
Comprehensive studies of continuous flow reversible addition–fragmentation chain transfer copolymerization and its application for photoimaging materials†
Jiyeong
Yeo‡
a,
Jihoon
Woo‡
 b,
Seungyeon
Choi
a,
Kiyoung
Kwon
b,
Jin-Kyun
Lee
*bc and
Myungwoong
Kim
*a
b,
Seungyeon
Choi
a,
Kiyoung
Kwon
b,
Jin-Kyun
Lee
*bc and
Myungwoong
Kim
*a
aDepartment of Chemistry and Chemical Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: mkim233@inha.ac.kr
bProgram in Environment and Polymer Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: jkl36@inha.ac.kr
cDepartment of Polymer Science and Engineering, Inha University, Incheon 22212, Republic of Korea
First published on 12th July 2022
Abstract
We performed thorough studies of reversible addition–fragmentation chain transfer (RAFT) copolymerization of industrially relevant functional monomers utilizable for chemically amplified resist systems using an industry-friendly solvent, propylene glycol monomethyl ether, in a continuous flow to gain deeper insights into its kinetics, reactivity, and applicability. Kinetic examinations reveals that the RAFT copolymerization in flow mode exhibits typical pseudo-first-order kinetics with a large increase in the apparent propagation rate constant up to approximately 10 times that of a conventional batch process. The products obtained at the same residence time exhibit outstanding homogeneity in molecular weight, dispersity, and composition. Furthermore, flow chemistry allows easy access to the reactivity ratio of eight different monomer pairs, which is validated by the comparison of the theoretically estimated compositions from the reactivity ratio values with the empirically determined compositions in a full conversion range. The applicability of flow copolymerization toward practical photolithography is confirmed by photolithographic positive tone pattern formation in a chemically amplified resist formulation including the flow-derived copolymer. Our comprehensive exploration provides fundamental insights into the nature of continuous flow RAFT copolymerization in terms of reactivity and kinetics, which further enables the simultaneous achievement of target complex copolymers with precise, uniform, and homogeneous control of structural and compositional parameters on a large scale, maximizing its potential capability to address the challenges in pioneering technologies such as photolithography toward the development of polymers for the patterns with single-nanometer digit dimensions.
1. Introduction
Precise control of copolymerization reactions to achieve the desired chemical structures and properties in a scalable manner has become increasingly important in both academia and industry. The modulation of the structural parameters, such as molecular weight and its distribution, composition, and chain sequence, allows the expansion of the application scope of copolymers in different pioneering technologies.1–4 A representative protocol allowing such control of polymerization is reversible deactivation radical polymerization (RDRP), an extensively employed approach to synthesize well-defined copolymers from a simple to highly complex architecture exhibiting a controlled molecular weight and dispersity.5 To successfully apply the polymerization methodology to the industry-scale production of copolymers as well as their reliable utilization in small lab-scale synthesis, the simultaneous control of the above-mentioned parameters with low batch-to-batch variability is required, which is one of the fundamental challenges in polymer chemistry and engineering.6,7The technological, scientific, and industrial interest in the continuous flow reaction process has grown rapidly owing to its great potential as a next-generation synthetic methodology. It provides several excellent advantages that can overcome the limitations of conventional batch reaction processes;8–10 these advantages include homogeneous reaction conditions that can be accurately and reliably controlled, effective prevention of batch-to-batch variability, improved heat transport owing to the high surface-to-volume ratio of a flow channel, low cost, high safety, and industrial scalability.11 This unique methodology has been applied for the production of a wide range of synthetic chemicals from small molecules to macromolecules. Particularly, polymerization in continuous flow mode has attracted increasing attention because it allows precise and reliable control of the structural parameters of polymers in a rapid manner.12,13 Moreover, it can be expanded to and applied in a variety of polymerization reactions, including step growth polymerization and different chain growth polymerization reactions to prepare large quantities of polymers.14,15
Herein, we demonstrate the comprehensive investigation of the continuous flow reaction for the reversible addition–fragmentation chain transfer (RAFT) copolymerization of model methacrylate, styrene, and maleimide monomers for the successful synthesis of complex polymers with high structural and compositional homogeneities and minimized batch-to-batch variability. The model monomers were selected for a copolymer in a chemically amplified resist (CAR), an important frontier material platform for the photolithographic fabrication of an integrated circuit chip on a semiconductor substrate using deep UV (DUV) and extreme UV (EUV) light.16,17 In a CAR, copolymers are used as matrices for attaining the light-mediated solubility change, etch resistance, and surface-adhesion capabilities; therefore, guaranteeing the structural homogeneity of the constituting copolymers is an important challenge in securing high performance of CARs. In particular, the importance of microstructural homogeneity, that is, uniform chain sequence and comonomer composition, has recently been emphasized in high-resolution photoresists used for the formation of patterns with dimensions equivalent to a single copolymer chain.18 Small variations in the parameters, which can be induced due to the batch-to-batch variability,19–23 can significantly affect the diffusion behavior of the generated acid and change in the solubility of copolymer chains in developing solvents, leading to the deterioration of the final lithographic performance in terms of parameters such as line edge roughness (LER), defect density, and sensitivity.24
The continuous flow copolymerization of model monomers (Fig. 1) was performed using an industry-friendly solvent, namely propylene glycol monomethyl ether (PGME), which has economic and environmental benefits.25 RAFT polymerization was chosen as a model protocol because it possibly allows the improvement of the pattern quality with the control of molecular weight and its distribution, interchain composition and comonomer sequence.26–31 For different combinations of monomers, copolymerization kinetics was thoroughly examined and was further extended to the determination of the reactivity ratio, which is a quantitative parameter not only for understanding the sequence in a copolymer chain, but also to predict the composition of the resulting copolymer (Fig. 1). Eventually, the RAFT copolymerization in continuous flow mode was a highly effective tool that allows high copolymerization efficiency, high structural and compositional homogeneities, and facile access to kinetic parameters, including the apparent propagation rate constant and reactivity ratio, making it applicable toward the development of various important copolymers. Finally, the resulting copolymers were modified and used to define the photo-pattern in the CAR system, highlighting the potential of the continuous flow reaction to achieve well-defined functional copolymers towards target applications.
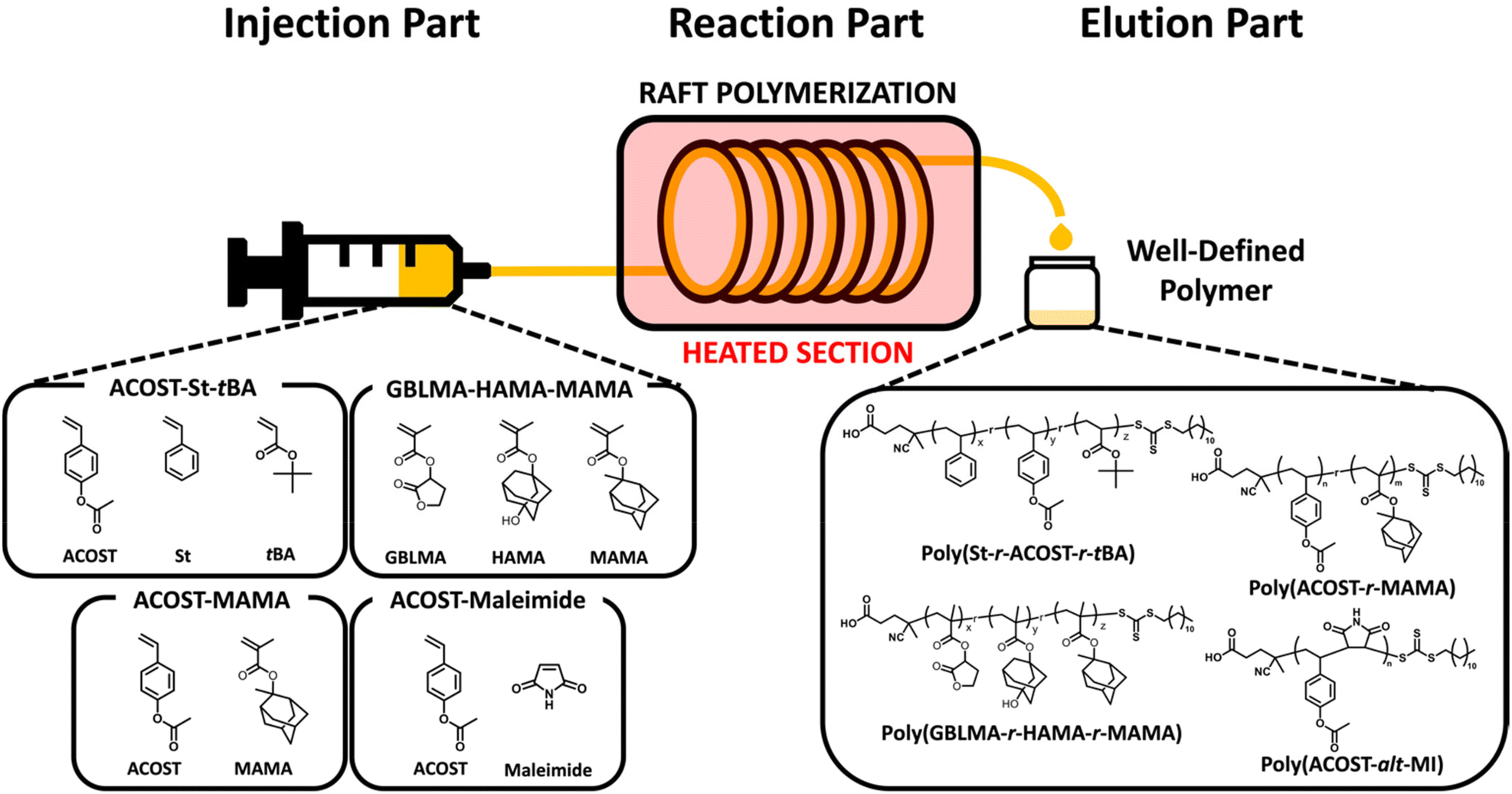 | ||
| Fig. 1 Schematic of the flow reactor, and structures of copolymers and monomers examined in this study. | ||
2. Experimental
2.1. Materials
4-Acetoxystyrene (ACOST), 2-oxotetrahydrofuran-3-yl methacrylate (GBLMA), 3-hydroxy-1-methacryloyloxyadamantane (HAMA), 2-methyl-2-adamantyl methacrylate (MAMA), 2,2′-azobis(2,4-dimethylvaleronitrile) (V-65), and N-hydroxynaphthalimide perfluoro-1-butanesulfonate (HNPFBS) were obtained from KISCO Inc. (Seoul, South Korea). 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDSTSP) was synthesized according to the literature.32 4,4′-Azobis(4-cyanovaleric acid) (ACVA; 98%), hexamethyldisilazane (HMDS; 99%), and tetrahydrofuran (THF; 99.9%) were purchased from Sigma Aldrich (Milwaukee, WI, USA). An aqueous tetramethylammonium hydroxide (TMAH) solution (2.38%; AZ 300 MIF developer) was purchased from Merck KGaA (Darmstadt, Germany). PGME (98%) and methyl alcohol (99.5%) were obtained from Daejung Chemicals & Metals (Siheung, South Korea) and Samchun Chemicals (Seoul, South Korea), respectively. Anisole (99%) and maleimide (MI; 98%) were purchased from Acros Organics (Geel, Belgium). Styrene (St; 99%) and tert-butyl acrylate (tBA; 98%) were obtained from Tokyo Chemical Industry (Tokyo, Japan) and purified by passing them through an alumina column to remove the inhibitor prior to polymerization. Aqueous ammonium hydroxide solution (28%) and 2,2′-azobis(2-methylpropionitrile) (AIBN) were purchased from Junsei Chemical (Tokyo, Japan); AIBN was recrystallized from methanol before use. Unless otherwise noted, the reagents were used without further purification.2.2. RAFT copolymerization in a continuous flow
The desired monomers (St, ACOST, tBA, GBLMA, HAMA, MAMA, and MI) were mixed with CDSTSP, an initiator (AIBN, ACVA, or V-65), and PGME in Schlenk flasks, followed by stirring for dissolution. The homogeneous solutions were degassed by three freeze–pump–thaw cycles. The molar ratio of CDSTSP to initiator was maintained at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all cases, and the amount of PGME was varied to attain a concentration of 10–90 wt%, depending on the monomer type. The flow reactor was designed to define three regions: injection, reaction, and elution (Fig. 1). A syringe containing the degassed reaction solution was installed on the syringe pump. The syringe was connected to a PTFE tube (inner diameter = 1/16 inch) through a Luer connector. The PTFE tube, whose length was set for the polymerization time of 3 h, was immersed in an oil bath at the desired temperature. If necessary, the length of the PTFE tube was varied to control the residence time, i.e. the polymerization reaction time. The PTFE tubing with a desired length was wound to have a diameter of approximately 4–5 cm so that it could fit inside an oil bath, and the rolled tubing was tied with plastic straps or cable ties. It was then immersed in an oil bath and pressed down with a round bottom flask filled with silicone oil to prevent floating. The position of the tubing roll was also kept approximately 1–2 cm away from the bottom to avoid direct contact to the hot surface. The reaction mixture was quickly injected into the reaction tube up to the oil-immersed part at 600 mL h−1 (typically less than 15 s). Immediately, the flow rate was changed and fixed at 1 mL h−1 (0.016 mL min−1). In the elution part, the resulting reaction solution was passed through the cooling section at ≈20 °C, subsequently dropped in a solvent for quantitative proton nuclear magnetic resonance (1H NMR) analysis to determine the polymerization conversion and copolymer composition, or dropped in a solvent (heptane for styrenic copolymers or methanol for methacrylate copolymers) for precipitation. The precipitate was collected via vacuum filtration, followed by drying in a vacuum oven at 100 °C (for heptane) or 30 °C (for methanol).
:1 in all cases, and the amount of PGME was varied to attain a concentration of 10–90 wt%, depending on the monomer type. The flow reactor was designed to define three regions: injection, reaction, and elution (Fig. 1). A syringe containing the degassed reaction solution was installed on the syringe pump. The syringe was connected to a PTFE tube (inner diameter = 1/16 inch) through a Luer connector. The PTFE tube, whose length was set for the polymerization time of 3 h, was immersed in an oil bath at the desired temperature. If necessary, the length of the PTFE tube was varied to control the residence time, i.e. the polymerization reaction time. The PTFE tubing with a desired length was wound to have a diameter of approximately 4–5 cm so that it could fit inside an oil bath, and the rolled tubing was tied with plastic straps or cable ties. It was then immersed in an oil bath and pressed down with a round bottom flask filled with silicone oil to prevent floating. The position of the tubing roll was also kept approximately 1–2 cm away from the bottom to avoid direct contact to the hot surface. The reaction mixture was quickly injected into the reaction tube up to the oil-immersed part at 600 mL h−1 (typically less than 15 s). Immediately, the flow rate was changed and fixed at 1 mL h−1 (0.016 mL min−1). In the elution part, the resulting reaction solution was passed through the cooling section at ≈20 °C, subsequently dropped in a solvent for quantitative proton nuclear magnetic resonance (1H NMR) analysis to determine the polymerization conversion and copolymer composition, or dropped in a solvent (heptane for styrenic copolymers or methanol for methacrylate copolymers) for precipitation. The precipitate was collected via vacuum filtration, followed by drying in a vacuum oven at 100 °C (for heptane) or 30 °C (for methanol).
2.3. Evaluation of the reactivity ratio
A series of reaction solutions consisting of two monomers with various feed ratios (fmonomer), CDSTSP, ACVA, and PGME were prepared and degassed prior to flow experiments. The feed ratios of various monomer pairs are listed in Table S1.† The solutions were subjected to copolymerization by a continuous flow reaction to achieve low conversion. The actual compositions of the copolymers (Fmonomer) were determined by quantitative 1H NMR spectroscopy of the resulting copolymer solutions obtained in the elution part of the flow reactor. The reactivity ratio values of the monomer pairs were obtained by analyzing the plots of the feed ratio and actual composition via nonlinear least square (NLS) fitting with the Mayo–Lewis equation, Fineman–Ross (FR) and Kelen–Tüdös (KT) methods. Details of all quantitative 1H NMR analyses are described in the ESI (Fig. S1–S10†).2.4. Post-polymerization modification
To remove the trithiocarbonate chain end group, the resulting copolymer obtained by RAFT polymerization and AIBN (20 molar equivalents of the copolymer) were dissolved in THF (50 times the copolymer weight), followed by refluxing for 19 h at 80 °C. The solution was cooled to room temperature and concentrated using a rotary evaporator. The solution was poured into excess methanol for precipitation. The solid was obtained as a white powder via vacuum filtration and dried under vacuum at 30 °C. For copolymers containing ACOST, deacetylation was performed after trithiocarbonate removal. To this end, the ACOST-containing copolymer was mixed with an aqueous ammonium hydroxide solution (70 molar equivalents of the acetyl group in the copolymer) and methanol (10 times the copolymer weight). The solution was stirred at room temperature for 18 h under a nitrogen atmosphere. The reaction solution was concentrated using a rotary evaporator, followed by the addition of a specific amount of THF. The solution was added to DI water and the resulting solid was collected by vacuum filtration and subsequently dried in a vacuum oven at 80 °C.2.5. Lithographic performance evaluation
Photoresist solutions were formulated using a synthesized resin, HNPFBS as a photoacid generator (PAG), and n-butyl acetate as a solvent. The concentration of the synthesized resin was 3 wt% in n-butyl acetate, and 5 wt% PAG (relative to the resin) was added. The solutions were filtered through a 0.2 μm PTFE syringe filter. Photoresist films were cast on HMDS-treated silicon wafers by spin-coating the solutions at 2000 rpm for 60 s, followed by soft baking at 130 °C for 60 s. The films were photo-patterned using I-line UV light (365 nm) illumination with the desired energy passed through a photomask. The films were subjected to post-exposure baking (PEB) at 140 °C for 90 s, followed by film development upon immersion in an AZ 300 MIF developer for 10 s and subsequent rinsing with DI water.2.6. Characterization
1H NMR spectra were recorded in CDCl3 using a Bruker Avance III 400 MHz spectrometer (Bruker, USA) or a JEOL JNM-ECZ400S 400 MHz spectrometer (JEOL, Japan). Size exclusion chromatography (SEC) was performed using a Thermo Scientific Ultimate 3000 system equipped with three columns (Styragel HR5, Styragel HR4, Styragel HR3; Waters) at a flow rate of 1 mL min−1 with THF as the eluent at 35 °C. The molecular weight information was acquired using a calibration curve constructed from 10 polystyrene (PS) standard samples (Mn = 1.2–1390 kg mol−1). Fourier transform infrared (FT-IR) and UV/vis absorption spectra were recorded on an IR Prestige-21 (Shimadzu, Japan) and UV-5100B (Metash, China). Optical microscopy images were obtained using a BX53MRF-S (Olympus, Japan).3. Results and discussion
3.1. Kinetics of continuous flow RAFT copolymerization
To investigate the fundamental kinetic aspects of continuous flow RAFT copolymerization, we chose two model copolymers for typical CAR systems, namely poly(St-r-ACOST-r-tBA) and poly(GBLMA-r-HAMA-r-MAMA), for KrF and EUV photolithography16,31,33–36 and ArF photolithography,30,36–39 respectively (Fig. 2). A common azo-type initiator (AIBN or ACVA) and a versatile and commercially available trithiocarbonate chain transfer agent (CDSTSP) covering a wide monomer scope were selected for copolymerization.40 PGME was chosen as the solvent because it is widely used in the production of chemicals for semiconductor manufacturing. For the initial kinetic assessment, RAFT copolymerization for poly(St-r-ACOST-r-tBA) and poly(GBLMA-r-HAMA-r-MAMA) was performed at [monomer]:[CDSTSP]:[ACVA] = 256:2:1 and 90:2:1 and PGME concentrations of 30 and 50 wt% to the mass of the solution, respectively. The feed ratios for the copolymerization were 0.25:0.60:0.15 for fSt:fACOST:ftBA and 0.40:0.20:0.40 for fGBLMA:fHAMA:fMAMA. The reaction temperature and maximum residence time were 75 °C and 3 h, respectively. The continuous flow reaction is a highly beneficial method for readily acquiring a polymerization kinetic dataset because the reaction time of the resulting eluted reaction mixtures during the first 3 h matches the residence time, allowing automatic sampling as a function of reaction time. Fig. 3a and c show the polymerization conversion determined by quantitative 1H NMR analyses of the acquired samples (see the ESI† for details). The total conversion of poly(St-r-ACOST-r-tBA) increased to ≈0.6, at a reaction time of 150 min. In the case of poly(GBLMA-r-HAMA-r-MAMA), the total conversion reaches ≈0.9 at 90 min, suggesting that the copolymerization of styrenic monomers is relatively slow. In addition, the reactivity of monomers during copolymerization can be roughly estimated. In Fig. 3a, the consumption rate of tBA is higher than that of the other two monomers, and that of ACOST is the lowest, indicating that the acrylate and protected styrene monomers exhibited the highest and lowest reactivities in the copolymerization. It has been shown that the radical-based polymerization of ACOST is slower than that of other styrenic monomers, which corresponds with our results.41 In the methacrylate copolymerization (Fig. 3c), the consumption rates of GBLMA and HAMA were similar, but that of MAMA was slightly lower, which is consistent with the rates of the batch RAFT copolymerization of GBLMA, HAMA, and 2-ethyl-2-adamantyl methacrylate (EAdMA), which are structurally similar.30
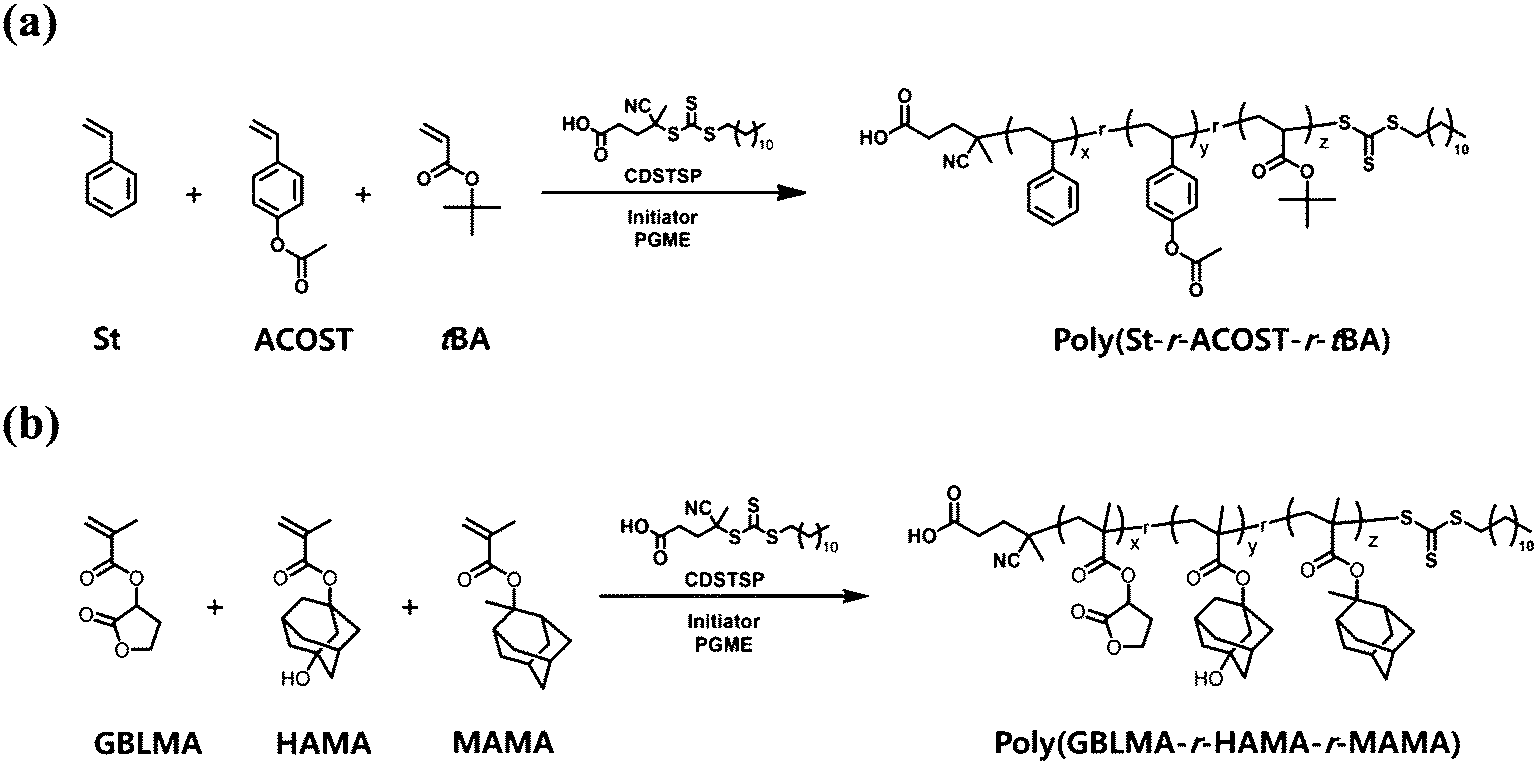 | ||
| Fig. 2 Continuous flow RAFT copolymerization of (a) poly(St-r-ACOST-r-tBA) and (b) poly(GBLMA-r-HAMA-r-MAMA). | ||
 | ||
| Fig. 3 Kinetic studies for continuous flow RAFT copolymerization. Plots showing (a) conversion for each monomer and total conversion, and (b) ln([M]0/[M]) as a function of reaction time for poly(St-r-ACOST-r-tBA), (c) conversion for each monomer and total conversion, and (d) ln([M]0/[M]) as a function of reaction time for poly(GBLMA-r-HAMA-r-MAMA). | ||
Based on the measurement of total conversion as a function of time, the kinetic plots of ln([M]0/[M]) versus copolymerization time were constructed (Fig. 3b and d). In a typical RDRP, the propagation rate (Rp) is given by the equation Rp = kp[P*][M] = kappp[M], where kp denotes the propagation rate constant, [P*] denotes the concentration of the active propagating species, and [M] denotes the concentration of the monomer at time t. Under steady state conditions, whereunder the concentration of the radical species is constant, the product of kp and [P*], defined as an apparent propagation rate constant (kappp) is also constant. This pseudo-first-order reaction yields the kinetic equation ln([M]0/[M]) = kapppt, where [M]0 denotes the initial monomer concentration. As shown in Fig. 3b and d, the ln([M]0/[M]) values for both copolymerization reactions linearly scale with the reaction time to yield the kappp values of 0.0047 ± 0.0002 and 0.0269 ± 0.0010 min−1, strongly suggesting that in the continuous flow RAFT copolymerization, the concentration of the growing radical does not change in the flow; hence, the RAFT polymerization occurs in flow mode despite the reaction geometry difference.42 Continuous flow polymerization is considerably faster than batch copolymerization. The RAFT polymerization of various methacrylates bearing adamantyl or γ-butyrolactone pendant groups in batch mode typically requires more than 10 h to reach a conversion of >0.9, and the estimated kappp is 0.0007–0.0017 min−1,30 which is approximately 10 times lower than the values obtained for continuous flow copolymerization. The acceleration of the propagation can be attributed to the high surface area-to-reaction volume ratio in the flow reactor channel, providing more efficient mixing of reactants and mass/heat transfer.43–45 With the initial kinetic assessment, we varied the copolymerization conditions, that is the reaction temperature, the type of initiator, and the amount of solvent, to further understand their effect on the copolymerization of St, ACOST, and tBA in a continuous flow reactor. The conversion as a function of residence time (Fig. 4a) was converted to the kinetic plot shown in Fig. 4b. With a conversion of ≈0.7, ln([M]0/[M]) shows a linear increase as a function of time. The extracted kappp values were 0.0077 ± 0.0006, 0.0053 ± 0.0010, 0.0049 ± 0.0013, and 0.0045 ± 0.0007 min−1 for the copolymerization at 85 °C (PGME 50 wt%, AIBN), the use of ACVA (75 °C, PGME 50 wt%), the use of AIBN (75 °C, PGME 50 wt%), and the variation in the amount of PGME to 60 wt% (75 °C, AIBN), respectively. These results strongly suggest that under various conditions, the copolymerization is still controlled in the same manner as the batch reaction.
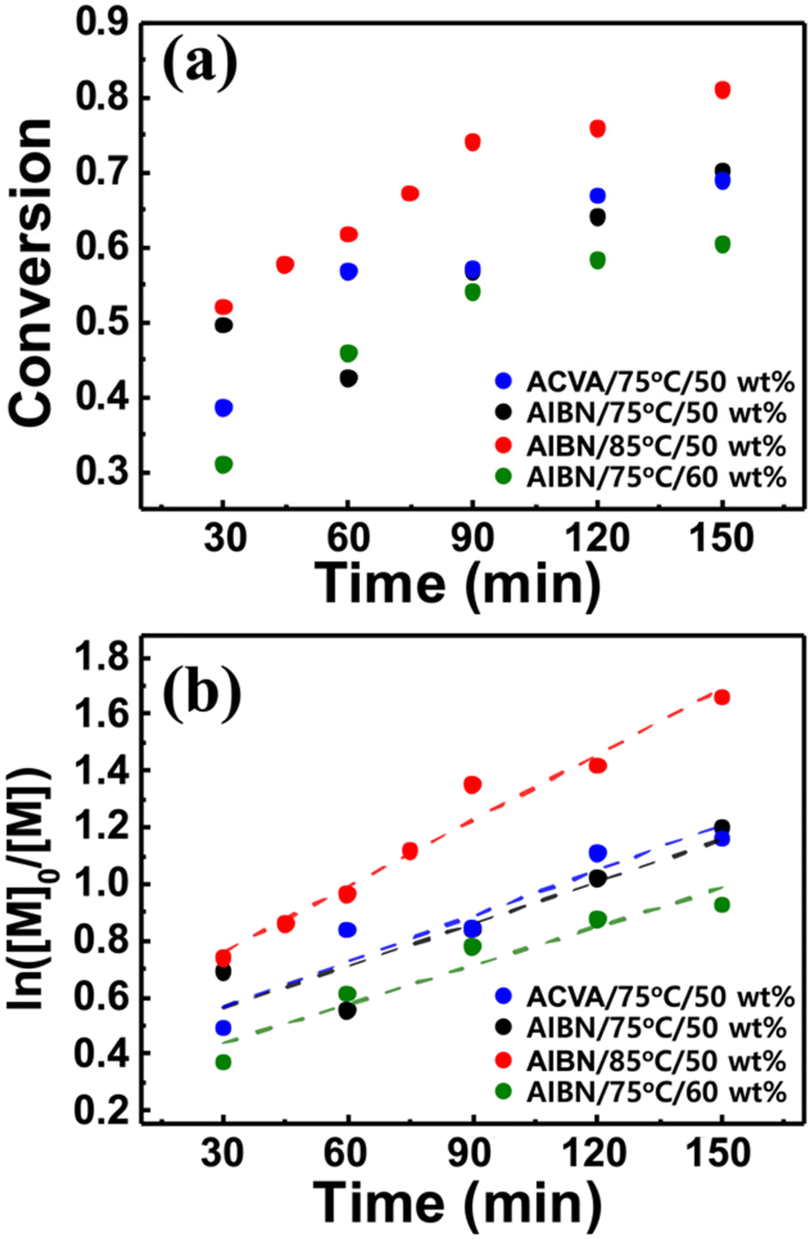 | ||
| Fig. 4 Kinetic studies for the effect of reaction conditions. Plots showing (a) total conversion, and (b) ln([M]0/[M]) as a function of reaction time under varied reaction conditions for poly(St-r-ACOST-r-tBA). | ||
Finally, we aimed to achieve poly(St-r-ACOST-r-tBA) with a Mw of ≈10.0 kg mol−1, which is in a typical molecular weight range for CAR copolymer resins. With systematic variations in the amount of PGME and the type of initiator (Table S2†), we observed that the resulting molecular weight increased when the amount of solvent was reduced, and the required temperature for ten hour half-life of the initiator decreased (ACVA: 69 °C; V-65: 51 °C).46,47 The representative SEC chromatograms and 1H NMR spectra in Fig. S11–S13,† and the analysis results in Tables S3 and S4† show that the molecular weight, dispersity, and composition of the samples synthesized with a residence time of 180 min were highly similar with a low coefficient of variation, confirming that the structural uniformity of the resulting copolymers is guaranteed by the continuous flow reaction.
3.2. Copolymerization kinetics of monomers relevant for the CAR in a continuous flow reaction
Each monomeric unit in the copolymers for CAR has its own purpose; hence, the composition of the units in the copolymer chains drastically affects the final lithographic performance in terms of parameters such as sensitivity, dissolution rate, and etching resistance. Furthermore, as the size of the pattern decreases on the single digit nanometer scale, the control of the chain structure at the molecular level should be emphasized. In this regard, understanding of the reactivity of propagating species in copolymerization, that is the reactivity ratio, is essential. This insight can be acquired by considering conventional copolymerization kinetics, which allows us to appreciate the sequence of monomer units along the copolymer chain, as well as to predict the composition of the resulting copolymer from the feed composition. The monomers illustrated in Fig. 1 are typically used in DUV/EUV lithographic applications and were investigated using continuous flow chemistry in the current study. By controlling the residence time of the flow copolymerization, we assessed the composition of the copolymer at low conversion in a precise and efficient manner as the residence time was controlled at the minute scale.22 On the basis of the kinetic studies described above, the feed amount dependences (fmonomer = 0.1–0.9) of the compositions of the resulting copolymers with different monomer pairs were determined by quantitative 1H NMR analysis using an internal standard. The [ACVA]:[CDSTSP] ratio was fixed at 2:1, and the copolymerization temperature was 75 °C.
The copolymerization kinetics of RDRPs are described using a conventional radical terminal model, wherein the reactivity of the propagating radical species depends on the nature of the terminal unit, rather than the chain length.48 Therefore, during RAFT copolymerization, different monomers exhibit different reactivities to activated radicals at the terminal units of the growing chains; hence, the resulting composition of the copolymer often varies in relation to the fed amount. This is generally described using reactivity ratios r12 and r21, which are defined as k11/k12 and k22/k21, where kxy denotes the rate constant of the reaction of the active radical on monomer x to monomer y. The reactivity ratio is typically determined experimentally by fitting the experimental dataset of actual compositions as a function of feed composition at low conversion (typically 0.04–0.15 in this study) using three methods: NLS fitting with the Mayo–Lewis equation,49 FR method,50 and KT method.51 It is noted that right after the reaction mixture exited the heating section, the tube was exposed to the room temperature environment for rapid cooling which is a benefit of the flow reaction, i.e., highly efficient heat transfer. In the conversion range of interest (0.04–0.15) where the possible compositional drift that may affect the estimation of reactivity ratio can be minimized,52,53 the conversion, kappp, and actual composition were not significantly varied compared to that obtained upon quenching with 0 °C cooling (Fig. S14†), which possibly disrupts the flow, e.g. the increase of viscosity in the high concentration range.
Fig. 5 shows the plots for the actual composition as a function of the fed amount with five different monomer pairs including two styrenic monomers, which are for the model copolymers utilizable for the KrF resist and EUV resist.16,33–36,54 The experimental datasets were effectively interpreted through NLS fitting with the Mayo–Lewis equation; the resulting reactivity ratio values are listed in Table 1. The fitting results with the FR and KT methods are shown and summarized in Fig. S15–S18 and Tables S5 and S6.† Among them, the values obtained from the NLS fitting are discussed, as they reliably represent the copolymerization behaviors.
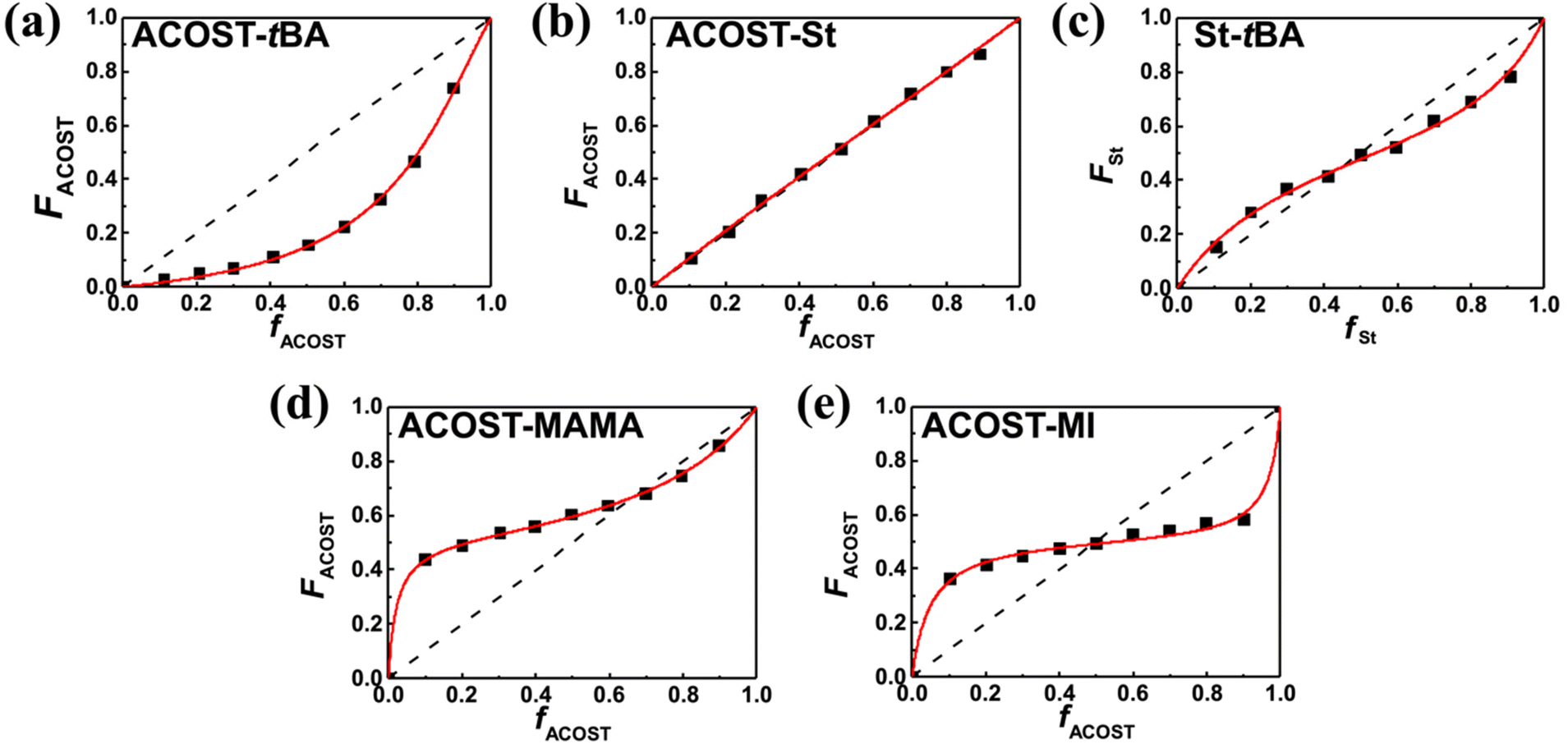 | ||
| Fig. 5 Plots to acquire reactivity ratios for the monomer pairs of (a) ACOST/tBA, (b) ACOST/St, (c) St/tBA, (d) ACOST/MAMA, and (e) ACOST/MI. The red lines are the NLS fitting results with the Mayo–Lewis equation. | ||
| Monomer A/monomer B | r AB | r BA |
|---|---|---|
| ACOST/MAMA | 0.53 ± 0.01 | 0.04 ± 0.00 |
| ACOST/tBA | 0.43 ± 0.03 | 7.12 ± 0.39 |
| ACOST/St | 0.98 ± 0.06 | 0.93 ± 0.06 |
| St/tBA | 0.35 ± 0.03 | 0.47 ± 0.03 |
| ACOST/MI | 0.06 ± 0.01 | 0.09 ± 0.01 |
First, the reactivity ratios for the copolymerization of MI with ACOST were close to zero, and the product was 0.005, indicating that alternating copolymerization occurred. The radical-based copolymerization of an MI-type monomer with a styrenic monomer typically results in an alternating copolymer owing to their electron-deficient and electron-rich structures,55 and the reported r12 and r21 values are typically less than 0.10 regardless of the solvent type.55–59 For example, the reactivity ratios for MI/St and MI/(N,N-diethyldithiocarbamyl)methylstyrene were 0.088 and 0.057,59 and 0.15 and 0.00,56 respectively, which correspond with our results to validate the copolymerization kinetics in a continuous flow. In addition, the photoresist resin in an alternating sequence, which has the potential to address patterning performance degradation owing to sequential inhomogeneity in the polymer chain,18 is attainable with continuous flow chemistry. Among the three monomer pairs for poly(St-r-ACOST-r-tBA), the ACOST/St pair, exhibiting minimal structural variation, showed a reactivity ratio close to unity, confirming that random copolymerization occurred. This result agrees with those of previous reports; the reactivity ratios were indistinguishable from unity in free radical suspension polymerization,60,61 and rACOST–St and rSt–ACOST in 1,4-dioxane were found to be 1.245 and 0.773 and 1.384 and 0.691 for free radical copolymerization and RAFT copolymerization, and the products of the reactivity ratios were 0.962 and 0.956, respectively. The copolymerization of tBA with St exhibits reactivity ratio values less than unity and, therefore, inclines for both units to prefer cross-polymerization rather than homopolymerization. This behavior was also observed in previous studies investigating free radical, nitroxide-mediated, and RAFT copolymerization reactions.26,62 Notably, ACOST/tBA exhibited a large deviation from unity, leading to a large compositional drift with a preference for cross-polymerization and homopolymerization, respectively. This result differs from the literature, in which rACOST–tBA is typically higher than unity and rtBA–ACOST is lower than unity.26,63,64 However, the observed reactivity is consistent with the results of the kinetic studies shown in Fig. 3a, in which the rate of conversion increase of tBA is significantly higher than that of ACOST, implying that tBA is more reactive than ACOST in copolymerization. Because the copolymerization kinetic results of other monomer pairs are found to be reasonable, the copolymerization behavior of this particular monomer pair is possibly affected by other parameters, such as temperature and the type of solvent, which possibly results in this deviation as it has been reported that the solvent polarity can largely alter the reactivity ratio,65,66 though further studies are necessary to understand the underlying chemical principle. For the last monomer pair in a styrenic monomer-containing entry, which is a useful ACOST/MAMA system for the EUV resist, both monomers show a tendency for cross-polymerization. Although the reported values for this pair are not available in the literature, the reactivity ratios for a similar pair, St/1-adamantyl methacrylate, were 0.62 and 0.54.67 The tendency for cross-polymerization is attributable to the considerable bulky pendant group affecting the reactivity.
The reactivity of the methacrylate monomers for poly(GBLMA-r-HAMA-r-MAMA), a model copolymer for the ArF resist, was also examined under continuous flow conditions. The resulting reactivity ratios of the three monomer pairs obtained by NLS fitting (Fig. 6) are listed in Table 2. When GBLMA was involved, all reactivity ratios are less than unity, indicating that cross-polymerization was preferred. The HAMA/MAMA system exhibits a different copolymerization behavior, in which HAMA predominantly forms a copolymer chain, and the MAMA unit is added to the chain.
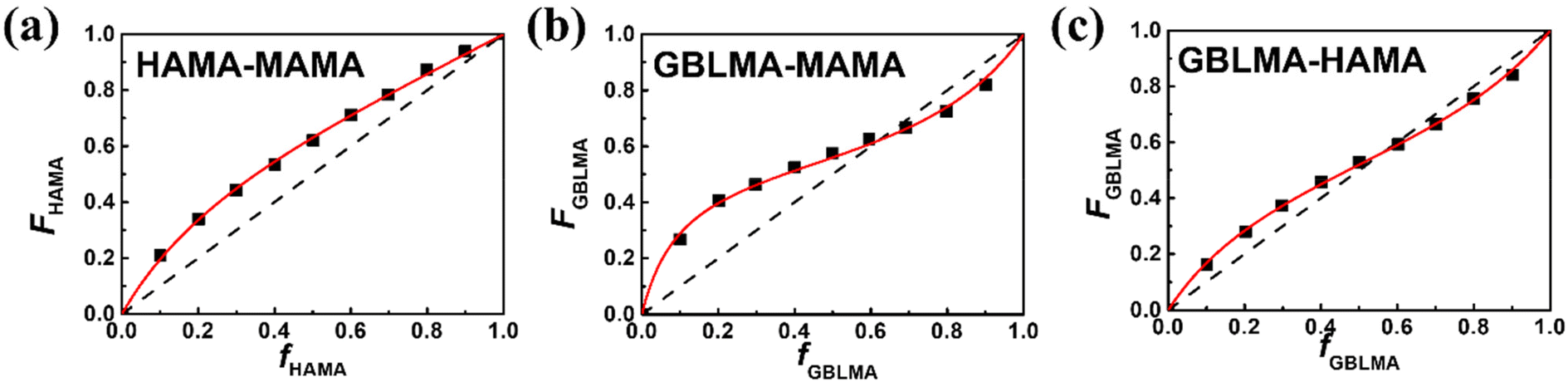 | ||
| Fig. 6 Plots to acquire reactivity ratios for the monomer pairs of (a) HAMA/MAMA, (b) GBLMA/MAMA, and (c) GBLMA/HAMA. The red lines are the NLS fitting results with the Mayo–Lewis equation. | ||
| Monomer A/monomer B | r AB | r BA |
|---|---|---|
| GBLMA/HAMA | 0.60 ± 0.02 | 0.48 ± 0.02 |
| MAMA/GBLMA | 0.18 ± 0.02 | 0.50 ± 0.03 |
| HAMA/MAMA | 1.42 ± 0.06 | 0.42 ± 0.02 |
The reactivity ratios were further used to predict the compositions of poly(GBLMA-r-HAMA-r-MAMA) over the full range of polymerization conversion. Hence, the instantaneous monomer feed composition (Mi), instantaneous terpolymer composition (Fi), and cumulative terpolymer composition (![[F with combining macron]](https://www.rsc.org/images/entities/i_char_0046_0304.gif) i) of the copolymer were predicted at a fixed feed ratio of 0.40:0.20:0.40 for GBLMA:HAMA:MAMA, by numerical analysis using the standard Alfrey–Goldfinger (AG) model, realized with the custom-made Python code.68–71 As shown in Fig. 7a, the consumption of GBLMA is accelerated although MAMA is consumed slowly and the consumption rate of HAMA is almost constant. The initial feed fraction of both GBLMA and MAMA was 0.40; however, because the reactivity of GBLMA was higher than that of MAMA, the GBLMA portion in the sequence of the formed polymer chains was considerably higher. At high conversions (>0.8), GBLMA is dominantly consumed, and at later stages, MAMA and HAMA are dominantly polymerized. These results strongly suggest that the chain formed at the initial stage has a blocky GBLMA domain, and the chain formed later possesses HAMA- and MAMA-rich domains, as reflected in the instantaneous terpolymer compositions in Fig. 7b. Fig. 7c shows the total composition of the cumulative terpolymer. The composition of the terpolymer at low conversion is comparable to that of Fi, that is a GBLMA-dominant composition; a similar tendency was observed as the conversion increased. Most importantly, in the continuous flow reaction, compositional changes during the RAFT copolymerization of GBLMA, HAMA, and MAMA at the same feed ratios were thoroughly observed as a function of conversion (open dots in Fig. 7c). To further validate these results, we synthesized poly(GBLMA-r-HAMA-r-MAMA) with a varied feed composition at low conversion, and the resulting terpolymer compositions were compared with the predicted compositions using the acquired reactivity ratios in Table 2. As can be observed in Table 3, the predicted compositions are highly comparable to the actual ones, strongly suggesting that the examined copolymerization kinetics are valid for accurately reporting the actual reactivities of the propagating species in continuous flow RAFT copolymerization. All empirical data correspond with the theoretical prediction of the estimated reactivity ratio, highlighting the significance of the current kinetic studies under continuous flow conditions.
i) of the copolymer were predicted at a fixed feed ratio of 0.40:0.20:0.40 for GBLMA:HAMA:MAMA, by numerical analysis using the standard Alfrey–Goldfinger (AG) model, realized with the custom-made Python code.68–71 As shown in Fig. 7a, the consumption of GBLMA is accelerated although MAMA is consumed slowly and the consumption rate of HAMA is almost constant. The initial feed fraction of both GBLMA and MAMA was 0.40; however, because the reactivity of GBLMA was higher than that of MAMA, the GBLMA portion in the sequence of the formed polymer chains was considerably higher. At high conversions (>0.8), GBLMA is dominantly consumed, and at later stages, MAMA and HAMA are dominantly polymerized. These results strongly suggest that the chain formed at the initial stage has a blocky GBLMA domain, and the chain formed later possesses HAMA- and MAMA-rich domains, as reflected in the instantaneous terpolymer compositions in Fig. 7b. Fig. 7c shows the total composition of the cumulative terpolymer. The composition of the terpolymer at low conversion is comparable to that of Fi, that is a GBLMA-dominant composition; a similar tendency was observed as the conversion increased. Most importantly, in the continuous flow reaction, compositional changes during the RAFT copolymerization of GBLMA, HAMA, and MAMA at the same feed ratios were thoroughly observed as a function of conversion (open dots in Fig. 7c). To further validate these results, we synthesized poly(GBLMA-r-HAMA-r-MAMA) with a varied feed composition at low conversion, and the resulting terpolymer compositions were compared with the predicted compositions using the acquired reactivity ratios in Table 2. As can be observed in Table 3, the predicted compositions are highly comparable to the actual ones, strongly suggesting that the examined copolymerization kinetics are valid for accurately reporting the actual reactivities of the propagating species in continuous flow RAFT copolymerization. All empirical data correspond with the theoretical prediction of the estimated reactivity ratio, highlighting the significance of the current kinetic studies under continuous flow conditions.
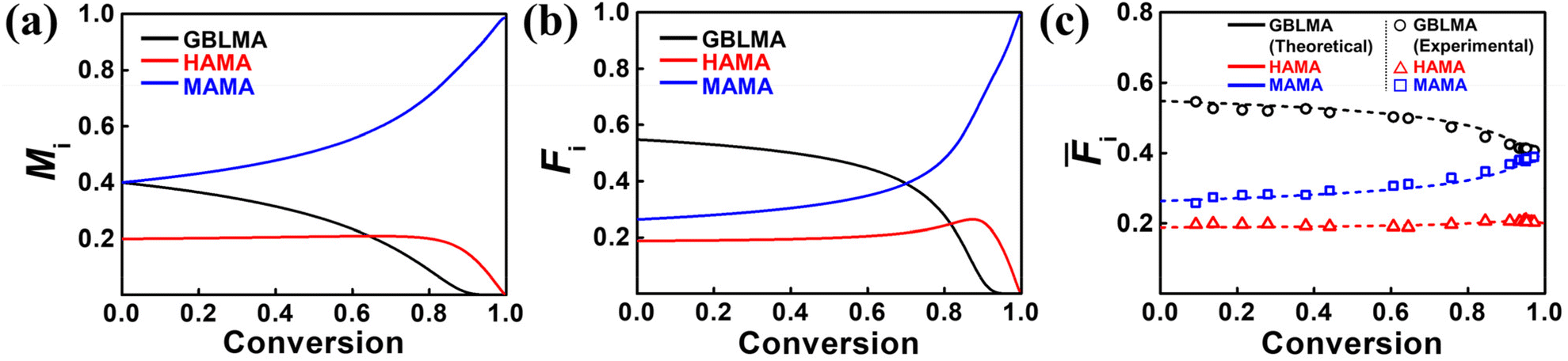 | ||
| Fig. 7 Predicted (a) instantaneous monomer compositions (Mi), (b) instantaneous terpolymer compositions (Fi), and (c) cumulative terpolymer compositions (i) for GBLMA (black line), HAMA (red line), and MAMA (blue line). The open dots in (c) represent empirically determined compositions with the variation of conversion. | ||
| Conversion | Feed composition | Actual composition | Predicted composition |
|---|---|---|---|
|
f
GBLMA:fHAMA:fMAMA |
F
GBLMA:FHAMA:FMAMA |
F
GBLMA:FHAMA:FMAMA |
|
| 0.137 | 0.40:0.20:0.40 |
0.53:0.27:0.20 |
0.51:0.28:0.21 |
| 0.070 | 0.20:0.40:0.40 |
0.35:0.26:0.39 |
0.33:0.27:0.40 |
| 0.154 | 0.40:0.30:0.30 |
0.45:0.23:0.32 |
0.49:0.21:0.30 |
| 0.111 | 0.50:0.20:0.30 |
0.58:0.20:0.22 |
0.60:0.19:0.21 |
| 0.126 | 0.30:0.30:0.40 |
0.46:0.27:0.26 |
0.47:0.27:0.26 |
3.3. Photo-induced catalytic acidolysis reactions of the copolymer synthesized in a continuous flow
To confirm the applicability of the continuous flow process, the synthesized copolymers were used to realize a CAR system. Poly(HSt-r-MAMA), a model copolymer for the EUV CAR,72–75 was obtained by synthesizing poly(ACOST-r-MAMA) in a continuous flow at a feed ratio fACOST:fMAMA of 0.8:0.2 and the subsequent post-polymerization modification process (Fig. 8). Under the same reaction conditions as described above, well-defined poly(ACOST-r-MAMA) was successfully synthesized (Mn = 5.1 kg mol−1, Đ = 1.19, FMAMA = 0.22). The chemical structure of poly(ACOST-r-MAMA) needed to be modified to be applicable to the photoresist owing to (i) the trithiocarbonate end group derived from the RAFT agent, and (ii) the acetoxy group, which should be returned to the phenolic hydroxyl group in the styrenic unit. The trithiocarbonate end group strongly absorbs in the UV light range (250–400 nm), which affects the sensitivity of the CAR system. In addition, the end group may affect the thin film properties when the molecular weight of the copolymer is considerably low. Therefore, the copolymer was subjected to terminal group removal by the reaction with an excess of the thermal radical generator, that is AIBN.76–79 The extent of the modification reaction was traced through UV/vis absorption spectroscopy (Fig. S19†),80,81 and the typical degree of removal was higher than 0.98 without any significant side reactions such as chain coupling (Fig. S20†) under the optimized reaction conditions described in the Experimental section. Then, the deacetylation reaction was performed by treatment with an aqueous ammonium hydroxide solution,42,82 and the complete reaction was confirmed by examining the signal of the acetoxy group in the 1H NMR and FT-IR spectra, shown in Fig. S21.†
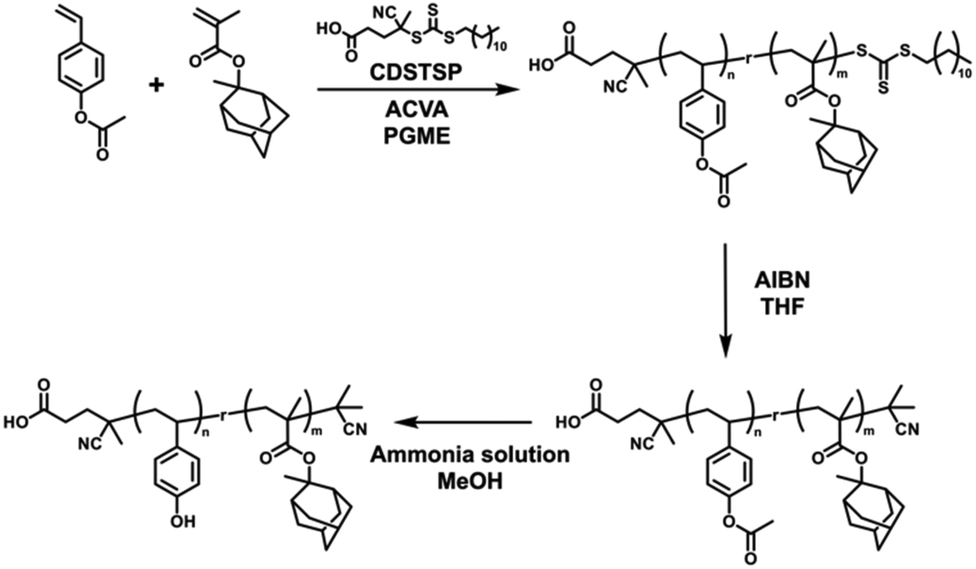 | ||
| Fig. 8 Synthesis of poly(ACOST-r-MAMA) and post-polymerization modifications to yield the poly(HSt-r-MAMA) copolymer. | ||
The resulting poly(HSt-r-MAMA) copolymer was further used to formulate a photoresist solution with HNPFBS as the PAG in n-butyl acetate. The illumination of UV light at 365 nm (40 mJ cm−2) on the deposited resist thin film through a photomask in contact, subsequent PEB (140 °C, 90 s), and development with a conventional aqueous base developer (immersion time: 10 s) led to the formation of a positive tone photo pattern with the smallest dimension of ≈3 μm with a simple and crude lithography setup (Fig. 9). The control sample, poly(HSt-r-MAMA) (corresponding poly(ACOST-r-MAMA): Mn = 4.7 kg mol−1, Đ = 1.13, FMAMA = 0.21), synthesized through conventional batch copolymerization also showed comparable results in the same post-exposure process, confirming the applicability of continuous flow copolymerization. Under the optimized conditions, the effect of the removal of the end group was further examined (Fig. S22†), and the photo-pattern generated with the copolymer without removing the terminal group exhibited poorer adhesion properties than those of the copolymer upon removal, highlighting the importance of the post-polymerization modification process.
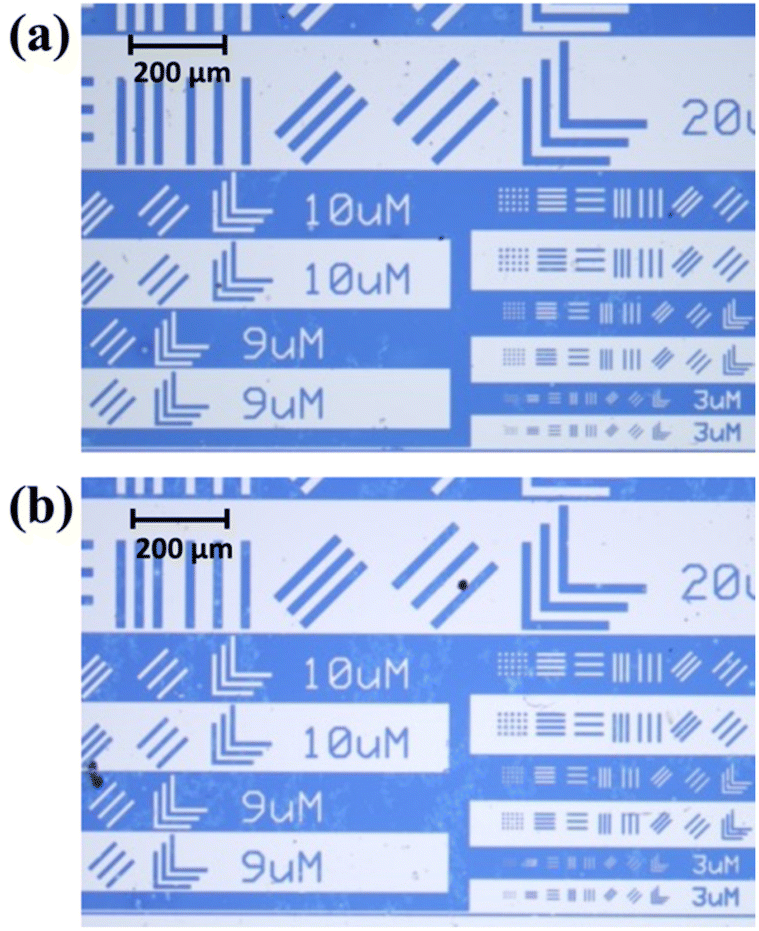 | ||
| Fig. 9 Optical microscopy images of the photo-patterns fabricated using photoresists consisting of the HNPFBS and poly(HSt-r-MAMA) copolymers synthesized by (a) batch copolymerization and (b) continuous flow copolymerization. | ||
5. Conclusion
Various aspects of continuous flow RAFT copolymerization of model monomers used in the CAR were investigated using PGME, which is widely used in different industrial fields because of its economic and environmental advantages. The fundamental copolymerization kinetics to synthesize poly(St-r-ACOST-r-tBA) and poly(GBLMA-r-HAMA-r-MAMA) were explored, and it was found that the RAFT copolymerization in flow also follows typical pseudo-first-order kinetics, exhibiting a largely increased kappp up to approximately 10 times that of conventional batch RAFT copolymerization processes. The copolymer samples obtained after a 3 h residence time showed a highly uniform molecular weight, dispersity, and composition, with a low coefficient of variation. The nature of the continuous flow reaction and thorough kinetic investigation with well-established quantitative analysis methods further allowed facile access to the reactivity ratios of eight monomer pairs that can be adopted for DUV/EUV lithographic applications. The estimated reactivity ratio values were compared with literature values and empirically validated by comparing the predicted compositions under specific feed conditions with the actual compositions of copolymers synthesized under flow conditions. Significantly, the reactivity ratio values were used to predict the compositions in a full range of polymerization conversions by numerical analysis using the standard Alfrey–Goldfinger (AG) model and corresponded well with the experimental studies. To elucidate the applicability of the copolymers in photolithography, poly(HSt-r-MAMA), a model copolymer for a CAR-type EUV photoresist, was obtained by RAFT copolymerization in flow mode and subsequent post-polymerization modifications. With a simple formulation with PAG, the copolymer was effective in forming a positive tone photo pattern with the smallest dimensions of ≈3 μm using the crude I-line lithography setup. Our comprehensive studies highlight the potential of continuous flow chemistry to facilitate the production of target complex copolymers with precise and uniform control of structural and compositional parameters on a large scale by allowing an in-depth understanding of the chemical nature of copolymerization reactions and furthermore, confirming the applicability of continuous flow RAFT copolymerization to pioneering technologies.Author contributions
J. Y. and J. W. contributed equally to this work. The manuscript was written based on the contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the Korea Evaluation Institute of Industrial Technology (KEIT), funded by the MOTIE of the Republic of Korea (Project No. 20010321). M. K. acknowledges the support of the National Research Foundation of Korea (NRF) grants (2021R1A2C1093999). We acknowledge the support by the Korea Institute for Advancement of Technology (KIAT) grant funded by the Korea Government (MOTIE) (P0008458, The Competency Development Program for Industry Specialist). The authors thank the staff of KISCO Inc. for generously providing the reagents and discussions.Notes and references
- B. Panganiban, B. Qiao, T. Jiang, C. DelRe, M. M. Obadia, T. D. Nguyen, A. A. A. Smith, A. Hall, I. Sit, M. G. Crosby, P. B. Dennis, E. Drockenmuller, M. O. de la Cruz and T. Xu, Science, 2018, 359, 1239–1243 CrossRef CAS PubMed.
- T. Jiang, A. Hall, M. Eres, Z. Hemmatian, B. Qiao, Y. Zhou, Z. Ruan, A. D. Couse, W. T. Heller, H. Huang, M. O. de la Cruz, M. Rolandi and T. Xu, Nature, 2020, 577, 216–220 CrossRef CAS PubMed.
- K. G. Cho, S. An, D. H. Cho, J. H. Kim, J. Nam, M. Kim and K. H. Lee, Adv. Funct. Mater., 2021, 31, 2102386 CrossRef CAS.
- J. Park, S. Jo, Y. M. Lee, G. Saravanakumar, J. Lee, D. Park and W. J. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 8060–8070 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- M. Rubens, J. Van Herck and T. Junkers, ACS Macro Lett., 2019, 8, 1437–1441 CrossRef CAS PubMed.
- S. M. Lee, K. H. Park, S. Jung, H. Park and C. Yang, Nat. Commun., 2018, 9, 1867 CrossRef PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- R. M. Myers, D. E. Fitzpatrick, R. M. Turner and S. V. Ley, Chem. – Eur. J., 2014, 20, 12348–12366 CrossRef CAS PubMed.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- S. V. Luis and E. Garcia-Verdugo, Flow Chemistry: Integrated Approaches for Practical Applications, Royal Society of Chemistry, London, 2019 Search PubMed.
- T. Junkers, Macromol. Chem. Phys., 2017, 218, 1600421 CrossRef.
- T. Iwasaki and J.-I. Yoshida, Macromolecules, 2005, 38, 1159–1163 CrossRef CAS.
- W. Shin, W. Ko, S.-H. Jin, T. Earmme and Y.-J. Hwang, Chem. Eng. J., 2021, 412, 128572 CrossRef CAS.
- A. Natalello, J. Morsbach, A. Friedel, A. Alkan, C. Tonhauser, A. H. E. Müller and H. Frey, Org. Process Res. Dev., 2014, 18, 1408–1412 CrossRef CAS.
- T. Manouras and P. Argitis, Nanomaterials, 2020, 10, 1593 CrossRef CAS PubMed.
- S.-Y. Moon and J.-M. Kim, J. Photochem. Photobiol., C, 2007, 8, 157–173 CAS.
- M. Wang, C.-T. Lee, C. L. Henderson, W. Yueh, J. M. Roberts and K. E. Gonsalves, J. Mater. Chem., 2008, 18, 2704–2708 RSC.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- A. M. Nightingale and J. C. Demello, Adv. Mater., 2013, 25, 1813–1821 CrossRef CAS PubMed.
- T. Junkers, J. Flow Chem., 2017, 7, 106–110 CrossRef CAS.
- M. H. Reis, C. L. Davidson and F. A. Leibfarth, Polym. Chem., 2018, 9, 1728–1734 RSC.
- C. Diehl, P. Laurino, N. Azzouz and P. H. Seeberger, Macromolecules, 2010, 43, 10311–10314 CrossRef CAS.
- S. H. Kang, V. M. Prabhu, B. D. Vogt, E. K. Lin, W.-L. Wu and K. Turnquest, Polymer, 2006, 47, 6293–6302 CrossRef CAS.
- Y. D. Chaniago, G. R. Harvianto, A. Bahadori and M. Lee, Process Saf. Environ. Prot., 2016, 103, 413–423 CrossRef CAS.
- Y. Guo, D. J. Hill, A. K. Whittaker, K. S. Jack and H. Peng, Macromolecules, 2015, 48, 3438–3448 CrossRef CAS.
- H. Catalgil-Giz, A. Giz, A. Alb, A. Öncül Koç and W. Reed, Macromolecules, 2002, 35, 6557–6571 CrossRef CAS.
- P. C. Tsiartas, L. L. Simpson, A. Qin, C. G. Willson, R. D. Allen, V. J. Krukonis and P. M. Gallagher-Wetmore, Proc. SPIE, 1995, 2438, 261–271 CrossRef CAS.
- G. Patsis, V. Constantoudis and E. Gogolides, Microelectron. Eng., 2004, 75, 297–308 CrossRef CAS.
- H.-S. Sohn, S.-H. Cha, W.-K. Lee, D.-G. Kim, H.-J. Yun, M.-S. Kim, B.-D. Kim, Y.-H. Kim, J.-W. Lee and J.-S. Kim, Macromol. Res., 2011, 19, 722–728 CrossRef CAS.
- M. M. Alam, H. Peng, K. S. Jack, D. J. Hill and A. K. Whittaker, Eur. Polym. J., 2020, 134, 109772 CrossRef CAS.
- K. Ponnusamy, R. P. Babu and R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1066–1078 CrossRef CAS.
- K. Arimitsu, M. Yonekura and M. Furutani, RSC Adv., 2015, 5, 80311–80317 RSC.
- D.-H. Lee, S.-J. Kim, D.-U. Choi, D. Kim, J. Kim and C.-H. Kim, Proc. SPIE, 2004, 5376, 565–574 CrossRef CAS.
- J. Choi, Y. C. Nho and S. K. Hong, Appl. Chem. Eng., 2006, 17, 158–162 CAS.
- S. Rangan, R. A. Bartynski, A. Narasimhan and R. L. Brainard, Int. J. Appl. Phys., 2017, 122, 025305 CrossRef.
- Q. Lou, M. A. Kishpaugh and D. A. Shipp, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 943–951 CrossRef CAS.
- Z. J. Wang, K. Wylie and M. Marić, Macromol. React. Eng., 2017, 11, 1600029 CrossRef.
- X. Hua, S. Engelmann, G. Oehrlein, P. Jiang, P. Lazzeri, E. Iacob and M. Anderle, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2006, 24, 1850–1858 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- H. Yoshioka, K. Yamaguchi and M. Kobayashi, Polym. J., 2019, 51, 627–636 CrossRef CAS.
- H. Li, J. Liu, X. Zheng, C. Ji, Q. Mu, R. Liu and X. Liu, J. Polym. Res., 2016, 23, 1–7 CrossRef.
- N. Zhu, W. Feng, X. Hu, Z. Zhang, Z. Fang, K. Zhang, Z. Li and K. Guo, Polymer, 2016, 84, 391–397 CrossRef CAS.
- J. Wang, X. Hu, N. Zhu and K. Guo, Chem. Eng. J., 2021, 420, 127663 CrossRef CAS.
- S. Kundu, A. S. Bhangale, W. E. Wallace, K. M. Flynn, C. M. Guttman, R. A. Gross and K. L. Beers, J. Am. Chem. Soc., 2011, 133, 6006–6011 CrossRef CAS PubMed.
- Y. Kotsuchibashi and R. Narain, Polym. Chem., 2014, 5, 3061–3070 RSC.
- M. Volz, P. Walther, U. Ziener and K. Landfester, Macromol. Mater. Eng., 2007, 292, 1237–1244 CrossRef CAS.
- J. Kim, J. Choe, D. Son and M. Kim, Bull. Korean Chem. Soc., 2019, 40, 1013–1019 CrossRef CAS.
- B. G. Manders, W. Smulders, A. M. Aerdts and A. M. van Herk, Macromolecules, 1997, 30, 322–323 CrossRef CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- T. Kelen, F. Tüdös and B. Turcsányi, Polym. Bull., 1980, 2, 71–76 CrossRef CAS.
- A. L. Holmberg, M. G. Karavolias and T. H. Epps III, Polym. Chem., 2015, 6, 5728–5739 RSC.
- J. Choe, W. J. Lee, H. G. Jang, Y. Song, J. H. Sim, J. Kim, K. Paeng and M. Kim, Polym. J., 2019, 51, 449–459 CrossRef CAS.
- H.-T. Oh, S.-H. Jung, K.-H. Kim, Y. Moon, D. H. Jeong, Y. Ku, S. Lee, B.-G. Park, J. Lee, C. Koh, H.-W. Kim and J.-K. Lee, RSC Adv., 2021, 11, 1517–1523 RSC.
- G.-Q. Chen, Z.-Q. Wu, J.-R. Wu, Z.-C. Li and F.-M. Li, Macromolecules, 2000, 33, 232–234 CrossRef CAS.
- K. Ishizu, C. Takashimizu, T. Shibuya and S. Uchida, Polym. Int., 2003, 52, 1010–1015 CrossRef CAS.
- F. Yao, Q. Liu, Z. Zhang and X. Zhu, Polymers, 2017, 9, 89 CrossRef PubMed.
- K. Nishimori, M. Sawamoto and M. Ouchi, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 367–375 CrossRef CAS.
- G. Van Paesschen and D. Timmerman, Macromol. Chem. Phys., 1964, 78, 112–120 CrossRef CAS.
- R. Arshady, G. Kenner and A. Ledwith, J. Polym. Sci., Part A: Polym. Chem., 1974, 12, 2017–2025 CAS.
- J. D. Quinn and R. A. Register, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2106–2113 CrossRef CAS.
- B. Lessard, A. Graffe and M. Marić, Macromolecules, 2007, 40, 9284–9292 CrossRef CAS.
- H. Ito, C. Dalby, A. Pomerantz, M. Sherwood, R. Sato, R. Sooriyakumaran, K. Guy and G. Breyta, Macromolecules, 2000, 33, 5080–5089 CrossRef CAS.
- H. Ito, G. Breyta, D. Hofer, R. Sooriyakumaran, K. Petrillo and D. Seeger, J. Photopolym. Sci. Technol., 1994, 7, 433–447 CrossRef CAS.
- L. A. Idowu and R. A. Hutchinson, Polymers, 2019, 11, 487 CrossRef PubMed.
- K. Liang and R. A. Hutchinson, Macromolecules, 2010, 43, 6311–6320 CrossRef CAS.
- A. Matsumoto, S. Tanaka and T. Otsu, Macromolecules, 1991, 24, 4017–4024 CrossRef CAS.
- A. J. Scott, V. A. Gabriel, M. A. Dubé and A. Penlidis, Processes, 2019, 7, 444 CrossRef CAS.
- K. R. Sharma, J. Encapsulation Adsorpt. Sci., 2013, 3, 77–92 CrossRef.
- N. Kazemi, T. A. Duever and A. Penlidis, Macromol. React. Eng., 2015, 9, 228–244 CrossRef CAS.
- N. S. Pujari, M. Wang and K. E. Gonsalves, Polymer, 2017, 118, 201–214 CrossRef CAS.
- D. R. McKean, W. D. Hinsberg, T. P. Sauer, G. Willson, R. Vicari and D. J. Gordon, J. Vac. Sci. Technol., B: Microelectron. Process. Phenom., 1990, 8, 1466–1469 CrossRef CAS.
- M. Wang, K. E. Gonsalves, M. Rabinovich, W. Yueh and J. M. Roberts, J. Mater. Chem., 2007, 17, 1699–1706 RSC.
- K. Gonsalves, M. Thiyagarajan, J. Choi, P. Zimmerman, F. Cerrina, P. Nealey, V. Golovkina, J. Wallace and N. Batina, Microelectron. Eng., 2005, 77, 27–35 CrossRef CAS.
- M. Thiyagarajan, K. Dean and K. E. Consalves, J. Photopolym. Sci. Technol., 2005, 18, 737–741 CrossRef CAS.
- D. Zhou, L.-W. Zhu, B.-H. Wu, Z.-K. Xu and L.-S. Wan, Polym. Chem., 2022, 13, 300–358 RSC.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- E. Oishi, M. Takamura and T. Takahashi, Polymers, 2021, 13, 4169 CrossRef CAS PubMed.
- T. McKenzie, L. D. M. Costa, Q. Fu, D. Dunstan and G. Qiao, Polym. Chem., 2016, 7, 4246–4253 RSC.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Polym. Chem., 2011, 2, 2074–2083 RSC.
- J. M. Nasrullah, S. Raja, K. Vijayakumaran and R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 453–461 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting figures, tables, and descriptions. See DOI: https://doi.org/10.1039/d2py00542e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |