
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanium dioxide-based anode materials for lithium-ion batteries: structure and synthesis
Huili Shia,
Chaoyun Shia,
Zhitong Jiaa,
Long Zhanga,
Haifeng Wangb and
Jingbo Chen *ac
*ac
aCollege of Chemistry and Chemical Engineering, Guizhou University, Guiyang, 550025, China
bCollege of Material and Metallurgy, Guizhou University, Guiyang, 550025, China
cCollaborative Innovation Center of Guizhou Province for Efficient Utilization of Phosphorus and Fluorine Resources, Guizhou University, Guiyang, 550025, China
First published on 23rd November 2022
Abstract
Lithium-ion batteries (LIBs) have high energy density, long life, good safety, and environmental friendliness, and have been widely used in large-scale energy storage and mobile electronic devices. As a cheap and non-toxic anode material for LIBs, titanium dioxide (TiO2) has a good application prospect. However, its poor electrical conductivity leads to unsatisfactory electrochemical performance, which limits its large-scale application. In this review, the structure of three TiO2 polymorphs which are widely investigated are briefly described, then the preparation and electrochemical performance of TiO2 with different morphologies, such as nanoparticles, nanowires, nanotubes, and nanospheres, and the related research on the TiO2 composite materials with carbon, silicon, and metal materials are discussed. Finally, the development trend of TiO2-based anode materials for LIBs has been briefly prospected.
1 Introduction
The development of advanced energy conversion systems such as Lithium-ion batteries (LIBs) is an important way to alleviate the world's energy shortage and global warming problems. LIBs with high power density and high energy density used at electric vehicles, energy storage systems and smart grids are increasingly demanding,1,2 but conventional LIBs are not sufficient to meet these demands, so new electrode materials with high capacity and low cost have become a hot research topic in recent years.3 As the most successful anode material in commercial LIBs at present, graphite has several fatal problem. Firstly, the operating voltage of graphite is below 0.2 V versus Li/Li+, which is close to the lithium deposition voltage. Lithium dendrites may be generated during the repeated charge and discharge process, resulting in internal short circuit and serious safety risks.4 Compared with graphite, TiO2 has a relatively high lithium insertion/extraction voltage of about 1.7 V, which can effectively avoid lithium dendrites. Secondly, the volume expansion of TiO2 during charge and discharge (<4%) is much smaller than that of graphite (10%). However the theoretical capacity of TiO2 is 335 mA h g−1, which is similar to that of graphite (372 mA h g−1).5 Therefore, TiO2 is one of the candidates materials to replace graphite for its excellent safety.6,7 However, as a wide bandgap semiconductor material, TiO2 has poor electronic conductivity (10−12 to 10−7 S cm−1) and low Li+ diffusion rate (10−15 to 10−9 cm2 s−1). To address these problems, current solutions are included: (1) Nanostructures designing to reduce ionic and electronic transport distances,8 which facilitates the electron/ion transport by increasing the contact area of electrolyte/electrode material. (2) Composites designing to take advantages of the high conductivity of carbon and metal materials and the high capacity of silicon materials, which improves the overall conductivity of electron and lithium by the synergistic effect, thereby improving the electrochemical performance. In this paper, the structure and performance of TiO2 are firstly briefly described, and then the research progress of TiO2 nanostructures designing and composites designing which are two aspects to enhance the electrochemical performance are reviewed, and finally, the development trend of TiO2 has further prospected.2 Crystal structure and lithium storage mechanism of TiO2
There are mainly three crystal types of TiO2: anatase, rutile and brookite.9 In addition, there are some rare crystal types, such as bronze. The crystal structures of anatase, rutile, and brookite are shown in Fig. 1.10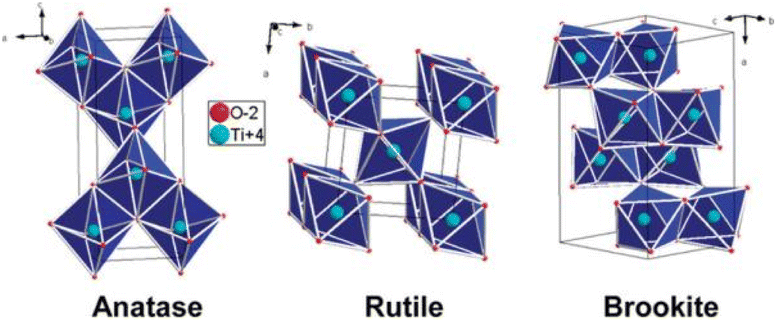 | ||
| Fig. 1 Anatase, rutile, and brookite crystal structure of TiO2.10 | ||
The basic composition of each crystal form is composed of an [TiO6]8− octahedron included of one Ti4+ and six O2−, and different connection modes and arrangement orders constitute different crystal structures of TiO2. The anatase phase is composed of [TiO6]8− octahedron with common edges, and its octahedra distortion is so significantly large that the symmetry is lower than that of the orthorhombic crystal system. The rutile phase is composed of [TiO6]8− octahedrons with common tops and common edges, and its octahedral distortion is smaller than that of anatase, and its symmetry is better than that of anatase. The brookite phase belongs to the orthorhombic system, which consists of six TiO2 to form a crystal cells. This crystal structure makes the brookite phase very unstable and therefore rare in nature. Table 1 lists the structure and lithium-storage properties of TiO2 polymorphs. Generally speaking, the lithium insertion process of titanium dioxide can be expressed by eqn (1):
| TiO2 + xLi+ + xe− → LixTiO2 | (1) |
| Polymorph | Crystal structure | Density (g cm−3) | Band gap energy (eV) | Unit cell (Å) | Li+ insertion (mol)/specific capacity (mA h g−1) | |
|---|---|---|---|---|---|---|
| Bulk | Nano | |||||
| Anatase | Tetragonal (I41/amd) | 3.79 | 3.20–3.23 | a = 3.79, c = 9.51 | 0.5/168 | 1.0/335 |
| Rutile | Tetragonal (P42/mnm) | 4.13 | 3.02–3.04 | a = 4.59, c = 2.96 | 0.1/34 | 0.85/285 |
| Brookite | Orthorhombic (Pbca) | 3.99 | 3.14–3.31 | a = 9.17, b = 5.46, C = 5.14 | 0.1/34 | 1.0/335 |
The lithium insertion process of anatase TiO2 can be divided into three regions (A, B, C) as shown in Fig. 2.11
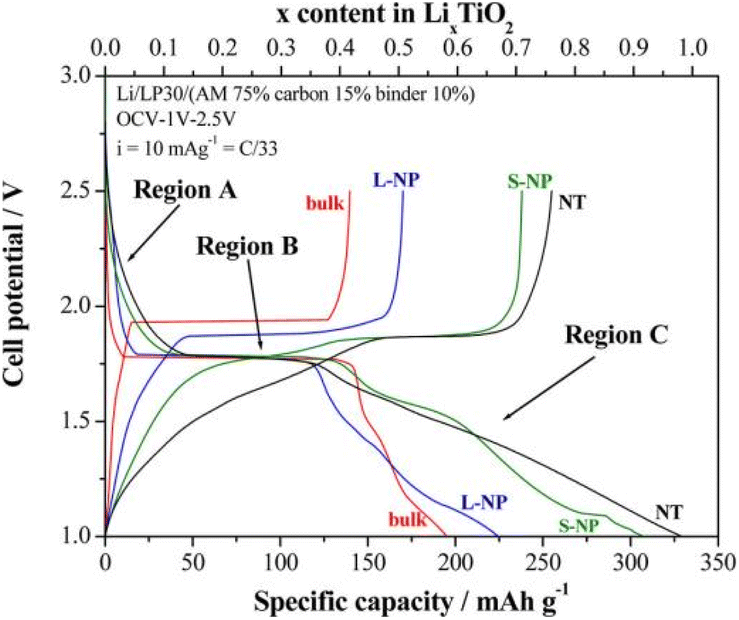 | ||
| Fig. 2 The voltage–capacity curves of anatase TiO2.11 | ||
Region A: The open-circuit voltage plunges to 1.8 V, and this region is considered as a small amount of Li+ embedded in anatase lattice (LixTiO2). In the bulk material, only 0.028 Li can be embedded, and reducing the particle size can increase the Li+ accommodation capacity. The lithium embedding coefficients of large and small nanoparticles are up to 0.05 and 0.12.
Region B: A plateau appears around 1.75 V, a typical two-phase mechanism. Li+ continues to be embedded in TiO2 lattice, and the Li-poor phase of tetragonal crystal system changes to the Li-rich phase of orthogonal crystal system, and the two phases coexist. For bulk materials, the lithium content combined with the platform is 0.39 Li/TiO2 formulation units.
Region C: The voltage decreases continuously and linearly below 1.7 V. The smaller the size of anatase crystal in this region, the larger the insertion amounts of Li+.
The lithium-storage mechanism of other crystal types is rarely reported. The insertion of Li+ in TiO2 rutile is highly anisotropic, preferentially occurring in the c-axis,12 tending to be one-dimensional, due to the large difference between the (ab-plane) a-axis and c-axis Li+ diffusion coefficients. c-Axis pore channels are narrow, with a radius of 4 nm for octahedral vacancies and 6 nm for Li+, so that only a small amount of Li+ can be embedded at room temperature. With the insertion of Li+, the diffusivity of Li+ in rutile decreases, further limiting the embedding of Li+. The process of Li+ embedding brookite TiO2 is also limited by structure, and the diffusion process is mainly along the direction of c-axis where the resistance is low. But the lithium storage mechanism is still unclear.
3 Pure nanostructured TiO2 materials
TiO2 materials have disadvantages such as low electrical conductivity, poor cycle performance and rate performance, however nanostructured TiO2 electrode materials have the advantages of large surface area and short ion diffusion distance,16 so the researchers are devoted to preparing various TiO2 nanomaterials. In terms of dimensions, TiO2 nanomaterials can be divided into zero-dimensional (TiO2 nanoparticles17,18), one-dimensional (TiO2 nanowires, TiO2 nanorods, TiO2 nanotubes, etc.19–23), two-dimensional (TiO2 nanosheets24–26), three-dimensional (TiO2 nanoflowers, TiO2 nanospheres, TiO2 nanotrees, etc.27–29). The studies find that the particle size, morphology, and surface area of nano-TiO2 have obvious effects on its electrochemical performance. When the particle size decreases, TiO2 nanomaterials have a larger active material/electrolyte contact area and shorter ion diffusion distance,30 which can effectively improve the rate performance and high-current charge–discharge performance. The unique morphology can alleviate the volume expansion during lithiation/delithiation processes, and improve the cycle stability. The larger surface area can increase active material/electrolyte contact area, and thinner wall thickness can reduce the diffusion distance of Li+, which is beneficial to improve the electrochemical performance.3.1 Zero-dimensional TiO2
Partheeban et al.38 used melamine as a soft template to prepare anatase phase TiO2 nanoparticles with an average size of 10–20 nm. It showed excellent electrochemical performance with an initial discharge capacity of 272 mA h g−1 at 1C, a slight decrease of about 240 mA h g−1 after 50 cycles, and still had a capacity of 208 mA h g−1 after 1000 cycles. Zhang et al.39 designed a two-step hydrothermal method for the preparation of brookite phase mesocrystalline TiO2 using amorphous titanate as a precursor and oxalic acid as a structural guide (see Fig. 3), which maintained a capacity of 105.7 mA h g−1 after 500 cycles at 5C. Ouardi et al.40 used titanium tetrisopropanol and sodium alginate as titanium precursor and template, respectively, and then calcined to prepare nano-sized TiO2. At 0.1C, TiO2 anode calcined at 300 °C and 450 °C had reversible capacities of 266 mA h g−1 and 275 mA h g−1 in the first cycle, respectively. Coulomb efficiencies are 70% and 75%, respectively. The cycling performances and capacities of TiO2 nanoparticlesis depicted in Table 2.
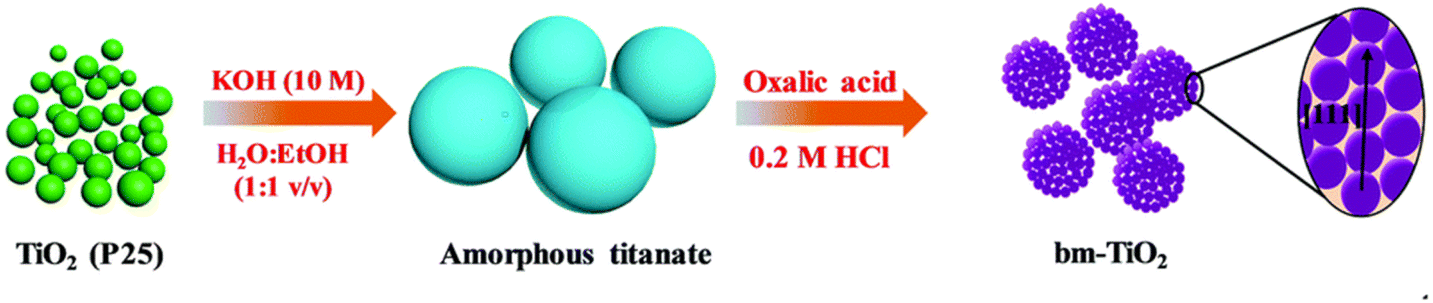 | ||
| Fig. 3 Preparation process of mesomorphic TiO2 with brookite phase.39 | ||
| Pm | Cd | Initial coulombic efficiency | Idc/mA h g−1 | Cn | Rc/mA h g−1 | Areal capacities (mA h cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| a Pm—preparation method, Tm—template method, Hm—hydrothermal method, Cd—current density, Ice—initial coulombic efficiency, Idc—initial discharge capacity, Cn—cycle numbers, Rc—remaining capacity, Ac—areal capacities. | |||||||
| Tm | 1C | 50% | 272 | 50 | 240 | 0.41 | 38 |
| Hm | 5C | 58% | — | 500 | 105.7 | 0.13 | 39 |
| Tm | 0.1C | 75% | 364 | 100 | 180 | — | 40 |
3.2 One-dimensional TiO2
Wang et al.45 prepared TiO2 nanowires containing anatase phase and a small amount of bronze phase by hydrothermal method with diameters of 40–80 nm and lengths of 400–1500 nm, and had a capacity of 280 mA h g−1 after 40 cycles at 140 mA g−1 with a coulombic efficiency close to 98%. Wu et al.46 used the hydrothermal method to prepare anatase TiO2 nanowires with initial discharge/charge capacities of 260 mA h g−1 and 224 mA h g−1 at 20 mA g−1, respectively, and the reversible capacity remained around 216 mA h g−1 in the 20th cycle. At a current density of 200 mA g−1, the discharge capacity was about 159 mA h g−1 at the 20th cycle, which showed good cycling stability. Wang et al.47 synthesized porous linear TiO2. And the synthesis process consisted of two steps, firstly, the synthesis of titanium ethanoate precursor nanowires by microwave-assisted solvothermal method, followed by the conversion of titanium ethanoate nanowires into porous TiO2 nanowires with diameters of about 0.4–2 μm and lengths of 2–20 μm, having a high surface area of 252 m2 g−1. Its unique nanoporous structure results in better electrolyte permeability and faster diffusion rates of electrons and Li+, as well as better buffering volume changes during charge/discharge cycles. The cycling performances and capacities of TiO2 nanowires is depicted in Table 3.
| Pm | L/nm | D/nm | Cd | Cn | Rc/mA h g−1 | Ref. |
|---|---|---|---|---|---|---|
| a Pm—preparation method, Hm—hydrothermal method, Sm—solvothermal method, L—length, D—diameter, Cd—current density, Cn—cycle numbers, Rc—remaining capacity. | ||||||
| Hm | 400–1500 | 40–80 | 140 mA g−1 | 40 | 280 | 45 |
| Hm | 250 | 20 | 200 mA g−1 | 20 | 159 | 46 |
| Sm | 2000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
400–2000 | 0.2C | 51 | 114.4 | 47 |
Seisenbaeva et al.52 prepared anatase phase nanorods, using a one-step hydrothermal method. It had homogeneous mesopores, high porosity, a surface area of about 300 m2 g−1, and initial charge/discharge capacities of 299 mA h g−1 and 225 mA h g−1 at C/15, respectively. Ji53 sintered titanium foil coated with sodium hydroxide solution at 800 °C to prepare TiO2 nanorods, which mainly consisted of rutile phase, with a width of about 500 nm and a thickness of about 5 μm. At the same time, they also studied the effect of different temperatures on TiO2 nanorods. Santhi et al.54 used a hydrothermal method to synthesize TiO2 nanorods at different pH value and it was showed that when pH = 7, the length of nanorods was about 300–350 nm and the diameter was about 70–100 nm. When the pH was increased to 9, the morphology was changed from rod-like structure to a platelet-like nanostructure.
Gong et al.60 used the anodic oxidation method to prepare TiO2 nanotubes with different diameters and lengths, and TiO2 nanotubes arrays with diameters ranging from 30 nm to 90 nm could be obtained by controlling the anodic oxidation voltage. Xu et al.61 used the hydrothermal method to prepare anatase TiO2 nanotubes with a diameter of about 10 nm and a length about 200–400 nm. At 36 mA g−1, the first charge–discharge capacities were 290 mA h g−1 and 238 mA h g−1, respectively. At the 20th cycle, the reversible capacity was still about 200 mA h g−1, and the coulomb efficiency is about 98%. Fasakin et al.62 obtained TiO2 nanotubes with average length, thickness, and diameter of 4 μm, 17 nm, and 20 nm, respectively. After stirring and hydrothermal treatment, the surface area of TiO2 nanoparticles increased from the original 8 m2 g−1 to 62 m2 g−1. The initial capacity of the original TiO2 nanoparticles at a current density of 36 mA g−1 was 98 mA h g−1, which decreased to 38 mA h g−1 at the 100th cycle. While the TiO2 nanotubes showed an initial capacity of about 123 mA h g−1 at a current density of 36 mA g−1 and showed a capacity of 142 mA h g−1 at the 100th cycle, which increased by about 17% relative to the initial value. The cycling performances and capacities of TiO2 nanotubes is depicted in Table 4.
| Pm | T/nm | Cd | Initial coulombic efficiency | Cn | Rc/mA h g−1 | Areal capacities (mA h cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| a Pm—preparation method, Hm—hydrothermal method, Tm—template method, Aom—anodic oxidation method, T—thickness, Cd—current density, Ice—initial coulombic efficiency, Cn—cycle numbers, Rc—remaining capacity, Ac—areal capacities. | |||||||
| Hm | — | 36 mA g−1 | 82% | 20 | 200 | 0.44 | 61 |
| Hm | 17 | 36 mA g−1 | 94% | 100 | 142 | 0.497 | 62 |
| Tm | 40 | 1C | 72.2% | 500 | 150 | 3.58 | 63 |
| Aom | 17 | 320 mA g−1 | — | 50 | 179 | — | 64 |
| Hm | 3–4.5 | 0.1C | 65% | 50 | 265 | 1.33 | 65 |
Zhuang et al.75 synthesized TiO2–B nanofibers with a specific surface area of 112 m2 g−1, high thermal stability and high crystallinity by steam thermal method. The initial capacity was 193.8 mA g−1, which dropped to 189.5 mA g−1 after 100 cycles at 1C, with a good capacity retention rate of 97.8%. Luo et al.76 synthesized unique MoO2-modified TiO2 nanofibers by electrospinning and layer-by-layer (LBL) self-assembly processes. It consisted of a TiO2 nanofiber core and a thin metal-like MoO2 nanolayer. MoO2-modified TiO2 nanofibers showed 514.5 mA h g−1 after 50 cycles at 0.2C. Zhu et al.77 used electrospinning and sintering processes to prepare nanofibers, which showed stable performance at the end of 800 cycles at 150 mA g−1, with a capacity retention of about 136 mA h g−1.
3.3 Two-dimensional TiO2
Liu et al.81 synthesized nanosheet-structured porous TiO2 with a thickness of 5–10 nm by the solvothermal method, with an initial discharge capacity of 216 mA h g−1 at a high current density of 10C and maintained at 200 mA h g−1 after 200 cycles with a capacity retention rate of 92.6%. Wang et al.82 used the multiple spin-coating methods on a fluorine-doped tin oxide substrate to prepare TiO2 nanosheets arrays. More importantly, the size and shape can be controlled and the nanosheets arrays can be arranged on the substrate in an orderly and horizontally homogeneous manner. Dai et al.83 successfully prepared ultrathin TiO2 nanosheets with a large surface area and fast electron transfer using a hydrothermal method with P123 as a soft template.
3.4 Three-dimensional TiO2
Zhang et al.85 synthesized homogeneous and hierarchical flower-like morphologies of TiO2 with a diameter of 124 ± 5 nm by solvothermal method followed by hydrogenation. And the petals of TiO2 nanoflowers consisted of nanosheets with a thickness of 10 ± 1.2 nm. After hydrogen reduction, Ti3+ or oxygen vacancies were introduced into the TiO2 nanoflowers structure. The hydrogenated TiO2 nanoflowers exhibited high capacity, excellent rate performance, and better cycling stability, with an initial discharge capacity of 179.6 mA h g−1 and the 200th discharge capacity of 177.1 mA h g−1 at 10C, with a capacity retention rate of 98.4% after 200 cycles. While the unhydrogenated TiO2 nanoflowers presented an initial capacity of 130.1 mA h g−1 at a high current density, which decreased to 121.4 mA h g−1 after 200 cycles with a capacity retention rate of 93.4%. Pavasupree et al.86 hydrothermally synthesized flower-like TiO2 with a mesoporous structure. And it was composed of slightly curved nanosheets with a width of about 50–100 nm and a thickness of a few nanometers (see Fig. 4). The average pore size of TiO2 nanosheets was about 3–4 nm, and the BET surface area and pore volume were about 642 m2 g−1 and 0.774 cm3 g−1, respectively. Gao et al.87 synthesized TiO2 nanoflowers by a hydrothermal synthesis route in saturated sodium chloride solution.
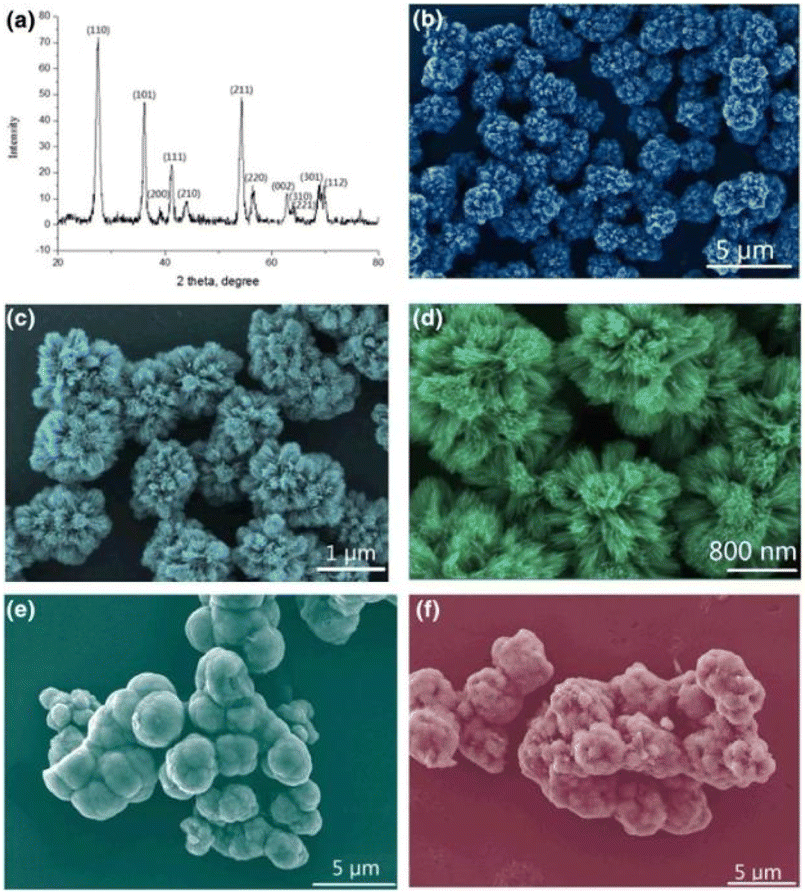 | ||
| Fig. 4 XRD pattern and FE-SEM images of TiO2 nanoflowers.87 | ||
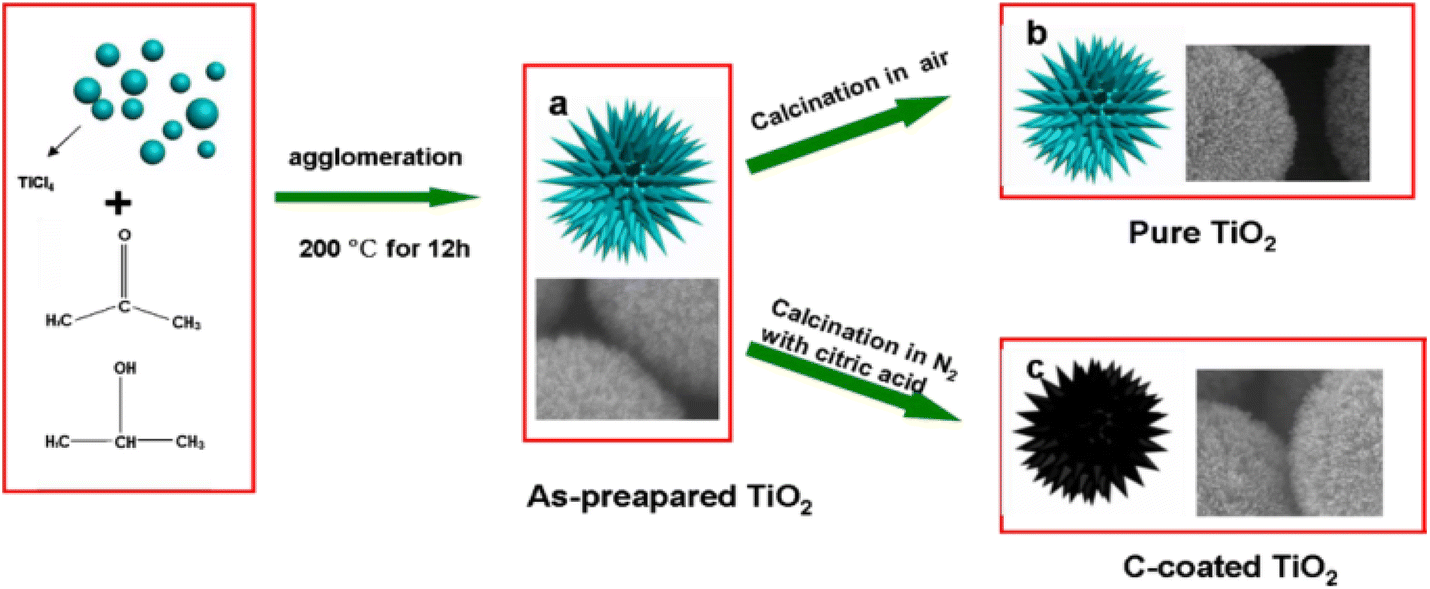 | ||
| Fig. 5 Schematic diagram of the growth mechanism of TiO2 microspheres.94 | ||
Zhen et al.89 synthesized hollow mesoporous TiO2 microspheres by a template-free method. It had diameters of about 200–300 nm, mesopore diameters of about 12 nm, microspheres walls of about 200 nm, surface area of 105 m2 g−1, the first discharge capacity of about 215 mA h g−1 at 2C, and a capacity of 132 mA h g−1 at 2C after 200 cycles. In addition, it also exhibited excellent cycling stability, with the capacity retention of 100 mA h g−1 at 5C after 800 cycles. Yue et al.96 synthesized mesoporous TiO2 microspheres by solvothermal method, and the TiO2 microspheres consisted of 6–8 nm nanocrystals with a uniform distribution of 4–8 nm mesopores on the surface. There was a reversible capacity of 180 mA h g−1 at a current density of 2C after 100 cycles. Zhang et al.97 prepared hollow porous TiO2 spheres with a thickness of about 450 nm by hydrothermal method (see Fig. 6). This unique hollow porous structure, which facilitated the transport of Li+, exhibited excellent electrochemical performance with a reversible capacity of 170 mA h g−1 at 0.6C after 150 cycles. Sunkara et al.98 prepared mesoporous TiO2 microspheres at a relatively low temperature using an acoustic-chemical synthesis method, which showed excellent cycling stability with a reversible capacity of 212.3 mA h g−1 after 100 charge–discharge cycles. The cycling performances and capacities of TiO2 hollow spheres is depicted in Table 5.
 | ||
| Fig. 6 Schematic illustration of the probable formation mechanism of hollow porous TiO2 microspheres.97 | ||
| Pm | D/nm | T/nm | Sa/m2 g−1 | Cd | Initial coulombic efficiency | Cn | Rc/mA h g−1 | Areal capacities (mA h cm−2) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Pm—preparation method, Tfm—template-free method, Sm—solvothermal method, Hm—hydrothermal method, Som—sonochemistry method, D—diameter, T—thickness, Sa—surface area, Cd—current density, Ice—initial coulombic efficiency, Cn—cycle numbers, Rc—remaining capacity, Ac—areal capacities. | |||||||||
| Tfm | 200–300 | 200 | 105 | 5C | — | 800 | 100 | 0.175 | 89 |
| Sm | 500–2000 | — | 118.1 | 2C | — | 100 | 180 | — | 96 |
| Hm | — | 450 | — | 0.6C | 85.6% | 150 | 170 | 0.24 | 97 |
| Som | 500 | — | 59.4 | 0.25C | 89% | 100 | 212.3 | 0.53 | 98 |
Wen et al.99 prepared TiO2 nanotrees by depositing ultrathin nanoribbons of mixed-phase anatase/bronzite TiO2 on an array of single-crystal anatase nanowires. The nanotrees branches were a few nanometers thick and 200–260 nm long, and had a discharge capacity of 330 mA h cm−2 in the second cycle at a current density of 0.1 mA cm−2 with stable capacity retention over the subsequent 50 charge/discharge cycles. Sun et al.100 prepared TiO2 nanotrees composed of nanorods, nanoribbons, and nanowires nanounits by a hydrothermal method (see Fig. 7), and the shape of TiO2 dendrimers was controlled by adjusting the precursor hydrolysis rate and surfactant. Compared with nanoparticles with smooth surfaces, the novel TiO2 nanostructures have higher surface area and excellent electrochemical performance.
 | ||
| Fig. 7 SEM images of 3D nanotree composed of (a–c) nanorods (d–f) nanoribbons (g–i) nanowires.100 | ||
3.5 Special shape
In addition, nanostructured TiO2 with morphologies such as microcone,102 candied fruit, sea urchin-like, and coral-like morphologies have also been studied, which have larger surface areas due to their unique morphologies and exhibit excellent electrochemical performance. The unique morphology is difficult to control in the synthesis process, and the exploration of synthetic methods with low cost, simple operation, and adjustable morphology will become the focus of future research.Alireza103 synthesized coral-like TiO2 by sol–gel method and investigated the effects of precursor hydrolysis rate, reaction time, type and concentration of acid, reaction temperature, and annealing temperature. The surface of the coral-like structure consists of nanoparticles with a cashew-like shape and a mesoporous structure with a surface area of up to 164 m2 g−1. Caruso et al.104 used a polymer gel template method to obtain wall thicknesses of about 100–150 nm and coral-like TiO2 with pore sizes ranging from 100 nm to several microns.
4 Construction of TiO2 composites
The poor ionic and electronic conductivity of TiO2 leads to a capacity far below the theoretical value and poor rate performance, which hinders its practical application in LIBs. Although the preparation of TiO2 nanomaterials with a high surface area exhibits good electrochemical performance, it is still difficult to meet the commercial demand. For this reason, it is usually necessary to compound TiO2 with other materials, and this review introduces the research progress of different types of TiO2 composites, mainly from the compounding of TiO2 with carbon, silicon, metals, and metal oxides.1054.1 TiO2 and carbon composite
Using the higher safety performance of TiO2 and the excellent electrical conductivity of carbon materials, compounding TiO2 with carbon materials is one of the development directions of TiO2 anode materials for LIBs.106–110 TiO2 compounded with carbon materials such as graphene, carbon nanotubes, and hard carbon can effectively improve the electrochemical performance.111,112 On the one hand, the introduction of carbon enhances the overall electrical conductivity of the electrode, and on the other hand, it suppresses the accumulation and agglomeration of TiO2 nanostructures, making it easier to diffuse for Li+ and electrons.113Zhang et al.114 used hollow porous pollen carbon microspheres as a backbone to load C@TiO2 nanoparticles. C@TiO2/3D pollen carbon (CTPC) composites with hollow porous structure were successfully prepared by a simple method (see Fig. 8). The results showed that the CTPC-700 (calcination temperature of 700 °C) composites exhibited excellent electrochemical performance with a high specific capacity of 148 mA h g−1 at 5C after 1000 cycles. Trang et al.115 prepared a composite material consisting of mesoporous TiO2 spheres and multi-walled carbon nanotubes by sol–gel and solvothermal methods, which exhibited excellent electrochemical performance. The first discharge capacity was 269 mA h g−1 at a current density of 1C, and after 100 cycles the capacity decreased by only 3.1%, with a capacity of about 260 mA h g−1. Lopez et al.116 synthesized a TiO2/carbon fiber composite with first discharge and charge capacities of 683 mA h g−1 and 356 mA h g−1 at 100 mA g−1, respectively, with a reversible capacity of 290 mA h g−1 after 100 cycles. Liang et al.117 prepared two-dimensional, ultrathin and mesoporous TiO2/reduced graphene using layer-by-layer assembly process. The synergistic effect of ultrathin mesoporous TiO2 and reduced graphene nanosheets significantly enhanced the ion diffusion and electronic conductivity. The capacity was maintained at 245 mA h g−1 at 1 A g−1 after 1000 cycles. Mondal et al.118 reported a large-scale preparation of hollow spherical TiO2/reduced graphene composites by aerosol-assisted spray drying method. The TiO2/reduced graphene spheres are porous and consist of 10–20 nm TiO2 particles dispersed on the surface of reduced graphene with a surface area of 86 m2 g−1. It has good electrochemical energy storage performance with a specific capacity up to 174.4 mA h g−1 at a current density of 188 mA g−1 after 200 cycles. The cycling performances and capacities of TiO2/C nanocomposite is depicted in Table 6.
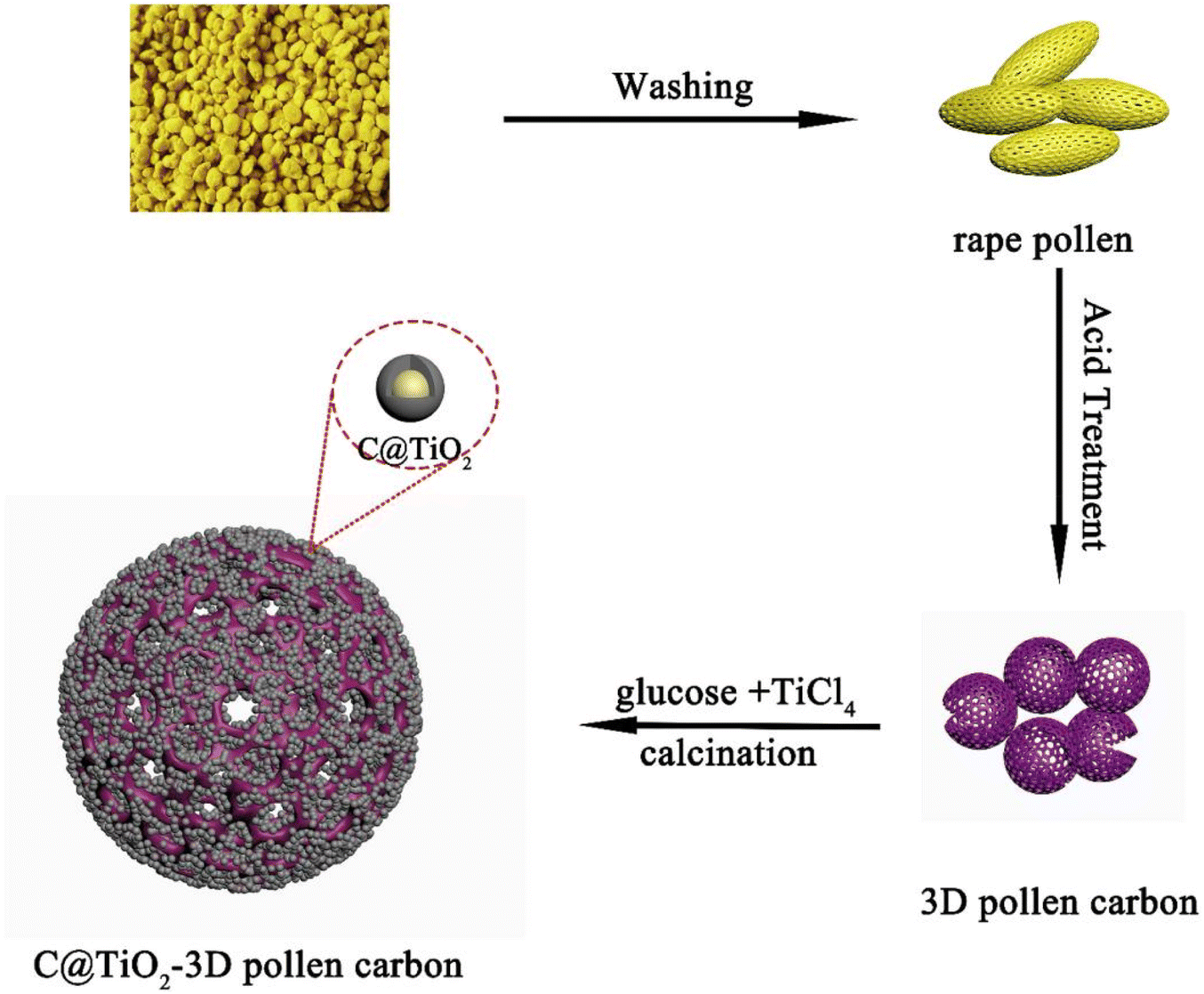 | ||
| Fig. 8 Schematical illustration of the synthetic process of C@TiO2/3D pollen carbon.114 | ||
4.2 TiO2 and silicon composite
Silicon materials have a high theoretical capacity of 4200 mA h g−1 and are abundant in the earth's crust,119 but the large bulk effect and low electrical conductivity lead to considerable challenges in related research. Silicon materials undergo structural damage during charge/discharge, resulting in lower reversible capacity, poor cycling stability, and rate performance. Although TiO2 has minimal volume change during charge/discharge, the theoretical capacity is low at 335 mA h g−1, which can be increased by compounding with silicon materials.120Fang et al.121 prepared a silicon–titanium core–shell structure consisting of silicon nanoparticles encapsulated in TiO2 hollow spheres using hydrolysis and magnesium thermal reduction (see Fig. 9), which maintained a capacity of 804 mA h g−1 after 100 cycles at 0.1C. This excellent cycling stability and high capacity can be attributed to the unique core–shell nanostructure and the synergistic effect between silicon and TiO2. Li et al.122 coated TiO2 on the surface of silicon nanoparticles by sol–gel method and maintained a reversible capacity of 1010.7 mA h g−1 after 100 cycles at a current rate of 0.1C. Excellent stability can be obtained at high current densities. Jia et al.123 prepared layered nanofiber TiO2/silica composites using natural cellulose material as a template, and their electrochemical performance improved with increasing titanium content in the composites. For composites with 54.3 wt% TiO2 and TiO2 nanoparticles of size 12 nm, at 200 mA g−1 current density, it has a capacity of 498.9 mA g−1 after 200 charge/discharge cycles. Zhou et al.124 prepared spherical TiO2/silica nanoparticles using the sol–gel method. It was shown that the optimal mass ratio of TiO2/silica was 1:0.15, the reversible capacity was 720.9 mA h g−1 at a current density of 100 mA g−1, and the capacity retention was 86.5% after 400 cycles, with a capacity of about 615.9 mA h g−1. Yan et al.125 successfully coated nano-Si with different contents of nano-TiO2 to form a new structure of pineapple. When the molar ratio of Si/TiO2 is 1:4, the cycle stability is the best. After 50 cycles at 0.1C, the specific capacity is 593 mA h g−1. The cycling performances and capacities of TiO2/Si nanocomposites is depicted in Table 7.
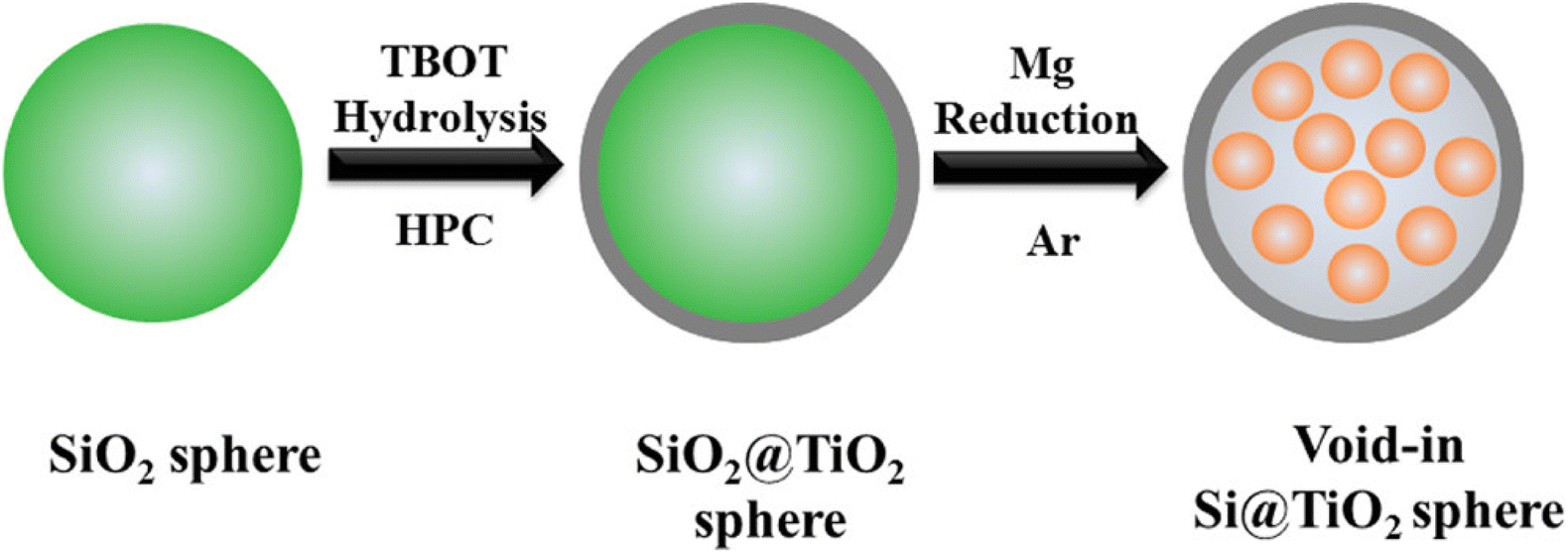 | ||
| Fig. 9 Schematic diagram of the preparation process of Si@TiO2 composite.121 | ||
| Cd | Initial coulombic efficiency | Cn | Rc/mA h g−1 | Areal capacities (mA h cm−2) | Tc/v | Ref. |
|---|---|---|---|---|---|---|
| a Cd—current density, Ice—initial coulombic efficiency, Cn—cycle numbers, Rc—remaining capacity, Ac—areal capacities, Tc—tested condition. | ||||||
| 0.1C | 53.1% | 100 | 804 | — | 0.01–2.5 | 121 |
| 0.1C | — | 100 | 1010.7 | — | 0–3 | 122 |
| 200 mA g−1 | 61.5% | 200 | 498.9 | 0.65 | 0.01–3 | 123 |
| 100 mA g−1 | 67.7% | 400 | 615.9 | 0.86 | 0.01–3 | 124 |
| 0.1C | 69.7% | 50 | 593 | — | 0–3 | 125 |
4.3 TiO2 and metal element/metal oxide composite
Using the good electrical conductivity of metals to enhance the capacity and cycling stability of electrodes is a common modification method for TiO2.126 In recent years, there have been many reports on the compounding of TiO2 with metal materials such as Ag, Cu, Fe, etc., and metal oxides such as SnO2,127 Sb2O3, ZnO, etc. to improve the electrochemical performance of the materials.Kacica et al.128 prepared tin dioxide composites with a TiO2 coating using the aerosol chemical vapor deposition method, and the SnO2/TiO2 electrode had a capacity of 497 mA h g−1 after 100 cycles at 2C current density. Compared to pure TiO2 materials, composite SnO2/TiO2 composites with highly oriented, nanostructured structures exhibited better performance. Fan et al.129 synthesized Cu-doped TiO2 nano hollow spheres with a capacity of 159.06 mA h g−1 at a high current density of 10C after 500 cycles. Ren et al.130 synthesized Ti3+ doped TiO2 nanoparticles with relatively uniform particles with an average size of 30 nm and a Ti3+ doping content of 4 wt%, with a capacity of 145.8 mA h g−1 at a current density of 200 mA g−1 after 50 cycles. Han et al.131 prepared Sb2O3/Sb@TiO2 composites with TiO2 films covering the surface of Sb2O3 and Sb elemental particles that were discontinuously distributed in the TiO2 film at a discharge capacity of 609 mA h g−1 could be achieved at a current density of 100 mA g−1. Gao et al.132 reported the preparation of porous Li4Ti5O12 (LTO)–TiO2 nanosheet arrays by hydrothermal method, which had a high initial discharge capacity of 184.6 mA h g−1 at 200 mA g−1 and only lost 8.3% of the specific capacity after 1000 cycles. Cao et al.133 prepared a thin layer of titanium dioxide by calcination of MnCO3 microspheres and thermal decomposition of tetrabutyl titanate, and prepared a layered structure MnO2/TiO2 composite (nano-MnO2@TiO2). After 200 cycles at 300 mA g−1, its capacity is 938 mA h g−1. Opra et al.134 synthesized vanadium-doped TiO2-B/anatase nanotubes by a simple one-step hydrothermal reaction. The material has a mesoporous structure with a specific surface area of 179.1 m2 g−1. After 100 cycles at 3000 mA g−1, the specific capacity is 133 mA h−1, which has good cyclability. The cycling performances and capacities of TiO2/metal and TiO2/metal oxide nanocomposites is depicted in Table 8.
| Tm | Cd | Initial coulombic efficiency | Cn | Rc/mA h g−1 | Areal capacities (mA h cm−2) | Tc/v | Ref. |
|---|---|---|---|---|---|---|---|
| a Tm—typical materials, Cd—current density, Ice—initial coulombic efficiency, Cn—cycle numbers, Rc—remaining capacity, Ac—areal capacities, Tc—tested condition. | |||||||
| SnO2/TiO2 | 2C | — | 100 | 497 | — | 0.05–1.5 | 128 |
| Cu/TiO2 | 10C | 90% | 500 | 159.06 | — | 0.01–3 | 129 |
| Ti3+/TiO2 | 200 mA g−1 | — | 50 | 145.8 | 0.15 | 1–3 | 130 |
| Sb2O3/Sb@TiO2 | 100 mA g−1 | 68.4% | 500 | 485 | — | 0.01–3 | 131 |
| Li 4Ti5O12–TiO2 | 200 mA g−1 | — | 100 | 172.4 | — | 1–3 | 132 |
| MnO2@TiO2 | 300 mA g−1 | 86% | 200 | 938 | — | 0.01–3 | 133 |
| V-doped TiO2-B/anatase | 3 A g−1 | 75% | 100 | 133 | 0.33 | 1–3 | 134 |
5 Summary and outlook
TiO2 is a promising anode material for LIBs, but its large-scale application is limited due to its poor electrical conductivity and low Li+ diffusion coefficient. In view of these problems, this paper reviews the research achievements of researchers to improve the electrochemical performance of TiO2 and promote their practical applications, including the preparation of nanomaterials with different morphologies and the preparation of composite materials with other materials. Although considerable progress has been made in improving the lithium storage performance of TiO2, there are still some fundamental issues that need to be addressed. For example, nanomaterials usually have a larger surface area, which leads to more pronounced side reactions, resulting in low coulombic efficiencies. In addition, TiO2 nanomaterials combining with other materials is the development direction of TiO2 anode materials, but the current preparation methods are complicated. As a result, low yields and high costs limit its commercial application. It is necessary to develop efficient and low-cost preparation methods. In conclusion, the exploration of TiO2 anode materials with the advantages of low cost, large capacity and good cycling performance will be the focus of future research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51764008, 51864012), the Natural Science Research Project of Education Department of Guizhou Province (No. QJJ[2022]001), the Science and Technology Plan of Guizhou Province (Qian Kehe [2021]-305).Notes and references
- H. S. Liu, Z. H. Bi, X.-G. Sun, R. R. Unocic, M. P. Paranthaman, S. Dai and G. M. Brown, Adv. Mater., 2011, 23, 3450–3454 CrossRef CAS PubMed.
- J. S. Chen and X. W. Lou, Small, 2013, 9, 1877–1893 CrossRef CAS PubMed.
- J. B. Chen, H. L. Zhao, N. Chen, X. C. Wang, J. Wang, R. Zhang and C. Q. Jin, J. Power Sources, 2012, 200, 98–101 CAS.
- S. B. Yang, X. L. Feng and K. Müllen, Adv. Mater., 2011, 23, 3575–3579 CrossRef CAS PubMed.
- S. I. Choi, E.-J. Jung, M. Park, H.-S. Shin, S. Huh and Y. S. Won, Appl. Surf. Sci., 2020, 508, 145237 CrossRef CAS.
- Q. H. Tian, D. Luo, X. W. Li, Z. X. Zhang, L. Yang and S.-I. Hirano, J. Power Sources, 2016, 313, 189–197 CrossRef CAS.
- Y. Cai, H.-E. Wang, S.-Z. Huang, M. F. Yuen, H.-H. Cai, C. Wang, Y. Yu, Y. Li, W.-J. Zhang and B.-L. Su, Electrochim. Acta, 2016, 210, 206–214 CrossRef CAS.
- V. Mathew, J. Gim, M. H. Alfaruqi, S. Kim, J. Song, J. P. Baboo, S. Kim, S. Park, D. Kim and J. Kim, J. Electrochem. Soc., 2015, 162, A1220–A1226 CrossRef CAS.
- Z. H. Chen, I. Belharouak, Y.-K. Sun and k. Amine, Adv. Funct. Mater., 2013, 23, 959–969 CrossRef CAS.
- D. Dambournet, I. Belharouak and K. Amine, Chem. Mater., 2010, 22, 1173–1179 CrossRef CAS.
- V. Gentili, S. Brutti, L. J. Hardwick, A. R. Armstrong, S. Panero and P. G. Bruce, Chem. Mater., 2012, 24, 4468–4476 CrossRef CAS.
- X. D. Yan, Z. H. Wang, M. He, Z. H. Hou, T. Xia, G. Liu and X. B. Chen, Energy Technol., 2015, 3, 801–814 CrossRef CAS.
- Y. Y. Zhang, Y. X. Tang, W. L. Li and X. D. Chen, ChemNanoMat, 2016, 2, 764–775 CrossRef CAS.
- V. Aravindan, Y.-S. Lee, R. Yazami and S. Madhavi, Mater. Today, 2015, 18, 345–351 CrossRef CAS.
- I. Ali, M. Suhail, Z. A. Alothman and A. Alwarthan, RSC Adv., 2018, 8, 30125 RSC.
- Y. K. Tang, L. Liu, X. C. Wang, D. Z. Jia, W. Xia, Z. B. Zhao and J. S. Qiu, J. Power Sources, 2016, 319, 227–234 CrossRef CAS.
- Wahyudiono, H. Kondo, M. Yamada, N. Takada, S. Machmudah, H. Kanda and M. Goto, ACS Omega, 2020, 5, 5443–5451 CrossRef CAS PubMed.
- C. M. Guo, Q. H. Tian and L. Yang, J. Alloys Compd., 2019, 776, 740–745 CrossRef CAS.
- T. P. B. Rajakaruna, C. P. Udawatte, R. Chandrajith and R. M. G. Rajapakse, ACS Omega, 2020, 5, 16176–16182 CrossRef CAS PubMed.
- Q. Qian, Y. Lin, Z. L. Xiong, P. H. Su, D. X. Liao, Q. Z. Dai, L. S. Chen and D. L. Feng, Electrochem. Commun., 2022, 135, 107201 CrossRef CAS.
- S. Pervez, D. Kim, C.-H. Doh, U. Farooq, A. Yaqub, J.-H. Choi, Y.-J. Lee and M. Saleem, Mater. Lett., 2014, 137, 347–350 CrossRef CAS.
- Y. X. Tang, Y. Y. Zhang, J. Y. Deng, J. Q. Wei, H. L. Tam, B. K. Chandran, Z. L. Dong, Z. Chen and X. D. Chen, Adv. Mater., 2014, 26, 6111–6118 CrossRef CAS PubMed.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 2336–2338 CrossRef.
- H. Bo, Y. Yong, X. Wang and C. Ge, ACS Appl. Mater. Interfaces, 2013, 5, 6285–6291 CrossRef PubMed.
- W. Zhang, W. D. Zhou, J. H. Wright, Y. N. Kim, D. Liu and X. C. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 7292–7300 CrossRef CAS PubMed.
- A. G. Dylla, P. H. Xiao, G. Henkelman and K. J. Stevenson, J. Phys. Chem. Lett., 2012, 3, 2015–2019 CrossRef CAS.
- Y. J. Ren, G. Q. Zhang, J. H. Huo, J. H. Li, Y. Liu and S. W. Guo, J. Alloys Compd., 2022, 902, 163730 CrossRef CAS.
- W. Dong, H. X. Li, J. H. Xi, J. X. Mu, Y. W. Huang, Z. G. Ji and X. Wu, J. Alloys Compd., 2017, 724, 280–286 CrossRef CAS.
- H. Huang, Z. Y. Yu, W. J. Zhu, Y. P. Gan, Y. Xia, X. Y. Tao and W. K. Zhang, J. Phys. Chem. Solids, 2014, 75, 619–623 CrossRef CAS.
- J. B. Chen, Y. W. Wang, X. M. He, S. M. Xu, M. Fang, X. Zhao and Y. M. Shang, Electrochim. Acta, 2014, 142, 152–156 CrossRef CAS.
- Z. L. Liu, L. Hong and B. Guo, J. Power Sources, 2005, 143, 231–235 CrossRef CAS.
- D. K. Muthee and B. F. Dejene, Mater. Sci. Semicond. Process., 2020, 106, 104783 CrossRef CAS.
- T. Y. Peng, D. Zhao, K. Dai, W. Shi and K. Hirao, J. Phys. Chem. B, 2005, 109, 4947–4952 CrossRef CAS PubMed.
- A. Mezni, N. B. Saber, M. M. Ibrahim, M. El-Kemary, A. Aldalbahi, P. Feng, L. S. Smiri and T. Altalhi, New J. Chem., 2017, 41, 5021–5027 RSC.
- D. M. Bao and Q. H. Tian, Chem. Phys. Lett., 2018, 709, 125–129 CrossRef CAS.
- P. Mathumba, A. T. Kuvarega, L. N. Dlamini and S. P. Malinga, Mater. Lett., 2017, 195, 172–177 CrossRef CAS.
- G. Y. Huang, S. M. Xu, Z. H. Xu, H. Y. Sun and L. Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 21325–21334 CrossRef CAS PubMed.
- T. Partheeban, T. Kesavan, A. Jithin, S. Dharaneshwar and M. Sasidharan, J. Solid State Electrochem., 2021, 25, 919–926 CrossRef CAS.
- W. F. Zhang, D. L. Shen, Z. W. Liu, N.-L. Wu and M. D. Wei, Chem. Commun., 2018, 54, 11491–11494 RSC.
- K. E. Ouardi, M. Dahbi, C. Hakim, M. O. Güler, H. Akbulut, A. E. Bouari and I. Saadoune, J. Appl. Electrochem., 2020, 50, 583–595 CrossRef.
- B. Zhao, F. Chen, H. Q. Liu and J. L. Zhang, J. Phys. Chem. Solids, 2011, 72, 201–206 CrossRef CAS.
- H. Yin, G. Q. Ding, B. Gao, F. Q. Huang, X. M. Xie and M. H. Jiang, Mater. Res. Bull., 2012, 47, 3124–3128 CrossRef CAS.
- M. Rodríguez-Reyes and H. J. Dorantes-Rosales, J. Sol-Gel Sci. Technol., 2011, 59, 658–661 CrossRef.
- E. C. Giraud, P. Mokarian-Tabari, D. T. W. Toolan, T. Arnold, A. J. Smith, J. R. Howse, P. D. Topham and M. A. Morris, ACS Appl. Nano Mater., 2018, 1, 3426–3434 CrossRef CAS.
- Y. F. Wang, M. Y. Wu and W. F. Zhang, Electrochim. Acta, 2008, 53, 7863–7868 CrossRef CAS.
- F. X. Wu, X. H. Li, Z. X. Wang, H. J. Guo, L. Wu, X. H. Xiong and X. J. Wang, Mater. Lett., 2011, 65, 1514–1517 CrossRef CAS.
- H. E. Wang, Z. G. Lu, L. J. Xi, R. G. Ma, C. D. Wang, J. A. Zapien and I. Bello, ACS Appl. Mater. Interfaces, 2012, 4, 1608–1613 CrossRef CAS PubMed.
- R. Govindaraj, N. Santhosh, M. Senthil Pandian and P. Ramasamy, J. Cryst. Growth, 2017, 468, 125–128 CrossRef CAS.
- Q. S. Zhang, J. Dispersion Sci. Technol., 2010, 31, 1358–1361 CrossRef CAS.
- J. Jin, S.-Z. Huang, J. Shu, H.-E. Wang, Y. Li, Y. Yu, L.-H. Chen, B.-J. Wang and B.-L. Su, Nano Energy, 2015, 16, 339–349 CrossRef CAS.
- X. P. Huang and C. X. Pan, J. Cryst. Growth, 2007, 306, 117–122 CrossRef CAS.
- G. A. Seisenbaeva, J.-M. Nedelec, G. Daniel, G. Tiseanu, V. Parvulescu, V. G. Pol, L. Abrego and V. G. Kessler, Chem.–Eur. J., 2013, 19, 17439–17444 CrossRef CAS PubMed.
- Y. J. Ji, Mater. Lett., 2013, 108, 208–211 CrossRef CAS.
- K. Santhi, M. Navaneethana, S. Harish, S. Ponnusamy and C. Muthamizhchelvan, Appl. Surf. Sci., 2020, 500, 144058 CrossRef CAS.
- A. Ranjitha, N. Muthukumarasamy, M. Thambidurai, D. Velauthapillai, S. Agilan and R. Balasundaraprabhu, Optik, 2015, 126, 2491–2494 CrossRef CAS.
- J.-H. Kim, K. Zhu, J. Y. Kim and A. J. Frank, Electrochim. Acta, 2013, 88, 123–128 CrossRef CAS.
- M. Moazeni, H. Hajipour, M. Askari and M. Nusheh, Mater. Res. Bull., 2014, 61, 70–75 CrossRef.
- F. Nasirpouri, I. Yousefi, E. Moslehifard and J.-K. Allafi, Surf. Coat. Technol., 2017, 315, 163–171 CrossRef CAS.
- X. Y. Zhao, Y. X. Zhu, Y. F. Wang, L. Zhu, L. B. Yang and Z. L. Sha, J. Nanomater., 2015, 201, 104193 Search PubMed.
- Z. H. Gong, Y. D. Hu, F. Gao, L. Quan, T. Liu, T. Gong and C. J. Pan, Colloids Surf., B, 2019, 184, 110521 CrossRef CAS PubMed.
- J. W. Xu, C. H. Jia, B. Cao and W. F. Zhang, Electrochim. Acta, 2007, 52, 8044–8047 CrossRef CAS.
- O. Fasakin, K. O. Oyedotun, M. Kebede, M. Rohwer, L. L. Roux, M. Mathe, M. A. Eleruja, E. O. B. Ajayi and N. Manyala, Energy Rep, 2020, 6, 92–101 CrossRef.
- Z. R. Wang, Y. L. Tong, L. Y. Dang, F. Gao and Q. Y. Lu, Chem.–Eng.J., 2019, 370, 1434–1439 CrossRef CAS.
- D. W. Liu, Y. H. Zhang, P. Xiao, B. B. Garcia, Q. F. Zhang, X. Y. Zhou, Y.-H. Jeong and G. Z. Cao, Electrochim. Acta, 2009, 54, 6816–6820 CrossRef CAS.
- G. S. Zakharova, C. Jahne, A. Popa, Ch. Taschner, Th. Gemming, A. Leonhardt, B. Buchner and R. Klingeler, J. Phys. Chem. C, 2012, 116, 8714–8720 CrossRef CAS.
- Y. Zhao, J. H. Yan, J. Y. Yu and B. Ding, Macromol. Rapid Commun., 2022, 2200740 CrossRef PubMed.
- S. Supothina, R. Rattanakam and S. Tawkaew, J. Nanosci. Nanotechnol., 2012, 12, 4998–5003 CrossRef CAS PubMed.
- Z.-J. Fan, J. Yan, T. Wei, G.-Q. Ning, L.-J. Zhi, J.-C. Liu, D.-X. Cao, G.-L. Wang and F. Wei, ACS Nano, 2011, 5, 2787–2794 CrossRef CAS PubMed.
- H. Han, T. Song, J.-Y. Bae, L. F. Nazar, H. Kim and U. Paik, Energy Environ. Sci., 2011, 4, 4532–4536 RSC.
- J. N. Wang, G. R. Yang, L. Wang, S. L. Wang, W. Yan and S. J. Ding, J. Alloys. Compd., 2019, 787, 1110–1119 CrossRef CAS.
- P. S. Kumar, V. Aravindan, J. Sundaramurthy, V. Thavasi, S. G. Mhaisalkar, S. Ramakrishna and S. Madhavi, RSC Adv., 2012, 2, 7983–7987 RSC.
- J.-W. Jung, C.-L. Lee, S. Yu and I.-D. Kim, J. Mater. Chem. A, 2016, 4, 703–750 RSC.
- S. Lee, J. Ha, J. Choi, T. Song, J. W. Lee and U. Paik, ACS Appl. Mater. Interfaces, 2013, 5, 11525–11529 CrossRef CAS PubMed.
- J. S. Cho, Y. J. Hong and Y. C. Kang, Chem.–Eur. J., 2015, 21, 11082–11087 CrossRef CAS PubMed.
- W. Zhuang, L. H. Lu, X. B. Wu, W. Jin, M. Meng, Y. D. Zhu and X. H. Lu, Electrochem. Commun., 2013, 27, 124–127 CrossRef CAS.
- W. Luo, X. L. Hu, Y. M. Sun and Y. H. Huang, J. Mater. Chem., 2012, 22, 4910–4915 RSC.
- P. N. Zhu, Y. Z. Wu, M. V. Reddy, A. S. Nair, B. V. R. Chowdari and S. Ramakrishna, RSC Adv., 2012, 2, 531–537 RSC.
- M.-C. Yang, Y.-Y. Lee, B. Xu, K. Powers and Y. S. Meng, J. Power Sources, 2012, 207, 166–172 CrossRef CAS.
- F. Y. Li, Y. Huang, H. L. Peng, Y. Cao and Y. Niu, Int. J. Photoenergy, 2020, 3617312 CAS.
- W. J. Yu, Y. M. Liu, N. Cheng, B. Cai, K. K. Kondamareddy, S. Kong, S. Xu, W. Liu and X.-Z. Zhao, Electrochim. Acta, 2016, 220, 398–404 CrossRef CAS.
- S. H. Liu, H. P. Jia, L. Han, J. L. Wang, P. F. Gao, D. D. Xu, J. Yang and S. N. Che, Adv. Mater., 2012, 24, 3201–3204 CrossRef CAS PubMed.
- J. Q. Wang, X. P. Zou, J. L. Zhu, J. Cheng, X. Bai, Y. J. Yao, C. C. Chang, X. Yu, B. Y. Liu, Z. X. Zhou, G. D. Li and D. Chen, Chem. Phys. Lett., 2021, 765, 138284 CrossRef CAS.
- Z. D. Dai, X.-Z. Song, F. Tang, X. L. Kang, S. H. Liu, H. Abe, S. Ohara and Z. Q. Tan, Micro Nano Lett., 2021, 16, 313–318 CrossRef CAS.
- P. Shende, P. Kasture and R. S. Gaud, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 413–422 CrossRef CAS PubMed.
- Z. H. Zhang, Z. F. Zhou, S. Nie, H. H. Wang, H. R. Peng, G. C. Li and K. Z. Chen, J. Power Sources, 2014, 267, 388–393 CrossRef CAS.
- S. Pavasupree, S. Ngamsinlapasathian, Y. Suzuki and S. Yoshikawa, Mater. Lett., 2007, 61, 2973–2977 CrossRef CAS.
- X. Gao, Y. Q. Li, W. Zeng, C. F. Zhang and Y. M. Wei, J. Mater. Sci. Mater. Electron., 2017, 28, 18781–18786 CrossRef CAS.
- S. Ri, H. G. Deng, L. H. Zhou, J. Hu, H. L. Liu and Y. Hu, Chin. J. Chem. Eng., 2014, 22, 1153–1161 CrossRef CAS.
- M. M. Zhen, K. F. Li, S.-Q. Guo, H. Z. Li and B. X. Shen, J. Alloys. Compd., 2021, 859, 157761 CrossRef CAS.
- S. Yoon and A. Manthiram, J. Phys. Chem. C, 2011, 115, 9410–9416 CrossRef CAS.
- H. Guo, T. T. Li, W. W. Chen, L. X. Liu, X. J. Yang, P. Wang and Y. C. Guo, Nanoscale, 2014, 6, 15168–15174 RSC.
- L. Yu, H. B. Wu and X. W. Lou, Adv. Mater., 2013, 25, 2296–2300 CrossRef CAS PubMed.
- G. Q. Zhang, H. B. Wu, T. Song, U. Paik and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1–5 CrossRef.
- L. Gao, X. R. Li, H. Hu, G. J. Li, H. W. Li and Y. Yu, Electrochim. Acta, 2014, 120, 231–239 CrossRef CAS.
- Y. X. Zhang, G. H. Li, Y. C. Wu and T. Xie, Mater. Res. Bull., 2005, 40, 1993–1999 CrossRef CAS.
- Y. Ma, G. Ji, B. Ding and J. Y. Lee, J. Mater. Chem., 2012, 22, 24380–24385 RSC.
- P. G. Zhang, C. Y. Zhang, A. J. Xie, C. Li, J. M. Song and Y. H. Shen, J. Mater. Sci., 2016, 51, 3448–3453 CrossRef CAS.
- S. Sunkara, N. Munichandraiah, K. B. R. Varma and S. A. Shivashankar, New J. Chem., 2016, 40, 7197–7203 RSC.
- W. Wen, J.-M. Wu, Y.-Z. Jiang, J.-Q. Bai and L.-L. Lai, J. Mater. Chem. A, 2016, 4, 10593–10600 RSC.
- Z. Q. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- X.-Y. Li, L.-H. Chen, J. C. Rooke, Z. Deng, Z.-Y. Hu, S.-Z. Wang, L. Wang, Y. Li, A. Krief and B.-L. Su, J. Colloid Interface Sci., 2013, 394, 252–262 CrossRef CAS PubMed.
- O. Rhee, G. Lee and J. Choi, ACS Appl. Mater. Interfaces, 2016, 8, 14558–14563 CrossRef CAS PubMed.
- A. Bahramian, Ind. Eng. Chem. Res., 2013, 52, 14837–14846 CrossRef CAS.
- R. A. Caruso, M. Giersig, F. Willig and M. Antonietti, Langmuir, 1998, 14, 6333–6336 CrossRef CAS.
- G. M. Peng, R. A. Du, Q. M. Peng, S. Q. Wu and C. L. Yu, Mater. Chem. Phys., 2018, 214, 34–40 CrossRef CAS.
- L. Ding, J. X. Chen, B. H. Dong, Y. N. Xi, L. Shi, W. Liu and L. X. Cao, Electrochim. Acta, 2016, 200, 97–105 CrossRef CAS.
- H. N. He, Q. M. Gan, H. Y. Wang, G.-L. Xu, X. Y. Zhang, D. Huang, F. Fu, Y. G. Tang, K. Amine and M. H. Shao, Nano Energy, 2018, 44, 217–227 CrossRef CAS.
- Z. R. X. Guan, X. X. Wang, T. T. Li, Q. Z. Zhu, M. Q. Jia and B. Xu, J. Mater. Sci. Technol., 2019, 35, 1977–1981 CrossRef CAS.
- H. Liu, W. Li, D. K. Shen, D. Y. Zhao and G. X. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 CrossRef CAS PubMed.
- Z. X. Huang, C. Zhao, R. P. Xu, Y. J. Zhou, R. P. Jia, X. W. Xu and S. J. Shi, J. Alloys. Compd., 2020, 857, 158048 CrossRef.
- Z. G. Fu, R. X. Li and Z. Kan, J. Solid State Electrochem., 2019, 23, 1779–1785 CrossRef CAS.
- Z. G. Luo, S. N. Liu, Y. S. Cai, S. Li, A. Pan and S. Q. Liang, Energy Technol., 2018, 6, 759–765 CrossRef CAS.
- J. Zhang, J. F. Ni, J. Guo and B. Q. Cao, New J. Chem., 2014, 38, 3722–3728 RSC.
- Y. F. Zhang, N. Zhang, J. Chen, T. Z. Zhang, W. Q. Ge, W. M. Zhang, G. Xie, L. P. Zhang and Y. H. He, J. Alloys Compd., 2020, 815, 152511 CrossRef CAS.
- N. T. H. Trang, Z. Ali and D. J. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 3676–3683 CrossRef PubMed.
- J. Lopez, R. Gonzalez, J. Ayala, J. Cantu, A. Castillo, J. Parsons, J. Myers, T. P. Lodge and M. Alcoutlabi, J. Phys. Chem. Solids, 2020, 149, 109795 CrossRef.
- Y. R. Liang, X. Xiong, Z. J. Xu, Q. B. Xia, L. Y. Wan, R. T. Liu, G. X. Chen and S.-L. Chou, Small, 2020, 16, 2000030 CrossRef CAS PubMed.
- A. Mondal, S. Maiti, K. Singha, S. Mahanty and A. B. Panda, J. Mater. Chem. A, 2017, 5, 23853–23862 RSC.
- D. Yan, Y. Bai, C. Y. Yu, X. G. Li and W. F. Zhang, J. Alloys Compd., 2014, 609, 86–92 CrossRef CAS.
- Z. Wei, R. S. Li, T. Huang and A. S. Yu, J. Power Sources, 2013, 238, 165–172 CrossRef CAS.
- S. Fang, L. F. Shen, G. Y. Xu, P. Nie, J. Wang, H. Dou and X. G. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 6497–6503 CrossRef CAS PubMed.
- J. Li, Y. Wang, Z. Y. Huang, K. Huang, X. Qi and J. X. Zhong, J. Mater. Sci. Mater. Electrons., 2016, 27, 12813–12819 CrossRef CAS.
- D. L. Jia, Y. H. Chen and J. G. Huang, ChemNanoMat, 2017, 3, 120–129 CrossRef CAS.
- Y. Zhou, S. Liu, F. Liu, T. Gao, K. Fu, A. Dou, M. Su and Y. J. Liu, Ionics, 2020, 26, 5349–5355 CrossRef CAS.
- D. Yan, Y. Bai, C. Y. Yu, X. G. Li and W. F. Zhang, J. Alloys Compd., 2014, 609, 86–92 CrossRef CAS.
- Q. C. Zhu, H. Hu, G. J. Li, C. B. Zhu and Y. Yu, Electrochim. Acta, 2015, 156, 252–260 CrossRef CAS.
- P. P. Zhang, S. S. Zhu, Z. S. He, K. Wang, H. Q. Fan, Y. Zhong, L. Chang, H. B. Shao, J. M. Wang, J. Q. Zhang and C.-N. Cao, J. Alloys Compd., 2016, 674, 1–8 CrossRef CAS.
- C. T. Kacica, L. Wang, T. Chadha and P. Biswas, Energy Technol., 2018, 6, 1966–1974 CrossRef CAS.
- Y. C. Fan, X. H. Chen, K. Zhang, J. Rong and X. H. Yu, J. Alloys Compd., 2020, 860, 158282 CrossRef.
- Y. Q. Ren, J. P. Li and J. Yu, Electrochim. Acta, 2014, 138, 41–47 CrossRef CAS.
- Q. G. Han, Y. L. Sheng, Z. W. Han, X. Li, W. Q. Zhang, Y. Li and X. Zhang, New J. Chem., 2020, 44, 13430–13438 RSC.
- L. Gao, S. H. Li, D. K. Hang, Y. Shen and M. K. Wang, J. Mater. Chem. A, 2015, 3, 10107–10113 RSC.
- Z. G. Cao, X. Q. Chen, L. D. Xing, Y. H. Liao, M. Q. Xu, X. P. Li, X. Liu and W. S. Li, J. Power Sources, 2018, 379, 174–181 CrossRef CAS.
- D. P. Opra, S. V. Gnedenkov, A. A. Sokolov, A. B. Podgorbunsky, A. Y. Ustinov, V. Y. Mayorov, V. G. Kuryavyi and S. L. Sinebryukhov, J. Mater. Sci. Technol, 2020, 54, 181–189 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |