
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA gold(I)-catalysed approach towards harmalidine an elusive alkaloid from Peganum harmala†
Solène Miaskiewicz,
Jean-Marc Weibel,
Patrick Pale and
Aurélien Blanc*
and
Aurélien Blanc*
Laboratoire de Synthèse, Réactivité Organiques et Catalyse, Institut de Chimie, UMR 7177 - CNRS, Université de Strasbourg, 4 Rue Blaise Pascal, 67070 Strasbourg, France. E-mail: ablanc@unistra.fr
First published on 21st September 2022
Abstract
Upon gold catalysis, the 2,3-dihydropyrrolo[1,2-a]indole motif, encountered in few but interesting bioactive natural products, was efficiently obtained from N-aryl 2-alkynylazetidine derivatives. In an attempt to apply this methodology to the synthesis of harmalidine, isolated from the seeds of Peganum harmala, advanced amino 2,3-hydropyrrolo[1,2-a]indol(one) derivatives were readily obtained in only 11 steps from but-3-yn-1-ol. While the reported structure of harmalidine could not be reached from these intermediates, a surprising 12-membered diimino dimer was isolated. Extensive comparison of the reported harmalidine NMR data to the experimental and calculated data of our synthetic molecules, harmaline or the synthetised N-methylharmaline show discrepancies with the proposed natural product structure.
Introduction
Traditional local uses and folk medicine often rely on plant extracts. The latter are a rich source of interesting natural products and numerous bioactive compounds have been isolated as such. The plant Peganum harmala (Fig. 1, right), common in semi-arid areas such as Sahara borders, the Middle East, or Central Asia, is a typical example. This plant was used by indigenous people to make a ‘magic’ beverage, which promotes hallucinogenic and psychoactive effects, but also as a treatment for asthma, diabetes, and rheumatism. Among the compounds identified from this plant, alkaloids named harmine, harmaline, harmalidine, harmalol, harmol and tetrahydroharmine (Fig. 1, top) are mostly responsible for the psychoactive activities. These substances belong to the β-carboline family, except for harmalidine which contains a 2,3-dihydropyrrolo[1,2-a]indole motif (Fig. 1, middle).1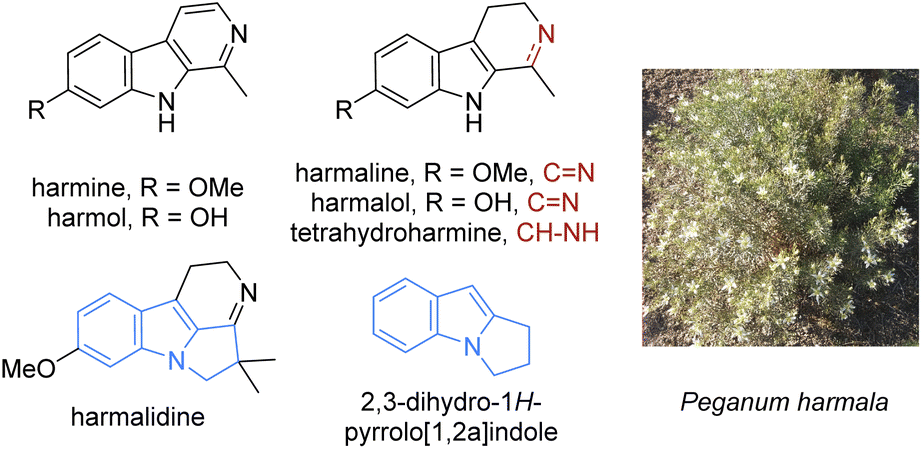 | ||
| Fig. 1 Main psychoactive harmala alkaloids and harmalidine structure including the 2,3-dihydropyrrolo[1,2-a]indole motif. | ||
Less common than other fused indolo-polycyclic motifs, pyrrolo[1,2-a]indole nevertheless represents the core of various interesting natural products. From chinese medicine were isolated isatisine A and some derivatives, including one exhibiting anti-HIV activity.2 Other pyrrolo[1,2-a]indole derivatives have been isolated from plants and used for treating infections, skin lesions, rheumatic pain3 or exhibiting filaricidal (polysin)4 or antimalarial (flinderoles)5 activities (Fig. 2). The pyrrolo[1,2-a]indole motif is also the core of a family of antibiotic and anticancer compounds named mitomycins A-K (Fig. 2).6–8 The importance and the variety of pyrrolo[1,2-a]indole natural product biological activities have induced several synthetic studies,9–11 especially in the mitomycin area.12–14
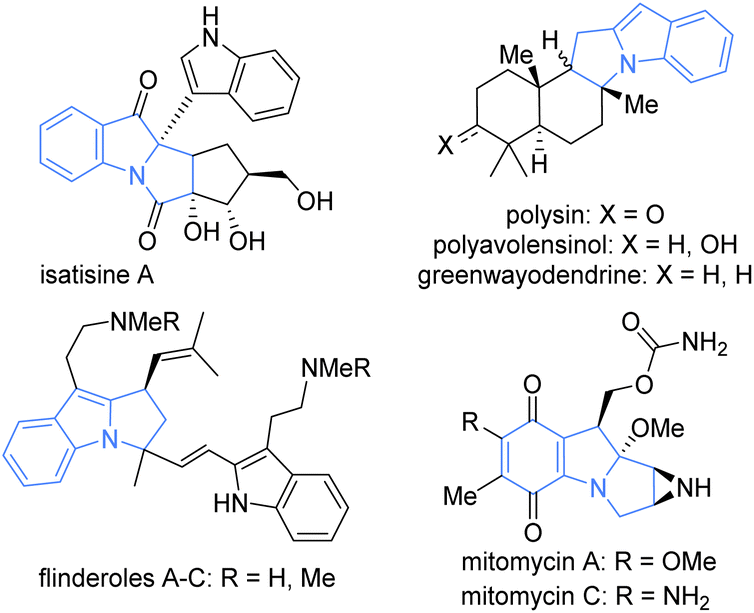 | ||
| Fig. 2 Natural products exhibiting the pyrrolo[1,2-a]indole motif. | ||
Engaged in gold-catalysed cascade reactions,15 we reasoned that these reactions could provide an alternative and rapid access to pyrrolo[1,2-a]indole derivatives (Scheme 1). In 2013, we reported a new route to such compounds based on the gold catalysed rearrangement of N-aryl-2-alkynylazetidines and we demonstrated its efficiency through the formal syntheses of the antibiotic 7-methoxymitosene and of a 5-HT2C receptor agonist.16
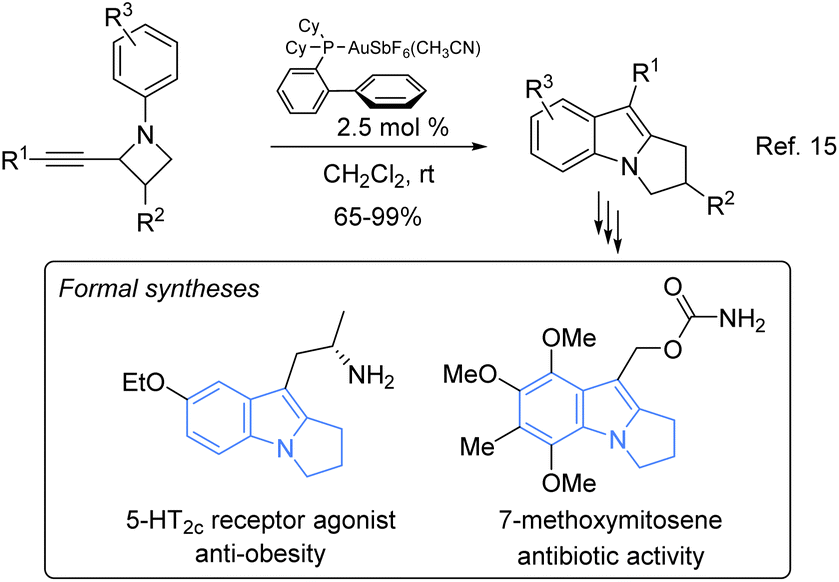 | ||
| Scheme 1 Rearrangement of N-aryl-2-alkynylazetidines catalysed by gold and its application to formal syntheses. | ||
In the present report, we described our effort towards the total synthesis of harmalidine based on this gold-catalysed reaction.
Applying this rearrangement to this context implies to start from the N-3-methoxyphenyl azetidine C, which should be obtained through the formal [2 + 2] condensation and reduction (Scheme 2).16,17 This would require the formation of the imine D from the aldehyde F and 3-methoxyaniline. Isobutyrate ester was selected, as the condensation of its enolate to imine D should allow introducing the gem-dimethyl unit present in the natural product structure. Upon rearrangement, the resulting product B must then be oxidized at a benzylic position to a ketone.18 The latter would serve for forming the fourth harmalidine cycle through an intramolecular aza-Wittig reaction from the azide A.
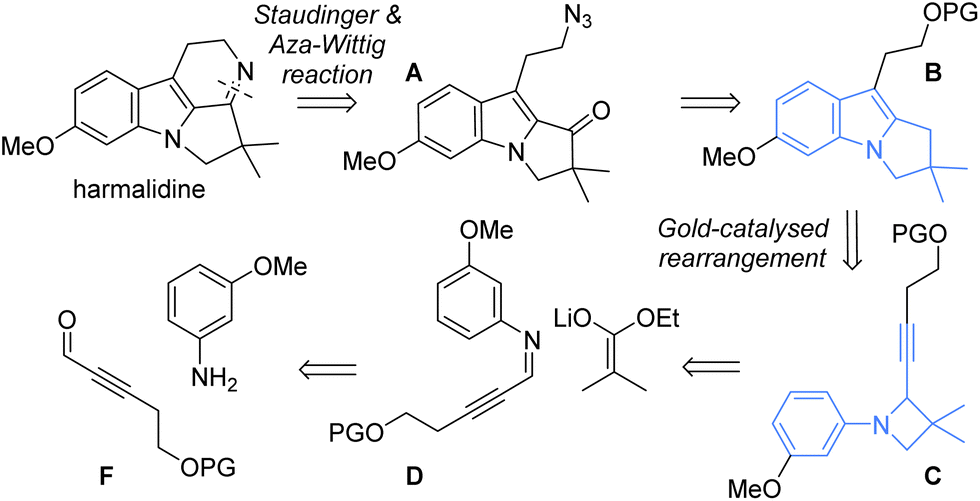 | ||
| Scheme 2 Our retrosynthesis of harmalidine involving our key gold-catalysed rearrangement of N-aryl azetidines to pyrrolo[1,2-a]indoles. | ||
Results and discussion
Synthesis of the key N-3-methoxyphenyl azetidine C
For convenience, we started the synthesis of C from the commercially available butyn-3-ol (Scheme 3). It was first protected as tert-butyldimethyl or diphenylsilyl ethers under classical conditions. The so-formed silyl butynyl ethers 1a, b were then formylated upon alkyne deprotonation and addition of dimethylformamide in THF at low temperature.16,17 The resulting aldehydes 2a, b were then converted to imines 3a, b by condensation with 3-methoxyaniline. The best and most convenient procedure at this stage was achieved by mixing both reagents in the presence of magnesium sulphate in ether at room temperature. The expected imines 3a, b were pure enough after filtration to be directly engaged in the next step.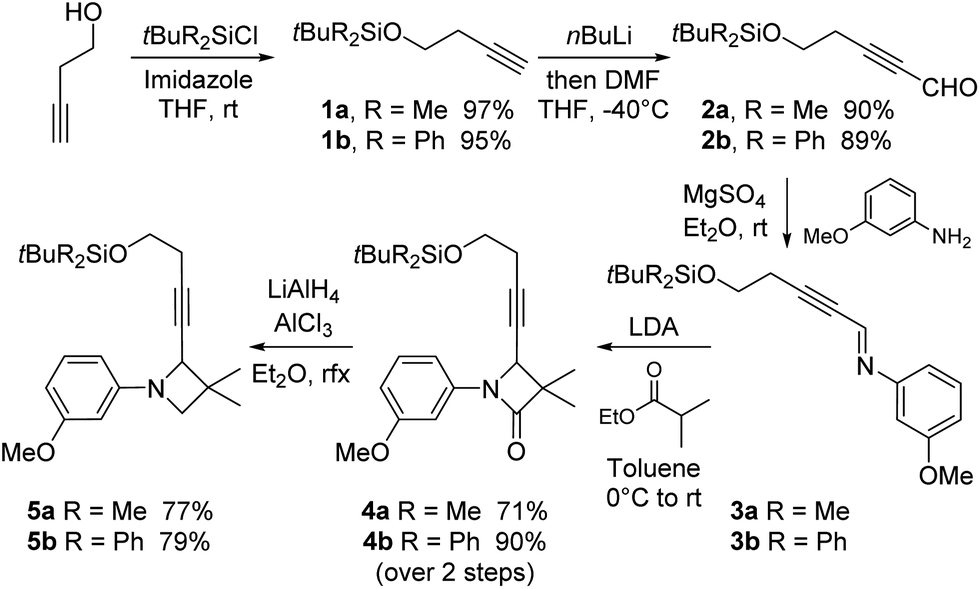 | ||
| Scheme 3 Synthesis of the key intermediate N-aryl azetidines. | ||
Upon addition to a lithium enolate solution derived from ethyl isobutyrate, these imines were efficiently converted to the expected N-3-methoxyphenyl gem-dimethyl azetidinones 4a, b in good to high yields. The latter were then reduced to the corresponding azetidines. The best conditions were those described by Ojima in the presence of in situ formed monochloroalane,19 providing the expected azetidines 5a, b (synthon C) within seconds in good to high yields, while avoiding opening side-reactions.
Gold-catalysed rearrangement: from N-3-methoxyphenyl alk-1-ynyldimethylazetidine C to pyrrolo[1,2-a]indole B
With these azetidines in hand, the gold-catalysed rearrangement was studied. Under our precedent optimized conditions,16 the expected 2,3-dihydropyrrolo[1,2-a]indoles were readily obtained but in modest yields due to some degradation. A brief condition screening revealed that 2-biphenyldicyclohexylphosphino-gold(I) hexafluoroantimonate was still the best catalyst but 1,2-dichloroethane (DCE) turned out to be better as solvent, while refluxing minimized degradation due to short reaction times. Excellent conversions and yields were thus achieved within 2 minutes under such conditions (Scheme 4). Rewardingly, this reaction could be performed in gram scale without compromising reaction time and yields.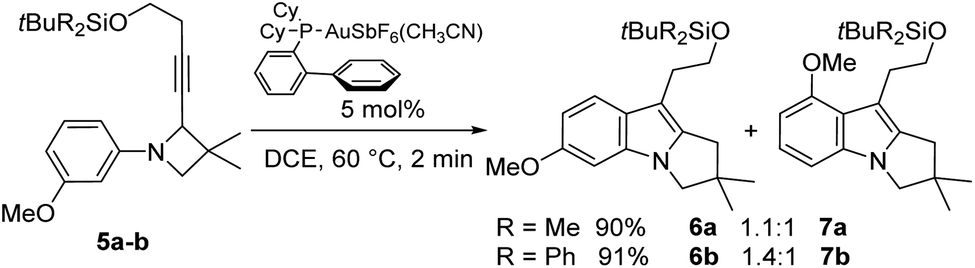 | ||
| Scheme 4 The gold-catalysed rearrangement of N-aryl azetidines to pyrrolo[1,2-a]indoles. | ||
As expected from an azetidine carrying on its nitrogen atom a 3-substituted phenyl group, two regioisomers were formed from both substrates 5a and 5b, but they were easily isolated by flash chromatography, respectively providing 6a (47%)/7a (43%) and 6b (53%)/7b (38%). Interestingly, the size of the alkynyl substituent seemed to play a favourable role in the corresponding regioselectivity. While an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was observed starting from the tert-butyldimethylsilyl substituted 5a, a slightly higher selectivity was achieved with the bulkier tert-butyldiphenylsilyl derivative 5b in favour of the desired regioisomer. It is worth noting that a similar effect of bulkiness on regioselectivity has already been observed in our preliminary studies.16
:1 ratio was observed starting from the tert-butyldimethylsilyl substituted 5a, a slightly higher selectivity was achieved with the bulkier tert-butyldiphenylsilyl derivative 5b in favour of the desired regioisomer. It is worth noting that a similar effect of bulkiness on regioselectivity has already been observed in our preliminary studies.16
Functional modifications of intermediate B
To advance towards the third key step of the proposed strategy, i.e. the intramolecular aza-Wittig reaction, the O-silylated pyrrolo[1,2-a]indole 6b must be converted to the azido derivative and the 2α position of the 2,3-dihydropyrrolo[1,2-a]indole motif must be selectively oxidised.The latter required specific conditions, which will be discussed in the next section. The former was readily achieved by a conventional desilylation, O-tosylation and azido substitution sequence (Scheme 5). It is nevertheless worth noting that the tosylation step required specific base and conditions to avoid direct in situ chlorination and degradation.
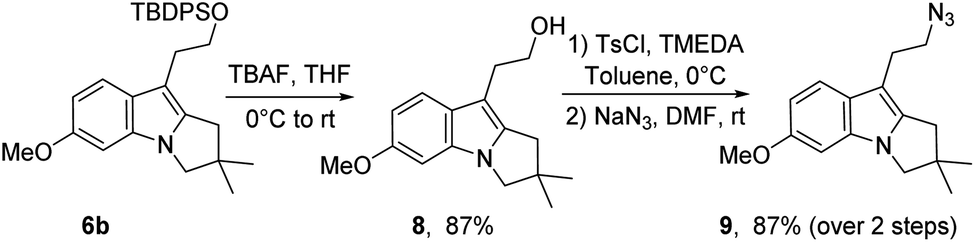 | ||
| Scheme 5 Functional group interchange of pyrrolo[1,2-a]indole 6. | ||
The selective oxidation at the 2α position of the 2,3-dihydropyrrolo[1,2-a]indole moiety proved much more tricky than expected. To study this critical step and set up appropriate conditions, the hexyl substituted 2,3-dihydropyrrolo[1,2-a]indole model compound 6c (Table 1: R = H, Fn = C4H9) was synthesized using the same strategy described in Schemes 3 and 4 (See ESI†).
|
||||||
|---|---|---|---|---|---|---|
| entry | Substrate | Reagents (equiv.) | Base | Time(min) | Alcohol 10 (%) | Ketone 11 (%) |
| a The model compound 6c was prepared according to the same gold catalysed strategy, see ESI. DPSO = diphenylsulfoxide. TFAA = trifluoroacetic anhydride. | ||||||
| 1 | 6ca, R = H Fn = C4H9 | DPSO (3) TFAA (3) | — | 45 | 27 (10a) | — |
| 2 | " | DMSO (3) | — | 1 | Trace | 88 (11a) |
| TFAA (3) | ||||||
| 3 | 6a, b or 9 | " | — | 1 | Degradation | |
| 4 | 6b | DMSO (6) | NEt3 (3) | 60 | 64 (10b) | — |
| TFAA (3) | ||||||
| 5 | 7b | DMSO (6) | NEt3 (6) | 15 | 60 (10c) | 29 (11c) |
| (COCl)2 (3) | ||||||
| 6 | 9 | " | NEt3 (6) | 120 | No reaction | |
| 7 | " | " | — | 15 | — | 87 (11d) |
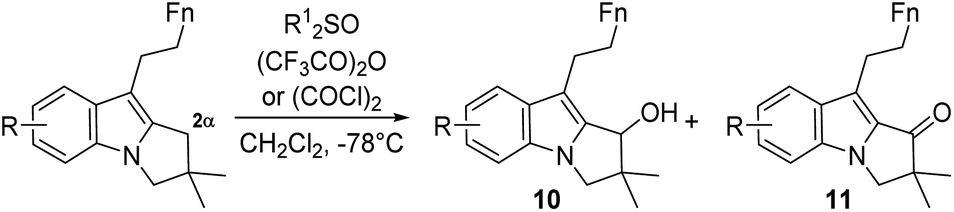
Applying to our model 6c the modified sulfoxide-based oxidation method (DPSO/TFAA) of indoles 2α position reported by Kawasaki et al.20 selectively afforded the alcohol 10a although in modest yield (entry 1). Therefore, the reaction conditions were adjusted by using DMSO, as less hindered sulfoxide than DPSO, leading efficiently within short time to ketone 11a (entry 2). Unfortunately, DMSO/TFAA oxidation conditions were deleterious with the more functionalized substrates 6a, b or 9 (Entry 3). Nevertheless, the alcohol 10b could be selectively formed from silylated compound 6b in the presence of triethylamine as base (entry 4). Shifting to Swern conditions (DMSO/(COCl)2/Et3N), the reaction gave a mixture of alcohol 10c and ketone 11c starting from the silylated compound 7b (entry 5). It is worth noticing that the alcohols 10 could not be correctly oxidised to the corresponding ketone in independent reactions with various reagents. However, such conditions were ineffective on azido derivative 9 as the starting material remain untouched (entry 6). Rewardingly, the formation of the expected azido ketone 11d was observed in very good yield without addition of base (entry 7).
Staudinger and aza-Wittig reactions
As first shown by Staudinger, azides can be converted to iminophosphoranes upon treatment with phosphine. After work-up, iminophosphoranes are hydrolysed to the corresponding amine and phosphine oxide.21 However, without water, iminophosphoranes could react with aldehydes or ketones, forming azaphosphetane intermediates, which decompose to imine and phosphine oxide, in a process analogue to the Wittig reaction.22With the azido 2,3-dihydropyrrolo[1,2-a]indolone 11d, such a sequence should directly provide the expected natural product (Scheme 6). However, and despite extensive investigation, no harmalidine could be detected whatever the conditions with different phosphines, including supported ones,23 and only the corresponding amine could be isolated, although in low to modest yields. These results revealed that the iminophosphorane intermediate was formed but could not react with the neighbouring ketone to form the azaphosphetane [2 + 2] intermediate.
 | ||
| Scheme 6 The expected Staudinger-aza-Wittig reaction towards harmalidine, highlighting the iminophosphorane and azaphosphetane intermediates. | ||
To solve this problem, we envisaged producing this amine by adjusting the Staudinger conditions to form the expected imine in a second step. The former could only be achieved by using the combination of a non-hindered and nucleophilic phosphine, i.e. the simple trimethylphosphine, with a mildly basic aqueous solution. Under these conditions, the amino 2,3-dihydropyrrolo[1,2-a]indolone 12 was isolated in good yield (Scheme 7). The structure of this compound was confirmed by X-ray diffraction of suitable crystals, grown after HPLC purification (see ESI†).‡.
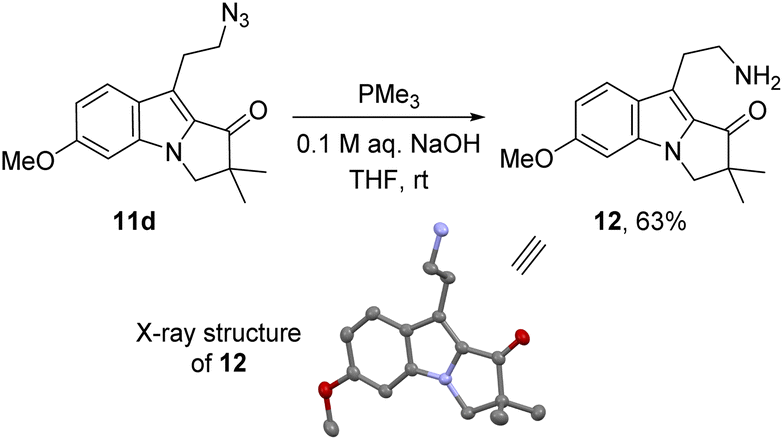 | ||
| Scheme 7 Formation of the amino pyrrolo[1,2-a]indolone 12 by a Staudinger reaction and its ellipsoid representation (hydrogen atoms have been omitted for clarity). | ||
The intramolecular cyclisation of this amino ketone was then attempted. However, and despite extensive experimentation (see Table 2 for selected examples), none of the expected cyclised product was detected. Either no transformation (e.g. entries 1–3) or extensive degradation (e.g. entries 4–5) was observed. Under a few conditions, an imine was isolated (entry 6), but its spectroscopic analysis, notably HR-MS, revealed a dimeric structure ascribed to the 12-membered diimino macrocycle 13 (see Table 2 Scheme). Attempts to avoid this dimerization were undertaken under higher dilution conditions, but only returned the starting material unchanged (entry 7).
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Conc. (mmol L−1) | Time (h) | Product |
| a No reaction.b Starting material recovered. MS: molecular sieves; PPTS: pyridinium para-toluenesulfonate. | |||||
| 1 | MS 4A | THF, rt | 10 | 24 | -a,b |
| 2 | NH4Cl | EtOH, 70 °C | 4 | 48 | -a,b |
| 3 | ArCOO.NEt3H | Tol, 70 °C | 10 | 24 | -a,b |
| 4 | TiCl4 | THF, rt | 10 | 24 | Degradation |
| 5 | Ti(OiPr)4 | THF, 80 °C | 0.2 | 24 | Degradation |
| 6 | cat. PPTS | PhH, 120 °C | 10 | 144 | Dimerb (13) |
| 55% | |||||
| 7 | cat. PPTS | PhH, 120 °C | 0.2 | 168 | -a,b |
It is interesting to note here that the NMR spectra of this dimer 13 did not fit with the data reported for the harmalidine natural product, although both were quite similar (see below).
These results as well as the aza-Wittig results may be due to the hindrance brought by the gem-dimethyl groups adjacent to the carbonyl group in 11d or 12. From the iminophosphorane derived from 11d, the [2 + 2] transition state towards the azaphosphetane intermediate may be too difficult to reach due to strong steric interaction with the gem-dimethyl groups, especially with large substituents around the phosphorous atom (Scheme 6). For the intramolecular imine formation from 12, the Bürgi-Dunitz angle required for the amine nucleophilic addition to the carbonyl group may be hampered again by this gem-dimethyl groups, but also by the strain induced by the rigidity of the 2,3-dihydropyrrolo[1,2-a]indolone motif. The latter effect would favour inter- rather than intra-molecular condensation, although the former would also be slowed down by the gem-dimethyl groups; and indeed, it took up to 6 days to get 55% yield of 13 (entry 6 in Table 2).
In response to this problematic imine formation, we envisaged a reverse cyclisation strategy via substitution of a leaving group at the terminal carbon side chain position by the nitrogen atom of an amine at the 2α position of the 2,3-dihydropyrrolo[1,2-a]indole moiety (Scheme 8, top). As amine can be directly introduced by oxy-amination at the 2α position (see below), cyclisation and deprotection would provide the reduced form of hamalidine, mentioned in the original discovery report.1 For this approach, we had to synthesized the chloroethyl 2,3-dihydropyrrolo[1,2-a]indole derivative 6d from azetidine 5a, because the introduction of halogenated group using classical methods from 8 consistently led to the formation of large amount of degradation products. The chlorinated but sensitive derivative 6d was finally available in 4 steps: TBDPS deprotection, tosylation, Finkelstein-type reaction and our gold-catalysed rearrangement (Scheme 8, bottom).
 | ||
| Scheme 8 Preparation of pyrrolo[1,2-a]indole 6d for reverse cyclisation strategy. | ||
The direct amination of the 2α position of 6d was performed using modified conditions of the oxidation reaction developed above (Table 1). In the presence of DMSO and oxalic chloride (1 equivalent each) and an excess of the desired amine, amination could occur (Table 3). From the silylated substrate 6b, the expected aminated 2,3-dihydropyrrolo[1,2-a]indole 14 derivatives could be obtained in high yield with benzylamine (14a, 78%), but in modest yield with allylamine (14b, 25%) together with degradation products for the latter (entry 1 vs. 2). Unfortunately, starting from the sensitive chloro compound 6d, the reaction with benzylamine only provided degradation products (entry 3), while with allylamine, another 2,3-dihydropyrrolo[1,2-a]indole unexpected derivative 15 was obtained, with a modest yield of 20% (entry 3 vs. 4). Extensive NMR investigation revealed that an N-allylcarbamate group was introduced at the 2α position, presumably via the formation of the corresponding alcohol derivative 10 (see Table 1). Attempts to advance 14a to a halogenated and/or cyclised intermediate only led to degradation. These results unfortunately precluded the full exploration of the envisaged reverse cyclisation strategy.
|
||||
|---|---|---|---|---|
| Entry | Substrate | RNH2 | Time (min) | Yield (%) |
| a Isolated along with 18% of remaining starting material.b isolated along with unidentified degradation products and starting material.c Degradation occurs. | ||||
| 1 | 6b (Fn = OTBDPS) | Benzylamine | 30 | 78a (14a) |
| 2 | 6b | Allylamine | 120 | 25b (14b) |
| 3 | 6d (Fn = Cl) | Benzylamine | 30 | -c |
| 4 | 6d | Allylamine | 120 | 20b (15) |
Surprises in NMR spectra
The present results raised questions about the structure reported for the isolated harmalidine natural product. Besides the problems associated to the intramolecular imine formation, differences were observed between the reported data and NMR data gained from our advanced intermediates 11d or 12 and the obtained dimer 13. Moreover, harmalidine has never been isolated again despite several studies on alkaloids extracts from Peganum harmala,24 nor synthesized.The harmalidine structure was mostly assigned by comparison with the harmaline structure, especially for attributing the methoxy position, while the position of the gem-dimethyl groups was assigned based on NMR spectra after catalytic hydrogenation of the natural product.1 However, the 1H and 13C NMR comparison was realised in two different deuterated solvents, i.e. DMSO-d6 and CDCl3 for respectively harmaline and harmalidine. To get a better comparison, and as harmaline is commercially available, we got it and recorded its NMR data in CDCl3 and checked its structure.
When these NMR data were compared to those of the advanced intermediates 11d and 12, the dimer 13, or harmaline in CDCl3, some discrepancies clearly showed up (Table 4). Indeed, the methoxyphenyl signals of the natural product were reported as a doublet of doublet at 6.80 ppm (1H, J = 8.9 and 1.8 Hz) and two doublets at 7.08 (1H, J = 1.8 Hz) and 7.42 ppm (1H, J = 8.9 Hz) attributed to H-10, H-12 and H-9, respectively.1 However, the aromatic AMX spin system for compounds 11d, 12 or the dimer 13 as well as for harmaline exhibited shielded H-12 signals ranging from 6.86 to 6.67 ppm. Similarly, the methylene protons H-15 were reported as a multiplet at 3.30 ppm, while these protons appeared as singlet shifted at 4.09 in 12 and 3.95 ppm in 13.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Position | Harmaline | Harmalidine1 | 13 | 12 | N-Methylharmaline | |||||
| 1Ha | 13Ca | 1Hb | 13Cb | 1H | 13C | 1H | 13C | 1H | 13C | |
| a Chemical shifts obtained from commercially available harmaline.b NMR data from ref. 1.c Values may be reversed.d Values may be reversed. | ||||||||||
| 1 (NH) | 8.03 br s | — | — | — | — | — | — | — | — | — |
| 2 | — | 128.4 | — | 126.3 | — | 129.5 | — | 130.8 | — | 125.6 |
| 3 | — | 157.0 | — | 161.3c | — | 168.4 | — | 198.1 (CO) | — | 162.3 |
| 5 | 3.83 m | 48.2 | 3.93 t | 42.8 | 4.26 m | 56.1 | 3.09 t | 43.2 | 3.64 t | 49.8 |
| 6 | 2.82 m | 19.5 | 3.11 t | 19.0 | 3.41 dd | 28.2 | 3.01 t | 28.9 | 3.01 t | 20.7 |
| 7 | — | 117.1 | — | 119.1 | — | 109.7 | — | 116.1 | — | 118.9 |
| 8 | — | 120.0 | — | 126.7d | — | 126.8 | — | 126.7 | — | 118.2 |
| 9 | 7.47 d | 120.8 | 7.42 d | 122.1 | 7.78 d | 122.0 | 7.59 d | 123.1 | 7.43 d | 120.9 |
| 10 | 6.83 dd | 111.0 | 6.80 dd | 115.0 | 6.84 dd | 111.3 | 6.81 dd | 113.0 | 6.80 dd | 111.0 |
| 11 | — | 158.2 | — | 144.5c | — | 158.0 | — | 159.0 | — | 158.4 |
| 12 | 6.86 d | 94.5 | 7.08 d | 94.0 | 6.69 d | 91.6 | 6.67 d | 91.7 | 6.74 d | 92.4 |
| 13 | — | 137.4 | — | 124.4d | — | 134.2 | — | 136.1 | — | 140.1 |
| 14 | 2.33 s | 22.0 | — | 39.7 | — | 48.6 | — | 54.4 | 3.11 s | 34.1 |
| 15 | — | — | 3.30 m | 43.0 | 3.95 s | 53.9 | 4.06 s | 50.1 | 4.07 s | 31.2 |
| 16 | — | — | 1.30 s | 27.0 | 1.42 s | 27.2 | 1.36 s | 24.9 | — | — |
| 17 | — | — | 1.30 s | 14.0 | 1.42 s | 27.2 | 1.36 s | 24.9 | — | — |
| OMe | 3.86 s | 55.6 | 3.85 s | 55.2 | 3.89 s | 55.6 | 3.86 | 55.6 | 3.89 s | 55.5 |
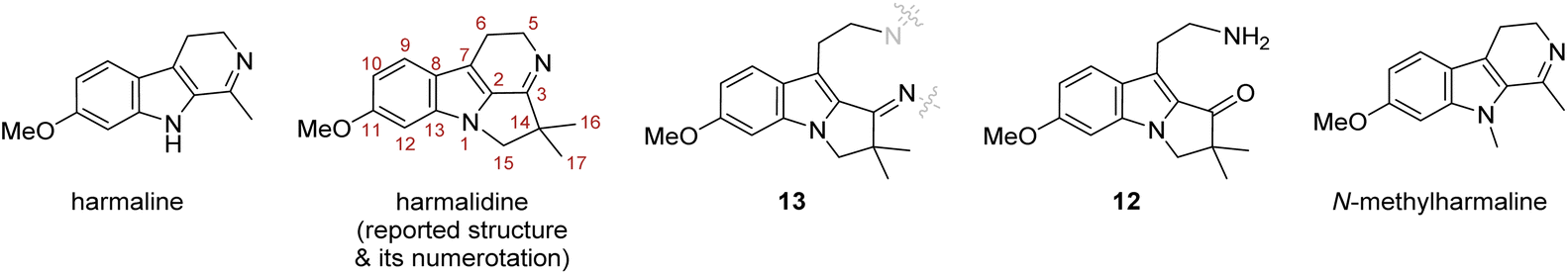
The analysis of the harmalidine 13C NMR data also revealed some inconsistencies. While the 13C NMR shifts for C-9, C-10 and C-12 are in agreement with those of various 6-methoxy pyrrolo[1,2-a]indole derivatives, the reported chemical shifts assigned to C-11 (C–OMe, δC 144.5 or 161.3 ppm) and C-13 (C–N, δC 124.4 or 126.7 ppm) are out of range compared to those of compounds 12, 13 or harmaline (Table 4) with δC values between 158 and 159 ppm and between 134 and 137 ppm respectively. Of note, if the δC 161.3 is attributed to C-11 and thus closer to the range, the remaining value δC 144.5 ppm would be assigned to the C-3 imine function but seems rather low compared to harmaline (δC 157.0 ppm) or 13 (δC 168.4 ppm). Furthermore, two different values were reported for the gem-dimethyl of the hydropyrrolyl part of the harmalidine (δC 14.0 and 27 ppm), while they seem to exhibit a single signal in 1H NMR. It is worth noting that one of these values is very similar to the one measured for dimer 13 (δC 27.0 vs. 27.2 ppm) and close to the N-indolic methyl of N-methyl harmaline (δC 31.2 ppm).
For further comparison, we prepared the N-methyl harmaline since it could be a structural alternative to the postulated harmalidine structure. To get this closely related compound, we modified the commercially available harmaline by methylation of the indolic nitrogen using iodomethane and NaH as base in DMF (See ESI†). Comparison of the 1H and 13C NMR data of N-methylharmaline25 showed again a shielded H-12 signal at 6.74 ppm compared to the reported value for harmalidine and a N-methyl signal at 4.07 ppm (H-15) in the same range of H-15 methylene of 12 and 13 (3.95–4.06 ppm). Furthermore, the C-3 imine is again different (δC 162.3 ppm) and closer to those of harmaline or 13 than to the reported harmalidine structure (see above).
These inconsistencies prompted us to compare experimental 13C NMR data of harmalidine and synthetic compound 13 with 13C predictions of harmalidine generated by computational chemistry (See Table S1 in ESI†). Indeed, computational prediction of NMR spectroscopic data have been used for both structure elucidation of new natural products and structural revisions.26 Nowadays, user-friendly accessible softwares are available and can be used to evaluate the quality of published 13C NMR.27 Based on the difference of experimental and values from Neural Network and HOSE-code 13C predictions,28 the same discrepancies were noticed for harmalidine (Table S1 in ESI†). Indeed, the major δC variations was localised on the gem-dimethyl hydropyrrolyl part of the harmalidine and on its aromatic C13, while the dimer 13 was in very good agreement with the harmalidine predicted values.
Nevertheless, a significant variation seems to occur at the methylene carrying the non-indolic nitrogen atom (position 5 in harmalidine numbering). This difference is lower in N-methyl harmaline (δC 49.8 vs. 42.8 ppm in the reported harmalidine structure), but the two methyl groups are now quite different (δC 31.3 and 34.1 vs. 14.3 and 27.0 ppm). Both N-methyl harmaline and the dimeric structure of 13 may thus not correspond to the actual harmalidine structure, despite the nice fit between experimental and calculated NMR values.
Noteworthy, the original isolation paper1 suggested another chemical structure for harmalidine, mostly from biosynthetic hypothesis. This proposed structure corresponds to an isomer in which the dimethyl groups are located at C-15 position instead of C-14. This proposal was nevertheless excluded upon mass analysis. 13C predictions of this isomer generated by computational chemistry was also in agreement with this conclusion (See Table S2 in ESI†). Therefore, the exact structure of this natural product remains so far elusive.
Conclusions
In this work, we demonstrated that our gold catalysed rearrangement of N-aryl-2-alkynylazetidines to pyrrolo[1,2-a]indoles could be extended to complex substrates and scale up to gram quantities. The synthesis of the psychoactive harmalidine was selected to illustrate the potential of this method. Although advanced amino 2,3-hydropyrrolo[1,2-a]indole or indolone intermediates could be readily obtained according to two routes in only 11 steps from but-3-yn-1-ol (14% overall yield), the natural product as reported could not be formed, but a dimeric compound was isolated. The latter exhibits an unusual pyrrolo[1,2-a]indole structure involved in a novel 12-membered diimino macrocycle. This structure and various NMR discrepancies between the reported natural structure and those gained through this work suggest revising the proposed structure but do not allow to conclude on the real natural product structure.Further work is currently underway to re-isolate harmalidine and reinvestigate its structure.
Author contributions
Solène Miaskiewicz: Investigation Jean-Marc Weibel: validation Patrick Pale: writing – original draft Aurélien Blanc: supervision, writing – review & editing, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the French Ministry of Research and the CNRS for financial support. S. M. thanks the French Ministry of Research for a PhD fellowship. The authors thank Dr Pierre de Frémont for crystallographic structure refinement of compound 12. The MCAT company is also acknowledged for providing gold catalyst precursors (https://www.mcat.de).Notes and references
- S. Siddiqui, O. Y. Khan, B. S. Siddiqui and S. Faizi, Harmalidine, a β-Carboline Alkaloid from Peganum harmala, Phytochemistry, 1987, 26, 1548 CrossRef CAS.
- J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Isatisine A, a Novel Alkaloid with an Unprecedented Skeleton from Leaves of Isatis indigotica, Org. Lett., 2007, 9, 4127 CrossRef CAS PubMed.
- (a) D. A. Okorie, Polyavolinamide, an Indolosesquiterpene Alkaloid from Polyathia suaveolens, Phytochemistry, 1981, 20, 2575 CrossRef CAS; (b) C. M. Hasan, T. M. Healey, P. G. Waterman and C. H. Schwalbe, Chemical Studies on the Annonaceae. Part 9. Indolosesquiterpene and Aporphine Alkaloids from Greenwayodendron (Polyalthia) suaveolens Stem Bark. X-ray Crystal Structure of Greenwayodendrin-3-one, J. Chem. Soc., Perkin Trans., 1982, 1, 2807 RSC.
- (a) I. Ngantchou, B. Nyasse, C. Denier, C. Blonski, V. Hannaert and B. Schneider, Antitrypanosomal Alkaloids from Polyalthia suaveolens (Annonaceae): Their Effects on Three Selected Glycolytic Enzymes of Trypanosoma Brucei, Bioorg. Med. Chem. Lett., 2010, 20, 3495 CrossRef CAS PubMed; (b) J. L. Poussett, A. Cave, A. Chiaroni and C. Riche, A novel Bis-indole Alkaloid. X-ray Crystal Structure Determination of Borreverine and its Rearrangement Product on Deacetylation, J. Chem. Soc., Chem. Commun., 1977, 261 RSC.
- L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn, V. M. Avery and A.-C. Flinderoles, Antimalarial Bis-indole Alkaloids from Flindersia Species, Org. Lett., 2009, 11, 329 CrossRef CAS PubMed.
- For a review, see: J.-C. Andrez, Mitomycins Syntheses: A Recent Update, Beilstein J. Org. Chem., 2009, 5, 33, DOI:10.3762/bjoc.5.33.
- (a) S. Wakaki, H. Marumo, K. Tomioka, G. Shimizu, E. Kato, H. Kamada, S. Kudo and Y. Fujimoto, Isolation of new Fractions of Antitumor Mitomycins, Antibiot. Chemother., 1958, 8, 228 CAS; (b) V. Lefemine, M. Dann, F. Barbatschi, W. K. Hausmann, V. Zbinovsky, P. Monnikendam, J. Adam and N. Bohonos, Isolation and Characterization of Mitiromycin and other Antibiotics Produced by Streptomyces Verticillatus, J. Am. Chem. Soc., 1962, 84, 3184 CrossRef; (c) A. Tulinsky, Structure of Mitomycin A, J. Am. Chem. Soc., 1962, 84, 3188 CrossRef CAS.
- (a) W. T. Bradner, Mitomycin C: A Clinical Update, Cancer Treat. Rev., 2001, 27, 35 CrossRef CAS PubMed , ; For the initially proposed but now proven mode of action, see:; (b) V. N. Oyer and W. Szybalski, Mitomycins and Porfiromycin; Chemical Mechanism of Activation and Cross-linking of DNA, Science, 1964, 145, 55 CrossRef CAS.
- For reviews on the synthesis of pyrrolo[1,2-a]indoles, see: (a) N. Monakhova, S. Ryabova and V. Makarov, Synthesis and Some Biological Properties of Pyrrolo[1,2-a]indoles, J. Heterocycl. Chem., 2016, 53, 685 CrossRef CAS; (b) Y. G. Shelke, P. E. Hande and S. J. Gharpure, Recent Advances in the Synthesis of Pyrrolo[1,2-a]indoles and their Derivatives, Org. Biomol. Chem., 2021, 19, 7544 RSC.
- A. Karadeolian and M. A. Kerr, Total Synthesis of (+)-Isatisine A, J. Org. Chem., 2010, 75, 6830 CrossRef CAS PubMed.
- (a) D. H. Dethe, R. D. Erande and A. Ranjan, Biomimetic Total Syntheses of Flinderoles B and C, J. Am. Chem. Soc., 2011, 133, 2864 CrossRef CAS; (b) R. M. Zeldin and F. D. Toste, Synthesis of Flinderoles B and C by a Gold-Catalyzed Allene Hydroarylation, Chem. Sci., 2011, 2, 1706 RSC.
- For leading references on mitomycin synthetic studies as well as total syntheses, see: (a) H. Namiki, S. Chamberland, D. A. Gubler and R. M. Williams, Synthetic and Biosynthetic Studies on FR900482 and Mitomycin C: An Efficient and Stereoselective Hydroxymethylation of an Advanced Benzazocane Intermediate, Org. Lett., 2007, 9, 5341 CrossRef CAS PubMed; (b) A. L. Williams, J. M. Srinivasan and J. N. Johnston, Synthesis of an Advanced Intermediate en Route to the Mitomycin Natural Products, Org. Lett., 2006, 8, 6047 CrossRef CAS PubMed; (c) R. S. Coleman, F.-X. Felpin and W. Chen, Mitomycin Synthetic Studies: Stereocontrolled and Convergent Synthesis of a Fully Elaborated Aziridinomitosane, J. Org. Chem., 2004, 69, 7309 CrossRef CAS PubMed; (d) S. J. Danishefsky and J. M. Schkeryantz, Chemical Explorations Driven by an Enchantment with Mitomycinoids - A Twenty Year Account, Synlett, 1995, p. 475 Search PubMed; (e) T. Fukuyama and L. Yang, Practical total synthesis of (±)-mitomycin C, J. Am. Chem. Soc., 1989, 111, 8303 CrossRef CAS; (f) T. Fukuyama, F. Nakatsubo, A. J. Cocuzza and Y. Kishi, Synthetic Studies toward Mitomycins. III. Total Syntheses of Mitomycins A and C, Tetrahedron Lett., 1977, 4295 CrossRef CAS.
- R. Peters, P. Waldmeier and A. Joncour, Efficient Synthesis of a 5-HT2C Receptor Agonist Precursor, Org. Proc. Res. Dev., 2005, 9, 508 CrossRef CAS.
- (a) J. S. Webb, D. B. Cosulich, J. H. Mowat, J. B. Patrick, R. W. Broschard, W. E. Meyer, R. P. Williams, C. F. Wolf, W. Fulmor, C. Pidacks and J. E. Lancaster, Structures of Mitomycins A, B, and C and Porfiromycin. Part I, J. Am. Chem. Soc., 1962, 84, 3185 CrossRef CAS; (b) J. S. Webb, D. B. Cosulich, J. H. Mowat, J. B. Patrick, R. W. Broschard, W. E. Meyer, R. P. Williams, C. F. Wolf, W. Fulmor, C. Pidacks and J. E. Lancaster, Structures of Mitomycins A, B, and C and Porfiromycin. Part II, J. Am. Chem. Soc., 1962, 84, 3187 CrossRef.
- (a) R. Pertschi, S. Miaskiewicz, N. Kern, J.-M. Weibel, P. Pale and A. Blanc, Gold(I)-Catalyzed Divergent and Diastereoselective Synthesis of Azepines by Ammoniumation/Ring-Expansion Reactions, Chem. Catal., 2021, 1, 129 CrossRef; (b) F. Sirindil, S. Golling, R. Lamare, J.-M. Weibel, P. Pale and A. Blanc, Synthesis of Indolizine and Pyrrolo[1,2-a]azepine Derivatives via a Gold(I)-Catalyzed Three-Step Cascade, Org. Lett., 2019, 21, 8997 CrossRef CAS PubMed; (c) F. Sirindil, J.-M. Weibel, P. Pale and A. Blanc, Total Synthesis of Rhazinilam through Gold-Catalyzed Cycloisomerization-Sulfonyl Migration and Palladium-Catalyzed Suzuki-Miyaura Coupling of Pyrrolyl Sulfonates, Org. Lett., 2019, 21, 5542 CrossRef CAS PubMed; (d) R. Pertschi, J.-M. Weibel, P. Pale and A. Blanc, Benzosultam Synthesis by Gold(I)-Catalyzed Ammonium Formation/Nucleophilic Substitution, Org. Lett., 2019, 21, 5616 CrossRef CAS; (e) S. Miaskiewicz, B. Gaillard, N. Kern, J.-M. Weibel, P. Pale and A. Blanc, Gold(I)-Catalyzed N-Desulfonylative Amination versus N-to-O 1,5-Sulfonyl Migration: A Versatile Approach to 1-Azabicycloalkanes, Angew. Chem., Int. Ed., 2016, 55, 9088 CrossRef CAS PubMed; (f) M. Hoffmann, J.-M. Weibel, P. de Fremont, P. Pale and A. Blanc, Gold(I)/(III)-Catalyzed Rearrangement of Divinyl Ketones and Acyloxyalkynyloxiranes into Cyclopentenones, Org. Lett., 2014, 16, 908 CrossRef CAS PubMed.
- N. Kern, M. Hoffmann, A. Blanc, J.-M. Weibel and P. Pale, Gold(I)-Catalyzed Rearrangement of N-Aryl 2-Alkynylazetidines to Pyrrolo[1,2-a]indoles, Org. Lett., 2013, 15, 836 CrossRef CAS PubMed.
- M. Journet, D. Cai, L. M. DiMichele and R. D. Larsen, Highly Efficient Synthesis of α,β-Acetylenic Aldehydes from Terminal Alkynes using DMF as the Formylating Reagent, Tetrahedron Lett., 1998, 39, 6427 CrossRef CAS.
- A. Nakamura and M. Nakada, Allylic Oxidations in Natural Product Synthesis, Synthesis, 2013, 45, 1421 CrossRef CAS.
- M. Yamashita and I. Ojima, Effective Route to Azetidines from Azetidin-2-ones with the Use of Hydroalanes as Specific Reducing Agents, J. Am. Chem. Soc., 1983, 105, 6339 CrossRef CAS.
- (a) M. Tayu, K. Higuchi, M. Inaba and T. Kawasaki, Sulfoxide-TFAA and Nucleophile Combination as new Reagent for Aliphatic C-H Functionalization at Indole 2α-position, Org. Biomol. Chem., 2013, 11, 496 RSC; (b) K. Higuchi, M. Tayu and T. Kawasaki, Active Thionium Species Mediated Functionalization at the 2α-Position of Indole Derivatives, Chem. Commun., 2011, 47, 6728 RSC.
- H. Staudinger and J. Meyer, Ueber neue Organische Phosphorverbindungen II. Phosphazine, Helv. Chim. Acta, 1919, 2, 635 CrossRef CAS.
- F. Palacios, C. Alonso, D. Aparicio, G. Rubiales and J. M. de los Santos, The aza-Wittig reaction. An Efficient Tool for the Construction of Carbon-Nitrogen Double Bonds, Tetrahedron, 2007, 63, 523 CrossRef CAS.
- (a) J. Kim and R. J. Thomson, Enantioselective Total Synthesis of the Osteoclastogenesis Inhibitor (+)-Symbioimine, Angew. Chem., Int. Ed., 2007, 46, 3104 CrossRef CAS PubMed; (b) S. Ayesa, B. Samuelson and B. Classon, A One-Pot, Solid-Phase Synthesis of Secondary Amines from Reactive Alkyl Halides and an Alkyl Azide, Synlett, 2008, 97 CAS.
- For recent examples, see: (a) Z.-N. Wu, N.-H. Chen, Q. Tang, S. Chen, Z.-C. Zhan, Y.-B. Zhang, G.-C. Wang, Y.-L. Li and W.-C. Ye, β-Carboline Alkaloids from the Seeds of Peganum harmala and Their Anti-HSV-2 Virus Activities, Org. Lett., 2020, 22, 7310 CrossRef CAS PubMed; (b) K.-B. Wang, D.-H. Li, Y. Bao, F. Cao, W.-J. Wang, C. Lin, W. Bin, J. Bai, Y.-H. Pei, Y.-K. Jing, D. Yang, Z.-L. Li and H.-M. Hua, Structurally Diverse Alkaloids from the Seeds of Peganum harmala, J. Nat. Prod., 2017, 80, 551 CrossRef CAS PubMed.
- R. N. Gupta and I. D. Spenser, 3,4-Dihydro-β-carbolines: I. The Alkylation of 1-Substituted Nβ-Alkyl-3,4-Dihydro-β- carbolines Anhydro Bases, Can. J. Chem., 1962, 40, 2041 CrossRef CAS.
- B. K. Chhetri, S. Lavoie, A. M. Sweeney-Jones and J. Kubanek, Recent Trends in the Structural Revision of Natural Products, Nat. Prod. Rep., 2018, 35, 514 RSC.
- (a) E. Jonas, S. Kuhn and N. Schlörer, Prediction of Chemical Shift in NMR: A Review, Magn. Reson. Chem., 2021 DOI:10.1002/mrc.5234; (b) W. Robien, ed. A. D. Kinghorn, H. Falk, S. Gibbons and J. Kobayashi, Springer International Publishing AG, 2017, pp. 137–215, Ch. 3.A Critical Evaluation of the Quality of Published 13C NMR Data in Natural Product Chemistry in Progress in the Chemistry of Organic Natural Products 105 Search PubMed.
- N. Haider and W. Robien, https://nmrpredict.orc.univie.ac.at/c13robot/robot.php.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2111055. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05685b |
| ‡ Crystallographic data for 12 have been deposited in the CCDC under accession number 2111055. |
| This journal is © The Royal Society of Chemistry 2022 |