
Achieving selective photocatalytic CO2 reduction to CO on bismuth tantalum oxyhalogen nanoplates†
Xiaoping
Tao‡
a,
Yi
Wang‡
ab,
Jiangshan
Qu
ab,
Yue
Zhao
a,
Rengui
Li
 *a and
Can
Li
*a
*a and
Can
Li
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Zhongshan Road 457, Dalian, 116023, China. E-mail: rgli@dicp.ac.cn; canli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, China
First published on 7th May 2021
Abstract
The photocatalytic conversion of carbon dioxide to fuels presents great promise for storing renewable energy and alleviating global warming. Herein, using the visible-light-responsive semiconductor bismuth tantalum oxyhalogen (Bi4TaO8X, X = Cl, Br) with suitable band structures, we realize the photocatalytic reduction of CO2 to selectively produce CO under visible light without introducing any sacrificial reagents. An isotope-labeling experiment clearly demonstrated that the produced CO originated from CO2 and, additionally, continuous water oxidation for O2 evolution was also detected during photocatalytic CO2 reduction. Further introducing crystal morphology modulation to prepare well-defined nanocrystals enables great enhancement of the photogenerated charge separation performance compared to that of irregular nanoparticles. Moreover, surface modification of the silver nanoparticles deployed as the CO2 reduction cocatalyst evidently facilitates the generation of intermediate species to promote the surface catalytic reaction. This work not only presents a potential semiconductor candidate for photocatalytic CO2 reduction, but it also provides a feasible strategy for designing artificial photosynthetic systems via combining morphology tailoring and suitable cocatalysts.
Artificial photosynthesis mimicking the process in green plants to realize the conversion and storage of solar energy and atmospheric CO2 gas into high-value-added carbon-containing compounds is of profound fundamental importance for coping with global warming and energy problems.1–3 However, the photocatalytic conversion of abundant CO2 using H2O as a proton source offers both promise and challenges, especially given the relatively inert nature of CO2 molecules and the multi-electron reaction process.4,5 Although the use of semiconductor-based photocatalysts for photocatalytic CO2 reduction has received increasing attention since the first pioneering work in 1978,6 the progress has been limited by poor charge separation and sluggish reaction kinetics. To achieve the CO2 reduction reaction using H2O as the proton source, the photogenerated charges in semiconductors must separate, transfer to the surface, and then react with the absorbed molecules, giving rise to the photoassisted splitting of H2O to release oxygen and produce carbon-containing compounds. Additionally, the thermodynamic requirements necessitate semiconductor photocatalysts with suitable band positions to ensure that the photoexcited electrons and holes are endowed with sufficient energy to reduce CO2 and oxidize H2O simultaneously.7
The challenges in photocatalytic CO2 conversion include not only the difficult activation of CO2 and the sophisticated reaction kinetics, but also the notoriously fast recombination and deficient separation of the photogenerated charge carriers of the photocatalysts.2 Endeavors in the past few decades have resulted in the development of strategies including surface decoration of suitable cocatalysts, size control, and surface defect engineering of stoichiometric photocatalysts to promote the efficiency and selectivity of photocatalytic CO2 reduction.8–10 For instance, the decoration of an atomic-cobalt-based cocatalyst on W18O49 nanowires enabled the acceleration of the electron transport and modified the energy configuration, resulting in impressive CO generation.11 It was reported that the assembly of a BiOBr photocatalyst with abundant oxygen vacancies could tailor the electronic band structure and capture photoinduced carriers to benefit the separation and stabilize the intermediates to reduce the activation energy barrier.12 Although progress has been made, the overwhelming majority of published reports so far have concentrated on CO2 reduction in the presence of various easily oxidized reagents including alcohols or amines to donate electrons, producing carbon-containing products at the cost of these sacrificial reagents. Even in the overall reduction of CO2 using H2O as the sole proton source, the detection of water oxidation is scarcely considered, despite the nonnegligible significance of water oxidation in photosynthesis.13 The water oxidation reaction to evolve oxygen or hydroxyl radicals in parallel with the CO2 reduction reaction is recognized as the bottleneck, with a sluggish kinetics and complex proton-coupled electron transfer process.14 Moreover, the detection of oxygen is even harder given the fact that the evolved oxygen and hydroxyl radicals tend to be adsorbed on the surface of photocatalysts and to oxidize the CO2-reduced intermediates and interrupt the reduction; consequently, few reported works have referred to the oxidation reaction so far. Reliable detection of water oxidation was reported on a SiC@MoS2 Z-scheme photocatalyst that could achieve the overall conversion of CO2 with H2O and yielded CH4 and a stoichiometric ratio of O2 at the same time.15 A fundamental reason for the challenging nature of the water oxidation reaction in the overall photocatalytic reduction of CO2 is the relatively inferior photocatalytic water oxidation ability of many semiconductors in terms of thermodynamics and kinetics.
Bismuth tantalum oxygen halogen (Bi4TaO8X, X = Cl, Br) is a class of typical Sillén–Aurivillius compounds with layered perovskite structures that have excellent light harvesting properties.16 We previously reported the fabrication of a series of Bi4TaO8X photocatalysts that are able to achieve photocatalytic hydrogen reduction and the oxygen evolution reaction, especially their decent performance in photocatalytic water oxidation under the irradiation of visible light.17,18 Furthermore, the intrinsic layered structure makes it feasible to achieve morphology tuning as well as efficient photogenerated charge separation. Although Bi4TaO8X photocatalysts possess unique crystalline structures and water oxidation capabilities, little research has focused on this series of materials to date, except one previous study on the conversion of CO2 to CH4 over this semiconductor, which did not mention the detection of water oxidation during the process.19 This shows the need for further insight into the exploration of photocatalytic CO2 reduction on these promising materials.
Herein, we report that the selective photocatalytic conversion of CO2 to CO can be realized on visible-light-responsive bismuth tantalum oxygen halogen under visible light irradiation, and that the oxidation of water to O2 can also be detected simultaneously. Two determining factors, morphology regulation, which significantly tunes the photogenerated charge separation process, and surface decoration of a silver cocatalyst, which promotes the absorption and generation of intermediates, together result in a great enhancement in photocatalytic CO2 reduction, thereby providing a feasible strategy to assemble efficient photocatalysts for the photoassisted reduction of CO2 into valuable chemicals.
The crystalline structure of bismuth tantalum oxygen halogen (Bi4TaO8X, X = Cl, Br) is shown in Fig. 1a. It is composed of slices of [Bi2O2] and layers of [TaO6] octahedra, intercalated with halogen atoms. The intrinsic layered structure is expected to form two-dimensional morphologies, which exhibit superiority in photogenerated charge separation and transfer in photocatalysis.20,21 The Bi4TaO8X was verified to have a band gap of approximately 2.6 eV with the conduction band and valence band located at approximately −0.9 and 1.7 eV vs. NHE (pH = 7), respectively, which could meet the thermodynamic requirements for water oxidation and the reduction of CO2 to multiple products including CO (Fig. 1b). Notably, Bi4TaO8X demonstrated good photocatalytic water oxidation capability under visible light irradiation, making it a candidate for the photocatalytic overall CO2 conversion reaction.
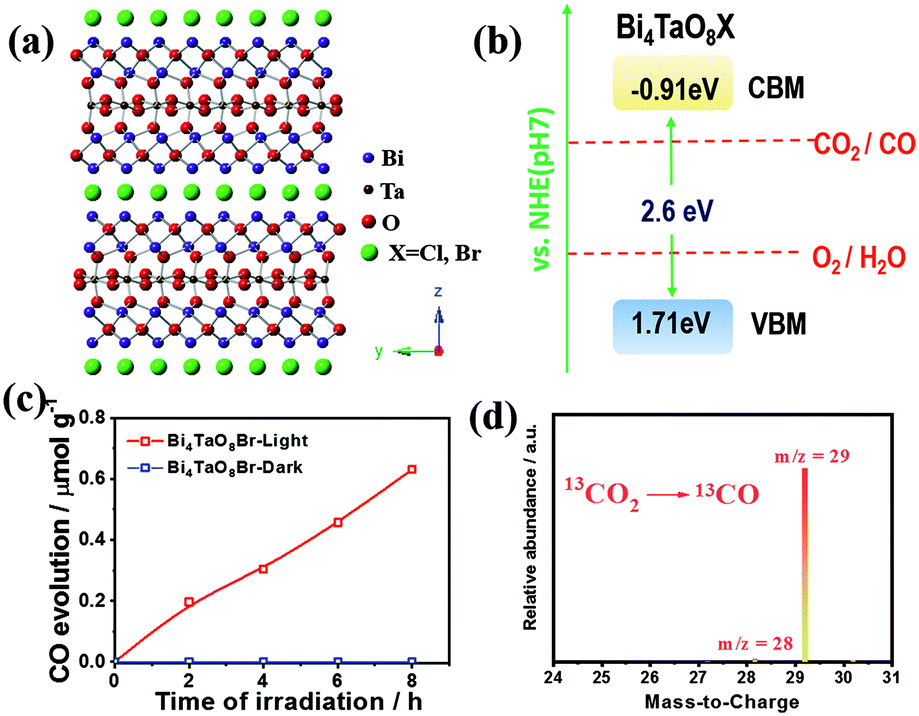 | ||
| Fig. 1 (a) The crystal structure of Bi4TaO8X (X = Cl, Br). (b) A schematic illustration of the band structure of Bi4TaO8X (X = Cl, Br). (c) The time course of photocatalytic CO2 reduction on Bi4TaO8Br. (d) GC-MS analysis of the generated CO using 13CO2 as the feedstock. | ||
Bi4TaO8X (X = Cl, Br) was synthesized via a traditional solid–state reaction, in which stoichiometric amounts of the precursors Bi2O3, Ta2O5, and BiOX were mixed and sealed in a vacuum quartz tube, followed by calcination at 973 K for 14 hours. Using the as-prepared Bi4TaO8Br as the photocatalyst, we found that surprisingly, photocatalytic CO2 reduction could be realized in the presence of H2O without introducing any sacrificial reagents, and CO was observed as the sole product with a selectivity of 100% and no other products being detected (Fig. 1c). To directly track the origin of the produced CO, an isotope labelling experiment using 13CO2 as the input was performed. As shown in Fig. 1d, the result of gas chromatography-mass spectrometry (GC-MS) analysis showed the production of 13CO, clearly evidencing that CO originated from the input CO2, although the photocatalytic activity was not high. Considering that the activity may be limited by the poor charge separation and transfer for Bi4TaO8Br, flux treatment was utilized in the preparation process, and the corresponding sample was labelled as Bi4TaO8Br–F. As shown in Fig. 2a and b, unlike the irregular Bi4TaO8Br nanoparticles, Bi4TaO8Br–F exhibits a nanoplate morphology with a diameter of ∼500 nm (Fig. S1†). XRD patterns of the two samples were consistent with the standard card for the pure phase and showed negligible differences between each other (Fig. S2†). Raman spectra were also collected, and no obvious variation was observed between the spectrum of the Bi4TaO8Br nanoparticles and that of the nanoplates, indicating no changes in the phase or bonding situation during the process of flux treatment (Fig. S3†). UV-visible spectra also depicted a comparable result in the light absorption range (Fig. S4†). However, as shown in Fig. 2c, when the photocatalytic CO2 reduction was conducted under light irradiation, the photocatalytic activity of the flux-treated Bi4TaO8Br nanoplates obviously showed enhanced photocatalytic reduction of CO2 to CO compared to the Bi4TaO8Br nanoparticles.
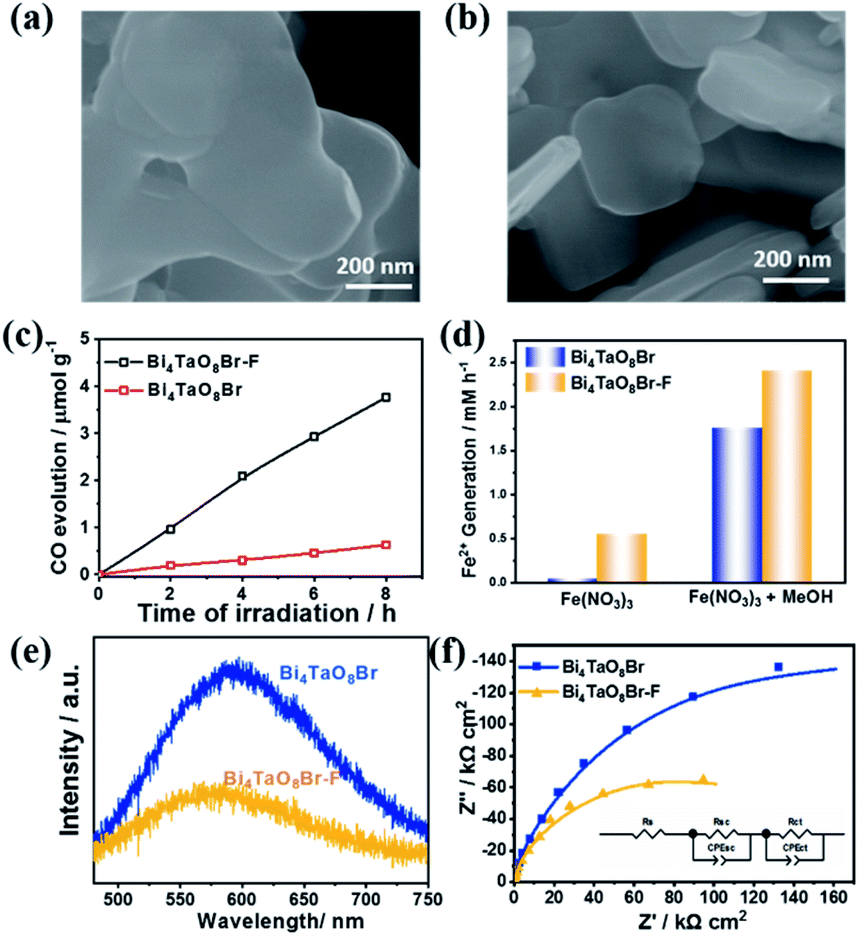 | ||
| Fig. 2 (a and b) SEM images of Bi4TaO8Br and Bi4TaO8Br–F. (c) The time course of photocatalytic CO2 reduction on Bi4TaO8Br and Bi4TaO8Br–F. (d) Photocatalytic reactions on Bi4TaO8Br and Bi4TaO8Br–F in the presence of Fe(NO3)3 and CH3OH. (e) Steady photoluminescence (PL) spectra of Bi4TaO8Br and Bi4TaO8Br–F at room temperature. (f) Electrochemical impedance spectra (EIS) of Bi4TaO8Br and Bi4TaO8Br–F and the corresponding equivalent circuit. | ||
To determine the influence of flux treatment on the photogenerated charge separation, both electron-consuming and hole-consuming reagents were introduced. It was assumed that both charge-consuming reactions were kinetically fast enough. We chose the photocatalytic conversion of Fe3+ to Fe2+ to probe the electrons and the oxidation of CH3OH molecules for capturing holes; the conversion of Fe3+ to Fe2+ can be used as an index for activity evaluation. As demonstrated in Fig. 2d, Bi4TaO8Br–F exhibited superior Fe3+ conversion performance in the presence of CH3OH as a result of the high activity of the oxidation half-reaction. The enhancement compared to Bi4TaO8Br implies that the charge separation efficiency of Bi4TaO8Br–F is higher than that of Bi4TaO8Br synthesized without flux treatment. Additionally, the water oxidation on the surface of Bi4TaO8Br–F is superior to that of Bi4TaO8Br in view of its higher conversion activity without CH3OH. Correspondingly, the steady photoluminescence (PL) spectrum of Bi4TaO8Br–F exhibits a significantly decreased intensity of radiative recombination compared to that of Bi4TaO8Br, implying more inhibited recombination over the same period (Fig. 2e). In addition, electrochemical impedance spectra (EIS) were also carried out to evaluate the charge separation properties for Bi4TaO8Br and Bi4TaO8Br–F. As can be seen in Fig. 2f, the fitting results showed smaller resistance values for Bi4TaO8Br–F in both the space charge region and the process of charge transfer from the semiconductor to the surface reactants compared to those of Bi4TaO8Br (Fig. S5†), indicating better charge separation and transfer properties in Bi4TaO8Br–F. Moreover, the charge separation efficiency calculated from the J–V curves in the presence of the hole acceptor further demonstrated the enhanced photogenerated charge separation in Bi4TaO8Br–F, which parallels the EIS and PL results (Fig. S6†). Similar results were also obtained for Bi4TaO8Cl and Bi4TaO8Cl–F (Fig. S7–S9†). The above results demonstrate that flux treatment greatly improves the photogenerated charge separation properties of Bi4TaO8X (X = Cl, Br).
As mentioned above, even if the photogenerated charge separation is satisfactory, the photocatalytic CO2 reaction is limited by the poor surface catalytic reaction. To promote the photocatalytic conversion of CO2 to CO, different kinds of surface cocatalysts (namely, Au, Ag, Pd, Ir, Rh and Cu) were loaded on the surface of Bi4TaO8Br–F, given its robust performance in electrocatalytic CO2 reduction.22,23 As shown in Fig. 3a, decoration with Au, Ag and Pd resulted in enhanced CO evolution compared to that of bare Bi4TaO8Br–F, while the others cocatalysts resulted in negative effects, which might be attributed to the mismatch between the semiconductor and the cocatalysts. Surprisingly, Ag was found to be the best cocatalyst for photocatalytic CO2 reduction on Bi4TaO8Br–F, and further optimization of the loading amount demonstrated a plateau in the CO evolution at a weight of 1%. Various oxide-based cocatalysts, namely, MnOx, CoOx, ZnO2, ZrO2, and MgO were also preliminarily introduced; unfortunately, none of these could work yet (Fig. S10 & S11†). Photocatalytic stability testing of Ag–Bi4TaO8Br–F was also carried out for more than three cycles with each reaction cycle being 4 hours (Fig. 3c). The result suggests that Ag–Bi4TaO8Br–F is able to realize the stable conversion of CO2 to CO, and only a slight decrease was observed after three cycling tests. Furthermore, the water oxidation product was also tracked during the photocatalytic CO2 reduction via a linkage with an online gas detection system. As expected, the appearance of an obvious O2 peak was observed via GC, and the intensity of the signal gradually increased with the reaction time, as shown in Fig. 3d. Although the amount of O2 is still very low and the production of CO could not be analysed simultaneously, the significant O2 peak strongly verified that the water oxidation half-reaction proceeded, which means the overall photocatalytic CO2 reduction with H2O could indeed take place on the semiconductor Ag–Bi4TaO8Br–F. Notably, given the low solubility of CO2 in water, all the photocatalytic experiments were conducted in a saturated KHCO3 solution; the photocatalytic performance in pure water was also demonstrated in Fig. S12.†
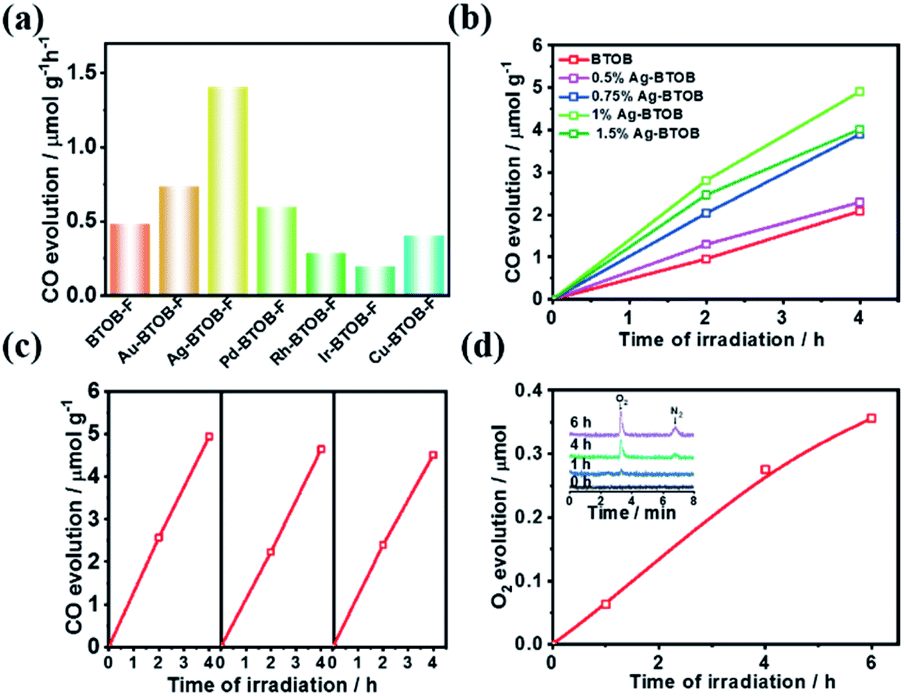 | ||
| Fig. 3 (a) The photocatalytic activity of CO2 reduction on Bi4TaO8Br–F loaded with different cocatalysts. (b) The time courses of photocatalytic CO2 reduction on Bi4TaO8Br–F loaded with different amounts of Ag. (c) Cycling tests of photocatalytic CO2 reduction on Ag–Bi4TaO8Br–F. (d) The time course of oxygen evolution during photocatalytic CO2 reduction on Ag–Bi4TaO8Br–F under UV light irradiation; inset: GC signals of O2 and N2 after different reaction times. | ||
The roles of the Ag cocatalyst in the photocatalytic reaction were then investigated to better understand the reaction mechanism. As can be seen in Fig. 4a, Ag nanoparticles with diameters of ∼5–20 nm were dispersed on the surface of the Bi4TaO8Br–F nanoplates (Fig. S13 & S14†). High-resolution transmission electron microscopy (HRTEM) provided great insight into the surface microstructure of Ag-loaded Bi4TaO8Br–F, as shown in Fig. 4b. The clear lattice fringes indicate the good crystallinity of both the cocatalyst and the semiconductor, where the lattice spacing of 0.233 nm is consistent with the (111) lattice plane of metallic Ag, while the observed lattice distance of 0.381 nm corresponds well with the (110) facet of Bi4TaO8Br. The Ag nanoparticles were in intimate contact with the semiconductor, which is conducive to better synergy. In addition, the chemical states of the Ag cocatalyst as well as the interaction between Ag and Bi4TaO8Br–F were investigated using XPS (X-ray photoelectron spectroscopy) (Fig. 4c). The XPS spectra of Bi, Ta and O showed slight shifts to higher bonding energy, which could be due to electron transfer from Bi4TaO8Br to Ag, implying intimate contact between Bi4TaO8Br and Ag (Fig. S15†).24 In addition, the fine spectrum of Ag showed the coexistence of peaks at 373.8 and 367.8 eV, which is due to spin–orbit splitting according to the literature, indicating the presence of the metallic state of Ag0, which agrees well with the HRTEM analysis.25,26 The Ag nanoparticles on the surface show some aggregation after the cycling reaction, and the structural characterizations of Ag–Bi4TaO8Br–F after the photocatalytic reactions indicate that it maintained similar phase and chemical states on the surface to the fresh material (Fig. S16–S18†). Furthermore, the influence of Ag on the CO2 absorption was also investigated, as shown in Fig. 4d, in which surface loading of the Ag cocatalyst resulted in obviously enhanced absorption of CO2 molecules compared to pristine Bi4TaO8Br, implying that the Ag on the surface could facilitate the process of CO2 absorption.
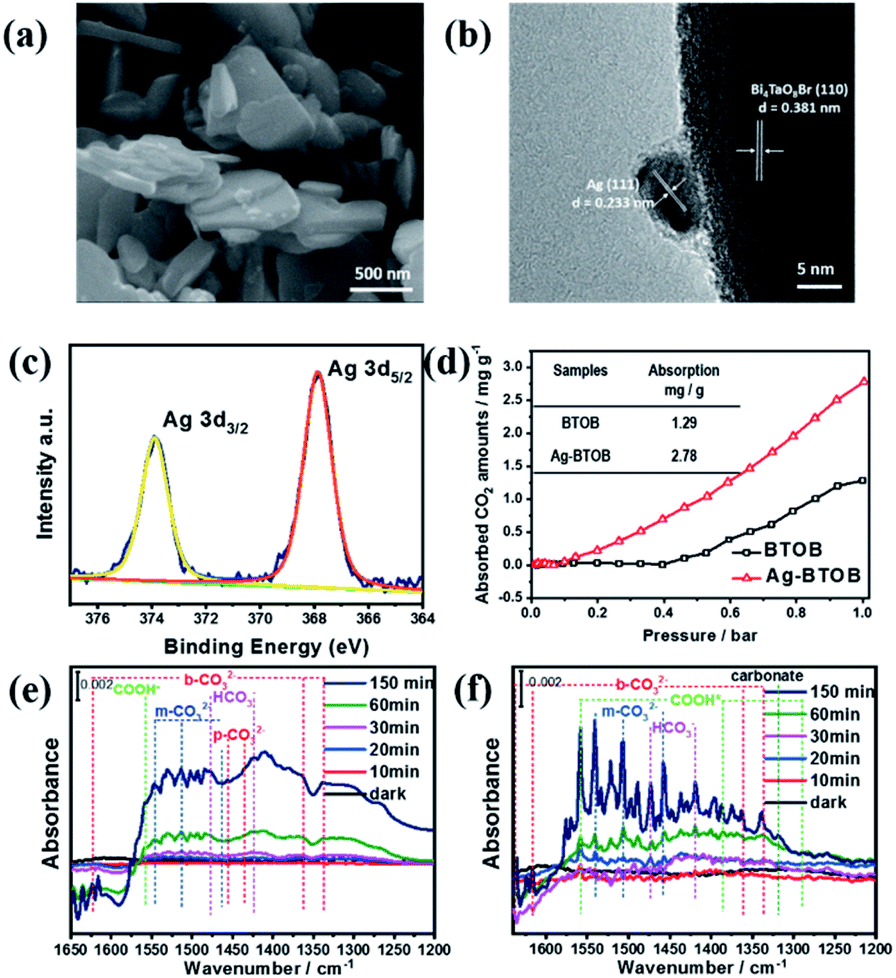 | ||
| Fig. 4 (a and b) SEM and HRTEM image of Bi4TaO8Br–F loaded with Ag. (c) The XPS fine spectrum of silver in Ag–Bi4TaO8Br–F. (d) The absorption curves of CO2 on Bi4TaO8Br–F and Ag–Bi4TaO8Br–F at room temperature. (e and f) In situ FTIR spectra of photocatalytic CO2 reduction on Bi4TaO8Br–F and Ag–Bi4TaO8Br–F. | ||
To explore the possible mechanism of CO2 conversion on Bi4TaO8Br, in situ Fourier Transform Infrared Spectrum (FTIR) was utilized to provide decisive evidence regarding the intermediate products and reaction pathways. Significant CO2 adsorption could be observed from the vibrational mode of (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 2295 and 2380 cm−1 (Fig. S19†). As shown in Fig. 4e and f, various carbon-containing reaction intermediates, including bicarbonate (HCO3−, 1473 and 1419 cm−1), bidentate carbonate (b-CO32−, 1338, 1363, 1616, and 1635 cm−1), and monodentate carbonate (m-CO32−, 1558, 1541, 1508 and 1457 cm−1) corresponding to adsorbed CO2 species were detected during the photocatalytic reaction, and all of these species gradually increased as the reaction proceeded. Compared to bare Bi4TaO8Br–F, the Ag-loaded Bi4TaO8Br–F demonstrated signals with stronger intensities within the same period, which could be attributed to the more intensive of CO2 absorption ability and activation on Ag–Bi4TaO8Br–F. Additionally, the peaks located at 1588, 1386 and 1288 cm−1 were assigned to the symmetric and antisymmetric OCO vibrations and CO stretching of *COOH, respectively,27,28 which is consistent with previous experimental and theoretical studies claiming that the conversion of CO2 to CO usually proceeds via a *COOH intermediate coordinated to the catalyst surface.29,30 As depicted in Fig. 4e and f, the Ag-loaded Bi4TaO8Br–F showed more significant surface intermediate signals compared to bare Bi4TaO8Br–F, which further certifies that the surface assembled with the Ag cocatalyst is favourable for CO2 activation (*COOH formation), thus promoting the overall conversion of CO2 to CO.
O) at 2295 and 2380 cm−1 (Fig. S19†). As shown in Fig. 4e and f, various carbon-containing reaction intermediates, including bicarbonate (HCO3−, 1473 and 1419 cm−1), bidentate carbonate (b-CO32−, 1338, 1363, 1616, and 1635 cm−1), and monodentate carbonate (m-CO32−, 1558, 1541, 1508 and 1457 cm−1) corresponding to adsorbed CO2 species were detected during the photocatalytic reaction, and all of these species gradually increased as the reaction proceeded. Compared to bare Bi4TaO8Br–F, the Ag-loaded Bi4TaO8Br–F demonstrated signals with stronger intensities within the same period, which could be attributed to the more intensive of CO2 absorption ability and activation on Ag–Bi4TaO8Br–F. Additionally, the peaks located at 1588, 1386 and 1288 cm−1 were assigned to the symmetric and antisymmetric OCO vibrations and CO stretching of *COOH, respectively,27,28 which is consistent with previous experimental and theoretical studies claiming that the conversion of CO2 to CO usually proceeds via a *COOH intermediate coordinated to the catalyst surface.29,30 As depicted in Fig. 4e and f, the Ag-loaded Bi4TaO8Br–F showed more significant surface intermediate signals compared to bare Bi4TaO8Br–F, which further certifies that the surface assembled with the Ag cocatalyst is favourable for CO2 activation (*COOH formation), thus promoting the overall conversion of CO2 to CO.
Based on the above results, a plausible mechanism for the photocatalytic conversion of CO2 to CO on Ag–Bi4TaO8Br–F was proposed (Scheme 1). Bi4TaO8Br was irradiated under visible light to generate electrons and holes with high energy, which could effectively separate and transfer to the surface due to the layer-structured properties with superior charge separation capability. The reduction of CO2 to CO on the surface usually consists of two elementary steps, the first of which is the reductive adsorption of CO2 on the catalyst surface involving a proton-coupled electron transfer process to form a *COOH intermediate, where * indicates the corresponding adsorption states of surface species. The adsorbed *COOH intermediate is further reduced to *CO via a concerted electron–proton transfer, and then desorbed from the catalyst surface. Ag has been shown to be an ideal electrocatalyst endowed with an appropriate adsorption strength for the key intermediate *COOH,31–33 and the cocatalyst on the surface with a relatively low Fermi level has also been proven to be capable of electron trapping and to further enhance the photogenerated charge separation,34 which is assumed to promote the reduction of CO2 to CO. Given the capability of Bi4TaO8Br for photocatalytic water oxidation, the water oxidation half-reaction to consume photogenerated holes is expected to take place on the surface of Bi4TaO8Br. Ultimately, the loading of the Ag cocatalyst promotes the CO2 reduction reaction on the surface, combined with morphology tuning, which promotes the process of photogenerated charge separation and transport, contributes to the realization of overall photocatalytic CO2 conversion on the Bi4TaO8Br photocatalyst.
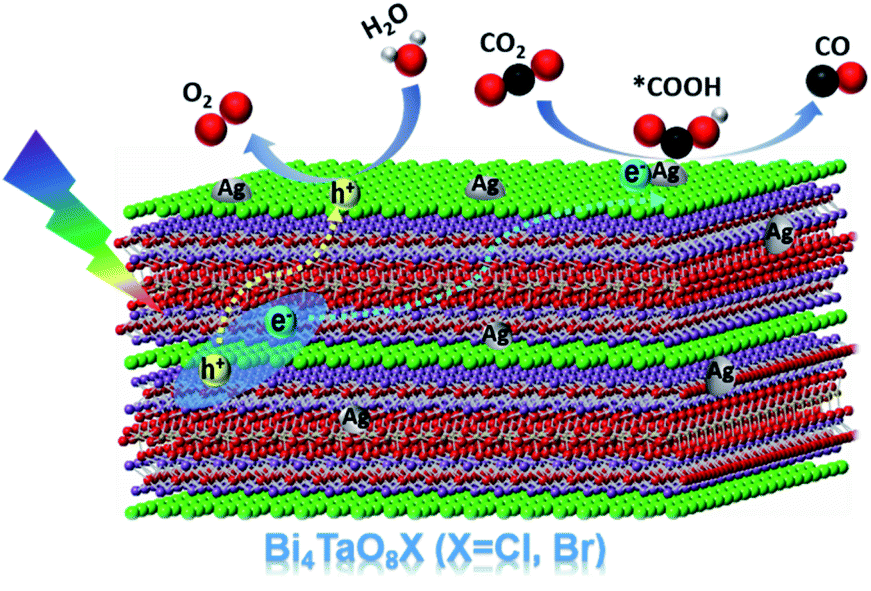 | ||
| Scheme 1 The proposed mechanism of photocatalytic CO2 reduction to CO on Bi4TaO8X. | ||
Conclusions
In conclusion, we have presented the achievement of the photocatalytic reduction of CO2 to CO on the semiconductor Bi4TaO8X (X = Cl, Br) using H2O as the electron donor under visible light irradiation. During the photocatalytic reduction of CO2 by the photogenerated electrons, the oxidation of H2O to O2 by photogenerated holes takes place simultaneously. Through the morphology modulation, the flux-treated nanoplates demonstrate greatly enhanced charge separation and transfer properties compared to irregular nanoparticles. Further decoration with the cocatalyst Ag was proven to be favourable for the adsorption and activation of CO2 and the generation of key intermediates, further promoting the overall CO2 reduction reaction. This work not only presents a potential semiconductor candidate for photocatalytic CO2 reduction, but also provides a feasible strategy combining morphology tailoring and appropriate cocatalysts in the hope of designing and recreating efficiently implemented artificial photosynthetic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22088102, 22090033) and Key Research Program of Frontier Sciences of Chinese Academy of Sciences (QYZDY-SSW-JSC023).References
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 Search PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 Search PubMed.
- D. Li, B. Cen, C. Fang, X. Leng, W. Wang, Y. Wang, J. Chen and M. Luo, New J. Chem., 2021, 45, 561–568 Search PubMed.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 21625–21649 Search PubMed.
- X. Li, S. Wang, L. Li, X. Zu, Y. Sun and Y. Xie, Accounts Chem. Res., 2020, 53, 2964–2974 Search PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 Search PubMed.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Solar RRL, 2020, 4, 1900546 Search PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 Search PubMed.
- D. Chen, X. Zhang and A. F. Lee, J. Mater. Chem. A, 2015, 3, 14487–14516 Search PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 Search PubMed.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. D. Lou, J. Am. Chem. Soc., 2021, 143, 2173–2177 Search PubMed.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She, J. Xia, H. Li and Z. Liu, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 Search PubMed.
- Y. Wang and T. He, J. Mater. Chem. A, 2021, 9, 87–110 Search PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 Search PubMed.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 Search PubMed.
- A. M. Kusainova, W. Zhou, J. T. S. Irvine and P. Lightfoot, J. Solid State Chem., 2002, 166, 148–157 Search PubMed.
- X. Tao, Y. Zhao, L. Mu, S. Wang, R. Li and C. Li, Adv. Energy Mater., 2018, 8, 1701392 Search PubMed.
- X. Tao, Y. Gao, S. Wang, X. Wang, Y. Liu, Y. Zhao, F. Fan, M. Dupuis, R. Li and C. Li, Adv. Energy Mater., 2019, 9, 1803951 Search PubMed.
- L. Li, Q. Han, L. Tang, Y. Zhang, P. Li, Y. Zhou and Z. Zou, Nanoscale, 2018, 10, 1905–1911 Search PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917 Search PubMed.
- D. Qin, Y. Zhou, W. Wang, C. Zhang, G. Zeng, D. Huang, L. Wang, H. Wang, Y. Yang, L. Lei, S. Chen and D. He, J. Mater. Chem. A, 2020, 8, 19156–19195 Search PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 Search PubMed.
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 Search PubMed.
- B. Zeng, S. Wang, Y. Gao, G. Li, W. Tian, J. Meeprasert, H. Li, H. Xie, F. Fan, R. Li and C. Li, Adv. Funct. Mater., 2021, 31, 2005688 Search PubMed.
- T. Takayama, H. Nakanishi, M. Matsui, A. Iwase and A. Kudo, J. Photochem. Photobiol., A, 2018, 358, 416–421 Search PubMed.
- G. B. Hoflund, J. F. Weaver and W. S. Epling, Surf. Sci. Spectra, 1994, 3, 151–156 Search PubMed.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 Search PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 Search PubMed.
- H. Xie, S. Chen, F. Ma, J. Liang, Z. Miao, T. Wang, H. L. Wang, Y. Huang and Q. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36996–37004 Search PubMed.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 Search PubMed.
- J. Chen, Z. Wang, H. Lee, J. Mao, C. A. Grimes, C. Liu, M. Zhang, Z. Lu, Y. Chen and S. P. Feng, Mater. Today Phys., 2020, 12, 100176 Search PubMed.
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 Search PubMed.
- Z. Wang, J. Fan, B. Cheng, J. Yu and J. Xu, Mater. Today Phys., 2020, 15, 100279 Search PubMed.
- F. Zhang, Y.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2020, 268, 118380 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02504j |
| ‡ These two authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |