
Small intestinal sampling capsule for inflammatory bowel disease type detection and management†
Sina
Nejati‡
 ab,
Jiangshan
Wang‡
bd,
Ulisses
Heredia-Rivera
ab,
Sotoudeh
Sedaghat
ab,
Ian
Woodhouse
ab,
Jay S.
Johnson
c,
Mohit
Verma
bde and
Rahim
Rahimi
*ab
ab,
Jiangshan
Wang‡
bd,
Ulisses
Heredia-Rivera
ab,
Sotoudeh
Sedaghat
ab,
Ian
Woodhouse
ab,
Jay S.
Johnson
c,
Mohit
Verma
bde and
Rahim
Rahimi
*ab
aSchool of Materials Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: rrahimi@purdue.edu
bBirck Nanotechnology Center, Purdue University, West Lafayette, IN 47907, USA
cUSDA-ARS Livestock Behavior Research Unit, West Lafayette, IN 47907, USA
dDepartment of Agricultural and Biological Engineering, Purdue University, West Lafayette, IN 47907, USA
eWeldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA
First published on 26th November 2021
Abstract
Although serum and fecal biomarkers (e.g., lactoferrin, and calprotectin) have been used in management and distinction between inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS), none are proven to be a differential diagnostic tool between Crohn's disease (CD) and ulcerative colitis (UC). The main challenge with laboratory-based biomarkers in the stool test is the inability to indicate the location of the disease/inflammation in the gastrointestinal (GI) tract due to the homogenous nature of the collected fecal sample. For the first time, we have designed and developed a battery-free smart capsule that will allow targeted sampling of inflammatory biomarkers inside the gut lumen of the small intestine. The capsule is designed to provide a simple and non-invasive complementary tool to fecal biomarker analysis to differentiate the type of IBD by pinpointing the site of inflammatory biomarkers secretion (e.g., small or large bowel) throughout the GI tract. The capsule takes advantage of the rapid change from an acidic environment in the stomach to higher pH levels in the small intestine to dissolve a pH-sensitive polymeric coating as a means to activate the sampling process of the capsule within the small intestine. A swelling polyacrylamide hydrogel is placed inside the capsule as a milieu to collect the sampled GI fluid while also providing the required mechanical actuation to close the capsule once the sampling is completed. The hydrogel component along with the collected GI fluid can be easily obtained from the capsule through the screw-cap design for further extraction and analysis. As a proof of concept, the capsule's performance in sampling and extraction of bovine serum albumin (BSA) and calprotectin – a key biomarker of inflammation – was assessed within the physiologically relevant ranges. The ratio of extracted biomarkers relative to that in the initial sampling environment remained constant (∼3%) and independent of the sampling matrix in both in vitro and ex vivo studies. It is believed that the demonstrated technology will provide immediate impact in more effective IBD type differential diagnostic and treatment strategies by providing a non-invasive assessment of inflammation biomarkers profile throughout the digestive tract.
Introduction
It is estimated that more than 1.6 million people in the United States and over 6.8 million people globally are affected by inflammatory bowel diseases (IBD) in the form of either Crohn's disease (CD) or ulcerative colitis (UC).1,2 Current evidence suggests that IBD is a result of a defective immune system in the host which appears to be caused by various genetic and environmental factors (e.g., smoking, diet, antibiotics, microbial dysbiosis, and appendectomy).3–9 In these patients the immune system responds excessively to harmless environmental triggers such as food, bacteria, and viruses, resulting in chronic and recurring inflammation in the gastrointestinal (GI) tract. In CD, inflammation can occur throughout the whole digestive tract, from the mouth to anus in the form of distributed patchy areas separated by healthy mucosa/tissue. Conversely, inflamed regions in UC are continuous and localized from the colon to the rectum.10Despite the distinctive morphological features, CD and UC patients often have very similar systemic symptoms which make their differential diagnosis one of the greatest diagnostic challenges in gastroenterology. While many symptoms depend on the severity of the disease and the patients age, both diseases often have many common presenting symptoms such as chronic diarrhea, abdominal pain, weight loss, fever, and fatigue. However, proper differential diagnosis between the type of IBD and level of activity are of great importance as each disease will require a specific management procedure including therapeutics, dietary restrictions, and surgical intervention.11
In recent years, there has been a tremendous effort towards a more specific targeted therapy that enables better distinction of the pathogenic mechanism within these diseases. The design and use of more of such disease-specific therapies will not only help more effective disease control but also help reduce many side effects of nonspecific immunosuppressants therapeutics (e.g., infection, and allergic reactions). For example, a type of immunosuppressant drug called methotrexate has shown higher efficacy in CD patients but insufficient in reducing inflammation in UC patients.12–16 In contrast, control studies on aminosalicylates have been shown to be more effective in mild to moderate UC as compared to CD patents.17–19 Therefore, there is a growing need for better and more clear distinction between CD and UC diseases as new disease specific therapies become more available in clinical practices. Among different diagnostic criteria, healing of the inflamed mucosa (mucosal healing) throughout the GI tract has gained a considerable level of acceptance as the measure of disease activity and is the therapeutic goal for clinical trials. Most of the clinical trials define mucosal healing as the absence of friability, erosion, and ulcer in all visible segments throughout the GI tract.20,21 Current procedure used for evaluating intestinal inflammation and mucosal healing is through visual examination of the upper and lower digestive system with endoscopy and colonoscopy, respectively.22,23 However, these procedures are invasive, unpleasant, time-consuming, costly, require skilled medical physicians and an irritative preliminary regimen, causing numerous challenges for frequent examinations.24,25 Moreover, these imaging tools are incapable of examining the entire GI tract, including the small intestine region in CD patients.24 More recently, advancement in medical devices and electronics have resulted in the development of a new class of smart capsule-based tools with a variety of chemical and gas sensing,26–30 as well as imaging technologies that could potentially open up new diagnostic possibilities. However, many of these systems are still not an alternative to endoscopy but a complementary test as they are unable to collect samples from the regions of interest in the GI tract.31–35 Recently introduced ingestible devices enable sampling from the gut,36–38 however, these devices are mostly vulnerable to contamination by the downstream liquid. While many endoscopic approaches can provide the opportunity to directly evaluate mucosal lesions throughout the GI tract, there is a substantial debate regarding the definition of mucosal healing and is often inadequate to be used as the single marker for determining the disease activity, as endoscopy only shows visible abnormalities rather than transmural healing.39 In this regard, for many patients with long-standing CD, mucosal healing may not always reflect healing of all layers of the tissue. In addition, patients at early stages of CD and UC may also have no clear signs of inflammation that are visible through endoscopy. Therefore, over the past decade many efforts have been conducted into identifying possible biomarkers as surrogates that could more objectively determine disease activity while also avoiding invasive endoscopy procedures.39 Among various indicators, fecal biomarkers (e.g., calprotectin and lactoferrin) have the advantage of being more specific to GI tract inflammation and being less invasive as compared to serological based biomarkers.40,41 Calprotectin, which is naturally released by activated neutrophils, represents more than 60% of the cytosolic proteins in granulocytes.42–44 Therefore, the elevated levels of these biomarkers in feces are directly proportional to the migration of neutrophils to the site of inflammation in the GI tract. However, they cannot be used to distinguish between CD and UC45 as they cannot indicate the location of the disease/inflammation in the GI tract due to the homogenous nature of the collected fecal sample. Thus, there is an unmet need for non-invasive means of targeted and representative sampling and monitoring of inflammatory biomarkers throughout the GI tract. Targeted sampling will enable a new opportunity by utilizing well understood inflammatory biomarkers inside the GI tract to differentiate CD from UC patients as well as managing their status.
To address this need, we introduce a non-invasive smart capsule device that allows targeted sampling of an inflammatory biomarker from the small intestine. By monitoring the inflammatory biomarkers sampled within the capsule along with excreted fecal biomarkers, one could rule out the localization of inflammation throughout the GI tract. This non-invasive approach will allow a new route towards employing well understood inflammatory biomarkers in differential diagnosis of CD and UC patients and realizing their current conditions.
Here, we describe the development and testing of a battery-free smart capsule which is composed of a 3D-printed casing with a superabsorbent hydrogel inside the capsule, which serves as a milieu for capturing the biomarkers of interest as well as providing the required actuation to enclose the capsule after the sampling is completed. The aperture on the capsule is covered with a pH sensitive enteric coating that delays the sampling process until the capsule enters the small intestine. Although the pH of each GI region can vary from person to person, there is a consistent and distinct pattern of increase in pH from the stomach to the small bowel (Fig. 1a). The capsule is designed to only open at certain pH levels that are distinctly associated with the small intestine. Once the capsule reaches the targeted location, the pH sensitive coating will dissolve, allowing GI fluid which contains the biomarkers of interest to enter the capsule. The dehydrated hydrogel will absorb the GI fluid entering the capsule and expand in volume and gradually move the PDMS disk that is bonded to the top end of the hydrogel. Upon full expansion, the PDMS disk will be pushed against the sampling aperture and seal the capsule before entering the large intestine (Fig. 1b). The natural peristaltic motion of the digestive system will allow the capsule to move throughout the GI tract without the need of any active battery powered components. The hydrogel component with the captured biomarkers can be removed from the capsule an extracted into a solution for further analysis using standard analytical tools (Fig. 1c). As a proof of concept, the sampling functionality of the smart capsule has been extensively validated in vitro and ex vivo with calprotectin as a widely used biomarker for IBD monitoring and management. However, future studies could also investigate the complementary use of this technology with other smart capsules equipped with different chemical and gas sensing capabilities for better differential diagnostics of IBS and IBD.26
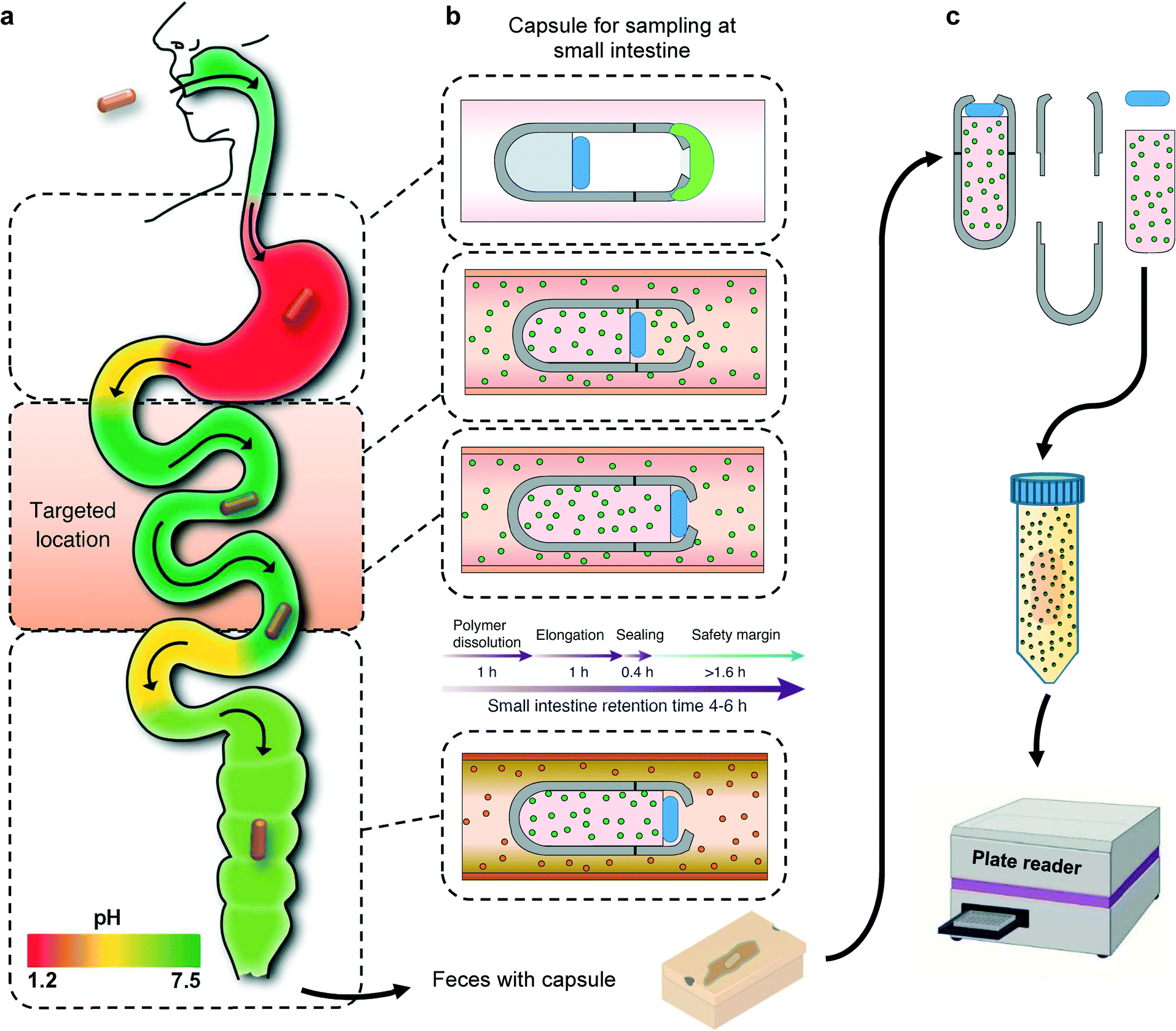 | ||
| Fig. 1 GI tract pH profile and capsule's sampling-extraction mechanism. (a) When a patient swallows the capsule, it is exposed to the saliva for less than 1 min with a pH 7, as it moves towards the stomach, the pH drastically drops to ∼3. As it further travels, the pH increases once it reaches the small intestine with an average pH of 6.8 and maximum of 7.4. The pH is then gently reduced when the capsule approaches the ileocecal valve and slightly increases as it continues its journey through the colon. (b) The capsule stays inactive as the enteric coating remains un-ionized in the acidic pH environment of the stomach. As the capsule enters the small intestine the pH-responsive coating will dissolve and allow the inflow of intestinal fluid into the capsule. Subsequently, the hydrogel in the capsule will swell by absorbing the intestinal fluid and push the PDMS disk towards the sampling aperture to seal the capsule before entering the large intestine. (c) After excretion, the hydrogel component is removed from the capsule and the sampled biomarkers are extracted into a solution for further analysis. | ||
Experimental
Smart sampling capsule fabrication
The housing including the body and cap were designed with SolidWorks (Dassault Systèmes) and the stereolithography technique (Form 2 by Formlabs, Formlabs Inc.) was utilized to 3D-print the housing with a layer thickness of 50 μm using a biocompatible resin (EN-ISO 10993-1:2009/AC: 2010, USP Class VI) purchased from Formlabs. After 3D-printing, the models were rinsed in 99% pure isopropyl alcohol (IPA) for 15 min to remove remnants and subsequently, post-cured in a UV photocuring device (Formlabs Inc.) for 60 min to complete the polymerization. The polydimethylsiloxane (PDMS) disk was fabricated using a standard 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of curing agent to silicone base (Dow Corning) and cured at 70 °C for 4 h. Subsequently, a circle of 6 mm in diameter was cut using a computer-controlled CO2 laser (10.6 μm wavelength) cutting and engraving system (PLS6MW, Universal Laser, Inc., Scottsdale, AZ).46–48 The PDMS disk was adhered to the hydrogel using a biocompatible adhesive (GelBond® PAG, Lonza).
:10 ratio of curing agent to silicone base (Dow Corning) and cured at 70 °C for 4 h. Subsequently, a circle of 6 mm in diameter was cut using a computer-controlled CO2 laser (10.6 μm wavelength) cutting and engraving system (PLS6MW, Universal Laser, Inc., Scottsdale, AZ).46–48 The PDMS disk was adhered to the hydrogel using a biocompatible adhesive (GelBond® PAG, Lonza).
pH-sensitive polymers
Five anionic polymers were employed to analyze their dissolution kinetic within different pH environments. Eudragit® L100-55 (poly methacrylic acid-co-ethyl acrylate) and Eudragit® L100 (poly methacrylic acid-co-methyl methacrylate) were kindly donated by Evonik (NJ, USA) with the dissolution pH threshold of 5.5 and 6.0, respectively. Two hydroxypropyl methylcellulose phthalate (HP-55) polymers were kindly donated by G. M. Chemie Pvt. Ltd (India) and Shin-Etsu Chemical Co. (Japan) both with the dissolution pH threshold of 5.5. Cellulose acetate phthalate (CAP) was kindly donated as well by G. M. Chemie Pvt. Ltd (India) with a pH threshold of 6.0. For L100-55 and L100, Eudragit® powder (3.8 g) was dissolved in IPA (9.5 g) and acetone (6.32 g), vortexed for 10 min followed by 1 h water bath sonication (Model 2510, Branson, Danbury, CT). Once the powder was fully dissolved, triethyl citrate (TEC) (0.38 g; Sigma-Aldrich) was added as a plasticizer and agitated for 2 min. For both Chemie and Shinetsu, powder (1.29 g) was dissolved in distilled water (3.716 g) and ethanol (14.864 g; Sigma-Aldrich) by vortexing for 15 min followed by 30 min bath sonication. Once fully dissolved, TEC (0.129 g) was added to the solution and agitated for 2 min. The CAP solution was prepared by dissolving CAP powder (1.44 g) in acetone (18 g) and triacetin (0.36 g; Sigma-Aldrich) through vortexing for 15 min followed by 30 min bath sonication.To obtain calibration curves and polymer dissolution plots, methylene blue (40 mg; Sigma-Aldrich) was added into each polymer solution. To achieve a crack-free thin film, doctor-blade coating technique (MSK-AFA-II, MTI, USA) was exploited. The solutions were separately spread onto an acrylic sheet substrate using doctor-blade and dried at 70 °C for 4 h. Gage blades (Precision Brand) were used for doctor-blading to form dried films with 50 μm thickness. Once films were completely dry, 10 mm diameter circles were cut using CO2 laser cutter machine and introduced into pH buffer solutions. Samples in triplicates were collected at 10 min intervals and UV-vis spectroscopy of samples was performed using BMG Clariostar microplate reader (BMG Labtech, Germany) at 665 nm corresponding to the peak wavelength of the methylene blue.
Hydrogel synthesis and characterization
Acrylamide (4.5 g; Sigma-Aldrich) and N,N′-methylenebisacrylamide (MBA) (0.15 g; Sigma-Aldrich) as the crosslinker were dissolved in 10 mL water by vortexing the solution for 3 min at room temperature using a Signature Digital Vortex Mixer 945303 (VWR, Radnor, PA, USA). The solution was degassed thoroughly by bubbling nitrogen gas for 10 min to displace dissolved oxygen as polymerization inhibitor. Ammonium persulfate (0.1 g; Sigma-Aldrich) was then added into the solution as the initiator. To fabricate hydrogels with proper dimensions, the pre-gel solution was transferred into PDMS mold cylinders in 150 μL volumes followed by placing the mold inside an isothermal oven overnight at 70 °C to form the fully cured acrylamide network. For the swelling ratio, hydrogels in triplicates were weighed and immersed in 25 ml buffer solutions of pH 6.8 and 7.4 at 37 °C inside an incubator with a shaking platform at 100 rpm for 36 h. Hydrogels were carefully removed from buffer solutions at 10 min intervals and the excess buffer on the outer surface was gently blotted by wiper followed by measuring their mass. Swelling ratio was defined as:where, Wt and Wi are the weights of swollen and dry hydrogels, respectively. To assess the swelling profile of the hydrogel in different complex media, we analyzed hydrogel swelling behavior in the filtered and unfiltered GI fluid obtained from the porcine small intestine.
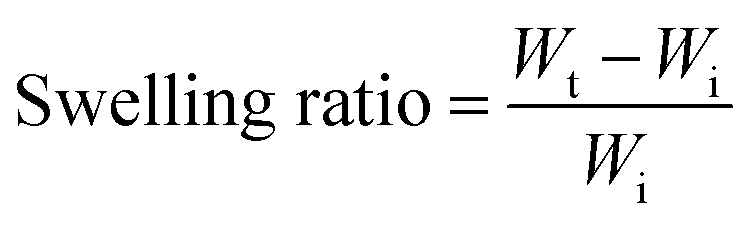
In order to assess the volumetric expansion, pictures of the hydrogel were taken at 10 min intervals and analyzed using ImageJ software. Moreover, to visualize the straight swelling of the hydrogel and to demonstrate the effect of GelBond® adhesive between hydrogel and the PDMS disk during swelling/hydration, two hydrogels with and without GelBond® adhesive were placed in a container filled with pH 6.8 buffer solution. The swelling direction of the hydrogel as well as proper attachment of the PDMS disk to the hydrogel were analyzed.
Furthermore, to assess whether the applied pressure from the hydrogel can effectively seal the capsule and overcome the intestinal back pressure, the hydrogel's compressive force during swelling was recorded using an Admet Tensile Tester. A capsule with a dry hydrogel inside was placed in a container while a probe attached to a 10 N load cell was set on top of the hydrogel. The container was then filled with pH buffer solutions while being stirred with an overhead mixer (Model 50006-03, Cole Parmer, IL, USA) at 100 rpm to mimic the disturbance in the GI tract environment. The generated force for the hydrogel was recorded over time and converted into pressure based on the hydrogel cross-sectional area.
Hydrogel protein sampling assessment
To visualize the diffusion of the sampled analytes and ensure effective absorption of proteins into the polymeric network structure of the hydrogel, we tested the ability of the hydrogels to absorb green fluorescent protein (GFP) as a surrogate with a molecular weight that is close to calprotectin. For this process, GFP (Sigma-Aldrich, St. Louis, MO) was diluted in PBS (Sigma-Aldrich, St. Louis, MO) to achieve 100 μg mL−1 GFP concentration. The solution was transferred to a microcentrifuge tube and centrifuged at 14000 × g for 1 min (Sorvall Legend Micro 21 Microcentrifuge, Thermo Fisher Scientific, Waltham, MA, USA) to precipitate any GFP aggregates. Next the hydrogels were introduced into the GFP supernatant for 4 h to qualitatively display the protein sampling efficiency throughout the hydrogel structure. The images were captured with a High Performance 2UV Transilluminator UVP (Upland, CA, USA) and a digital inverted microscope (AMG EVOS fl, Bothell, WA).
Furthermore, to monitor the GFP extraction behavior of the hydrogels, they were exposed to 4 mL of PBS for 2 h and the fluorescence analysis (excitation max at 488 nm and emission max at 510 nm) was carried out at 30 min intervals via BMG Clariostar microplate reader. At each time point, 100 μL in triplicates were transferred into a UV-transparent 96-well microplate purchased from Corning (NY, USA) and the fluorescence was measured.
BSA sampling and extraction characterization
Bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO) was diluted in PBS (Sigma-Aldrich, St. Louis, MO) twofold serially from 10 to 0.625 mg mL−1 for a total of 5 concentrations. Next fully assembled sampling capsules with no enteric coating were immersed into BSA solutions for 4 h at 100 rpm 37 °C. Subsequently, the capsules were retrieved, disassembled, and introduced into PBS media for 2 h extraction. To verify the existence of BSA in the extraction PBS solution, the bicinchoninic acid (BCA) assay was employed to detect the protein content. 25 μL of each standard samples and unknown samples were added into a microplate well followed by an addition of 200 μL working reagent. After incubation at 37°C for 30min, the plate was cooled to room temperature and then the absorbance at 562nm was measured via BMG Clariostar microplate reader.
Calprotectin sampling and extraction characterization
Calprotectin (MyBioSource, San Diego, CA) was reconstituted in Tris buffer (20 mM Tris, 100 mM NaCl, pH 7.4) to a concentration of 1 mg mL−1. The protein solution was further diluted into four different concentrations. For sampling, fully assembled capsules were immersed into 4 mL of each concentration for 4 h while the vials being agitated at 100 rpm 37 °C. The capsules were then opened, and the content was transferred into 4 mL Tris buffer for 2 h extraction at 100 rpm agitation. The measurement of calprotectin was an enzyme-linked immunosorbent assay (ELISA) analyzed using the BÜHLMANN fCAL ELISA according to the standard manufacturer's protocol (4–240 ng mL−1). The extraction buffer was loaded onto a 96-well plate coated with a capture antibody. After 30 min incubation and a washing step, a detection antibody conjugated to horseradish peroxidase (HRP) was added, where the detection antibodies attached to the calprotectin. After incubation and washing, tetramethylbenzidine (TMB) was added (blue color formation) followed by addition of a stop solution (change to yellow color). The absorption was determined at 450 nm using BMG Clariostar microplate reader.In vitro sampling and leakage test
For this test six, capsules with previously described fabrication process were introduced into a series of dyed buffer solutions for different durations including pH 7 (green – 1 min), pH 3 (red – 2 h), pH 6.8 (blue – 4 h), and pH 7.4 (colorless – 18 h) which corresponded to the pH of mouth, stomach, small intestine, and colon, respectively. At each exchange point between buffer solutions, the capsules were rinsed and inspected for signs of enteric coating dissolution. To evaluate any sign of fluid exchange between the capsule and the environment, one of the capsules was removed from the study group and opened to examine hydrogel swelling and dye uptake.49 Any indication of fluid exchange and leakage from the capsule into the test solution environments were analyzed by collecting aliquots of approximately 10 ml and recording the UV-visible spectra using BMG Clariostar microplate reader. The optical absorption spectra were obtained before the capsule was introduced into the test solution and after the capsule was removed from the test solution at predetermined time points.In vitro BSA and calprotectin sampling and extraction
To study the capsule efficiency in sampling biomarkers in more physiologically relevant conditions, two in vitro experiments were carried out. Similar to the in vitro sampling and leakage experiment, different buffer solutions were used to mimic distinct sections throughout the GI track. However, in this study the dyed buffer solutions were replaced with clear buffers. To simulate inflammation in the small intestine, the representing buffer solution (pH 6.8) was spiked with BSA and calprotectin as the biomarkers of interest to be sampled. To assess the capsules effective sampling and extraction, two sets of in vitro experiments with different amounts of BSA and calprotectin in the small intestine model were conducted. At the end of the experiments, the capsules were retrieved from the in vitro setup and the swollen hydrogels were removed from the capsules. For the first set of experiments with BSA as a sampling analyte of interest, the extraction procedure was performed by placing the retrieved hydrogels into separate PBS containing vials for 2 h under continuous agitation. In the second set of experiments, the calprotectin was extracted from the hydrogels by immersing them into Tris buffer for 2 h. Extracted value from each hydrogel was analyzed through ELISA. Beside capsule's sampling efficiency, the performance of free hydrogels was also assessed in sampling BSA inside filtered and unfiltered GI fluid at 37 °C at 100 rpm. The GI fluid was filtered using Whatman 1113-090 quantitative filter papers. The BSA concentration of sampling environment was 10 mg mL−1 and the extracted concentration was analyzed by BCA protein assay.Attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) using a PerkinElmer spectrum 100 FTIR spectrometer (MA, USA) and Raman spectroscopy using a Thermo Scientific DXR™ xi Raman Imaging Microscope (MA, USA) equipped with a 532 nm laser was performed to assess any possible chemical interaction between hydrogel polymer matrix and targeted biomolecules. For this test, the FTIR and Raman spectra of as prepared and dried hydrogels was compared with hydrated hydrogels in fluids containing 10 mg mL−1 BSA and 200 ng mL−1 calprotectin solutions for 4 h.
To assess the effective penetration of the sampled analyte into the hydrogel matrix, all ATR-FTIR and Raman spectra were collected from the central section of the hydrogel by cutting the swollen hydrogels with a sharp razor blade.
Ex vivo calprotectin sampling
To further assess the sampling performance of the capsule in complex media, we conducted an ex vivo study. For this experiment, freshly prepared porcine small intestine was employed in which the intestine was dissected into two pieces of ∼7 cm in length, followed by injection of two different calprotectin concentrations into the intestinal pieces, separately. Notably, the two testing environment concentrations were chosen such that both normal and elevated calprotectin levels were used. The fully assembled capsules were then introduced into the small intestine pieces while agitating at 100 rpm for 4 hours. Subsequently, the capsules were retrieved, opened, and the content was transferred into Tris buffer for analyte extraction while being agitated at 100 rpm for 2 hours at 37 °C inside an incubator. Finally, the calprotectin concentration in the buffer was measured through ELISA assay.Results and discussion
Sampling capsule design and working principle
The smart sampling capsule is composed of four elements: a pH-sensitive biodegradable enteric coating, a casing, a gas-permeable PDMS disk, and a fast-swelling hydrogel. We utilized a pH-sensitive polyacid polymer to withstand the low pH gastric environment where the device is supposed to remain inactive. Upon exposure to the targeted location of the small intestine where the average pH reaches to 6.8, the enteric coating disintegrates allowing the intestinal fluid to enter the device. At this point, the hydrogel swelling due to the fluid absorption helps to drive the PDMS disk towards the opening aperture and as the swelling is further proceeded, more force to the PDMS guarantees the effective sealing to avoid any potential downstream contamination. Owing to this mechanism, the sampling is solely performed in the small intestine which enables capturing the inflammation biomarkers to identify the IBD type as CD is only present in the small bowel. Assuming the required time for the polymer dissolution, hydrogel elongation, and complete sealing of the capsule is ∼2.4 h, a minimum extra time of 1.6 h is considered to compensate in case any unwanted parameters including low intestinal fluid content or lower pH values at the sampling site prolonged the site selective sampling process. The screw-cap design of the capsule allows for easy disassembly and removal of the swollen hydrogel. The captured biomarkers within the hydrogel matrix can be simply extracted into a solution and further analyzed using standard laboratory equipment (Fig. 1).Fig. 2a illustrates all the capsule components. To assemble all parts, first the PDMS disk was attached to the top surface of the dehydrated hydrogel using a biocompatible adhesion (GelBond® PAG, Lonza) and placed inside the 3D-printed body of the capsule. Next the 3D-printed cap covered with the pH sensitive enteric coating is screwed and threaded onto the body of the capsule. The small volume/length of dehydrated hydrogel will create a gap between the PDMS disk and the sampling aperture which will allow an easy flow of fluid into the capsule once the aperture has opened. The screw design of the capsule enables a quick and easy assembly/disassembly of the capsule. Fig. 2b presents an optical image of the disassembled device with an approximate reservoir volume of 430 μl (6.5 mm × 13 mm, D × H), the dehydrated hydrogel (3 mm × 9.5 mm, D × H) along with the PDMS disk (5.5 mm × 2 mm, D × H). The sampling aperture (4 mm in diameter) on the cap was optimized to facilitate the fluid flow during the sampling process. The assembled capsule shown in Fig. 2c pinpoints the outer diameter of 9 mm and the length of 15 mm, which is smaller than PillCam™ both in diameter and length (11 mm × 26 mm).34
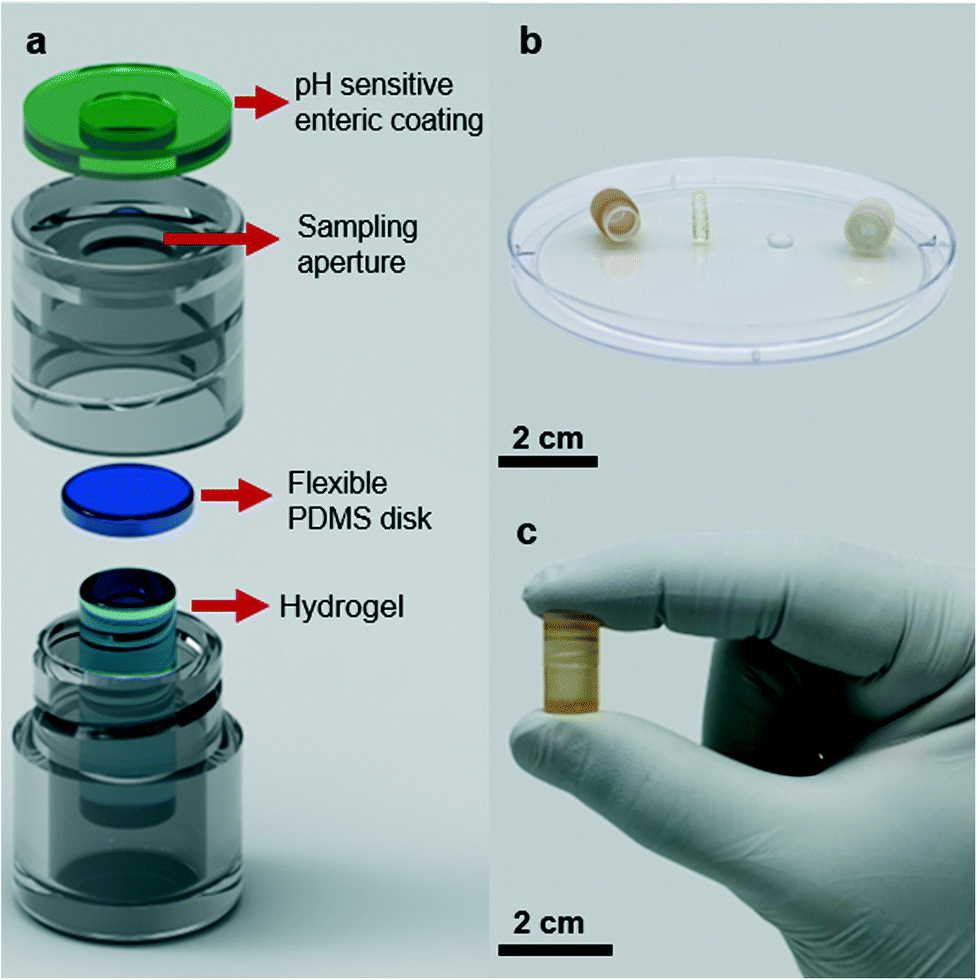 | ||
| Fig. 2 Smart sampling capsule components. (a) Schematic of the overall designed capsule composed of pH sensitive enteric coating, housing, flexible PDMS disk, and hydrogel. (b) Disassembled capsule components on a Petri dish. (c) Picture of the assembled sampling capsule. | ||
pH-sensitive enteric coatings
In order to identify the appropriate enteric coating on the capsule that will dissolve upon entering the small intestine, we investigated a range of different pH-responsive polymers in different pH environments. These polymers which contain carboxylic acid groups (R–COOH), remain un-ionized in acidic pH condition of the stomach but deprotonate and become ionized at higher pH of the small bowel, hence the repulsion produced by negative charges causes the swelling and final disintegration of the polymers.50 The polymer release profiles in pH of the stomach (pH 1.2 for fast and pH 3.0 for fed modes) as well as the small intestine (pH 6.8) are shown in Fig. 3. The dissolution profiles of the organic polymers in pH 1.2 (Fig. 3a) suggest that all organic polymers remained intact, except for Chemie and Shinetsu with a dissolution of 34% and 28%, respectively. These two polymers showed similar degradation in pH 3.0 with a relatively similar trend, whereas L100-55, CAP, and L100 showed a slow dissolution of 10%, 14%, and 14% after 2 h, respectively (Fig. 3b). Conversely, in pH 6.8 all organic formulations except L100 with a dissolution of 32%, were fully dissolved within a time course of 2 h (Fig. 3c). The dissolution order of the organic enteric coatings can be elaborated by pKa and backbone structure.51 For instance, L100 which possesses higher pKa values displays higher pH thresholds, and thus partially dissolved in pH 6.8. Additionally, the water insoluble backbone of CAP results in slower dissolution compared to Chemie and Shinetsu.52 Overall, the dissolution order for the enteric coating formulations was Chemie ≈ Shinetsu > L100-55 > CAP > L100. For further capsule experiments, L100-55 was used as the enteric coating due to its rapid dissolution (1 h) in the intestinal pH and insignificant dissolution in acidic environment preventing any inflow towards the capsule in the stomach.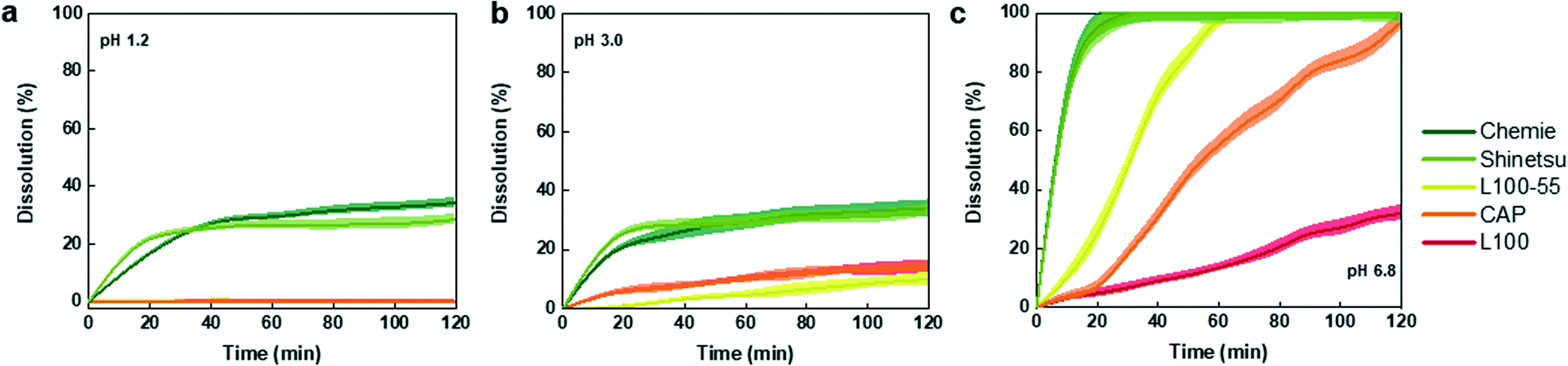 | ||
| Fig. 3 Polymer dissolution profiles in different pH buffer solutions. (a) Eudragit polymers (L100-55 and L100) along with CAP remained completely intact in the pH 1.2, whereas Chemie and Shinetsu started to partially disintegrate after 20 min. (b) Minor dissolution from CAP and Eudragit polymers in pH 3.0. (c) All polymers except L100 were fully dissolved in pH 6.8 within 2 h. | ||
Hydrogel characterization
At first, we investigated the swelling kinetics of the hydrogel by measuring the swelling ratio and swelling speed. Fig. 4a depicts a picture of a fully dried hydrogel beside a swollen hydrogel that was placed in a pH 6.8 buffer solution containing methylene blue dye at 37 °C for 4 h. The results of swelling ratio, as shown in Fig. 4b, implies the high diffusivity of water into preexisting or dynamically formed gaps among hydrogel chains before it approaches a plateau at 14 h. The profiles of the hydrogel swelling in filtered and unfiltered GI fluid are identical to swelling in pH 6.8 and 7.4 buffer solutions (Fig. S6b†), which further confirmed that swelling profile of the hydrogel is independent from the sampling environment composition. Beside weight-based swelling experiment, assessment of hydrogel elongation is essential to determine the exact time when the PDMS disk blocks the capsule. Our findings from elongation study showed a rapid increase in length within 60 min for sealing the sampling aperture via the PDMS disk (Fig. 4c). At this point, as the hydrogel is lengthwise confined in the capsule reservoir, it continues to apply more force towards PDMS disk as the swelling is not halted, hence it competes with the intestinal pressure. The compression force profile confirms that over the course of 2 h, the applied compression force exerted from the hydrogel exceeded 1.7 N over an area of ∼7 mm2, in which after force–pressure conversion, we expected the hydrogel to have enough force to overcome the maximum intestinal backpressure.53Fig. 4d demonstrates that the hydrogel could effectively surpass the maximum GI pressure after ∼20 min from the time it touched the PDMS disk leading to a perfect capsule sealing and fluid exchange prevention. For all characterizations, two pH buffer solutions of 6.8 and 7.4 displayed consistently similar trend suggesting the identical hydrogel behavior towards various pH range throughout the small intestine. Additionally, the swelling direction of the hydrogel inside pH 6.8 buffer solution and the effect of GelBond® adhesion results between PDMS disk and the hydrogel are shown in Fig. S4.† Fig. S4a† shows, two hydrogels with and without GelBond® adhesives between the PDMS disk and the hydrogel surface. To ensure proper fixture of the hydrogels to the bottom of the container GelBond® was used followed by gently adding the buffer solution to the container. After addition of pH 6.8 buffer, both PDMS disks were still attached to the hydrogel (Fig. S4b†). However, only after 1 h of hydrogel swelling, the PDMS disk with no GelBond®, dropped beside the hydrogel (Fig. S4c†), whereas the GelBonded PDMS disk was strongly adhered to the hydrogel surface. Moreover, the straight swelling of the hydrogel with minimum sign of distortion is clearly displayed in Fig. S4c.†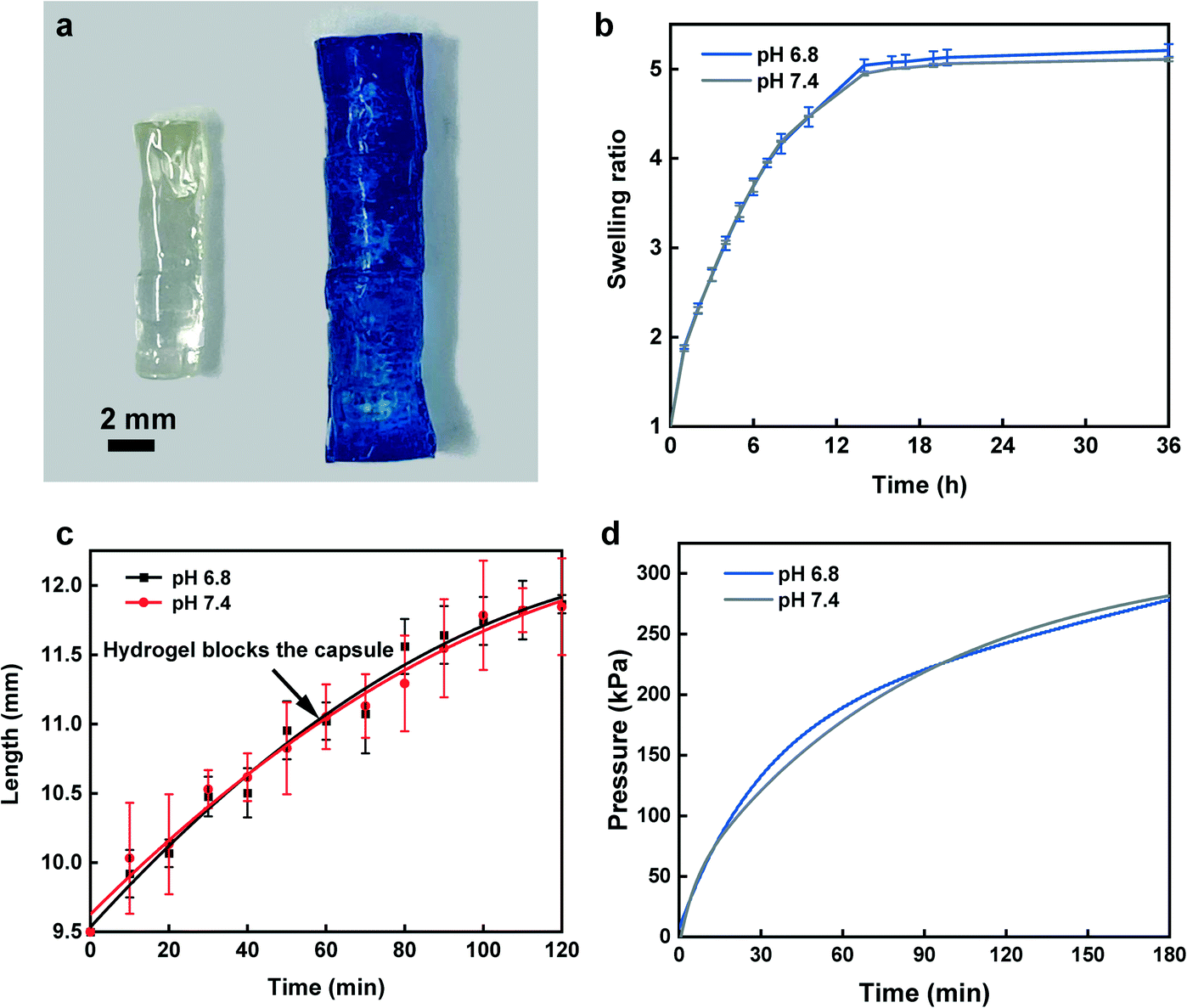 | ||
| Fig. 4 Hydrogel characterization. (a) Shows a fully dry hydrogel beside a 4 h-swollen hydrogel. (b) High-speed and high-swelling ratio of the hydrogel. (c) Hydrogel elongation over time verifies that within 60 min of swelling, it can rapidly swell to move the PDMS disk to touch the sampling aperture. (d) Compression force profile of the hydrogel indicates once it touches the PDMS disk, it only takes ∼20 min to surpass the maximum intestinal pressure. All (b–d) imply that pH variability within the small intestine, has no considerable effect on the hydrogel performance. Error bars represent s.d (n = 3). | ||
Protein sampling and extraction
To evaluate the sampling performance of the smart capsule, we tested three different proteins with relatively similar size and molecular mass including GFP, BSA, and calprotectin (inflammation biomarker). GFP was selected to visually display the protein is captured within the hydrogel composition. Due to the large hydrogel pore size, GFP with the average diameter of 5 nm, could readily find a path to enter the hydrogel matrix. Fig. 5a shows the colors of the hydrogels being exposed to deionized (DI) water and GFP solution under UV illuminator. Microscopic images taken from the same hydrogels also verify no color change in the sample submerged in DI water (Fig. 5b) compared to the one exposed to the GFP solution for 2 h (Fig. 5c). In addition to the protein sampling, it is also important to measure how the proteins are extracted from the hydrogel over time. Hence, we introduced the GFP loaded hydrogel into PBS and measured its fluorescence intensity overtime. The results showed an increase in fluorescence intensity within the first 30 min of extraction (Fig. 5d). These results show the need that a minimum of 30 minutes is required for the captured proteins within the hydrogel matrix to release into the extraction solution. This fast protein extraction is attributed to the large pores within the hydrogel structure during the polymerization which facilitates the extraction process.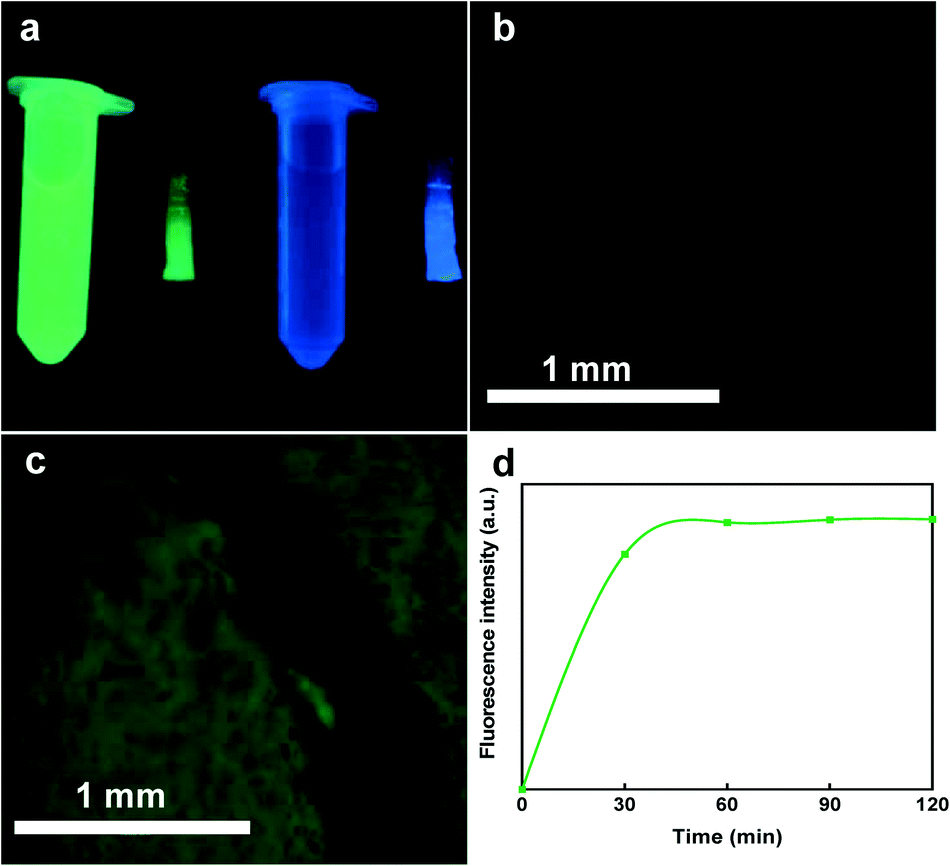 | ||
| Fig. 5 GFP sampling and extraction. (a) Depicts two microcentrifuges filled with GFP and DI along with corresponding hydrogels being exposed to the relevant solutions for 2 h. Successful GFP sampling was demonstrated by comparing the control sample exposed to DI (b) with the one exposed to GFP solution for 2 h (c). The extraction profile (d) suggests how fast the protein is released from the GFP loaded hydrogel. The fundamental of easy sampling and extraction is attributed to the large void space in the net-like structure of the hydrogel in comparison to the size of the protein. | ||
The second protein which was studied to ensure the capsule sampling-extraction efficiency was BSA with an average size of ∼7 nm. Fig. 6a illustrates UV-vis absorption of 1:10 diluted testing environment concentrations with the absorbance peaks at 562 nm. To obtain a calibration curve of sampling-extraction of BSA, capsules in triplicates with no pH-sensitive polymer coating were introduced into various testing environment concentrations for 4 h while being agitated at 100 rpm to mimic the peristalsis motion. Once the sampling was done, the capsules were then retrieved, opened, and introduced into PBS for 2 h for the extraction. The total concentration of BSA in the extraction solution was determined using BCA assay (Fig. 6b and c).
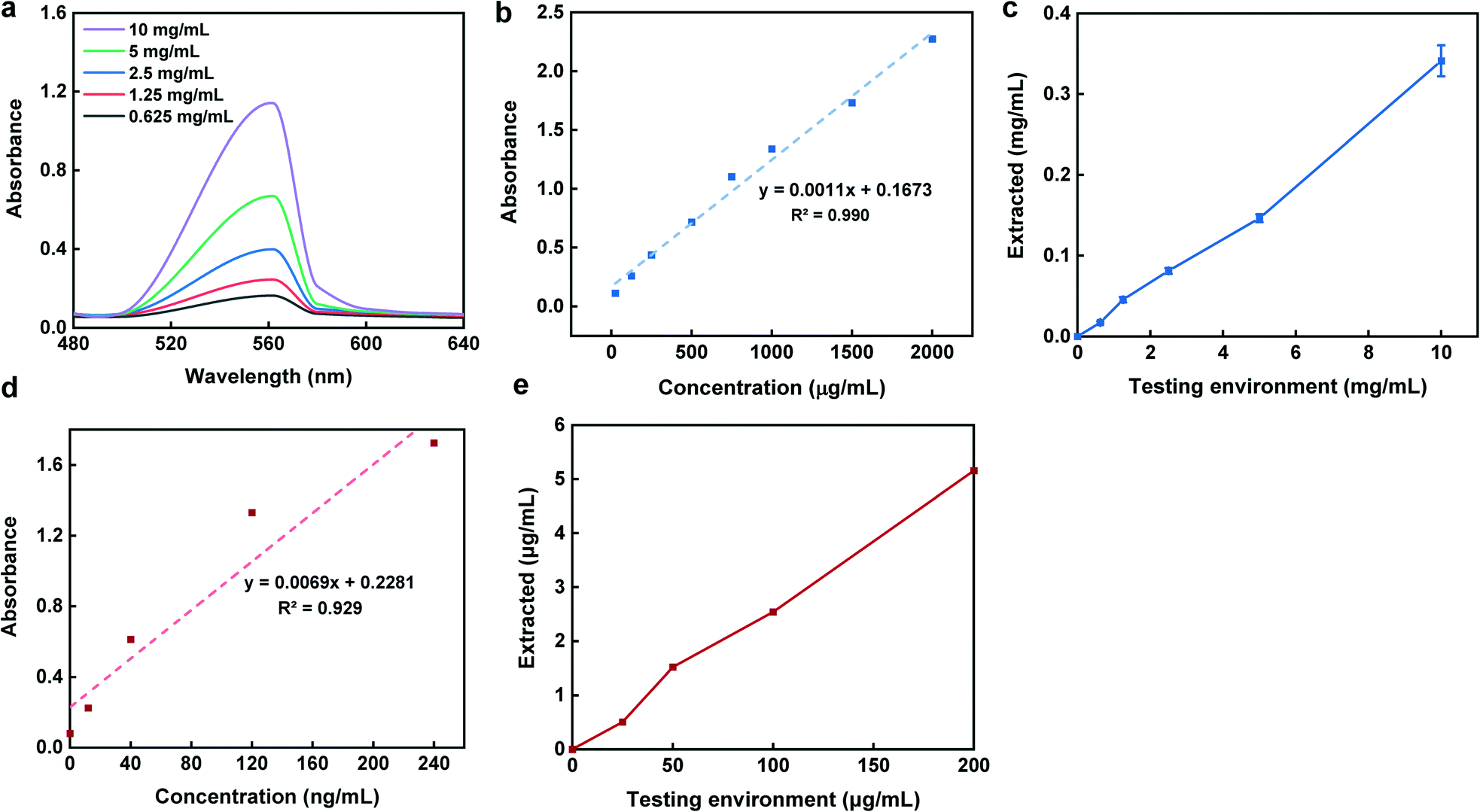 | ||
| Fig. 6 BSA and calprotectin sampling-extraction results. (a) UV-vis absorbance of BSA testing environment with different concentrations showing the absorbance peaks at 562 nm. (b) Standard calibration curve for BSA testing solutions. (c) Extracted BSA from sampling capsule vs. testing environment BSA concentrations. (d) Standard calibration curve for calprotectin testing solutions. (e) Extracted calprotectin from sampling capsule vs. testing environment calprotectin with least square regressions of R2 = 0.994 and R2 = 0.995, and E values of 3.2% and 2.6%, respectively. Dashed lines are least square regressions. Error bars represent s.d (n = 3). | ||
In the calprotectin experiment, which was the third tested protein, similar steps were followed for the sampling, however, for the extraction, the capsule content was buffer exchanged into Tris buffer for 4 h while agitated at 100 rpm. The comparison of the extracted concentrations with that of testing environment based on the calprotectin standard curve (Fig. 6d), is shown in Fig. 6e. As shown in Fig. 6c and e, there was a linear trend between the extracted and testing environment concentrations for both BSA and calprotectin. Additionally, the capsule sampling efficiency parameter (E) defined as the ratio between the extracted concentration compared to the testing environment concentration was approximately 3.2% and 2.6% for BSA and calprotectin, respectively. The linear correlation between the concentrations as well as similar BSA and calprotectin E values suggest a reliable sampling mechanism and ability to accurately predict the concentration of biomarkers in the small intestine from the retrieved concentration of analyte in the extracted solution.
Also, the results of BSA sampling efficiency associated with free swelling hydrogels are shown in Fig. S5.† The BSA E values of hydrogels in filtered and unfiltered GI fluid were 2.8 and 3% (Fig. S5†), respectively which are consistent with the BSA E value obtained from the capsule sampling efficiency (Fig. 6c). The two filtered and unfiltered fluid types may represent two diet conditions. Therefore, the performance results convey that sampling procedure is diet independent and can effectively sample from the complex GI fluid environment.
The ATR-FTIR spectrum for the as prepared and dried hydrogel is shown in the Fig. S1.† Hydrogel spectrum shows characteristic peaks associated with polyacrylamide hydrogel structure including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and asymmetric valence vibration for CH2 group at 1644 cm−1 and 2927 cm−1, respectively. Besides, the N–H stretching observed at 3300–3400 cm−1corresponds to the crosslinking bridge between the amide groups through the hydrogel matrix.54,55 Fig. S2† represents comparative ATR-FTIR spectra for the dry hydrogel and swollen hydrogels that were immersed inside solutions containing BSA and calprotectin. The hydrogels immersed in the BSA and calprotectin solutions display clear peaks for NH2, N–H, and C–N vibrations which also well matched the FTIR spectra peaks of the solution that contained BSA and calprotectin. The NH2, N–H, and C–N vibration at about 1600, 1550, and 1320 cm−1 are associated with amide types I, II, and III, respectively.56,57 While the dry hydrogel shows weak amide I and II vibration peaks for the acrylamide structure, addition of BSA and calprotectin resulted in stronger vibrations for all amide types. This enhanced intensity of the amide groups and appearance of the amide III peak in the swollen hydrogels further confirmed effective sampling of these proteins into the hydrogel structure. Furthermore, after protein sampling, it is clear that the vibrational peaks in the hydrogel are solely associated with either proteins or polyacrylamide suggesting no chemical interaction between hydrogel and the proteins. Raman analysis was also performed for further investigation of the possible interaction between the hydrogel and the protein molecules. Raman spectra show the characteristic peaks for polyacrylamide structure including the NH2 bending peak at about 850 cm−1, CO2 bending at 1450 cm−1, and primary amide NH2 rock band at 1105 cm−1 (Fig. S3†).58
O stretching vibration and asymmetric valence vibration for CH2 group at 1644 cm−1 and 2927 cm−1, respectively. Besides, the N–H stretching observed at 3300–3400 cm−1corresponds to the crosslinking bridge between the amide groups through the hydrogel matrix.54,55 Fig. S2† represents comparative ATR-FTIR spectra for the dry hydrogel and swollen hydrogels that were immersed inside solutions containing BSA and calprotectin. The hydrogels immersed in the BSA and calprotectin solutions display clear peaks for NH2, N–H, and C–N vibrations which also well matched the FTIR spectra peaks of the solution that contained BSA and calprotectin. The NH2, N–H, and C–N vibration at about 1600, 1550, and 1320 cm−1 are associated with amide types I, II, and III, respectively.56,57 While the dry hydrogel shows weak amide I and II vibration peaks for the acrylamide structure, addition of BSA and calprotectin resulted in stronger vibrations for all amide types. This enhanced intensity of the amide groups and appearance of the amide III peak in the swollen hydrogels further confirmed effective sampling of these proteins into the hydrogel structure. Furthermore, after protein sampling, it is clear that the vibrational peaks in the hydrogel are solely associated with either proteins or polyacrylamide suggesting no chemical interaction between hydrogel and the proteins. Raman analysis was also performed for further investigation of the possible interaction between the hydrogel and the protein molecules. Raman spectra show the characteristic peaks for polyacrylamide structure including the NH2 bending peak at about 850 cm−1, CO2 bending at 1450 cm−1, and primary amide NH2 rock band at 1105 cm−1 (Fig. S3†).58
The Raman spectra further confirm that no new peak or shift appeared after exposing the hydrogel to the protein solutions indicating that the molecules are not chemically bonding to the hydrogel structure.
In vitro dye sampling and leakage test
Fig. 7a shows various dyed buffer solutions representing different GI tract sections including mouth, stomach, small intestine, and colon with the relevant pH conditions of pH 7 (green), 3 (red), 6.8 (blue), and 7.4 (colorless), respectively. Visual inspections of the enteric coating clearly showed that the capsule remained close after 1 min and 2 h retention time within the mouth (green buffer) and stomach (red buffer) conditions. In addition, the test capsules that were disassembled after exposure to the stomach and mouth conditions showed no uptake of the fluid within the hydrogel which further confirmed that the capsule did not collect any fluid within the upper regions of the GI tract (mouth and stomach) (Fig. 7b). However, the capsules exposed to the small intestine (blue buffer) conditions, showed complete dissolution of the enteric coating with full hydration of the hydrogel and visible uptake of blue dye within its matrix (Fig. 7c). Fig. 7d–f show changes in the UV absorption spectra that were collected from the buffer solutions before introducing the capsules and after they were removed at predetermined timepoints.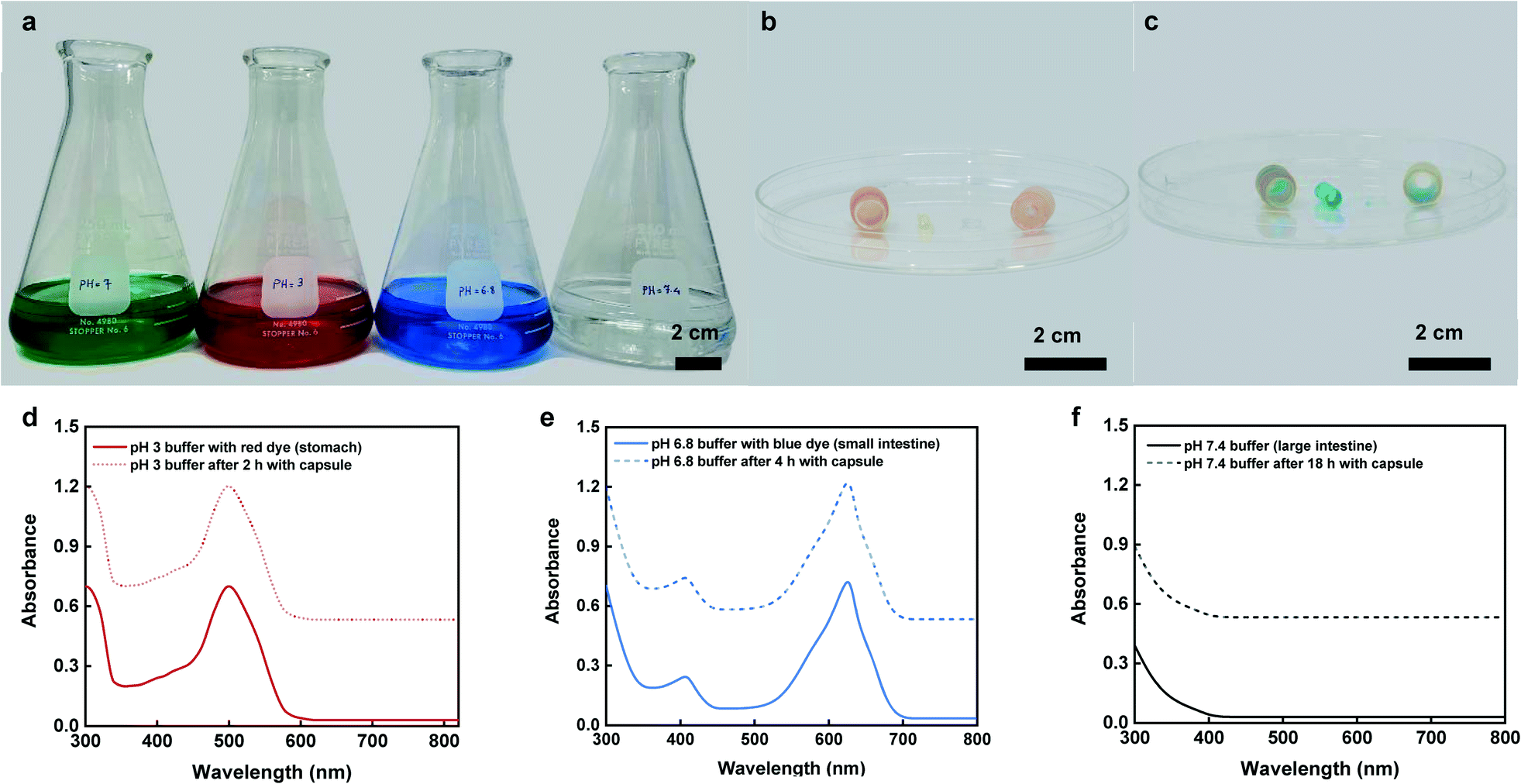 | ||
| Fig. 7 In vitro dye sampling and leakage. (a) Four flasks containing various pH buffer solutions simulate the pH changes along the GI tract using different colored dyes: saliva pH 7 (green), stomach pH 3 (red), small intestine pH 6.8 (blue), and colon pH 7.4 (colorless). (b) Shows when a capsule was retrieved and opened after 2 h of residence in the stomach (red buffer). (c) Shows the opened capsule after 4 h of exposure to the small intestine (blue buffer) (d–f) UV-vis absorbance spectra of buffer solutions of pH 3 (with red dye), pH 6.8 (with blue dye), and pH 7.4 (colorless) before and after the capsules were retained. | ||
The collected spectra from stomach (red buffer), and small intestine (blue buffer) conditions showed obvious peaks at around 498 nm and 630 nm that were associated with colors red and blue, respectively. There was no noticeable change in the spectra after exposing the capsules to these solutions. Moreover, the UV absorption spectra collected from the final large intestine (clear buffer) conditions showed no detectable peak before and after exposing the capsule to the solution (Fig. 7f). These results confirm the targeted opening and sampling of the capsule within the small intestinal region and proper sealing of the capsule without any sign of leakage or contamination while transitioning in lower sections of the GI tract (e.g., large intestine).
In vitro and ex vivo protein sampling
The BSA results determined by BCA assay and UV-vis spectroscopy (Fig. 8a) exhibited similar extracted concentrations compared to those performed in the buffer (Fig. 6c and e). This similarity in the extracted concentrations suggests the enteric coating was dissolved as expected which resulted in an on-time hydrogel swelling as well as capsule sealing and finally sample capturing performance of the device. In addition, E value was calculated as 3.2% which is consistent with the value achieved from the buffer experiment. For calprotectin also, the in vitro extracted concentrations obtained from the normal calprotectin levels of testing environments (<100 μg mL−1) match the extracted concentrations of the buffer experiment. Moreover, calprotectin's E value (2.6%) was consistent with that of obtained in the buffer experiment, again supporting the reliability of capsule sampling performance (Fig. 8b). | ||
| Fig. 8 In vitro sampling of BSA and calprotectin. Extracted concentrations of both (a) BSA and (b) calprotectin in vitro match the extracted concentrations of buffer practice which was carried out with considering no pH simulation throughout the GI tract. The E values for BSA and calprotectin were still 3.2% and 2.6%, respectively. Error bars represent s.d (n = 3). | ||
The ex vivo sampling-extraction of calprotectin (Fig. 9a–c) was carried out to validate that capsules performance in complex and more physiologically relevant conditions. In practical clinical use, it is envisioned that by measuring the level of extracted inflammation biomarkers from the hydrogel and utilizing the E value (2.6%) conversion factor, one can predict the level of the inflammation biomarkers within the small intestine. In order to validate this assumption, the capsule was placed into two different ex vivo conditions containing low (113 μg mL−1) and high levels (745.5 μg mL−1) of calprotectin to simulate both normal and inflamed conditions in the small intestine. Extracted concentration of calprotectin from the capsules placed in the simulated normal and inflamed conditions were 3.85 μg mL−1 and 27.5 μg mL−1, respectively. By using the E value (2.6% conversion factor), the estimated concentrations of calprotectin in the two test environments were 148.1 and 1057.7 μg mL−1, suggesting a normal and inflamed conditions in the small intestine, respectively. The ex vivo experiment further confirms the reliable performance of the capsule in effective sampling and ability to demonstrate reasonable prediction levels of inflammation within the small intestine region of the GI tract.
 | ||
| Fig. 9 Ex vivo calprotectin sampling and extraction. (a) A segment of small intestine filled with calprotectin loaded intestinal contents with two capsules inside for sampling. (b) Shows capsule retrieval for further extraction. (c) Depicts disassembled capsule with sampled intestinal contents into Tris buffer for 2 h of extraction. (d) Extracted concentrations of two testing environment concentrations with normal and elevated calprotectin concentrations shows consistent E values of 3.4% and 3.6%. | ||
Conclusions
Although fecal calprotectin is commonly used as a non-invasive biomarker for IBD diagnosis, it is unable to differentiate the IBD types (between CD and UC). To the best of our knowledge, this is the first study that shows the use of a non-invasive approach towards IBD type detection through site-selectively sampling of calprotectin in the small intestine. Our ingestible capsule allowed simple sampling of GI fluid within the small intestine without the need of any complex procedures such as sedation, ionizing radiation, or air insufflation. The capsule is made of a 3D printed container that holds superabsorbent hydrogel that provides the required sampling environment as well as the mechanical actuation for closing the capsule after a sampling is completed. The captured calprotectin within the hydrogel is simply retrieved by removing hydrogel from the capsule and placing it in an extraction solution. The capsule's performance in effective sampling of calprotectin was evaluated in three different conditions; i) buffer solutions, ii) in vitro GI-simulated environment, and iii) ex vivo porcine small intestine. Our findings from sampling-extraction experiments indicate a linear correlation between the concentration of extracted calprotectin and its concentration in the sampling environment. This demonstrates the sampling reliability of the capsule and its independence of the sampling environment composition. As a result, our sampling device accompanied with the standard fecal calprotectin assay will enable a promising way not only to diagnose the IBD and differentiate its type, but also to monitor the patient status as a cost-effective disease management tool in the future. We believe that this sampling capsule can potentially open new horizons to different applications in GI tract related disease diagnostics and treatment.Author contributions
R. R., M. V., S. N., and J. W. designed the experiments, S. N. conducted the bibliographic review and capsule fabrication. J. W. and S. N. carried out the protein experiments and enteric coating dissolution tests. U. H. and S. N. designed the 3D model and modified SLA printing. S. S. carried out ATR-FTIR and Raman spectroscopy and analyzed the data. I. W. and S. N. performed the hydrogel characterizations. J. J. provided the porcine intestine for ex vivo testing. S. N. wrote the first draft and prepared the figures. All authors reviewed the manuscript. R. R. and M. V. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Eli Lilly & Company. We thank Thiti Premrudeepreechacharn and Aidan Papay for their assistance on pH-sensitive polymer tests.References
- S. Alatab, S. G. Sepanlou, K. Ikuta, H. Vahedi, C. Bisignano, S. Safiri, A. Sadeghi, M. R. Nixon, A. Abdoli, H. Abolhassani, V. Alipour, M. A. H. Almadi, A. Almasi-Hashiani, A. Anushiravani, J. Arabloo, S. Atique, A. Awasthi, A. Badawi, A. A. A. Baig, N. Bhala, A. Bijani, A. Biondi, A. M. Borzì, K. E. Burke, F. Carvalho, A. Daryani, M. Dubey, A. Eftekhari, E. Fernandes, J. C. Fernandes, F. Fischer, A. Haj-Mirzaian, A. Haj-Mirzaian, A. Hasanzadeh, M. Hashemian, S. I. Hay, C. L. Hoang, M. Househ, O. S. Ilesanmi, N. J. Balalami, S. L. James, A. P. Kengne, M. M. Malekzadeh, S. Merat, T. J. Meretoja, T. Mestrovic, E. M. Mirrakhimov, H. Mirzaei, K. A. Mohammad, A. H. Mokdad, L. Monasta, I. Negoi, T. H. Nguyen, C. T. Nguyen, A. Pourshams, H. Poustchi, M. Rabiee, N. Rabiee, K. Ramezanzadeh, D. L. Rawaf, S. Rawaf, N. Rezaei, S. R. Robinson, L. Ronfani, S. Saxena, M. Sepehrimanesh, M. A. Shaikh, Z. Sharafi, M. Sharif, S. Siabani, A. R. Sima, J. A. Singh, A. Soheili, R. Sotoudehmanesh, H. A. R. Suleria, B. E. Tesfay, B. Tran, D. Tsoi, M. Vacante, A. B. Wondmieneh, A. Zarghi, Z. J. Zhang, M. Dirac, R. Malekzadeh and M. Naghavi, Lancet Gastroenterol. Hepatol., 2020, 5, 17–30 CrossRef.
- U. Mahadevan, C. Robinson, N. Bernasko, B. Boland, C. Chambers, M. Dubinsky, S. Friedman, S. Kane, J. Manthey, J. Sauberan, J. Stone and R. Jain, Inflammatory Bowel Dis., 2019, 25, 627–641 CrossRef.
- T. Sairenji, K. L. Collins and D. V. Evans, Prim. Care, 2017, 44, 673–692 CrossRef.
- D. S. Shouval and P. A. Rufo, JAMA Pediatr., 2017, 171, 999–1005 CrossRef.
- S. Danese, M. Sans and C. Fiocchi, Autoimmun. Rev., 2004, 3, 394–400 CrossRef CAS.
- A. Vedamurthy and A. N. Ananthakrishnan, Gastroenterol. Hepatol., 2019, 15, 72 Search PubMed.
- D. C. Baumgart and S. R. Carding, Lancet, 2007, 369, 1627–1640 CrossRef CAS.
- J. H. Cho and C. Abraham, Annu. Rev. Med., 2007, 58, 401–416 CrossRef CAS.
- P. R. Mangan, L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb and C. T. Weaver, Nature, 2006, 441, 231–234 CrossRef CAS.
- G. P. Christophi, R. Rong, P. G. Holtzapple, P. T. Massa and S. K. Landas, Inflammatory Bowel Dis., 2012, 18, 2342–2356 CrossRef PubMed.
- G. E. Tontini, M. Vecchi, L. Pastorelli, M. F. Neurath and H. Neumann, World J. Gastroenterol., 2015, 21(1), 21 CrossRef.
- D. Laharie, A. Reffet, G. Belleannée, E. Chabrun, C. Subtil, S. Razaire, M. Capdepont and V. de Ledinghen, Aliment. Pharmacol. Ther., 2011, 33, 714–721 CrossRef CAS.
- B. G. Feagan, R. N. Fedorak, E. J. Irvine, G. Wild, L. Sutherland, A. H. Steinhart, G. R. Greenberg, J. Koval, C. J. Wong and M. Hopkins, N. Engl. J. Med., 2000, 342, 1627–1632 CrossRef CAS.
- J. W. McDonald, Y. Wang, D. J. Tsoulis, J. K. MacDonald and B. G. Feagan, Cochrane Database Syst. Rev., 2014,(8), CD003459 Search PubMed.
- S. Saibeni, S. Bollani, A. Losco, A. Michielan, R. Sostegni, M. Devani, G. Lupinacci, L. Pirola, C. Cucino, G. Meucci, G. Basilisco, R. D. Incà and S. Bruno, Dig. Liver Dis., 2012, 44(2), 123–127 CrossRef CAS PubMed.
- S. Willot, A. Noble and C. Deslandres, Inflamm. Bowel Dis., 2011, 17(12), 2521–2526 CrossRef PubMed.
- A. Dignass, J. O. Lindsay, A. Sturm, A. Windsor, J. Colombel, M. Allez, G. D. Haens, A. D. Hoore, G. Mantzaris, G. Novacek, T. Öresland, W. Reinisch, M. Sans, E. Stange, S. Vermeire, S. Travis and G. Van Assche, J. Crohns Colitis, 2012, 6, 991–1030 CrossRef.
- J. P. Gisbert, M. Chaparro and F. Gomollón, World J. Gastroenterol., 2011, 17(30), 3467 CrossRef PubMed.
- W. C. Lim, Y. Wang, J. K. MacDonald and S. Hanauer, Cochrane Database Syst. Rev., 2016,(7), CD008870 Search PubMed.
- E. R. Paine, Gastroenterol. Rep., 2014, 2, 161–168 CrossRef PubMed.
- P. S. Dulai, B. G. Levesque, B. G. Feagan, G. D'Haens and W. J. Sandborn, Gastrointest. Endosc., 2015, 82, 246–255 CrossRef PubMed.
- C. M. Spiceland and N. Lodhia, World J. Gastroenterol., 2018, 24(35), 4014 CrossRef.
- S. Mazzuoli, F. W. Guglielmi, E. Antonelli and M. Salemme, Dig. Liver Dis., 2013, 45, 969–977 CrossRef PubMed.
- M. Schneider, J. Höllerich and T. Beyna, World J. Gastroenterol., 2019, 25, 3538–3545 CrossRef.
- E. Rondonotti, C. Spada, S. Adler, A. May, E. J. Despott, A. Koulaouzidis, S. Panter, D. Domagk, I. Fernandez-Urien, G. Rahmi, M. E. Riccioni, J. E. Van Hooft, C. Hassan and M. Pennazio, Endoscopy, 2018, 50, 423–446 CrossRef PubMed.
- K. Kalantar-Zadeh, K. J. Berean, N. Ha, A. F. Chrimes, K. Xu, D. Grando, J. Z. Ou, N. Pillai, J. L. Campbell and R. Brkljača, Nat. Electron., 2018, 1, 79–87 CrossRef.
- K. Kalantar-Zadeh, N. Ha, J. Z. Ou and K. J. Berean, ACS Sens., 2017, 2, 468–483 CrossRef CAS PubMed.
- G. Cummins, D. E. Yung, B. F. Cox, A. Koulaouzidis, M. P. Y. Desmulliez and S. Cochran, Expert Rev. Gastroenterol. Hepatol., 2017, 11, 1119–1134 CrossRef CAS.
- P. Mojaverian, Drug Dev. Res., 1996, 38, 73–85 CrossRef CAS.
- A. Nemiroski, M. Ryou, C. C. Thompson and R. M. Westervelt, Lab Chip, 2015, 15, 4479–4487 RSC.
- E. S. Boroff and J. A. Leighton, Tech. Gastrointest. Endosc., 2015, 17, 5–11 CrossRef.
- C. M. Caffrey, O. Chevalerias, C. O'Mathuna and K. Twomey, IEEE Pervasive Comput., 2008, 7, 23–29 Search PubMed.
- A.-M. Singeap, C. Stanciu and A. Trifan, World J. Gastroenterol., 2016, 22, 369–378 CrossRef CAS PubMed.
- N. Hosoe, K. Takabayashi, H. Ogata and T. Kanai, J. Dig. Endosc., 2019, 31, 498–507 CrossRef PubMed.
- C. Spada, C. Hassan, M. Campanale and G. Costamagna, Tech. Gastrointest. Endosc., 2015, 17, 19–23 CrossRef.
- J. Cui, X. Zheng, W. Hou, Y. Zhuang, X. Pi and J. Yang, Telemed. e-Health, 2008, 14, 715–719 CrossRef.
- H. Rezaei Nejad, B. C. M. Oliveira, A. Sadeqi, A. Dehkharghani, I. Kondova, J. A. M. Langermans, J. S. Guasto, S. Tzipori, G. Widmer and S. R. Sonkusale, Adv. Intell. Syst., 2019, 1, 1900053 CrossRef.
- J. F. Waimin, S. Nejati, H. Jiang, J. Qiu, J. Wang, M. S. Verma and R. Rahimi, RSC Adv., 2020, 10, 16313–16322 RSC.
- O. M. Nardone, R. Cannatelli, D. Zardo, S. Ghosh and I. Marietta, Ther. Adv. Gastroenterol., 2019, 12, 1756284819863015 CrossRef.
- G. Pineton de Chambrun, P. Blanc and L. Peyrin-Biroulet, Expert Rev. Gastroenterol. Hepatol., 2016, 10, 915–927 CAS.
- F. G. C. E. Penna, R. M. Rosa, P. F. S. Da Cunha, S. C. S. De Souza and M. D. L. De Abreu Ferrari, BMC Gastroenterol., 2020, 20, 1–10 CrossRef PubMed.
- A. Moniuszko, A. Wiśniewska and G. Rydzewska, Przegl. Gastroenterol., 2013, 8, 275–283 Search PubMed.
- A. Derkacz, P. Olczyk and K. Komosinska-Vassev, Dis. Markers, 2018, 2018, 7451946 CrossRef.
- F. S. Lehmann, E. Burri and C. Beglinger, Ther. Adv. Gastroenterol., 2015, 8, 23–36 CrossRef.
- E. L. Barnes and R. Burakoff, Inflammatory Bowel Dis., 2016, 22, 2956–2965 CrossRef PubMed.
- I. Woodhouse, S. Nejati, V. Selvamani, H. Jiang, S. Chittiboyina, J. Grant, Z. Mutlu, J. Waimin, N. S. Abutaleb, M. N. Seleem and R. Rahimi, ACS Appl. Bio Mater., 2021, 4, 5405–5415 CrossRef CAS.
- S. Nejati, S. A. Mirbagheri, J. Waimin, M. E. Grubb, S. Peana, D. M. Warsinger and R. Rahimi, J. Environ. Chem. Eng., 2020, 8(5), 104–109 CrossRef.
- S. Sedaghat, S. Nejati, L. H. Bermejo, Z. He, A. M. Alcaraz, A. Roth, Z. Li, V. G. Pol, H. Wang and R. Rahimi, J. Mater. Chem. C, 2021, 9(42), 14997–15010 RSC.
- M. Ashford and J. Fell, J. Drug Targeting, 1994, 2, 241–257 CrossRef CAS PubMed.
- R. C. Rowe, P. Sheskey and M. Quinn, Handbook of pharmaceutical excipients, Libros Digitales-Pharmaceutical Press, 2009 Search PubMed.
- S. S. Ozturk, B. O. Palsson, B. Donohoe and J. B. Dressman, Pharm. Res., 1988, 5, 550–565 CrossRef CAS PubMed.
- M. Davis, I. Ichikawa, E. J. Williams and G. S. Banker, Int. J. Pharm., 1986, 28, 157–166 CrossRef CAS.
- G. Li, Y. Li, H. Hu, P. Lan and X. He, Text. Res. J., 2013, 83, 2129–2141 CrossRef.
- A. M. Dumitrescu, G. Lisa, A. R. Iordan, F. Tudorache, I. Petrila, A. I. Borhan, M. N. Palamaru, C. Mihailescu, L. Leontie and C. Munteanu, Mater. Chem. Phys., 2015, 156, 170–179 CrossRef CAS.
- S. Sedaghat, M. M. Ahadian, M. Jafarian and S. Hatamie, Ind. Eng. Chem. Res., 2019, 58, 10341–10351 CrossRef CAS.
- G. Dravecz, T. Z. Jánosi, D. Beke, D. Á. Major, G. Károlyházy, J. Erostyák, K. Kamarás and Á. Gali, Phys. Chem. Chem. Phys., 2018, 20, 13419–13429 RSC.
- A. Bouhekka and T. Bürgi, Appl. Surf. Sci., 2012, 261, 369–374 CrossRef CAS.
- P. Larkin, Infrared and Raman spectroscopy: principles and spectral interpretation, Elsevier, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00451d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |