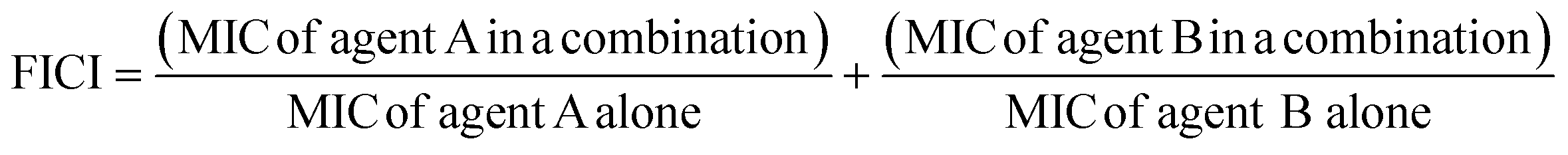DOI:
10.1039/D1MD00249J
(Research Article)
RSC Med. Chem., 2022,
13, 79-89
Discovery of FtsZ inhibitors by virtual screening as antibacterial agents and study of the inhibition mechanism†
Received
25th July 2021
, Accepted 7th November 2021
First published on 15th November 2021
Abstract
Inhibition of bacterial cell division is a novel mechanistic action in the development of new antimicrobial agents. The FtsZ protein is an important antimicrobial drug target because of its essential role in bacterial cell division. In the present study, potential inhibitors of FtsZ were identified by virtual screening followed by in vivo and in vitro bioassays. One of the candidates, Dacomitinib (S2727), shows for the first time its potent inhibitory activity against the MRSA strains. The binding mode of Dacomitinib in FtsZ was analyzed by docking, and Asp199 and Thr265 are thought to be essential residues involved in the interactions.
Introduction
In recent years, antimicrobial resistance (AMR) has received international concern because of its increasing threats to public health.1 Many conventional antibiotics have become ineffective because of the overuse of antibiotics leading to the evolution of antibiotic-resistant bacteria and superbugs.2 There is an urgent need to discover new and effective antibacterial agents to combat the AMR problem.
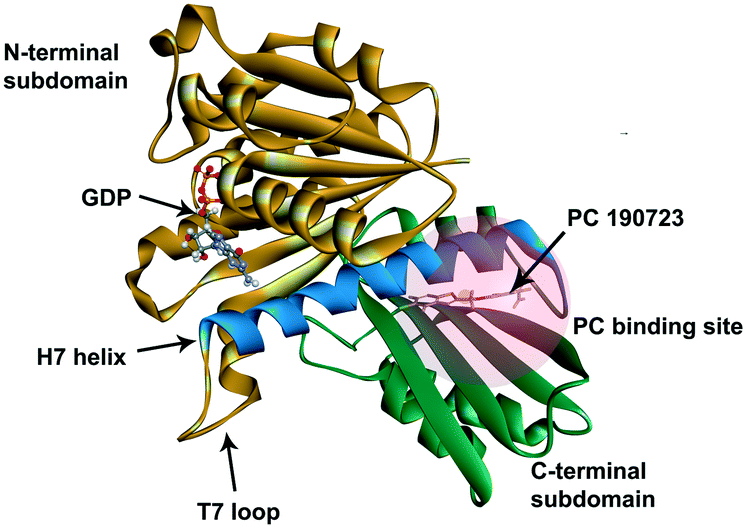
Because of the essential role of the FtsZ protein (filamentous temperature-sensitive mutant Z) in bacterial cell division, it has become an attractive target in the development of new antibiotics.3–7 Cell division, a crucial step in bacterial reproduction, is orchestrated by a large protein complex called the divisome, which localizes at the middle of the dividing cells.8,9 The assembly and functions of the divisome are dependent on FtsZ, which is the first protein recruited to the future division site.10–12 FtsZ assembles into a ring-like structure (Z-ring) at the division site, offering a platform for the positioning of the downstream proteins and controls the constriction of the cell membrane by continuous remodeling.13,14 Disturbing the cell division process will prevent bacterial reproduction, so inhibiting FtsZ is thought to be a promising approach to combat antibiotic resistance.15,16 FtsZ is considered a good target for new antibiotic discovery for the following reasons: (1) effective inhibition of the activity of FtsZ can stop the cell division and then trigger cell enlargement and subsequent lysis;15,16 (2) FtsZ is conserved in most bacteria but absent in eukaryote cells and only shares 40% to 50% sequence identity to tubulin.16,17 The crystal structure of FtsZ is available in the PDB database (PDB ID: 2VAM, 2VXY, 3VPA, 4DXD, and 5H5G), which is helpful in computer-aided drug design;18–22 (3) FtsZ has two druggable pockets: the nucleotide binding site, where GTP or GDP binds to, and the PC binding site, where a known FtsZ inhibitor, PC 190723,22 binds to (Fig. 1).16 Many researchers have made significant contributions to the developments of inhibitors of FtsZ, and several effective inhibitors have been discovered. Examples include natural products [e.g., sanguinarine23 and berberine24], synthetic GTP analogies25 and 3-methoxy benzamide derivatives [e.g., PC190723 (ref. 22)]. Nevertheless, there is still a long way to develop effective and safe FtsZ inhibitors for clinical applications.16,26
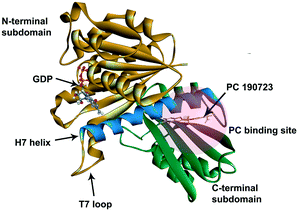 |
| | Fig. 1 The crystal structure of S. aureus FtsZ (PDB ID: 4DXD). The C-terminal domain and the N-terminal domain are in green and brown, respectively. The H7 helix is in blue. The PC binding site was marked in a semi-transparent red region. | |
Virtual screening (VS) is a powerful tool for searching hit molecules in drug discovery.27 In virtual screening, compounds in libraries are docked to the active site (the defined pocket) of the receptor in silico.28 After docking a ligand to the receptor, the best conformation of the ligand can be searched by optimizing parameters continuously and calculating the binding energy of the complex in the platform of the screening software.29 By comparing the docking scores of many molecules in the receptor, the ones with higher scores are supposed to have relatively strong affinities to the active site and they are selected for further studies.
In developing new inhibitors of FtsZ, B. subtilis and E. coli are commonly used as model bacteria for Gram-positive and Gram-negative strain respectively. B. subtilis has a rod-shaped morphology, which can be easily observed under a microscope, especially when it becomes filamentous. Therefore, the effects of the inhibitors of FtsZ on the morphologies are usually studied on B. subtilis by microscopic methods. Gram-negative strains are intrinsically more resistant to antibiotics than Gram-positive strains due to the difference in cell envelope structure of the two bacterial strains. In Gram-positive strains, the cell envelope consists of an inner cell membrane and a thick peptidoglycan layer. However, In Gram-negative strains, the envelope consists of an inner membrane, an outer membrane and a thin layer of peptidoglycan between the two membranes. The outer membrane prevents many antibiotics from penetrating into the cell, although some hydrophilic antibiotics can cross the membrane through porins.30
In the present study, virtual screening was used to discover new inhibitors of FtsZ from compound libraries. The compound Dacomitinib (S2727) was found to be a potential inhibitor of FtsZ, which was subsequently confirmed by in vivo and in vitro bioassays. Our experimental results indicate that this compound is a good antimicrobial agent against MRSA strains. In addition, the mechanical action of the compound against FtsZ was investigated.
Materials and methods
Virtual screening
Screening of inhibitors of FtsZ was carried out by Discovery Studio 2016 (BIOVIA). The crystal structure of S. aureus FtsZ (SaFtsZ) (PDB ID: 4DXD)18 retrieved from RCSB Protein Data Bank and the compound libraries (Selleck Company) were prepared using the built-in receptor and ligand preparation protocols, respectively. The hot spot, the PC binding site in FtsZ, was defined from the site recorded in the PDB file, and it was highlighted with a semi-transparent red sphere, as shown in Fig. 1. LibDock and CDOCKER are two built-in algorithms and they were for screen potential inhibitors of FtsZ. All settings in LibDock were set as default and calculated poses ranked by LibDock Scores. CDOCKER utilized a CHARMm-based molecular dynamics scheme to dock ligands into the PC binding site of FtsZ. After docking, different poses of ligands in the PC binding site were ranked by the CDOCKER energy and the CDOCKER interaction energy. The CDOCKER Energy represents the CHARMm energy, the sum of interaction energy plus the ligand strain. The CDOCKER interaction energy, usually used to compare different ligands in one protein model after molecular docking simulation, is the interaction energy alone for each final pose. A lower CDOCKER interaction energy indicates that the ligand binds more strongly to the binding site of the receptor. The compound library screened in this work was “All DMSO (Dimethyl sulfoxide) Compounds Library (Library A)” containing 313 compounds from the Selleck Company.
Antibacterial assay
Studies on the antibacterial activities of compounds were based on the CLSI (Clinical and Laboratory Standards Institute) guidelines.31 The test bacterial strains in their glycerol stocks were streak on LB (Luria-Bertani) agar plates at 37 °C overnight. A single colony from the plate was inoculated into 5 mL of MHB (Müller-Hinton Broth) (Sigma-Aldrich) or CAMHB (Cation adjusted Müller-Hinton Broth) (Sigma-Aldrich) and grown overnight at 37 °C with shaking at 250 rpm. Methicillin-resistant S. aureus (MRSA) or methicillin-sensitive S. aureus (MSSA) were inoculated into CAMHB. S. aureus BAA 41, S. aureus BAA 44, S. aureus ATCC 43300, S. aureus ATCC 33591, S. aureus ATCC 33592, and S. aureus BAA 1720 are MRSA strains. B. subtilis 168 and E. coli ATCC 25922 (E. coli) were cultured in MHB. The overnight cell culture was diluted by 10 times into a fresh medium and incubated in a shaker. After 2 hours of incubation, the absorbance of the cell culture at 600 nm (A600) was measured, and the concentration of the culture was diluted to 5 × 106 CFU mL−1. A 10 μL-portion of cells was added into each well of a 96-well microplate. All test compounds were dissolved in DMSO and two-fold serial dilutions were carried out. The concentration of DMSO in each well was maintained at 1%. After incubating the plate at 37 °C for 18 hours, the A600 of each well was recorded using a iMark™ microplate reader (Bio-rad). Each experiment was performed in duplicate. The MIC (minimum inhibitory concentration) is defined as the lowest concentration of a compound necessary to inhibit visible growth (A600 < 0.1).32
Bactericidal assay
Studies on the bactericidal activities of compounds were also based on the CLSI (Clinical and Laboratory Standards Institute) guidelines.31 The cell culture preparation and the dilution of the compounds were conducted as described in the antibacterial assay above. A 3 mL portion of the cell culture (5 × 105 CFU mL−1) was incubated with the test compounds at different concentrations or 1% DMSO as the control and cultured overnight at 37 °C with shaking at 250 rpm. After overnight incubation, the cell culture (500 μL) was diluted by 10 times or more when necessary and samples were plated onto each Trypticase Soy Agar (TSA) plate and incubated at 37 °C for 24 hours. The number of colonies that survived on the agar plates was counted. The minimum bactericidal concentration (MBC) was defined as the lowest concentration of an antibacterial agent required to reduce the viability of the initial bacterial counts by ≥ 99.9%.33 As for the time-kill assays, a 100 μL portion of cell culture was collected at different time intervals, diluted (when necessary), and spread out on the agar plate. The colonies were counted after 24 h of incubation at 37 °C.
Checkerboard assay
The synergistic effects of the test compounds with methicillin were assessed using the checkerboard assay.34,35 The test compounds were diluted in two-fold serial dilutions across a 96-well microplate, while methicillin was diluted similarly down the assay plate. Two columns of the assay plate were left for the untreated cells. 5 × 105 CFU mL−1 bacterial suspension was then added into each well to 100 μL. The whole 96-well plate was incubated at 37 °C for 16–24 hours. After incubation, the A600 value of each well was measured using the iMarkTM microplate reader.
Polymyxin B nonapeptide (PMBN) (Sigma-Aldrich, St. Louis, MO) was used in studying the antimicrobial activity of test compounds against E. coli ATCC 25922.36 The procedures for the dilution of test compounds in a 96-well plate were followed as described before. Then, PMBN was added to each well to a final concentration of 20 μg mL−1 before bacterial suspension (5 × 105 CFU mL−1) was added.
Phase-contrast microscopy
The overnight culture of B. subtilis 168 was diluted ten times into 5 mL of the fresh media and incubated for another 2 hours. The density of the cells was then adjusted to A600 = 0.01, and the cell culture was supplemented with the test compounds at different concentrations. After 4 h of incubation at 37 °C, cells were harvested, and a 1 μL portion of the resuspended cells in LB was added on a 1.2% agarose pad and then covered with a cover glass.37 The morphologies of B. subtilis 168 were observed under a TiE2 microscope (Nikon), and the cell length was measured using ImageJ.38
Z-ring visualization in B. subtilis cells
B. subtilis 168 carrying the IPTG (isopropyl b-D-1-thiogalactopyranoside) inducible green fluoresent protein labelled FtsZ (BsFtsZ-GFP) plasmid was available in our laboratory.37B. subtilis was grown in 5 mL of LB medium overnight with 30 μg mL−1 chloramphenicol at 37 °C. On the next day, the culture was back diluted to A600 = 0.01. The expression of FtsZ-GFP was then induced with 40 μM IPTG, and the cells were treated with test compounds at 37 °C with shaking for four hours. B. subtilis 168 cells were then collected by centrifugation and resuspended in PBS (phosphate-buffered saline). A 1 μL portion of cells was mounted on an agarose pad and covered with a coverslip. Those slides were observed using a Nikon super-resolution microscope (N-SIM).
GTPase activity assay
S. aureus FtsZ (SaFtsZ) was overexpressed and purified as described before.39 The GTPase activity of SaFtsZ was measured using a malachite green phosphate assay kit (BioAssay Systems) according to the protocol provided by the manufacturer. A 3 μM portion of SaFtsZ was incubated with each compound subject to serial dilutions to 63.2 μL of 50 mM Mops buffer (4-morpholinepropanesulfonic acid buffer, pH 6.5) at room temperature for 30 min. In the control group, the protein was incubated with 1%(v/v) DMSO·MgCl2 (5 mM) and KCl (200 mM) were then supplemented, and the reaction was started by the addition of 250 μM GTP. After incubation at 37 °C for 10 min, a 20 μL portion of the working reagent, provided in the kit, was added into this 96-well microplate under incubation at 37 °C for 30 min for color development. The free phosphate was quantified by measuring A600 by the microplate reader (Bio-rad).
Isothermal titration calorimetry
A MicroCal PEAQ-ITC instrument (Malvern Panalytical Company) was used to study the thermodynamic parameters in the binding between the test compounds and SaFtsZ. SaFtsZ was dialyzed against Tris-HCl buffer (20 mM Tris-HCl, 150 mM NaCl, pH 7.5), and the test ligands were dissolved in the same dialysis buffer to minimize the heat changes arising from the mismatch between the two portions of solution during the titration. The concentration of DMSO was kept at 5% in both the cell and the syringe. In a typical titration, at the time interval of every 4 seconds, a 2 μL portion of the test compound at 200 μM was titrated into the sample cell containing 20 μM SaFtsZ, and 150 seconds was then allowed to maintain a zero-temperature difference between the reference and the sample cell. The total number of injections was 19. The data were analyzed using the MicroCal PEAQ-ITC Analysis Software (Malvern Panalytical Company).
Cytotoxicity test
MTT (Invitrogen) assay was applied to test the cytotoxicity of S2727 against HFF1 and HepG2 (purchased from American Type Culture Collection, ATCC). Cells were counted and diluted to 5000 cells in 100 μL of the medium. Diluted cells were transferred into a 96-well tissue culture plate and allowed to grow overnight to adhere to the plate. Different concentrations of S2727 were added to incubate cells for up to 3 days. Then, the treated cells were mixed with 10 μL of 5 μg mL−1 water-soluble MTT regents, and the plate was incubated at 37 °C for 4 hours. Only live cells could reduce MTT into purple formazan. After incubation, acidified SDS (0.01 N HCl in 10% SDS) was added to dissolve formazan, and absorbance at 570 nm was measured by a plate reader. Cells treated with DMSO only was used as a negative control.
Results and discussion
Hit compound screening tests
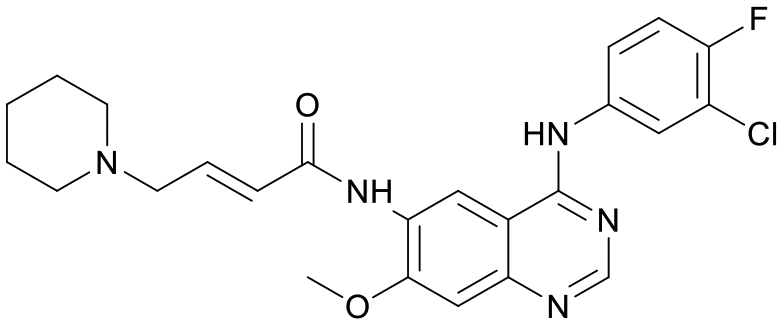
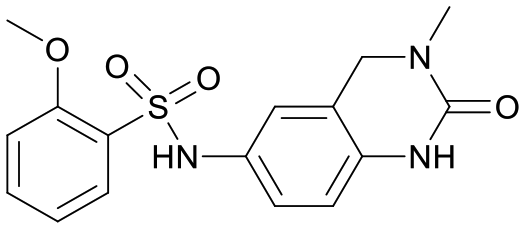
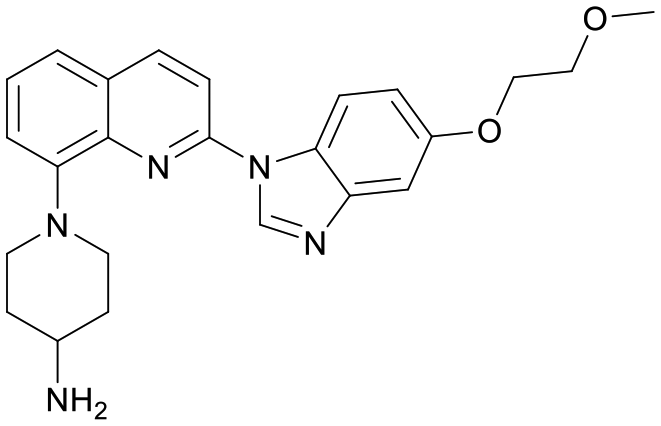
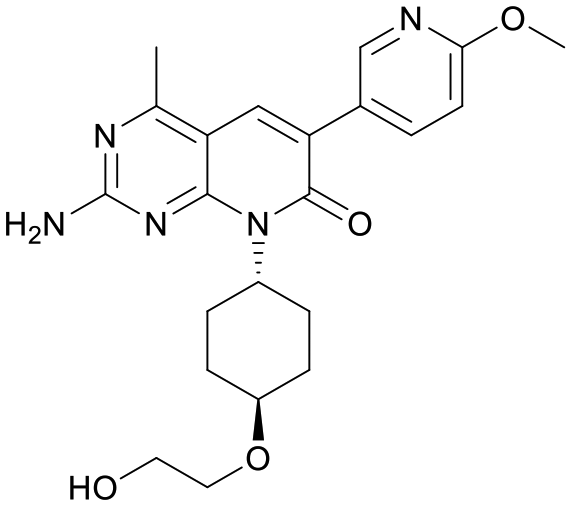
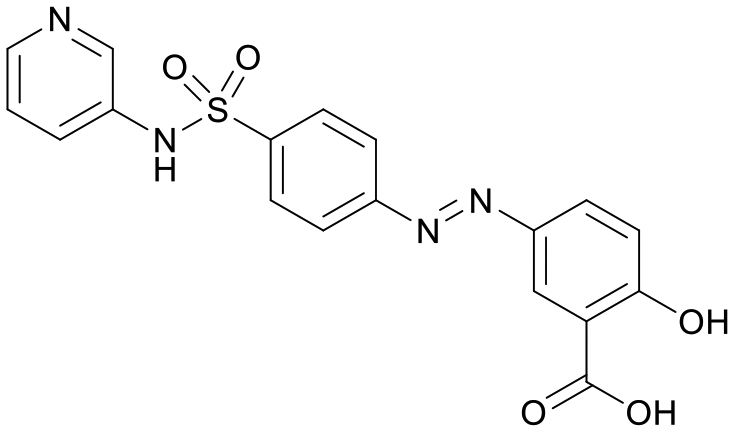
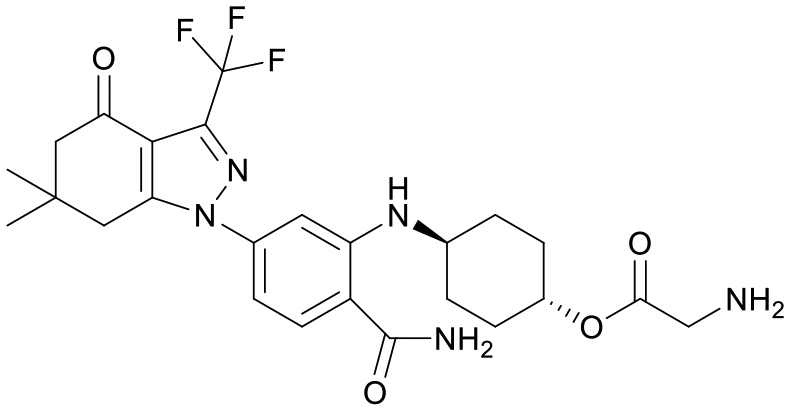
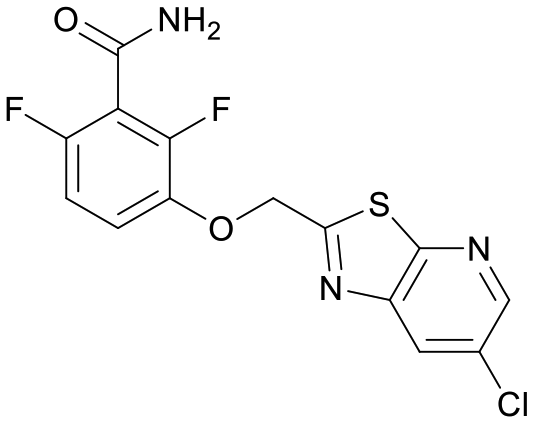
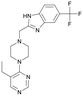
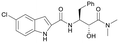
We used LibDock for the initial virtual screening and found that the LibDock score of PC190723 to SaFtsZ was 143.70, which was used as the reference for selecting potential inhibitors against FtsZ. Library A was screened in a high throughput manner; compounds with LibDock scores higher than 100 were purchased for the following bioassays (Table 1). The minimum inhibitory concentrations of the selected compounds were comprehensively evaluated against the three common bacterial strains (B. subtilis 168, S. aureus 29213, and E. coli 25922) to screen out the compounds with antimicrobial activities. Among the 10 potential inhibitors, only S2727, S2163, and S2717 were found to kill B. subtilis 168 and S. aureus 29213 (Table 2), while other compounds were unable to kill B. subtilis or S. aureus at 128 μg mL−1. S2727 killed B. subtilis 168 at 16 μg mL−1 and S. aureus 29213 at 32 μg mL−1. S2163 is a less potent inhibitor than S2727 because a higher concentration (128 μg mL−1) of S2163 was required to kill the gram-positive bacterial strains. The MIC of S2717 against B. subtilis was 16 μg mL−1, whereas the growth of S. aureus 29213 could not be inhibited even at a much higher concentration (128 μg mL−1 of S2717). PC190723 inhibited the growth of both B. subtilis 168 and S. aureus 29213 at 0.5 μg mL−1. Neither of the test compounds nor PC190723 could inhibit the growth of E. coli ATCC 25922 at a MIC less than 128 μg mL−1, presumably because of their weak permeability to the bacterial periplasm.40 The calculated CDOCKER and the CDOCKER interaction energy (Table 3) of the three compounds (S2727, S2163, and S2717) with the hot spot in the crystal structure of SaFtsZ from the molecular CDOCKER simulation indicate that all three compounds can bind with the protein. When comparing the CDOCKER interaction energies (representing the energies of the ligands) of S2727 and PC190723, which were −54.70 kcal mol−1 and −50.40 kcal mol−1, respectively, the CDOCKER interaction energy of S2727 in FtsZ is lower than that of PC190723 in the same protein. This implies that S2727 has a higher affinity with FtsZ as compared to PC190723. As for S2163 and S2717, these two compounds have less affinity with FtsZ than PC190723.
Table 1 Top 10 compounds with higher Libdock scores (over 100) and PC190723
| Compounds |
Scores |
Structures |
| S2727 |
147.29 |
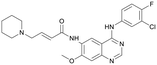
|
| S2163 |
154.63 |
|
| S2717 |
159.64 |
|
| S2777 |
133.86 |
|
| S1216 |
143.76 |
|
| S2581 |
132.75 |
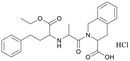
|
| S1536 |
148.22 |
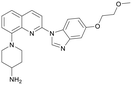
|
| S2743 |
150.75 |
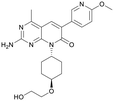
|
| S1576 |
170.06 |
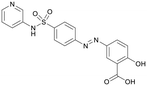
|
| S2656 |
179.38 |
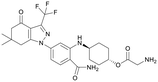
|
| PC190723 |
143.70 |
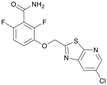
|
Table 2 Antimicrobial activities of the potential inhibitors of FtsZ. Minimum inhibitory concentrations of selected potential inhibitors against B. subtilis 168, S. aureus 29213, and E. coli 25922 are shown in this table
| Compounds |
MIC (μg/mL) |
|
B. subtilis 168 |
S. aureus 29213 |
E. coli25922 |
| S2727 |
16 |
32 |
>128 |
| S2163 |
128 |
128 |
>128 |
| S2717 |
16 |
>128 |
>128 |
| S2777 |
>128 |
>128 |
>128 |
| S1216 |
>128 |
>128 |
>128 |
| S2581 |
>128 |
>128 |
>128 |
| S1536 |
>128 |
>128 |
>128 |
| S2743 |
>128 |
>128 |
>128 |
| S1576 |
>128 |
>128 |
>128 |
| S2656 |
>128 |
>128 |
>128 |
| PC190723 |
0.5 |
0.5 |
>128 |
Table 3 The CDOCKER energies and the CDOCKER interaction energies of the potential inhibitors of FtsZ with antimicrobial activities
| Compounds |
CDOCKER energy (kcal/mol) |
CDOCKER interaction energy (kcal/mol) |
| S2727 |
−15.83 |
−54.70 |
| S2163 |
−32.29 |
−44.84 |
| S2717 |
−34.47 |
−47.39 |
| PC190723 |
−27.21 |
−50.40 |
The antimicrobial activities against the MRSA strains
The MIC values of S2727, S2163, S2717, PC190723, and methicillin against the clinically relevant bacterial strains, methicillin-resistant S. aureus strains (MRSA), are shown in Table 4. A high concentration of methicillin, in the range of 256 to 1024 μg mL−1, is required to kill the six MRSA strains. PC190723 is able to inhibit the growth of all the MRSA strains at 1–2 μg mL−1. Both S2727 and S2163 show antimicrobial activities against the six MRSA strains. The MIC values of S2727 and S2163 against the six bacterial strains are in the range of 32 to 64 μg mL−1. Because S2717 exerts no significant antimicrobial activities against the MRSA strains (except for S. aureus ATCC 43300), we did not further study this compound.
Table 4 Antimicrobial activities of the four compounds and methicillin (meth) against MRSA strains
| MRSA |
MIC (μg/mL) |
| Meth |
S2727 |
S2717 |
S2163 |
PC190723 |
| BAA 41 |
1024 |
32 |
>128 |
64 |
0.5 |
| BAA 44 |
512 |
64 |
>128 |
64 |
1 |
| ATCC 43300 |
256 |
32 |
32 |
32 |
1 |
| ATCC 33591 |
512 |
64 |
>128 |
64 |
1 |
| ATCC 33592 |
256 |
64 |
>128 |
64 |
2 |
| BAA 1720 |
256 |
64 |
>128 |
64 |
1 |
The combined effects of inhibitors and PMBN against E. coli
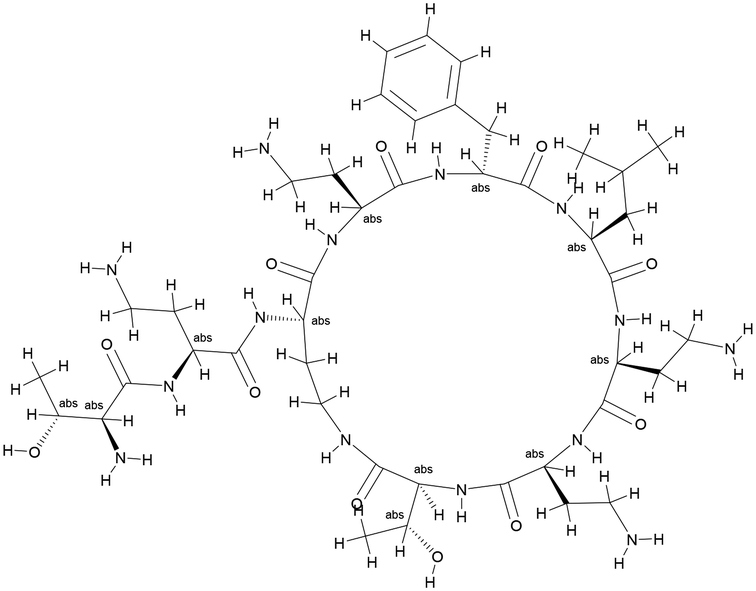
Although FtsZ is highly conserved in both Gram-negative and Gram-positive bacteria, neither our potential FtsZ inhibitors nor PC190723 show significant antimicrobial activities against E. coli, presumably due to the outer membrane of gram-negative bacterial strains effectively hindering the penetration of these compounds.41 Polymyxin B is an antibiotic isolated from Bacillus polymyxa36 and its derivative PMBN (polymyxin B nonapeptide) (Fig. 2) can enhance the permeability of the outer membrane of Gram-negative strains.36 Antimicrobial activities of these potential inhibitors of FtsZ supplemented with PMBN on E. coli are shown in Table 5. Results show that neither PMBN nor the potential inhibitors can kill E. coli when they are tested separately. However, when 20 μg mL−1 PMBN was supplemented, S2163 killed E. coli at 64 μg mL−1, while S2727 and PC190723 killed E. coli at 32 μg mL−1. These results support the hypothesis mentioned above that the reduced membrane penetration may be a reason that the selected inhibitors have no antimicrobial activities against the Gram-negative strain, E. coli 25922.
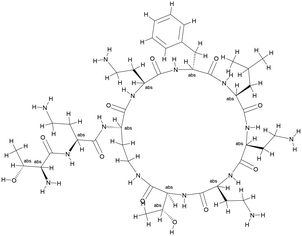 |
| | Fig. 2 The structure of polymyxin B nonapeptide (PMBN). | |
Table 5 Antimicrobial activities of the inhibitors of FtsZ against E. coli. The synergistic effects of S2727, S2163, and PC190723 with Polymyxin B nonapeptide (PMBN) against E. coli 25922. The concentration of PMBN is 20 μg mL−1
| Compounds |
MIC (μg/mL) |
| −PMBN |
+PMBN |
| PMBN |
>256 |
— |
| S2727 |
>128 |
32 |
| S2163 |
>128 |
64 |
| PC190723 |
>128 |
32 |
Synergistic effects with methicillin
The fractional inhibitory concentration indexes (FICIs) of S2727, S2163 or PC190723 with methicillin were measured to study the synergistic effects between the three compounds and methicillin (Table 6). The FICI is calculated using the formula shown below:42
FICI <0.5 indicates a strong synergistic effect between the two test agents. When the FICI is in the range of 0.5–1, the two agents are likely to have partial synergistic effects. If the index is >1, there are additive effects between the two test agents.43 The FICIs of the combined use of S2727 or PC190723 with methicillin are 0.50 and 0.52, respectively, indicating that both S2727 and PC190723 have partial synergistic effects with methicillin. The FICI of S2163 with methicillin is >1. Therefore, there is no synergistic effect between S2163 with methicillin. The partial synergistic effect may be caused by the fact that both the target of S2727 (or PC190723) (i.e., FtsZ) and that of methicillin (i.e., penicillin-binding proteins) participate in the process of cell divisions. In the process of the cell division, the first step requires the participation of FtsZ and the final step is the synthesis of bacterial cell walls, which involves the biosynthesis of peptidoglycans.44 Inhibition of the function of FtsZ is likely to exert a weakening effect on the cell wall synthesis since the Z-ring functions as a platform for the recruitment of downstream proteins.
Table 6 Synergistic effects of S2727, S2163 and PC190723 with methicillin (meth) against S. aureus BAA 41
| Compounds |
S. aureus BAA 41 |
| Meth alone |
1024 |
| S2727 alone |
128 |
| S2727 in a combination |
64 |
| Meth in a combination |
4 |
| FICI |
0.50 |
| S2163 alone |
128 |
| S2163 in a combination |
128 |
| Meth in a combination |
1 |
| FICI |
>1 |
| PC190723 alone |
1 |
| PC190723 in combination |
0.5 |
| Meth in combination |
4 |
| FICI |
0.52 |
Effects on cell morphology of B. subtilis
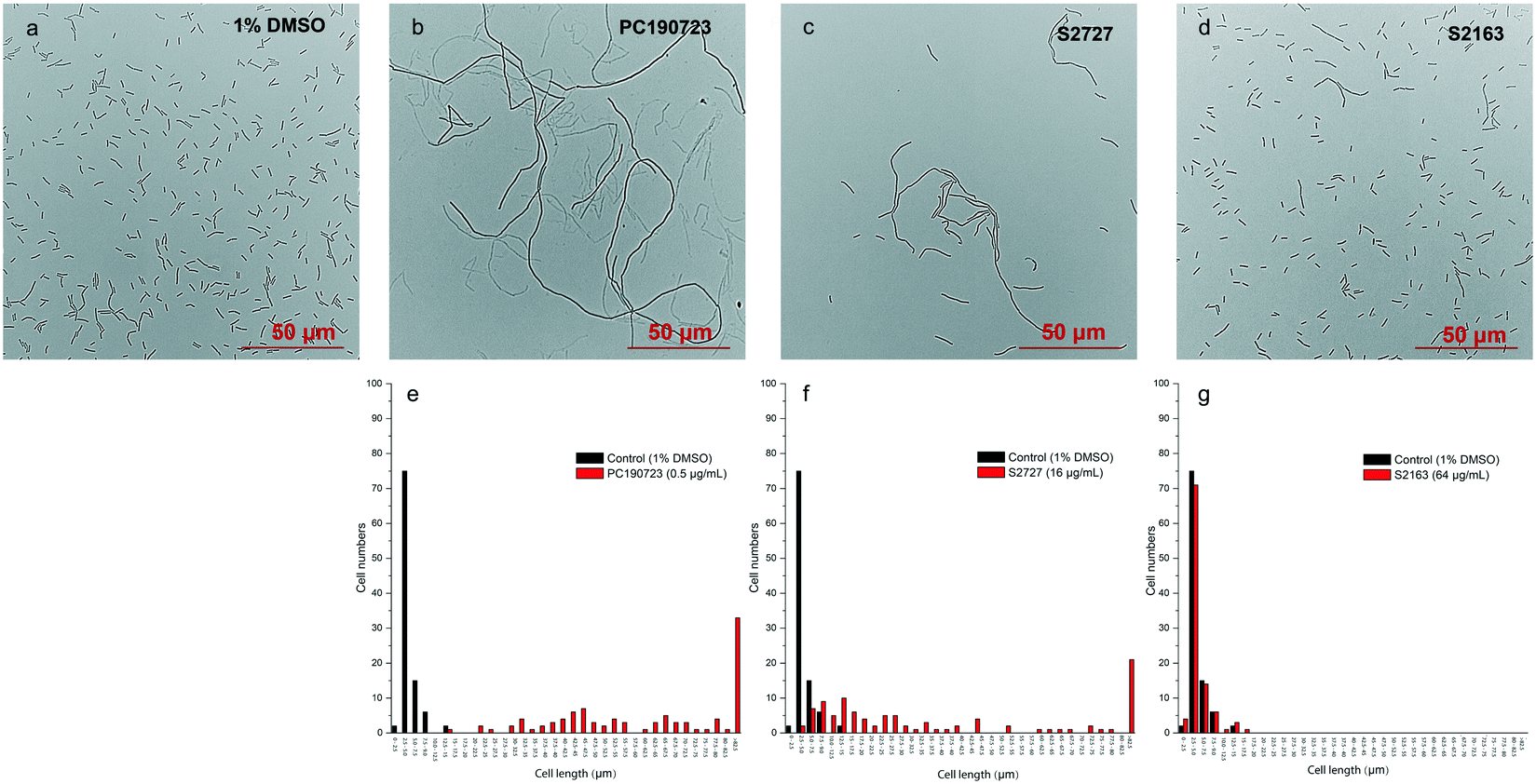
Inhibitors of FtsZ can disrupt the functions of FtsZ in cell division, disturbing the formation of Z-ring and then lead to filamentous cells. Such cells can be easily observed by microscopy.22 The effects of S2727, S2163, and PC190723 on the cell morphology of B. subtilis 168 are shown in Fig. 3. When B. subtilis 168 was cultured with 1%(v/v) DMSO only (Fig. 3a), the cells showed the typical rod and short morphologies. However, the cells were elongated after treating with 0.5 μg mL−1 PC190723 (Fig. 3b) or 16 μg mL−1 S2727 (Fig. 3c) for 4 h. However, the elongation of B. subtilis cells triggered by S2163 at 64 μg mL−1 was not obvious (Fig. 3d). In order to quantify the elongation effects caused by the potential inhibitors, the cell lengths of 100 cells removed from the cell culture treated with DMSO or the potential inhibitors of FtsZ were measured using ImageJ, and the distributions of cell lengths were plotted. In the control group [1%(v/v) DMSO], the lengths of 75% of the cells are between 2.5 and 5 μm, and the average length is 4.5 μm. After treating with PC190723 (Fig. 3e), the distribution of cell lengths is close to a normal distribution and 33% of cells are elongated to more than 82.5 μm with an average length of 135.8 μm, which is almost 30-fold longer than the normal cells. S2727 also induces cell elongation slightly (Fig. 3f), and the average cell lengths are 51.6 μm (around 12-fold longer). However, the distribution of the bacterial cells treated with S2163 is similar to that of the bacterial cells treated with DMSO only, and this result is consistent with that shown in Fig. 3g. Considering the fact that the elongation of B. subtilis after treated with S2163 was hardly seen, S2163 appears to have little effect on FtsZ because the filamentous morphologies are the major effect caused by the inhibitors of FtsZ. The elongation of the bacterial cells after treating with S2727 indicates that S2727 is likely to inhibit bacterial divisions by disrupting the function of FtsZ.
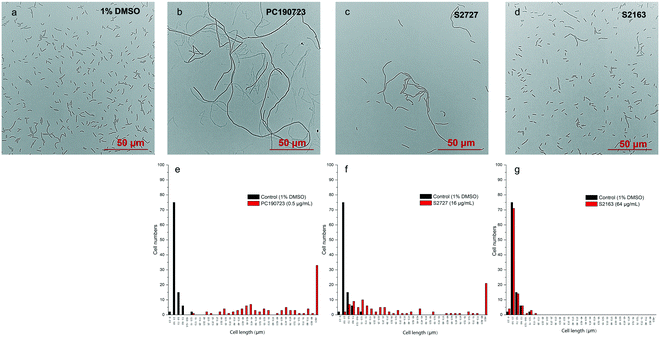 |
| | Fig. 3 The effects of the potential inhibitors of FtsZ on B. subtilis 168 cell morphologies. B. subtilis 168 were treated with a) 1%(v/v) DMSO; b) 0.5 μg mL−1 PC190723; c) 16 μg mL−1 S2727; d) 64 μg mL−1 S2163. The photos were taken under a phase-contrast microscope. Comparison of the cell length distributions of the total 100 cells treated by 1%(v/v) DMSO and the potential inhibitors were analysed and plotted using ImageJ: 1%(v/v) DMSO with e) 0.5 μg mL−1 PC190723 or f) 16 μg mL−1 S2727 or g) 64 μg mL−1 S2163. Scale bar = 50 μm. | |
Z-ring formation
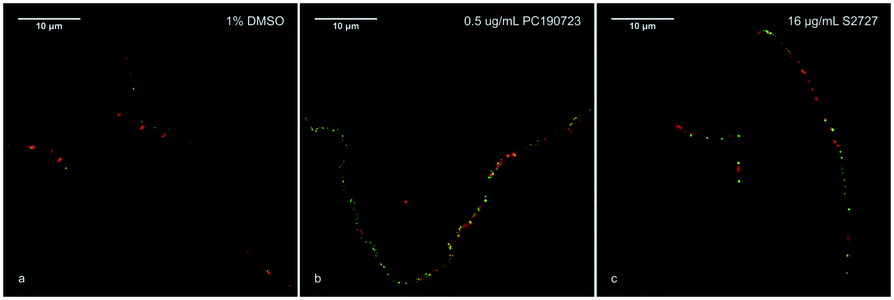
According to previous studies, inhibitors of FtsZ can delocalize the assembly of FtsZ.22,45 To further confirm the target of S2727 is FtsZ, the effects of S2727 on the assembly of FtsZ-GFP in B. subtilis were observed using a super-resolution microscope (Nikon). In bacterial cells treated with 1%(v/v) DMSO, a green band or a condensed spot located at the division site, where the cell membrane was also formed and this was indicated by the red fluorescence signal, while unpolymerized FtsZ cannot be found at other positions (Fig. 4a). With PC190723 as the positive control, in those cells treated with 0.125 μg mL−1 PC190723 or 8 μg mL−1 S2727, FtsZ-GFP complexes cannot assemble into a Z-ring in the middle of cells and are dispersed in the elongated cells (Fig. 4b and c). Thus, both PC190723 and S2727 can delocalize the FtsZ-GFP complex and prevent the monomers of FtsZ from forming the platform for the cell division.
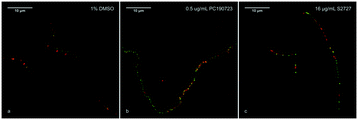 |
| | Fig. 4 The effects of the potential inhibitors of FtsZ on the Z-ring formation. B. subtilis cells were treated with a) 1%(v/v) DSMO; b) 0.5 μg mL−1 PC190723; c) 16 μg mL−1 S2727. The cell membrane was stained with the membrane dye FM 5-96 (red). Scale bar = 10 μm. | |
Time-kill kinetic assays and MBC tests
The bactericidal activities of S2727 and PC190723 against S. aureus BAA 41 (over 24 h) were determined by the time-kill kinetic assay (Fig. 5). When the bacterial cells were treated with PC190723 at 1 × MIC or 2 × MIC, cells were just inhibited and can grow when they were spread on agar plates. When cells were treated with 4 × MIC and 8 × MIC, the starting log(CFU mL−1) was reduced to nearly 0 after 24 h of treatment. S2727 exhibits bactericidal activity against BAA 41 at 256 μg mL−1 (8 × MIC) after 4 h of treatment. At 1, 2 and 4 × MIC of S2727, the bacterial cells were also recovered from the execrable living environment. The minimal bactericidal concentrations (MBC) of PC190723 and S2727 against BAA 41 were 2 μg mL−1 (4 × MIC) and 256 μg mL−1 (8 × MIC), respectively, because no observable colonies could be found on agar plates. A ratio of MBC and MIC over 32 demonstrates that the test bacteria is tolerant to the test compound.46 The ratios of PC190723 and S2727 against BAA 41 are 2 and 8, respectively. Consequently, BAA 41 is sensitive to PC190723 and S2727, and their antibacterial activities against methicillin resistant strains are consistent with their bactericidal modes of action.
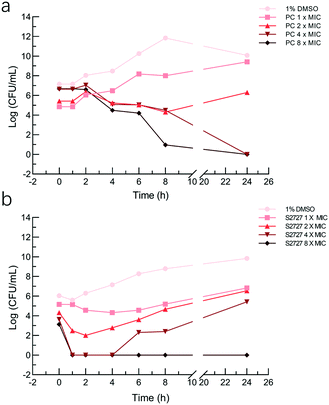 |
| | Fig. 5 Time-kill curves of S. aureus BAA41 after treating with PC190723 and S2727. Cells treated with 1%(v/v) DMSO served as the control. a) Cells were treated with PC190723 at 2 × MIC and 8 × MIC. b) Cells were treated with S2727 at 2 × MIC, 4 × MIC, and 8 × MIC, respectively. The MIC values of PC190723 and S2727 against S. aureus BAA 41 were 1 μg mL−1 and 32 μg mL−1, respectively. | |
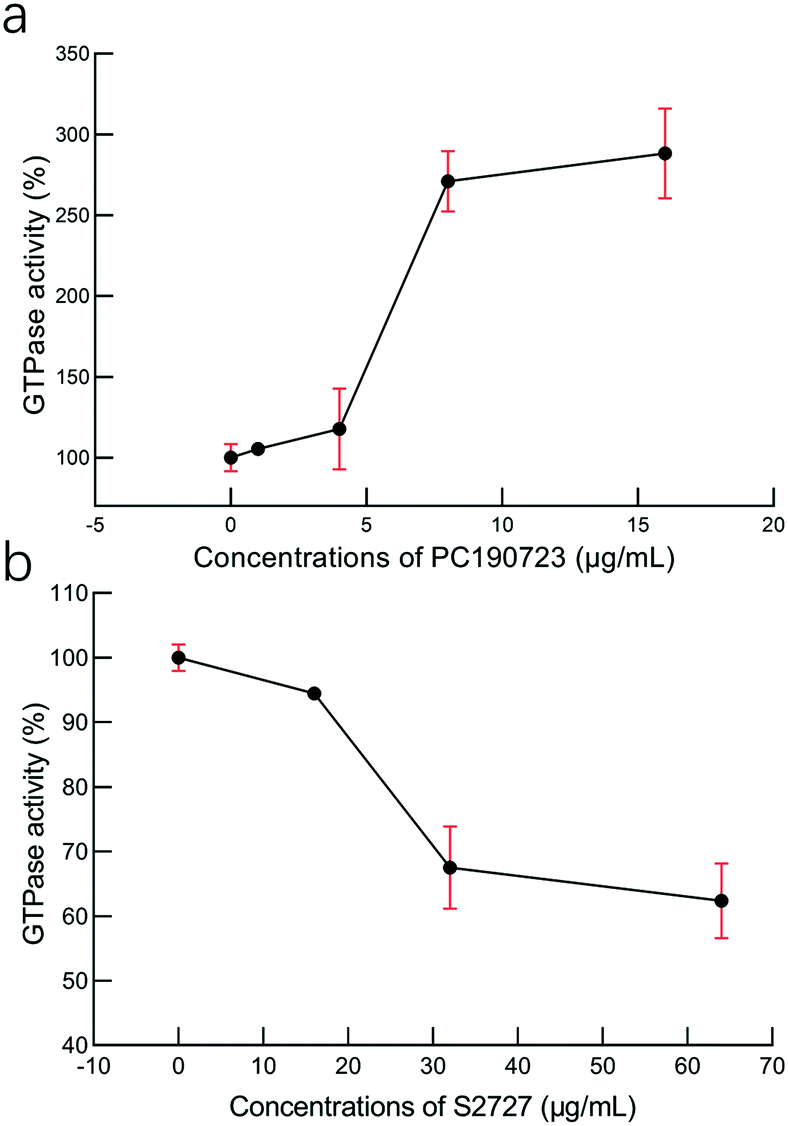
Effects on the GTPase activity
To study the interactions between S2727 and SaFtsZ, effects of S2727 and PC190723 (as the control) on the GTPase activity of SaFtsZ were measured, and changes in the GTPase activity of SaFtsZ were plotted (Fig. 6). We found that PC190723 enhances the GTPase activity in a dose-dependent manner, which is consistent with the result of a previously published work47 as well as documents supplied by Merck, which indicates that this compound is an activator of GTPase activity.48 When SaFtsZ was treated with PC190723 at 1 μg mL−1, its GTPase activity increases to 105.4 ± 3.5%, as compared to the GTPase activity treated with DMSO alone (100 ± 8.3%). 8 and 16 μg mL−1 PC190723 enhances the GTPase activity to 271.1 ± 18.8% and 288.2 ± 27.7%, respectively. However, at 64 μg mL−1, S2727 decreases the GTPase activity of SaFtsZ to 63 ± 6%. Apparently, S2727 exhibits an opposite effect on GTPase activity against SaFtsZ, as compared to PC190723, although its inhibitory effect is weaker. Both suppression and enhancement on the GTPase activities of SaFtsZ are regarded as inhibition of FtsZ activity. This is because polymerization of FtsZ mediated by GTP is a dynamic process. When the GTPase activity of FtsZ is enhanced by PC 190723, the transition slope from monomer to polymer is decreased and then the assembly cooperativity of FtsZ is reduced.47 When the GTPase activity of FtsZ is inhibited by S2727, the polymerization rate of FtsZ is reduced. Therefore, both decreasing and increasing the GTPase activity of FtsZ would disturb the dynamic process and then inhibit the cell division process.
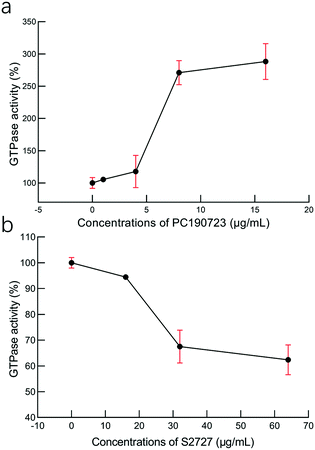 |
| | Fig. 6 The effects of (a) PC190723 and (b) S2727 on the GTPase activity of S. aureus FtsZ. The error bars are shown in red (n = 3 measurements). | |
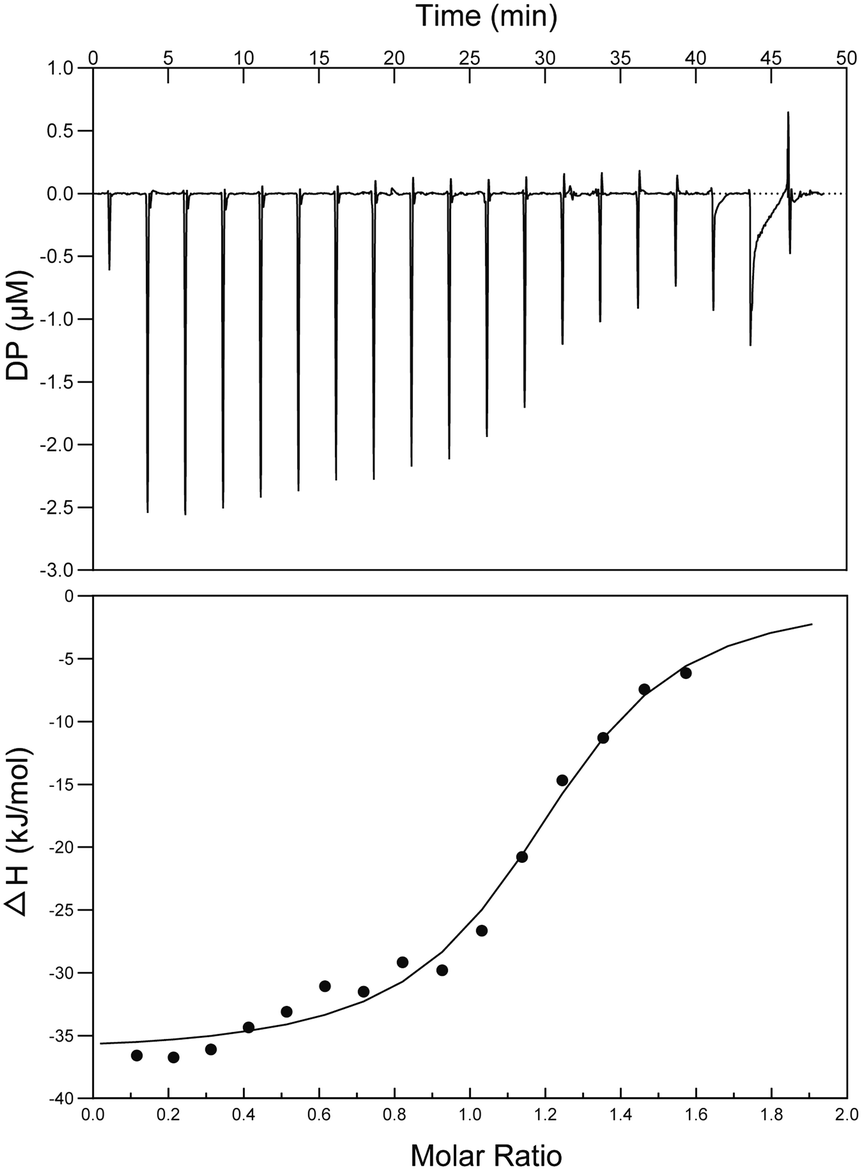
Binding affinity studies
Interactions between S2727 and SaFtsZ were characterized by isothermal titration calorimetry (ITC) (Fig. 7). When analyzing the ITC data, ‘one set of sites’ was selected as the fitting model. The dissociation constant (Kd) was determined by an analyzing software (the MicroCal PEAQ-ITC Analysis Software). The dissociation constants (Kd) S2727 with SaFtsZ were 0.6 ± 0.11 μM. The binding between S2727 and SaFtsZ is demonstrated.
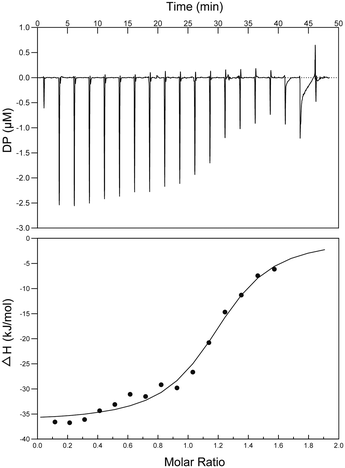 |
| | Fig. 7 ITC titration curve of SaFtsZ with S2727. The top picture is the raw ITC signals resulting fromS 2727 binding to SaFtsZ in 20 mM Tris-HCl, 150 mM NaCl, pH 7.5, at 25 °C. The curve in the bottom picture was the integrated ITC signals of S2727 binding to SaFtsZ in the same buffer. The dissociation constants (Kd) S2727 with SaFtsZ were 0.6 ± 0.11 μM. | |
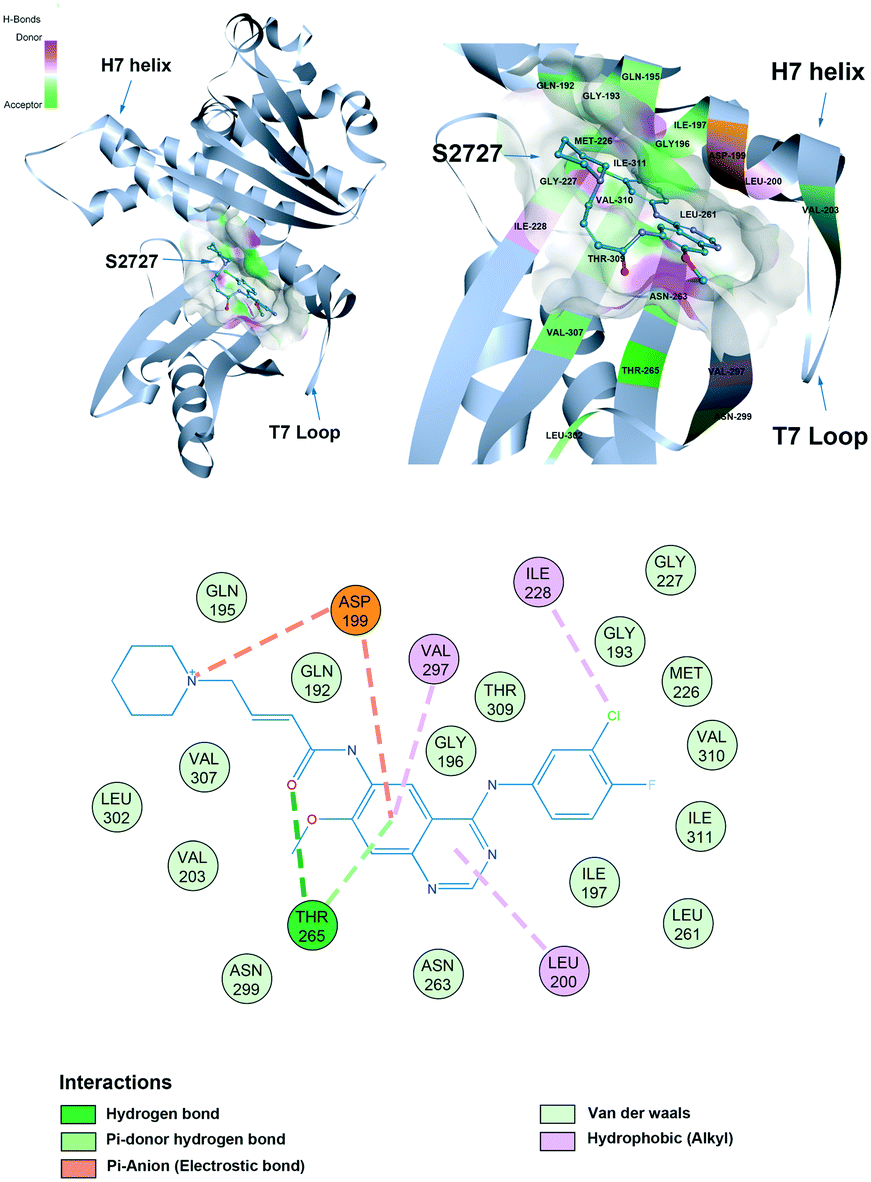
Proposed binding mode of S2727 in FtsZ
The proposed binding mode of S2727 in S. aureus FtsZ (Fig. 8) and that of PC190723 in S. aureus FtsZ (ESI,†Fig. 1) are compared to investigate whether there is difference in the binding mode of the two inhibitors.
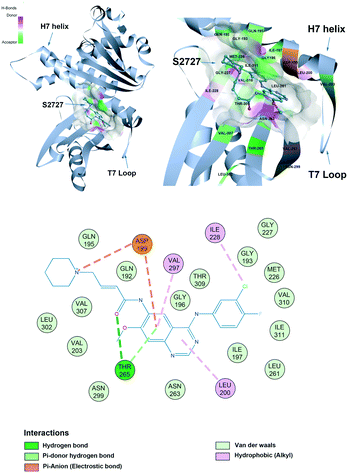 |
| | Fig. 8 The proposed binding mode of S2727 in the hot spot of S. aureus FtsZ (PDB ID: 4DXD). The mode was built using discovery studio (DS) by docking S2727 to the same pocket of PC190723, which was a cleft below helix 7 and surrounded by a T7 loop with four parallel β-strands. The interactions between S2727 and the amino acids involved in the binding site are shown in a 2-dimensional format. Colors of balls and dash lines represent different interactions between amino acids and S2727. | |
The binding mode of PC190723 in FtsZ presented by DS was in accordance with the crystal structure (PDB ID: 4DXD). PC190723 consists of two chemical groups: benzamide and triazolopyridine. Both groups have essential interactions with FtsZ. The amid hydrogens on the benzamide form hydrogen bonds with the carbonyl group on the side chain of Asn263 and the carbonyl group on the main chain of Val207. The amide group on the main chain of Leu209 also stabilize PC190723 by forming hydrogen bonds. The 2-fluoro groups on the benzamide moiety is located in a hydrophobic core formed by Leu209 and Leu200, while the hydrophobic environment for the 6-fluoro group is formed by Val203 with Val297. Another moiety of PC190723, triazolopyridine, is buried in the hydrophobic cleft between the Helix 7 and the C-terminal subdomain; Gly193, Gly196, Ile197, Ile224, Thr309, Ile311, and Met226 are essential for maintaining this hydrophobic environment for the stabilization of PC190723 in FtsZ.
S2727 has three groups: a piperidine group, a quinazoline group, and a benzene group. In the proposed binding model with SaFtsZ (Fig. 8), the benzene group is inserted into the pocket and surrounded by a hydrophobic environment formed by Gly193, Met226, Ile197, Ile228, Val310, and Ile311. The quinazoline group resides in another hydrophobic core which is formed by Leu200, Val203, Leu209, Asn263, Val297, and Asn299. Asp199 and Thr265 stabilize the position of S2727 by forming hydrogen bonds with the linker between the piperidine group and the quinazoline group. Thr265 and are essential for the binding of S2727 to FtsZ. Asp199 can form π-anion interactions with the quinazoline group and a salt bridge with the piperidine group. Thr265 can form a hydrogen bond with the carbonyl of the linker of S2727.
To summarize, in the crystal structure of SaFtsZ, Val207, Leu209, and Asn263 play essential roles in stabilizing PC190723 in the pocket of SaFtsZ (ESI,†Fig. 1). In the proposed binding model of S2727 with SaFtsZ, Thr265 and Asp199 are more likely to be crucial. Other amino acids, albeit with weaker interactions, may also be necessary for stabilizing the compounds in the pocket. For example, a previous study showed that when Gly196 in FtsZ was mutated to Ser196, bacteria developed resistance to PC190723; Gly196 could only form a carbon-hydrogen bond with PC190723, which is considered to be a weak hydrogen bond,18 but the G196S mutation also changed the spatial structure of the PC pocket.
Therefore, although S2727 was designed to bind to the PC pocket, the binding modes of the two compounds in the hot spot are still expected to be different.
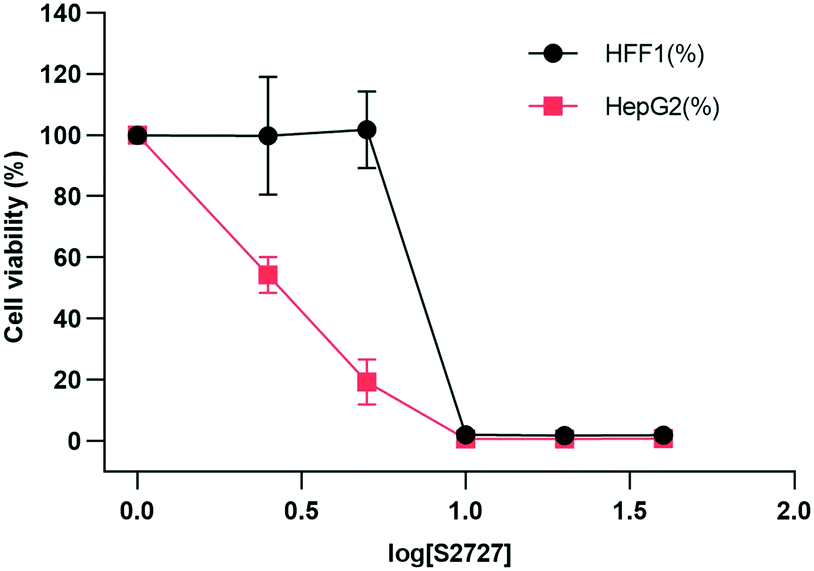
Cytotoxicity
The cytotoxicity of S2727 on both human skin cells and liver cancer cells is tested (Fig. 9). The viabilities of cells after treatment with S2727 were measured and calculated, and their relationships with S2727 were fitted with the log(agonist) vs. response functions in GraphPad Prism.
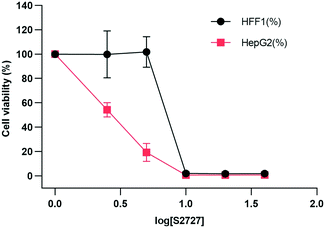 |
| | Fig. 9 Toxicity of S2727 over human skin cells and liver cancer cells. HFF1 is the human skin cell, and HepG2 is the liver cancer cells, which are shown in black and red, respectively. | |
When a human skin cell line is treated with 8.4 μg mL−1 S2727, 50% of cells are killed. When liver cancer cells are treated with S2727, the IC50 is 2.45 μg mL−1. The minimum inhibitory concentration of S2727 is 16 μg mL−1 against B. subtilis 168 and 32–64 μg mL−1 against S. aureus strains. S2727 is quite toxic to human cells. This is not surprising as Dacomitinib is an inhibitor of epidermal growth factor receptor and is used to treat non-small-cell lung cancer.49 Reducing its cytotoxicity is necessary if this compound is to be used as a template for further modification.
Conclusions
To summarize, results from in vivo and in vitro studies demonstrate that S2727 can serve as an inhibitor against FtsZ. Data from the in vivo study reveals that S2727 possesses antimicrobial activities against B. subtilis, MSSA, and MRSA strains. When using PMBN to increase the permeability of the outer membrane of the gram-negative strain, both S2727 and PC190723 exert inhibitory effects on the growth of E. coli. In addition, S2727 causes B. subtilis cells to be elongate and FtsZ to be delocalized as PC190723 does. These suggest that S2727 is an effective inhibitor towards FtsZ. In vitro studies on the GTPase activity assays and the ITC tests have revealed the binding of S2727 to SaFtsZ. Based on the proposed binding mode of S2727 in FtsZ, S2727 is likely to reside in the PC binding pocket formed by the H7 helix and four parallel β-strands; Asp199 and Thr265 are expected to interact with S2727 through hydrogen bond formations and π-anion interactions.
Comparing the antimicrobial potency, inhibition effect, and binding affinity of S2727 with PC190723 towards FtsZ protein, increasing the water solubility of S2727 may enhance its antimicrobial activity. In the proposed binding mode of S2727 with FtsZ, Asp199 and Thr265 are essential for contributing a high binding affinity of S2727 to FtsZ. Therefore, maintaining the interactions of S2727 with these two amino acids is of great importance. Further development of antibacterial agents based on the scaffold of S2727 should also consider its cytotoxicity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Innovation and Technology Commission, the Ministry of Science and Technology of China (BBXZ), and the Research Committee of The Hong Kong Polytechnic University. We thank the support of the University Research Facilities on Life Sciences and Chemical and Environmental Analysis of The Hong Kong Polytechnic University. KYW acknowledged the support from the Patrick S. C. Poon endowed professorship.
References
- C. Llor and L. Bjerrum, Ther. Adv. Drug Saf., 2014, 5, 229–241 CrossRef PubMed.
- R. Laxminarayan, D. Sridhar, M. Blaser, M. Wang and M. Woolhouse, Science, 2016, 353, 874–875 CrossRef CAS.
- D. Awasthi, K. Kumar and I. Ojima, Expert Opin. Ther. Pat., 2011, 21, 657–679 CrossRef CAS PubMed.
- X. Li and S. Ma, Eur. J. Med. Chem., 2015, 95, 1–15 CrossRef.
- C. Schaffner-Barbero, M. Martín-Fontecha, P. Chacón and J. M. Andreu, ACS Chem. Biol., 2012, 7, 269–277 CrossRef CAS.
- A. M. Veselinović, A. Toropov, A. Toropova, D. S. Đorđević and J. B. Veselinović, New J. Chem., 2018, 42, 10976–10982 RSC.
- J. Wang, A. Galgoci, S. Kodali, K. B. Herath, H. Jayasuriya, K. Dorso, F. Vicente, A. González, D. Cully and D. Bramhill, J. Biol. Chem., 2003, 278, 44424–44428 CrossRef CAS.
- S. Du and J. Lutkenhaus, Mol. Microbiol., 2017, 105, 177–187 CrossRef CAS PubMed.
- A. W. Bisson-Filho, Y. P. Hsu, G. R. Squyres, E. Kuru, F. Wu, C. Jukes, Y. Sun, C. Dekker, S. Holden, M. S. V. Nieuwenhze, Y. V. Brun and E. C. Garner, Science, 2017, 355, 739–743 CrossRef CAS PubMed.
- P. Gamba, J.-W. Veening, N. J. Saunders, L. W. Hamoen and R. A. Daniel, J. Bacteriol., 2009, 191, 4186–4194 CrossRef CAS PubMed.
- C. Ortiz, P. Natale, L. Cueto and M. Vicente, FEMS Microbiol. Rev., 2016, 40, 57–67 CrossRef CAS PubMed.
- W. Margolin, Nat. Rev. Mol. Cell Biol., 2005, 6, 862–871 CrossRef CAS PubMed.
- L. Romberg and P. A. Levin, Annu. Rev. Microbiol., 2003, 57, 125–154 CrossRef CAS PubMed.
- S. G. Addinall and J. Lutkenhaus, J. Bacteriol., 1996, 178, 7167–7172 CrossRef CAS PubMed.
- R. L. Lock and E. J. Harry, Nat. Rev. Drug Discovery, 2008, 7, 324–338 CrossRef CAS PubMed.
- K. D. Kusuma, M. Payne, A. T. Ung, A. L. Bottomley and E. J. Harry, ACS Infect. Dis., 2019, 5, 1279–1294 CrossRef CAS PubMed.
- R. Mahendran, F. J. Jenifer, M. Palanimuthu and S. Subasri, Indian J. Sci. Technol., 2011, 4, 141–146 CrossRef CAS.
- C. M. Tan, A. G. Therien, J. Lu, S. H. Lee, A. Caron, C. J. Gill, C. L. Jacob, L. B. Perdomo, J. M. Monteiro, P. M. Pereira, N. L. Elsen, J. Wu, K. Deschamps, M. Petcu, S. Wong, E. Daigneault, S. Kramer, L. Z. Liang, E. Maxwell, D. Claveau, J. Vaillancourt, K. Skorey, J. Tam, H. Wang, T. C. Meredith, S. Sillaots, L. W. Jarantow, Y. Ramtohul, E. Langlois, F. Landry, J. C. Reid, G. Parthasarathy, S. Sharma, A. Baryshnikova, K. J. Lumb, M. G. Pinho, S. M. Soisson and T. Roemer, Sci. Transl. Med., 2012, 4, 126ra135 Search PubMed.
- M. A. Oliva, D. Trambaiolo and J. Löwe, J. Mol. Biol., 2007, 373, 1229–1242 CrossRef CAS.
- T. Matsui, J. Yamane, N. Mogi, H. Yamaguchi, H. Takemoto, M. Yao and I. Tanaka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1175–1188 CrossRef CAS PubMed.
- J. Fujita, R. Harada, Y. Maeda, Y. Saito, E. Mizohata, T. Inoue, Y. Shigeta and H. Matsumura, J. Struct. Biol., 2017, 198, 65–73 CrossRef PubMed.
- D. J. Haydon, N. R. Stokes, R. Ure, G. Galbraith, J. M. Bennett, D. R. Brown, P. J. Baker, V. V. Barynin, D. W. Rice, S. E. Sedelnikova, J. R. Heal, J. M. Sheridan, S. T. Aiwale, P. K. Chauhan, A. Srivastava, A. Taneja, I. Collins, J. Errington and L. G. Czaplewski, Science, 2008, 321, 1673–1675 CrossRef CAS.
- T. K. Beuria, M. K. Santra and D. Panda, Biochemistry, 2005, 44, 16584–16593 CrossRef CAS PubMed.
- P. N. Domadia, A. Bhunia, J. Sivaraman, S. Swarup and D. Dasgupta, Biochemistry, 2008, 47, 3225–3234 CrossRef CAS.
- T. Läppchen, A. F. Hartog, V. A. Pinas, G. J. Koomen and T. D. Blaauwen, Biochemistry, 2005, 44, 7879–7884 CrossRef.
-
R. Khan and S. Ray, Phytochemistry: An in-silico and in-vitro Update, Springer, New York City, 2019, Chater 7, pp. 109–132 Search PubMed.
- G. Schneider, Nat. Rev. Drug Discovery, 2010, 9, 273–276 CrossRef CAS.
- B. K. Shoichet, Nature, 2004, 432, 862–865 CrossRef CAS PubMed.
- E. Lionta, G. Spyrou, D. K. Vassilatis and Z. Cournia, Curr. Top. Med. Chem., 2014, 14, 1923–1938 CrossRef CAS PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS.
-
CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard. M7-A6, Wayne, Pensylvania, USA, 6th ed., 2003, vol. 23 Search PubMed.
- M. Loose, K. G. Naber, A. Coates, F. M. E. Wagenlehner and Y. Hu, Front. Microbiol., 2020, 11, 54 CrossRef PubMed.
- P. C. Taylor, F. D. Schoenknecht, J. C. Sherris and E. C. Linner, Antimicrob. Agents Chemother., 1983, 23, 142–150 CrossRef CAS PubMed.
- G. P. Wormser and Y. W. Tang, Clin. Infect. Dis., 2005, 41, 577–577 CrossRef.
- D. Amsterdam, Clin. Infect. Dis., 2015, 60, 1146–1147 Search PubMed.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, J. Med. Chem., 2000, 43, 3085–3092 CrossRef CAS.
- N. Sun, F. Y. Chan, Y. J. Lu, M. A. C. Neves, H. K. Lui, Y. Wang, K. Y. Chow, K. F. Chan, S. C. Yan, Y. C. Leung, R. Abagyan, T. H. Chan and K. Y. Wong, PLoS One, 2014, 9, e97514 CrossRef.
-
C. Rueden, C. Dietz, M. Horn, J. Schindelin, B. Northan, M. Berthold and K. Eliceiri, ImageJ Ops [Software], 2021, https://imagej.net/Ops Search PubMed.
- F. Y. Chan, N. Sun, M. A. C. Neves, P. C. H. Lam, W. H. Chung, L. K. Wong, H. Y. Chow, D. L. Ma, P. H. Chan, Y. C. Leung, T. H. Chan, R. Abagyan and K. Y. Wong, J. Chem. Inf. Model., 2013, 53, 2131–2140 CrossRef CAS PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, 2009, 1794, 808–816 CrossRef CAS.
- K. Haranahalli, S. Tong and I. Ojima, Bioorg. Med. Chem., 2016, 24, 6354–6369 CrossRef CAS PubMed.
- J. Meletiadis, S. Pournaras, E. Roilides and T. J. Walsh, Antimicrob. Agents Chemother., 2010, 54, 602–609 CrossRef CAS.
- Y. Sakagami, M. Iinuma, K. G. N. P. Piyasena and H. R. W. Dharmaratne, Phytomedicine, 2005, 12, 203–208 CrossRef CAS PubMed.
- P. D. Stapleton and P. W. Taylor, Sci. Prog., 2002, 85, 57–72 CrossRef CAS PubMed.
- P. Domadia, S. Swarup, A. Bhunia, J. Sivaraman and D. Dasgupta, Biochem. Pharmacol., 2007, 74, 831–840 CrossRef CAS PubMed.
- M. M. Traczewski, B. D. Katz, J. N. Steenbergen and S. D. Brown, Antimicrob. Agents Chemother., 2009, 53, 1735–1738 CrossRef CAS PubMed.
- D. E. Anderson, M. B. Kim, J. T. Moore, T. E. O'Brien, N. A. Sorto, C. I. Grove, L. L. Lackner, J. B. Ames and J. T. Shaw, ACS Chem. Biol., 2012, 7, 1918–1928 CrossRef CAS PubMed.
- N. L. Elsen, J. Lu, G. Parthasarathy, J. C. Reid, S. Sharma, S. M. Soisson and K. J. Lumb, J. Am. Chem. Soc., 2012, 134, 12342–12345 CrossRef CAS.
- S. C. M. Lau, U. Batra, T. S. K. Mok and H. H. Loong, Drugs, 2019, 79, 823–831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00249j |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Yik-Hong
Fung
,
Yuan-Yuan
Zheng
,
Yu-Wei
Chen
,
Yik-Hong
Fung
,
Yuan-Yuan
Zheng
,
Yu-Wei
Chen