with the soft GPE. Namely, the soft GPE stands at a better position than the rigid one. Details of galvanostatic charging/discharging curves under different current densities are shown in Fig. 3c. When the current densities were varied from 20 to 200 A g−1, the specific capacity of the device with a rigid GPE attenuated severely. At the current density of 200 A g−1, a value of 74.4 mA h g−1 was returned (60% of the specific capacity at 20 A g−1), whereas the specific capacity of the device with a soft GPE remained rather constant at the level of 122 mA h g−1. Moreover, as the current density increased, a severe downward shift in discharging plateaus and upward shift in charging plateaus were observed in the device with a rigid GPE rather than that with a soft one. The saturation voltages (defined as the maximum cut-off voltage to ensure maximum coulombic efficiency (>95%)) of the two devices at different current densities are grouped in Fig. 3d. It was found that the device using a rigid GPE had higher saturation voltages, especially under harsh current conditions. For instance, the device with a rigid GPE needed a high charging voltage of 4.6 V at 300 A g−1. In comparison, a smaller value of 4.4 V was required for the device with a soft GPE even after the current density was accelerated to 1000 A g−1. In other words, the device with a rigid GPE required an extra amount of voltage to overcome the energy barrier for charge injections. The disparity caused by GPE membranes is further plotted under ultrahigh rate operations (as shown in Fig. 3e). In these experiments, the discharging rates were fixed at a moderate rate to allow the ions to completely de-intercalate from graphene layers25 (sphere: ic = idc; circle: ic ≠ idc = 20 A g−1; for detailed curves see Fig. S9†). Since a relatively slow decay in specific capacity is preferred, the charging current density in the device with a soft GPE can be further pushed compared with that with a rigid one. When charging at a fast rate of 800 A g−1, a specific capacity of 95.8 mA h g−1 was released from the device with a soft GPE. It only took 0.45 s (8000C; a rate of nC corresponds to charge in 1/n h) to get fully charged. When the charging rate was further increased to 1000 A g−1 (15100C, 0.24 s charging time), the device still kept a specific capacity of 63.4 mA h g−1.
Even when the device (with a soft GPE) was operated under these high current conditions, the battery still exhibited clear voltage plateaus (Fig. 3c; see Fig. S10† for more curves). As such, a high discharging voltage with a large current density will result in a high energy and power density at the same time. For instance, at the current density of 20 A g−1, a high energy density of 217 W h kg−1 and a power density of 35 kW kg−1 were released (calculated by integrating discharge curves and using the mass of the graphene cathode). Since the voltage plateau varied very little over the current density range of 20 to 100 A g−1, when the current density was increased to 100 A g−1, the energy density of the device remained at 194 W h kg−1, along with a substantial increase of power density to 158 kW kg−1. Furthermore, when the current density was increased to 500 A g−1, the power density can be as high as 469 kW kg−1 (with an energy density of 96 W h kg−1).
Effect of high-flux operation on aluminum plating
Fig. 4a shows the specific capacities of the device with a soft GPE at current densities ranging from 20 A g−1 (122 mA h g−1) to 500 A g−1 (101.3 mA h g−1), and back to 20 A g−1 (134 mA h g−1). Notably, a series of high-flux operations above had no obvious negative impact on the battery; rather, the solid-state platform was “upgraded” (higher specific capacity and better coulombic efficiency; Fig. 4a and S11†). This upgrade can be further manifested from the CV test. Here, a CV curve (before high-flux operation) was obtained from a battery that has undergone 250 charge–discharge cycles at a relatively low current density of 5 A g−1, where the device was activated to achieve high coulombic efficiency (Fig. S12†). Another CV curve (after high-flux) was obtained from the battery that has undergone a round of high-rate operations (as Fig. 4a). As shown in Fig. 4b, the latter curve (after high-flux operations) exhibited smaller oxidation potentials but higher reduction potentials than the former one (before high-flux operations). Major oxidation and reduction peaks shifted from 2.52 V and 2.05 V to 2.36 V and 2.09 V. This indicated that the device was upgraded by high-flux cycles, which consumed a lower amount of energy in charging but released more energy in discharging.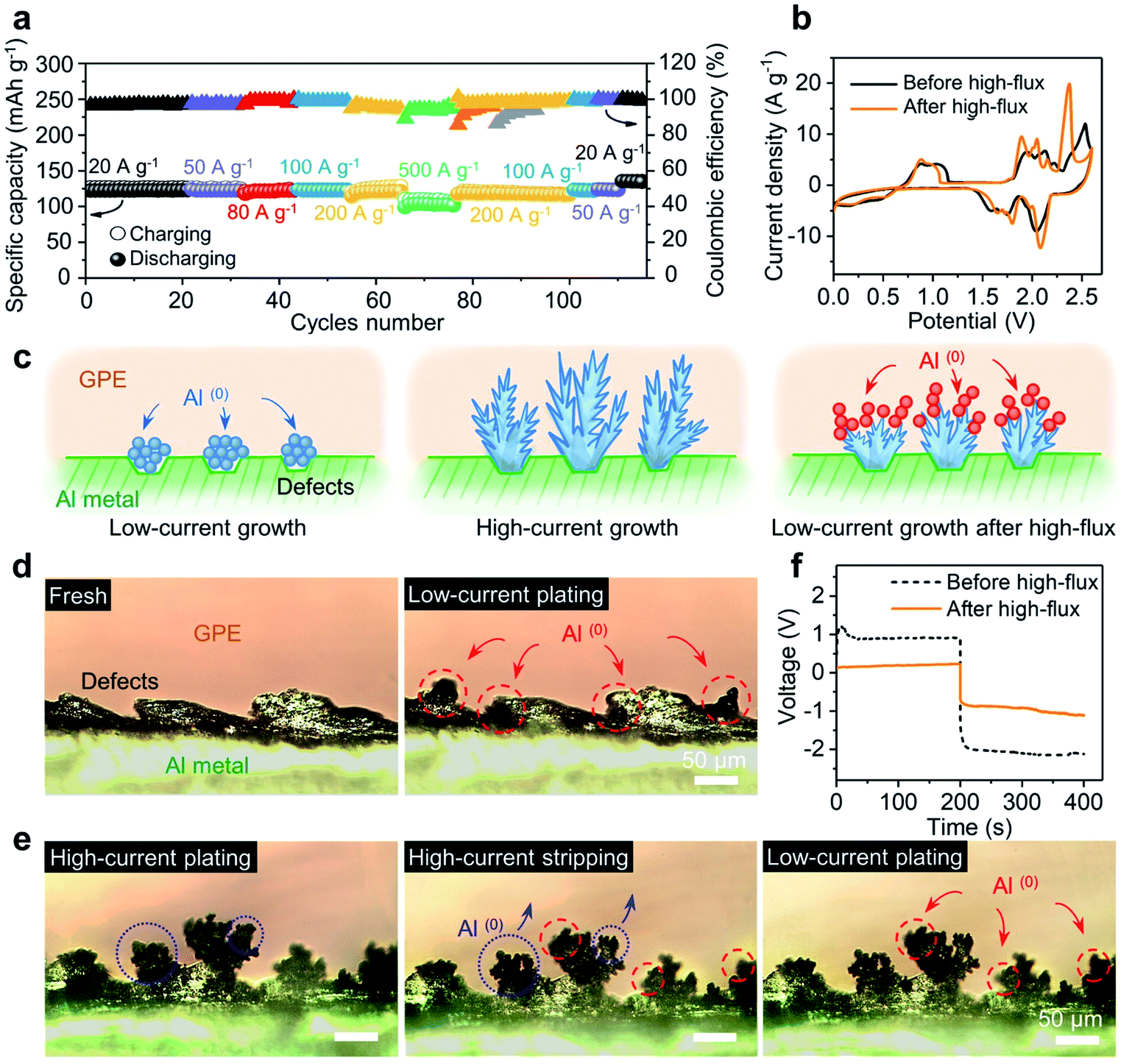 | ||
| Fig. 4 Dendrites on the aluminum–GPE interface promote aluminum plating. (a) Specific capacity and coulombic efficiency at different current densities (ic = idc = 20–500 A g−1). (b) High flux operations played a vital role in redox potentials. (c) Illustrated deposition of Al from a GPE membrane. (d) and (e) Cross-sectional images (OM) of the Al electrode next to a GPE membrane, where under low-current plating newly formed Al(0) spheres grew on uneven sites (d) and tiny leaves grew on dendritic branches after a high-flux treatment (e). (f) Polarization voltage for Al plating with a GPE membrane before and after high-flux. | ||
Fig. 4c illustrates the possible interpretation for this battery “upgrade”. Once the injection current is greatly increased (under high-flux operations), each nucleation spot could trigger an explosive growth of fresh aluminum, by producing dendrites (Fig. 4c-middle). Since the dendrites do not usually disappear uniformly after oxidation or stripping, there will always be some incomplete dendrites or partial structures left after stripping. The large surface area from these residues will likely make them favored nucleation sites for subsequent Al deposition (Fig. 4c-right) instead of pristine surface defects at Al metal (Fig. 4c-left). A symmetric Al/GPE/Al cell was designed to verify the above hypothesis. The process of Al plating and stripping was in situ tracked and recorded by a digital camera mounted on an optical microscope (OM). As shown in Fig. 4d, the pristine Al foil exhibited uneven edges due to rough mechanical cutting. Noticeable changes after low-current plating (i = 0.07 mA cm−2) were marked inside dashed circles (red color). In particular, newly formed Al(0) spheres preferred to grow on the uneven side of the edge. This is consistent with the common belief that the growth along defect sites will reduce the surface energy of the system.25 When plating was operated under a high-current flux (i = 2.8 mA cm−2), Fig. 4e reveals a different profile for Al(0). Simply, lush trees were observed on the edge, similar to the features back in Fig. 4c-middle. Next, when this electrode was under stripping, branches in the dendrites became thinner, with much fewer leaves left on them (highlighted inside the blue dotted circles; see Fig. S13† for the top-view of the charging/discharging process). When low-current plating is allowed again, these remaining branches became the nucleation sites for newly deposited aluminum. Tiny leaves grown on top of those branches were observed (highlighted inside the red dashed circles). Clearly, the size of those leaves was smaller than that of original spheres on rough edges. As the incomplete stripping produced a much higher surface area, it is not surprising to see these smaller features. In a sense, a wide range of Al nucleation sites was created to facilitate the plating at the electrode–GPE interface. Quantitatively, this facilitation is captured in Fig. 4f, where the polarization voltage for Al plating before/after the high-flux (i = 2.8 mA cm−2) under a same flux of current (i = 0.07 mA cm−2) was recorded. It is clear that the polarization voltage from a dendrite-grown electrode (after high-flux operation) is much smaller than that from a dendrite-absent surface (before high-flux operation) (see Fig. S14† for the reduction of resistance by high-flux). This observation validates our hypothesis that high-flux operations can modify the electrode interface and finally upgrade battery performances.
Upgrade role of high-flux treatment
The high-flux treatment along the electrode–GPE interface has provided a new avenue to lower the energy request and achieve excellent performances for solid-state batteries. As shown in Fig. 5e, the saturation voltages of a battery before and after high-flux at different current densities were grouped. Roughly, the charging voltage linearly increases with the rise in current density. A drop in surge voltage as high as 0.3 V was observed after high-flux treatment (see Fig. S15† for different GPE membranes). In other words, reduction of this extra voltage can therefore be correlated with a lowered energy barrier. Fig. 5b shows a more intuitive comparison between the charge and discharge curves, where the charge curve was shifted down but the discharge curve moved upward. In a nutshell, high-flux treatment not only lowered the energy barrier in charging, but also improved the energy release in discharging. The increase of voltage plateaus in discharging implies an increase in the potential difference between the two electrodes (cathode vs. anode). As the cathode (3D graphene) is operated on the basis of intercalation and de-intercalation of mono-complexes (AlCl4−) only,21 this solitary pathway leads to a fixed potential change on the cathode side, regardless of the injection rate of current. In contrast, on the anode side, varieties of those negatively charged aluminum complexes could have participated in the redox reactions in different ways.25 This will change the potential difference (voltage output) of the battery from reactions eqn (1) and (2).| Al3Cl10− + 2Al2Cl7− + 3e ↔ Al(0) + 6AlCl4− | (1) |
| 4Al2Cl7− + 3e → 7AlCl4− + Al(0) | (2) |
 | ||
| Fig. 5 Influence of high-flux treatment on the solid-state Al-ion battery. (a) Reduced saturation voltages were observed for the device receiving a high-flux treatment. (b) Downward shifting in charging and upward shifting in discharging observed after high-flux operations. (c) Potential for possible reductions at the electrodes (eqn (1) and (2) dominated before and after high-flux respectively). (d) Densities of discharging energy and power increase after the high-flux treatment (ic = idc = 20–200 A g−1). (e) Stable cycling performance of the solid-state Al-ion battery (ic = idc = 10 A g−1). (f) Specific capacities under high current densities for our work and state-of-the-arts. | ||
An estimate in the energy of these two equations did reveal a higher reduction potential from eqn (1) than that from eqn (2) (Fig. 5c and Table S1† for details). It is worth mentioning that this increase in potential difference is beneficial to obtain more energy/power density from the batteries. As shown in Fig. 5d, an energy density of up to 237 W h kg−1 (vs. 217 W h kg−1 before high-flux) was achieved at 20 A g−1. Moreover, at a high current density of 200 A g−1, the energy density jumped from 160 to 190 W h kg−1 (>18%) and power density from 263 to 308 kW kg−1 (>17%). Since the dendrites in the full cell are not as prominent as in electroplating (overcharging), this high-flux treatment did not drag the cyclability of the Al-ion battery. As shown in Fig. 5e, a stable operation was observed when the device was cycled 20000 times (at 10 A g−1). The devices from this work exhibit a battery-level capacity and energy density while achieving supercapacitor-level power density, thus far surpassing those of published solid-state Al-ion batteries31–35 (Fig. 5f and Table S2†). Their excellent electrochemical performances, especially high-rate capability and ultralong cycle life, will push the boundary of ultrafast charging in solid-state energy storage platforms. We attribute these superior performances to three factors: (1) an interpenetrated electron and ion pathway with a short transport distance (3D graphene cathode); (2) fast ion transport in the bulk electrolyte (high ionic conductivity and low activation energy (0.064 eV; Fig. S16†) of the GPE); and (3) a low energy request from those negatively charged aluminum complexes when high-rate operations were allowed (via the regulation of the dendrites).
Conclusions
In summary, we have designed and fabricated a solid-state Al-ion battery with both high power and energy densities. A free-standing gel polymer electrolyte (GPE) with an ionic conductivity in the magnitude of 10−2 S cm−1 was used to construct the Al/GPE/3D graphene battery. Optimizing the softness of the GPE enables the formation of Al dendrites with fractal branches. This lowered the energy request for high-rate operations and also provided augmented active sites for charging. The final solid-state battery exhibited a specific capacity of 122 mA h g−1 (in the range of 20–200 A g−1; 161–1610C), an ultrafast charging rate (15100C; 1000 A g−1; 0.24 s charging time) and an ultralong cycle life (20000 cycles at 10 A g−1). A high-flux operation was found to be essential in improving the device behavior, e.g., to reduce the voltage surge (0.3 V) and to increase the energy and power density in output (>17%). Thus, an impressive energy (237.1 W h kg−1 at 20 A g−1) and power density (468.8 kW kg−1 at 500 A g−1) are respectively obtained. We envision that this work has the potential to be extended to other solid-state battery systems (i.e., dual-ion, zinc and potassium batteries) as energy storage sources for hybrid vehicles, emergency facilities, and wind/solar energies.
Author contributions
X. S.: conceptualization; methodology; validation; formal analysis; investigation; data curation; writing – original draft; visualization. T. S.: conceptualization; methodology; validation; formal analysis; data curation; writing – review & editing; visualization. W. Z.: conceptualization; resources; supervision; project administration; funding acquisition; writing – review & editing. L. T.: conceptualization; methodology; validation; formal analysis; writing – review & editing; visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (Grant No. 2018YFA0702800), National Natural Science Foundation of China (Grant No. U1837205), and Fundamental Research Funds for the Central Universities (Grant No. DUT16ZD214, DUT19ZD101 and DUT19JC32). L. T. gratefully acknowledges the financial support from the University of Nebraska (Nebraska Research Initiative) and Nebraska Center for Energy Science Research.References
- A. Manthiram, X. W. Yu and S. F. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1701169 CrossRef PubMed.
- L. Fan, S. Y. Wei, S. Y. Li, Q. Li and Y. Y. Lu, Adv. Energy Mater., 2018, 8, 1702657 CrossRef.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2020, 120, 6820 CrossRef CAS PubMed.
- Y. Y. Liu, Y. Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540 CrossRef.
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599 CrossRef CAS PubMed.
- M. Dirican, C. Yan, P. Zhu and X. Zhang, Mater. Sci. Eng., R, 2019, 136, 27 CrossRef.
- Y. Lu, L. Li, Q. Zhang, Z. Q. Niu and J. Chen, Joule, 2018, 2, 1747 CrossRef CAS.
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, ACS Energy Lett., 2016, 1, 678 CrossRef CAS.
- B. Zahiri, A. Patra, C. Kiggins, A. X. B. Yong, E. Ertekin, J. B. Cook and P. V. Braun, Nat. Mater., 2021, 20, 1392 CrossRef CAS PubMed.
- C. Yu, S. Ganapathy, E. Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1086 CrossRef PubMed.
- L. L. Liu, X. G. Qi, S. J. Yin, Q. Q. Zhang, X. Z. Liu, L. M. Suo, H. Li, L. Q. Chen and Y. S. Hu, ACS Energy Lett., 2019, 4, 1650 CrossRef CAS.
- K. Fu, Y. H. Gong, G. T. Hitz, D. W. McOwen, Y. J. Li, S. M. Xu, Y. Wen, L. Zhang, C. W. Wang, G. Pastel, J. Q. Dai, B. Y. Liu, H. Xie, Y. G. Yao, E. D. Wachsman and L. B. Hu, Energy Environ. Sci., 2017, 10, 1568 RSC.
- S. Randau, D. A. Weber, O. Kotz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffee, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259 CrossRef CAS.
- Z. Z. Zhang, Q. H. Zhang, J. A. Shi, Y. S. Chu, X. Q. Yu, K. Q. Xu, M. Y. Ge, H. F. Yan, W. J. Li, L. Gu, Y. S. Hu, H. Li, X. Q. Yang, L. Q. Chen and X. J. Huang, Adv. Energy Mater., 2017, 7, 1601196 CrossRef.
- Q. Lu, Y. B. He, Q. Yu, B. Li, Y. V. Kaneti, Y. Yao, F. Kang and Q. H. Yang, Adv. Mater., 2017, 29, 1604460 CrossRef PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- W. Raza, F. Z. Ali, N. Raza, Y. W. Luo, K. H. Kim, J. H. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441 CrossRef CAS.
- J. Yan, Q. Wang, T. Wei and Z. J. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J. F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564 CrossRef CAS PubMed.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 324 CrossRef CAS PubMed.
- T. Leisegang, F. Meutzner, M. Zschornak, W. Munchgesang, R. Schmid, T. Nestler, R. A. Eremin, A. A. Kabanov, V. A. Blatov and D. C. Meyer, Front. Chem., 2019, 7, 268 CrossRef CAS PubMed.
- S. Liu, P. Wang, C. Liu, Y. Deng, S. Dou, Y. Liu, J. Xu, Y. Wang, W. Liu, W. Hu, Y. Huang and Y. Chen, Small, 2020, 16, e2002856 CrossRef PubMed.
- M. L. Agiorgousis, Y. Y. Sun and S. B. Zhang, ACS Energy Lett., 2017, 2, 689 CrossRef CAS.
- X. Shen, T. Sun, L. Yang, A. Krasnoslobodtsev, R. Sabirianov, M. Sealy, W. N. Mei, Z. Wu and L. Tan, Nat. Commun., 2021, 12, 820 CrossRef CAS PubMed.
- S. Takahashi, L. A. Curtiss, D. Gosztola, N. Koura and M. L. Saboungi, Inorg. Chem., 1995, 34, 2990 CrossRef CAS.
- M. Kotobuki, L. Lu, S. V. Savilov and S. M. Aldoshin, J. Electrochem. Soc., 2017, 164, A3868 CrossRef CAS.
- S. F. Song, M. Kotobuki, F. Zheng, Q. B. Li, C. H. Xu, Y. Wang, W. D. Z. Li, N. Hu and L. Lu, Solid State Ionics, 2017, 300, 165 CrossRef CAS.
- T. Schoetz, O. Leung, C. P. de Leon, C. Zaleski and I. Efimov, J. Electrochem. Soc., 2020, 167, 040516 CrossRef CAS.
- X. G. Sun, Y. Fang, X. Jiang, K. Yoshii, T. Tsuda and S. Dai, Chem. Commun., 2016, 52, 292 RSC.
- Z. J. Yu, S. Q. Jiao, S. Li, X. D. Chen, W. L. Song, T. Teng, J. G. Tu, H. S. Chen, G. H. Zhang and D. N. Fang, Adv. Funct. Mater., 2019, 29, 1806799 CrossRef.
- Z. J. Yu, S. Q. Jiao, J. G. Tu, W. L. Song, H. P. Lei, H. D. Jiao, H. S. Chen and D. N. Fang, J. Mater. Chem. A, 2019, 7, 20348 RSC.
- Z. Liu, X. Wang, Z. Liu, S. Zhang, Z. Lv, Y. Cui, L. Du, K. Li, G. Zhang, M. C. Lin and H. Du, ACS Appl. Mater. Interfaces, 2021, 13, 28164 CrossRef CAS PubMed.
- I. Kim, S. Jang, K. H. Lee, Y. Tak and G. Lee, Energy Storage Mater., 2021, 40, 229 CrossRef.
- Z. Huang, W. L. Song, Y. Liu, W. Wang, M. Wang, J. Ge, H. Jiao and S. Jiao, Adv. Mater., 2021, 2104557 Search PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105 CrossRef CAS PubMed.
- A. F. Blanchard and P. M. Wootton, J. Polym. Sci., 1959, 34, 627 CrossRef CAS.
- P. Shinde, A. N. Ahmed, M. K. Nahian, Y. Peng and R. G. Reddy, ECS Trans., 2020, 98, 129 CrossRef CAS.
- T. T. M. T. N. Oyama, Electrochim. Acta, 2001, 46, 1201 CrossRef.
- T. Vicsek, Fractal Growth Phenomena, World Scientific, Singapore 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1–S16 and Tables S1, S2. See DOI: 10.1039/d2ta00630h |
| This journal is © The Royal Society of Chemistry 2022 |