
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlating axial and equatorial ligand field effects to the single-molecule magnet performances of a family of dysprosium bis-methanediide complexes†
Lewis R.
Thomas-Hargreaves
 ,
Marcus J.
Giansiracusa
,
Matthew
Gregson
,
Emanuele
Zanda
,
Felix
O'Donnell
,
Ashley J.
Wooles
,
Nicholas F.
Chilton
* and
Stephen T.
Liddle
*
,
Marcus J.
Giansiracusa
,
Matthew
Gregson
,
Emanuele
Zanda
,
Felix
O'Donnell
,
Ashley J.
Wooles
,
Nicholas F.
Chilton
* and
Stephen T.
Liddle
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: steve.liddle@manchester.ac.uk; nicholas.chilton@manchester.ac.uk
First published on 2nd March 2021
Abstract
Treatment of the new methanediide–methanide complex [Dy(SCS)(SCSH)(THF)] (1Dy, SCS = {C(PPh2S)2}2−) with alkali metal alkyls and auxillary ethers produces the bis-methanediide complexes [Dy(SCS)2][Dy(SCS)2(K(DME)2)2] (2Dy), [Dy(SCS)2][Na(DME)3] (3Dy) and [Dy(SCS)2][K(2,2,2-cryptand)] (4Dy). For further comparisons, the bis-methanediide complex [Dy(NCN)2][K(DB18C6)(THF)(toluene)] (5Dy, NCN = {C(PPh2NSiMe3)2}2−, DB18C6 = dibenzo-18-crown-6 ether) was prepared. Magnetic susceptibility experiments reveal slow relaxation of the magnetisation for 2Dy–5Dy, with open magnetic hysteresis up to 14, 12, 15, and 12 K, respectively (∼14 Oe s−1). Fitting the alternating current magnetic susceptibility data for 2Dy–5Dy gives energy barriers to magnetic relaxation (Ueff) of 1069(129)/1160(21), 1015(32), 1109(70), and 757(39) K, respectively, thus 2Dy–4Dy join a privileged group of SMMs with Ueff values of ∼1000 K and greater with magnetic hysteresis at temperatures >10 K. These structurally similar Dy-components permit systematic correlation of the effects of axial and equatorial ligand fields on single-molecule magnet performance. For 2Dy–4Dy, the Dy-components can be grouped into 2Dy–cation/4Dy and 2Dy–anion/3Dy, where the former have almost linear C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) DyC units with short average DyC distances, and the latter have more bent CDyC units with longer average DyC bonds. Both Ueff and hysteresis temperature are superior for the former pair compared to the latter pair as predicted, supporting the hypothesis that a more linear axial ligand field with shorter M–L distances produces enhanced SMM properties. Comparison with 5Dy demonstrates unusually clear-cut examples of: (i) weakening the equatorial ligand field results in enhancement of the SMM performance of a monometallic system; (ii) a positive correlation between Ueff barrier and axial linearity in structurally comparable systems.
DyC units with short average DyC distances, and the latter have more bent CDyC units with longer average DyC bonds. Both Ueff and hysteresis temperature are superior for the former pair compared to the latter pair as predicted, supporting the hypothesis that a more linear axial ligand field with shorter M–L distances produces enhanced SMM properties. Comparison with 5Dy demonstrates unusually clear-cut examples of: (i) weakening the equatorial ligand field results in enhancement of the SMM performance of a monometallic system; (ii) a positive correlation between Ueff barrier and axial linearity in structurally comparable systems.
Introduction
Lanthanide (Ln) Single Molecule Magnets (SMMs) are of burgeoning interest due to their potential applications in high density storage and quantum computing.1 Following the discovery that single Ln-ions can function as effective SMMs,2 there has been a huge development in the field.3 Ln-based SMMs have been amenable to systematic improvement by optimisation of the crystal field (CF) generated by the coordination environment in order to best stabilise the most magnetic projections of the spin–orbit coupled total angular momentum (mJ states).4 This approach has permitted design of large barriers to magnetisation reversal, Ueff, over which magnetic relaxation occurs with an Arrhenius-like exponential temperature dependence, and thus larger Ueff values should lead to slower magnetic relaxation at a given temperature. This is well established for DyIII, where near-linear coordination environments stabilise the mJ = |±15/2〉 ground state.1b,3c,5 The preparation and computational investigation of prepared compounds has been instrumental in reinforcing and developing the theory behind SMMs, and yet very few studies have explicitly probed the correlation between axial and equatorial CF effects.5gThe systematic effect of equatorial donors has been shown in polymetallic cyclopentadienyl systems by descending the group 15/16 elements,6 but the only monometallic examples are that of substitution of a chloride for a bromide in [Dy(bbpen)X] (where X = Cl, Br; H2bbpen = N,N-bis(2-hydroxybenzyl)-N,N-bis(2-methylpyridyl)ethylenediamine) and [Dy(Mes*O)2(THF)2X] (where X = Cl, Br and I and Mes* = 2,4,6-tri-tert-butylphenyl).3b,3l There have also been further investigations into the effect of ligand properties on magnetic performance7 with Pc ((C6H4C2N)4N4),4c CpR systems,8 and a recent extensive study of axiality in pentagonal bipyramidal alkoxide SMMs, as particular highlights.9 Each of these studies demonstrated the effect of increased axial donor strength on magnetic properties, however none have specifically correlated the effect of axial linearity within a series of comparable systems. Furthermore, a recent study demonstrated that equatorial sulfur donors enabled Ueff barriers as high as 638 K, with computational investigations showing that heavier group 16 elements would likely further increase the barrier.10 To date, the effect of axial linearity on magnetic performance has only ever been modelled computationally or observed as a general qualitative trend for incomparable systems.4e
A recent breakthrough has been the advent of dysprosocenium cations [Dy(CpR)(CpR’)]+ (Cp = cyclopentadienyl), which have Ueff values ranging from 1760 to 2217 K and record zero field cooled (ZFC)/field cooled (FC) (TB1), hysteresis measurement (TH), and 100 second relaxation (TB2) blocking temperatures of 52, 80, and 67 K, respectively.3d,8,11 The vastly improved SMM properties of the dysprosocenium cations are thought to be due to the constrained vibrational modes of the five-membered Cp rings,3d,3j,8,12 suggesting more rigid ligand environments are beneficial. Indeed, some of us have recently suggested that quantum tunnelling of the magnetisation (QTM), which is responsible for fast relaxation at zero field and has been a blight on Ln-based SMMs, could be enhanced by flexible ligand environments, and thus ligand rigidity seems key to improving performance.3c,3d,12c,13
There are now an increasing number of Ln SMMs in the literature with Ueff barriers over 1000 K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3b–d,5d,8,9,11,14 but few systematic magneto-structural studies of coordination geometry.3l,8 Whilst this suggests that the current level of understanding of SMM behaviour is effective, methodical testing and evaluation of coordination geometry is required to develop the properties of SMMs to be functional at practical temperatures. This is particularly important for compounds that are not part of the successful families of dysprosocenium cations or pentagonal bipyramidal SMMs if the scope of the field is to be systematically expanded.
3b–d,5d,8,9,11,14 but few systematic magneto-structural studies of coordination geometry.3l,8 Whilst this suggests that the current level of understanding of SMM behaviour is effective, methodical testing and evaluation of coordination geometry is required to develop the properties of SMMs to be functional at practical temperatures. This is particularly important for compounds that are not part of the successful families of dysprosocenium cations or pentagonal bipyramidal SMMs if the scope of the field is to be systematically expanded.
We have been attempting to prepare DyIII SMMs that feature large Ueff barriers as well as rigid multi-dentate ligands. In previous work we constructed an SMM with trans-methanediide (formally C2−) donors supported with neutral imido donors in the equatorial plane, viz. [Dy(NCN)2][K(18C6)(THF)2] (a-Dy) (NCN = {C(PPh2NSiMe3)2}2−, 18C6 = 18-crown-6 ether), which has Ueff = 721 and 813 K with TB1/TH = 10 K (TB2 not measured).5c In order to improve this system, we proposed analogues of NCN where the equatorial imido donors were replaced with softer sulfur donors, therefore reducing the strength of the equatorial interaction. Here, we report a structurally similar ligand SCS = {C(PPh2=S)2}2− where the NSiMe3 groups have been replaced with softer S donors, and this family of molecules permits a systematic magneto-structural investigation and correlation of axial/linear and equatorial ligand field effects on the SMM performances of these complexes. Our combined experimental and computational investigation reveals that this replacement increases Ueff on the order of 40%, and increases to the TB behaviour by several K. Furthermore, a positive correlation between linearity and Ueff barrier is specifically demonstrated here for the first time within comparable systems.
Results
Synthesis
The SCS ligand has been used extensively throughout the d- and f-blocks, including previous reports of bis-SCS Ln complexes of Sm and Tm.15 Those compounds were prepared by reaction of SCS–Li2 with LnI3THF3.5 (Ln = Sm, Tm) via salt elimination. However, in order to introduce more modular variation of the alkali metal, since this was anticipated to provide greater opportunities for the structural variations needed to underpin a magneto-structural correlation study, we adapted the alkane elimination route previously used in our NCN work to the synthesis of the bis-SCS Ln complexes reported here, Scheme 1.5c Accordingly, treatment of LnI3THF3.5 with three equivalents of KCH2Ph and a sub-stoichiometric amount of SCSH2 produced the heteroleptic methanediide-methanide complex [Dy(SCS)(SCSH)(THF)] (1Dy), which was isolated as colourless crystals in 90% yield with toluene as a byproduct. A sub-stoichiometric amount of SCSH2 ensures the clean isolation of pure 1Dy since any unreacted SCSH2 has a very similar solubility to 1Dy and is thus impracticable to separate during work-up. | ||
| Scheme 1 Synthetic routes to the bis-methanediide complexes 1Ln–4Ln, Ln = Dy, Y, Gd. The synthesis of 5Dy is very similar, utilising H2C(PPh2NSiMe3)2 instead of H2C(PPh2S)2 and dibenzo-18-crown-6 ether as the auxiliary ligand. | ||
With 1Dy secured, the target bis-methanediide derivatives were prepared by deprotonation with MCH2Ph reagents (M = Na, K) in the presence of auxiliary ethers (DME or 2,2,2-cryptand), yielding colourless crystalline [Dy(SCS)2][Dy(SCS)2(K(DME)2)2] (2Dy), [Dy(SCS)2][Na(DME)3] (3Dy) and [Dy(SCS)2][K(2,2,2-cryptand)] (4Dy) in isolated crystalline yields of 81, 73, and 72%, respectively.
For completeness and to aid characterisation, we prepared diamagnetic 1Y–4Y which are largely isostructural to their Dy-analogues except for 4Y which contains a molecule of coordinated THF that is not present in 4Dy. The 31P NMR spectra are particularly diagnostic in these systems, with methanide and methanediide resonances observed for 1Y at ∼33 and ∼14 ppm, respectively, whilst compounds 2Y, 3Y, and 4Y all demonstrate a single methanediide resonance at ∼14 ppm indicating equivalent SCS ligands in solution on the NMR timescale. The magnetic data of 2Dy–4Dy are modelled well by the ab initio calculations (vide infra), suggesting minimal influence of intermolecular forces on the magnetic behaviour of these complexes and thus dilution studies were not required. Complexes 1Y–4Y are therefore not discussed any further but details are included in the ESI for completeness.
Since Dy is not amenable to electron paramagnetic resonance (EPR) studies, we prepared 3Gd from 1Gd so that the isotropic Gd ion in 3Gd could be probed to determine its crystal field parameters spectroscopically as a proxy to the Dy-congeners.
Lastly, for further comparisons, the bis-methanediide complex [Dy(NCN)2][K(DB18C6)(THF)(toluene)] (5Dy, NCN = {C(PPh2NSiMe3)2}2−, DB18C6 = dibenzo-18-crown-6 ether) was prepared; its synthesis largely followed the same strategy as the preparations of 3Dy, 4Dy, and a-Dy and is unremarkable.
Solid state structures
In order to verify the formulations of the complexes reported here we determined their solid-state structures by X-ray diffraction; the structures of 2Dy–4Dy are shown in Fig. 1–3 and Table 1 and details of the structural determinations of 1Dy, 3Gd, 5Dy, and 1Y–4Y can be found in the ESI. Compounds 3Dy and 4Dy comprise 6-coordinate C2S4-coordinated Dy complexes as discrete anions, while 2Dy exhibits two discrete Dy-components as a separated ion pair (hereafter referred to as 2Dy–anion and 2Dy–cation). The 7-coordinate 1Dy precursor has an additional THF coordinated to Dy, and so the resulting C2S4O-coordination diverges from the axial, linear coordination environment that is sought. For 5Dy, the steric bulk of SiMe3 groups on the NCN ligand prevents coordination of group 1 metals and orthogonally locks the two methanediides to give a C2N4-coordinated Dy ion. The main difference of 5Dy compared to a-Dy is the K-crown component, but as will be seen these two complexes are magnetically different.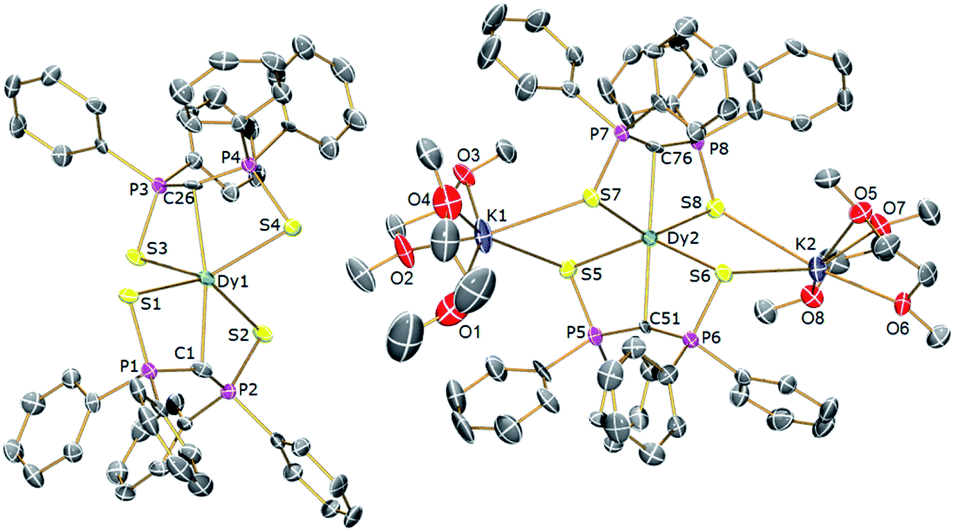 | ||
| Fig. 1 Solid-state structures of 2Dy at 150 K with selective labelling and displacement ellipsoids set at 40% probability. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
 | ||
| Fig. 2 Solid-state structures of 3Dy at 150 K with selective labelling and displacement ellipsoids set at 40% probability. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
 | ||
| Fig. 3 Solid-state structures of 4Dy at 150 K with selective labelling and displacement ellipsoids set at 40% probability. Hydrogen atoms and minor disorder components are omitted for clarity. | ||
| 2Dy–anion | 2Dy–cation | 3Dy | 4Dy | 5Dy | |
|---|---|---|---|---|---|
| CDyC/° |
166.1(3) | 178.6(2) | 164.01(11) | 176.03(11) | 176.45(9) |
| CDy/Å |
2.432(7), 2.409(8) | 2.415(7), 2.390(7) | 2.449(3), 2.407(3) | 2.381(4), 2.387(3) | 2.434(6), 2.431(6) |
Typically, bis-SCS Ln complexes exhibit CLnC angles of ∼166°; which is most likely due to only partial transfer of electron density from the methanediide centre to the Ln ion, which tends to produce trigonal pyramidalised carbon centres rather than trigonal planar ones.15 This is clearly observed in 3Dy which shows different CDy bond lengths of 2.407(3) and 2.449(3) Å, with the latter tending towards a methanide geometry around the carbon centre. This is also demonstrated by the greater Dy–S–C–S torsion angle of 23° for the longer CDy bond in comparison to 16° for the shorter bond, with the same effect observed for 2Dy–anion. In contrast, the 2Dy–cation and 4Dy both have near linear CDyC angles of 178.6(2)° and 176.03(11)°, respectively. In the case of 2Dy–cation, this is explained by the coordination of K ions to a S atom from each of the Dy(SCS)2 moieties, which locks the two SCS ligands into an orthogonal arrangement. A similar effect from the SiMe3 groups is observed in 5Dy (CDyC angle 176.45(9)°), which sterically locks the BIPM ligands orthogonally to each other. However, 4Dy also displays a large CDyC angle (176.03(11)°), despite the apparent lack of K-coordination or interlocking, and this is most likely due to crystal packing effects fortuitously producing the desired geometry. As expected, the C–DyC angle in 7-coordinate 1Dy is far from linear at 142.76(15)° due to the additional coordinating THF.
The 1Dy Dy–C and DyC bond lengths display a clear distinction between the methanide (2.757(4) Å) and methanediide distances (2.326(5) Å). The SCS ligand is able to form a short CDy bond in this case due to the elongation of the weaker methanide ligand trans to it, which reduces steric repulsion between the ligands and allows the methanediide ligand to approach closer to the metal. Comparing the DyC bond lengths of 2Dy–cation (2.391(7) and 2.415(7) Å) and 2Dy–anion (2.409(8) and 2.432(7) Å) we find that they are statistically indistinguishable, despite the difference in CDyC bond angle. The DyC bond lengths of 3Dy are comparable to that of 2Dy–anion, however, possibly due to a further 2° deviation from linearity, a longer DyC bond is now statistically distinguishable (2.449(3) Å vs. 2.407(3) Å). The DyC bond lengths in 4Dy (2.381(4) and 2.387(3) Å) are remarkably symmetric in comparison to the structures in this series, and are shorter on average than those in 3Dy, but statistically indistinguishable to those in 2Dy–cation.
For 5Dy, the CDy bond lengths are significantly longer than those in 4Dy, both of which have a CDyC angle of around 176°. This can be attributed to a result of the steric bulk of the NCN ligand compared to SCS, which can be intuitively recognised as preventing ligands trans to one another from forming closer contacts with the metal centre. However, shorter DyC distances in 4Dy compared to 5Dy could also be due to reduced donation of electron density from S to Dy in 4Dy compared to N to Dy donation in 5Dy. All Dy–S bond lengths are within the range of 2.74–2.87 Å and whilst those of 1Dy are longer on average, there is otherwise no clear trend. As expected, the Dy–N bonds in 5Dy are much shorter at 2.462(2)–2.516(2) Å.
EPR spectroscopic characterisation
Low temperature EPR spectroscopy is an excellent probe of paramagnetic ground states and local symmetry resulting from CF effects. Unfortunately, transitions involving the well-isolated ground mJ = |±15/2〉 Kramers doublet of high-performance Dy SMMs are not possible, and therefore we examined isostructural 3Gd to probe the axiality of the CF. The EPR spectrum at Q-band frequency (33.95491 GHz) for 3Gd at 5 K, Fig. 4, gives a highly featured spectrum that was modelled using Hamiltonian eqn (1) in PHI16a with g = 1.983, |D| = 0.11 cm−1, and |E| = 0.0085 cm−1 (|E/D| = 0.08). The sign of the parameters is not obvious from simulation of the spectrum and thus only the magnitudes are reported here.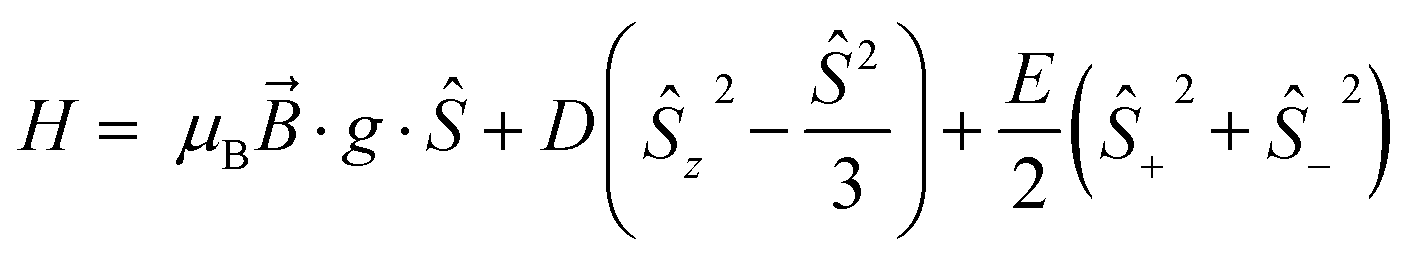 | (1) |
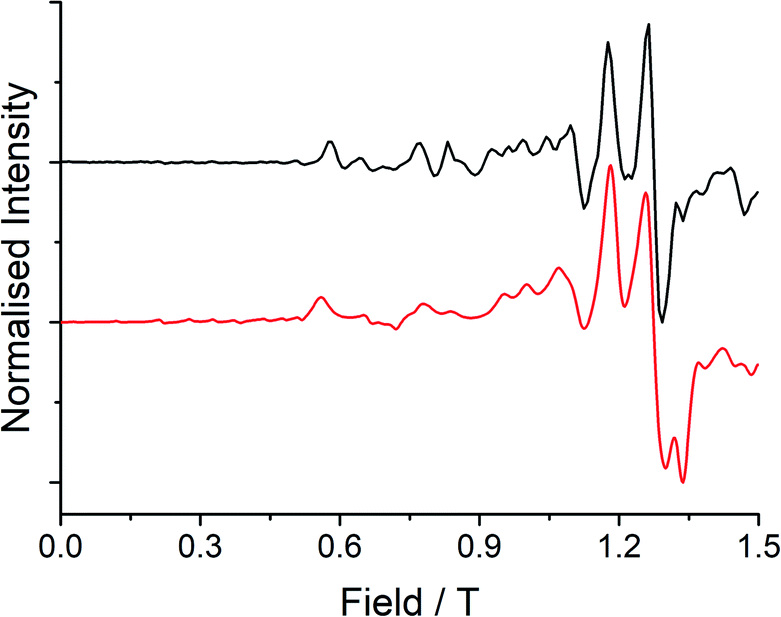 | ||
| Fig. 4 Q-band (33.95491 GHz) EPR spectrum (black line) with simulation (red line) at 5 K of a powdered sample of 3Gd restrained in eicosane. | ||
Observation of a large zero-field splitting (ZFS) for 3Gd is unsurprising given the strongly anisotropic electronic structure of 3Dy (vide infra), however D is somewhat smaller and |E/D| is somewhat larger (i.e. more rhombic) than for [Gd(Cpttt)2][B(C6F5)4], which has |D| = 0.3347 cm−1 and |E| = 0.01629 cm−1 (|E/D| = 0.05).12c This corresponds well with the magnetic results obtained here for 3Dy that show a smaller Ueff than for [Dy(Cpttt)2][B(C6F5)4] (vide infra).3d
Magnetometry
Variable temperature magnetic susceptibility measurements performed on 2Dy approach the room temperature χMT value predicted for two non-interacting DyIII ions (28.3 cm3 mol−1 K), while the data at room temperature for 3Dy–5Dy are consistent with a single DyIII ion (Fig. S15†). In all cases a subtle decrease in χMT with reducing temperature is observed, arising from the depopulation of excited CF states, and an abrupt drop at the lowest temperatures is due to magnetic blocking. Alternating Current (AC) susceptibility measurements16b reveal frequency dependent peaks in the out-of-phase component (χ′′) for 2Dy–5Dy, Fig. 5a, e, i, and Fig. S16–S19,† indicating slow relaxation of the magnetisation, thus classifying all four compounds as SMMs.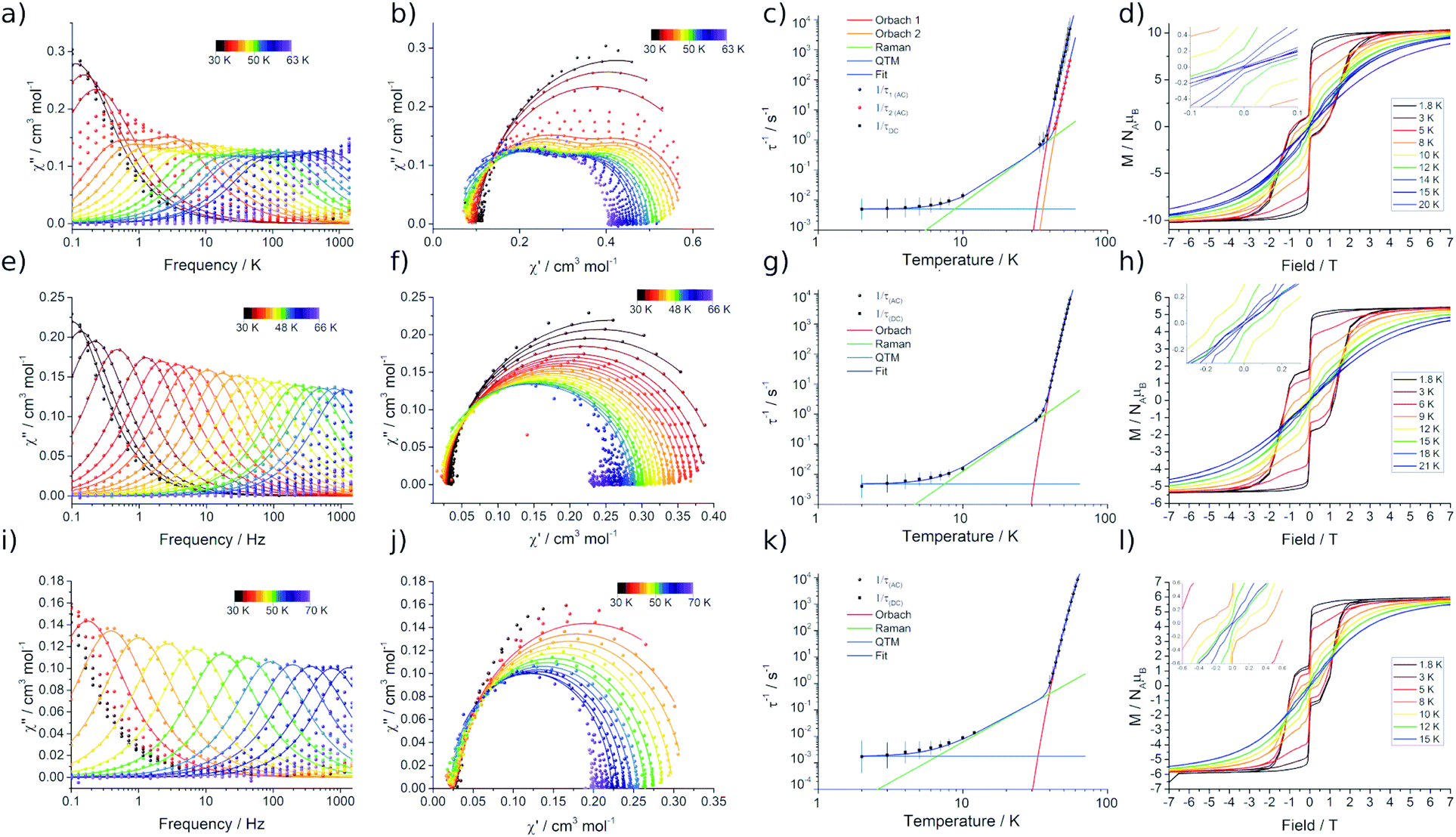 | ||
| Fig. 5 Alternating-current susceptibility (with generalised Debye model fits), Cole–Cole data (data points as coloured dots and fitted curves as lines, same Debye model fits), fitted relaxation data, and magnetic hysteresis measurements (sweep rate of ∼14 Oe s−1, insets show zoom-ins at zero field) for 2Dy–4Dy. (a) 2Dy 30–63 K (34–38, 43–55 K), (b) 2Dy from 30–63 K, (c) 2Dy, (d) 2Dy 1.8–20 K, (e) 3Dy 30–66 K (32–55 K), (f) 3Dy 30–66 K, (g) 3Dy, (h) 3Dy 1.8–15 K, (i) 4Dy 30–70 K (40–62 K), (j) 4Dy 30–70 K, (k) 4Dy, (l) 4Dy 1.8–21 K. | ||
Compound 2Dy displays irregular-shaped peaks in the out-of-phase AC susceptibility, which clearly resolves into two peaks in a Cole–Cole plot at higher temperatures (Fig. 5b, Tables S3 and S4†). Fitting the AC data with the generalised Debye model reveals two distinct relaxation processes above 43 K and a single relaxation process between 34 and 38 K. For both 3Dy and 4Dy, Fig. 5f and j, only one peak in the out-of-phase AC susceptibility is observed (Tables S6 and S7†). Interestingly, the behaviour for 5Dy is different to its isomer a-Dy; multiple relaxation pathways were observed for a-Dy, and yet only a single relaxation process is observed for 5Dy (Fig. S20, Table S10†).5c
In order to probe magnetic relaxation rates at lower temperatures, we also performed direct current (DC) magnetisation decay measurements in zero field. In all cases we find near mono-exponential decay at higher temperatures (>6 K) and a trend towards slightly multi-exponential decay (stretch parameter ca. 0.7) at 2 K (Fig. S21–S24; Tables S5, S8, S9 and S11†). The temperature dependence of the magnetic relaxation rates for 2Dy–5Dy all display three characteristic regimes; at high temperatures there is an exponential (and for 2Dy there are two) relaxation process, at intermediate temperatures there is a power-law process, and at the lowest temperatures there is a near temperature independent process. We assign these three processes to Orbach, Raman and QTM mechanisms, respectively, and the data were fitted with eqn (2) and (3); here we convert the distribution (α) and stretch (β) parameters from the generalised Debye model into estimated standard deviations (esds).17 We suggest that the two distinct exponential regions for 2Dy owe to each of the discrete molecules within the structure, 2Dy–anion and 2Dy–cation, as seen in other multimetallic Dy SMMs.18 Subsequently, we fit the data using two exponential terms, eqn (3). This contrasts to our previous report on a-Dy where two relaxation processes were observed to originate from the single unique DyIII centre as a result of two distinct relaxation pathways involving the 3rd and 4th excited states.5c Fitting these data, Fig. 5c, g, and k, gives the parameters in Table 2. All Ueff values for the SCS SMMs are around ∼1000 K, and that for 5Dy is substantially lower at ∼750 K; we note that this value is very close the average of the two Ueff values found previously for a-Dy (av. Ueff = 767 K).
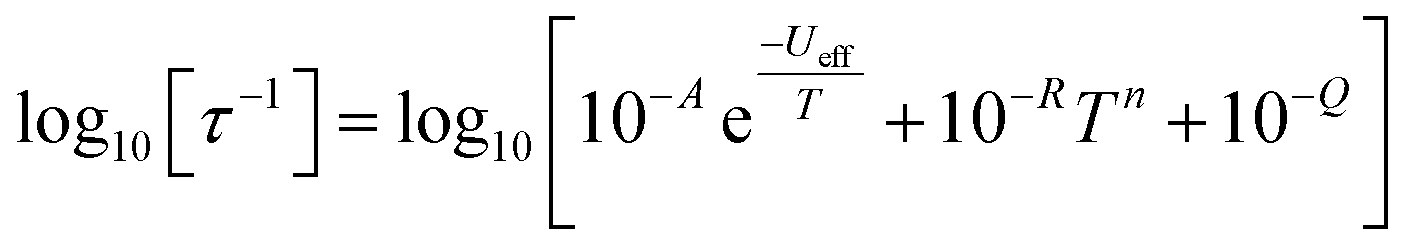 | (2) |
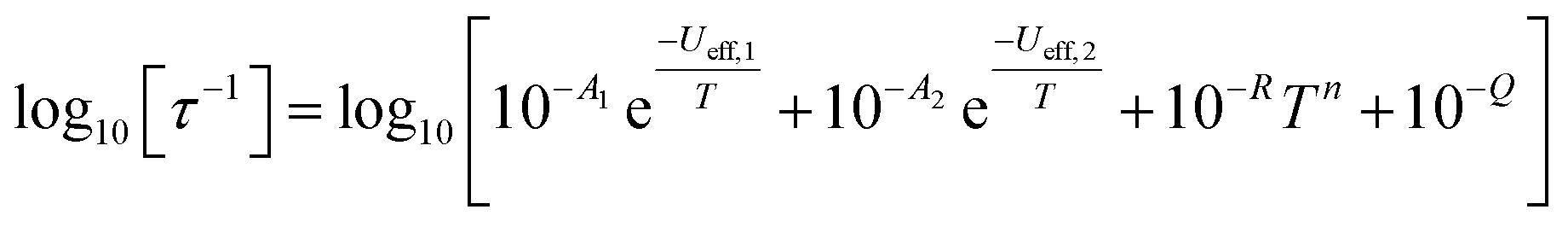 | (3) |
| Sample | U eff (K) | τ 0 (s) | C (s−1 K−n) | n | τ QTM (s) | T H (K) | T B1 (K) | T B2 (K) |
|---|---|---|---|---|---|---|---|---|
| a The fitting of the relaxation data using eqn (2) and (3) gives the relationships τ0 = 10A (s), C = 10−R (s−1 K−n), and τQTM = 10Q (s), where errors are reported in the exponents. | ||||||||
| 2Dy (1) | 1160(21) | 10–11.79(18) | 10–5.56(69) | 3.50(55) | 102.29(16) | 14 | 12 | 8 |
| 2Dy (2) | 1069(129) | 10–12.1(12) | ||||||
| 3Dy | 1015(32) | 10–11.82(29) | 10–5.53(46) | 3.54(32) | 102.26(14) | 12 | 11 | 8 |
| 4Dy | 1109(70) | 10–11.69(56) | 10–5.39(93) | 3.20(84) | 102.65(25) | 15 | 13 | 12 |
| 5Dy | 757(39) | 10–11.53(45) | 10–6.22(24) | 4.49(22) | 101.90(4) | 12 | 10 | — |
| a-Dy (1) | 721(1) | 1.11(3) | 3.01(7) × 10–11 | 8 | — | 10 | 10 | — |
| a-Dy (2) | 813(1) | 0.565(20) | 3.55(10) × 10−9 | 6 | — | |||
In order to define the blocking temperatures of these molecules, we performed TB1, TH, and TB2 measurements, Fig. 5d, h and l. Owing to the samples being moisture sensitive, the magnetic measurements are performed on samples sealed in borosilicate NMR tubes. In 2Dy, bifurcation is observed in ZFC/FC susceptibility below TIRREV = 30 K, indicating out-of-equilibrium behaviour, with a peak in the ZFC measurement observed at TB1 = 12 K (Fig. S25†). This large TIRREV may be an artefact of delayed temperature equilibration at the sample: for the field-cooling measurement, this would result in a higher true temperature and therefore lower signal than the equilibrium χ value when collecting this cooling cycle (and vice versa). Therefore, ZFC/FC measurements are also reported with a slower sweep rate allowing longer temperature equilibration between each measurement, subsequently shifting the peak position to lower temperature due to the longer waiting times TB1-slow = 8 K (Fig. S25,† ∼0.38 K min−1 (fast) and ∼0.031 K min−1 (slow)). For 2Dy, a relaxation rate of 100 seconds is found at TB2 = 8 K. Magnetic hysteresis loops collected with a sweep rate of ca. 15 Oe s−1 are open below TH = 14 K (Fig. 5d).
ZFC/FC measurements for 3Dy and 4Dy show separation at TIRREV = 12 and 15 K, respectively. Peaks are present in the ZFC measurements at TB1 = 11 and 13 K (Fig. S26 and S27,†TB1-slow = 7.5 and 9 K with the slower sweep rate), hysteresis loops are open to TH = 12 and 15 K (sweep rate ∼14 Oe s−1) and TB2 = 8 and 12 K for 3Dy and 4Dy, respectively. For 5Dy we find ZFC/FC separation below 13 K with a peak in the ZFC measurement at TB1 = 10 K (TB1-slow = 7.5 K) (Fig. S28†), and open hysteresis loops below 12 K (sweep rate ∼14 Oe s−1, Fig. S29†). As this molecule has a τQTM value of 79 s, a 100 s blocking temperature cannot be defined. For all hysteresis measurements we observe a step at zero field indicating fast relaxation due to QTM.
Ab initio calculations
We performed complete active space self-consistent field spin–orbit (CASSCF-SO) calculations for 2Dy–5Dy, employing the single crystal XRD atomic coordinates in all cases (see Methods), Fig. 6. A similar CF-splitting of the ground 6H15/2 multiplet is observed for the anionic and cationic molecules in 2Dy, which were performed as separate calculations. The [Dy(SCS)2K2(DME)4]+ cation has a slightly greater CF splitting compared to the [Dy(SCS)2]− anion (Tables S12 and S13†). In both cases we observe a mJ = |±15/2〉 ground state with principal axis directed towards one of the carbene bonds (deviation in angle to the equivalent carbene bonds with ∠gz-DyC76 is 0.552° for 2Dy–cation and ∠gz-DyC1 is 1.832° for 2Dy–anion, Table S19†). The first two excited states are also highly axial, while the 3rd excited state is highly mixed. Examination of the excited Kramers doublets and the average cartesian magnetic moment transition probabilities17 between all states reveals that relaxation via the Orbach mechanism would most likely occur through the 3rd excited state in both cases, which are calculated at 1196 and 1033 K, for [Dy(SCS)2K2(DME)4]+ and [Dy(SCS)2]−, respectively. These energies are in excellent agreement with the Ueff values determined from AC susceptibility for 2Dy (Fig. 3), and suggest that Ueff,1 = 1160(21) K corresponds to the [Dy(SCS)2K2(DME)4]+ cation while Ueff,2 = 1069(129) K corresponds to [Dy(SCS)2]−; note the midpoint of the experimental parameters is in good agreement, despite the large uncertainty due to the significant esds in the AC data.
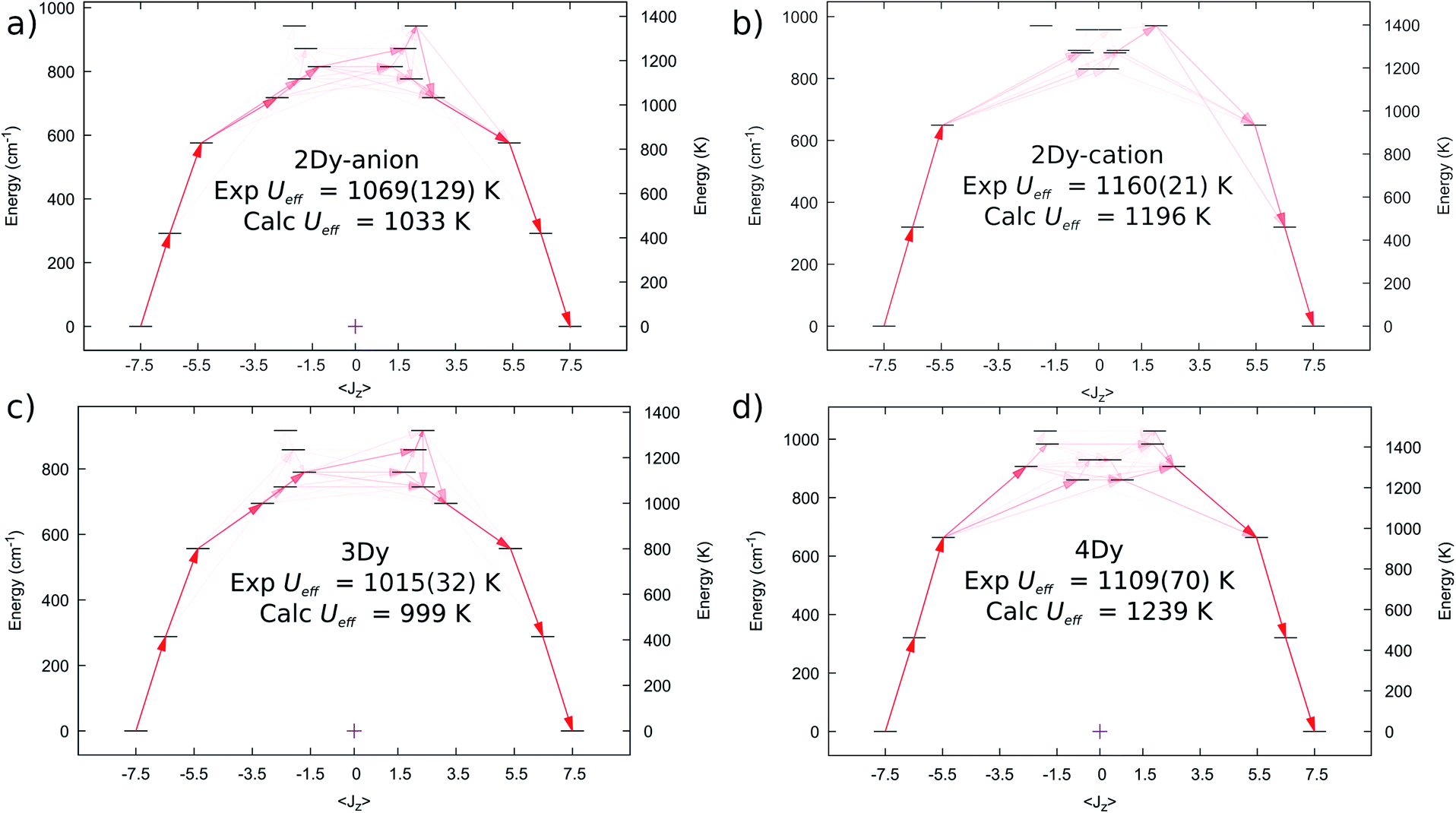 | ||
| Fig. 6 Computed ab initio crystal field diagrams for 2Dy–4Dy. (a) 2Dy–anion. (b) 2Dy–cation. (c) 3Dy. (d) 4Dy. | ||
The electronic structure of 3Dy similarly shows strong stabilisation of the large mJ projections of the DyIII ion (Fig. 6c, Table S14†), and suggests that Orbach relaxation is most likely to occur via the 3rd or 4th excited states (999 K or 1072 K, Fig. 6c), which is in good agreement with the experimental value of Ueff = 1015(27) K. Meanwhile, CASSCF-SO calculations suggest that 4Dy has the largest CF splitting of all the SCS analogues, with the 3rd excited state (where relaxation via the Orbach mechanism is favoured, Fig. 6d) predicted at 1239 K (Table S15†), higher than the experimental Ueff = 1109(70) K.
Calculations for 5Dy show almost pure mJ states for the first three Kramers doublets with a highly mixed 3rd excited doublet at 693 K (Table S16†). However, inspection of the average Cartesian magnetic moment transition probabilities (Fig. S31†) suggests that there is a favourable Orbach relaxation pathway via the 5th excited doublet, with 70%|±9/2〉 at 838 K, having the highest transition probability out of the 2nd excited doublet with 96%|±11/2〉. This would suggest a larger barrier than observed experimentally (Ueff = 757(39) K), however there is likely relaxation via the 3rd and 4th excited doublets at 693 and 793 K; indeed, the average energy of these three doublets is 774 K.
CASSCF-SO calculations of 3Gd predict the ZFS parameters of the system (Table S18†). Recall we were unable to determine the sign of D or E from the spectra, however CASSCF-SO predicts D = −0.078 and E = −0.010 cm−1, which are close to the experimental magnitudes of |D| = 0.11 cm−1 and |E| = 0.0085 cm−1, and thus we suggest both D < 0 and E < 0 here.
Discussion
The local coordination environment is responsible for the CF splitting of the J = 15/2 multiplet of DyIII and the origin of the Ueff energy barrier to magnetic relaxation. We have previously shown that classical electrostatics dominate the magnetic anisotropy for DyIII complexes,4d and thus simple parameters such as bond lengths and angles for the charged donor atoms have a significant impact on the Ueff barrier. Due to the dominant influence of the two near-linear trans-disposed C2− donor atoms of the bis-methanediide motif, the lowest three Kramers doublets for 2Dy–5Dy are almost pure |±15/2〉, |±13/2〉, and |±11/2〉 states quantised along the principal axes of the ground state doublets (the deviation angle of these axes are given in Table S19†), and the third excited state is highly mixed in all cases. The geometrical differences between the structures seem to have a small influence on the composition of these states, although, the main effect is changes in their energies (Tables S12–S16†).The four SCS complexes can be grouped into two pairs: the first consisting of 2Dy–cation and 4Dy, which have the largest CDyC angles (∼176–179°) and the shorter av. DyC bonds (2.38–2.40 Å), and the second consisting of 2Dy–anion and 3Dy that have the smaller CDyC angles (∼164–166°) and longer av. DyC bonds (2.42–2.43 Å), Table 1. Based on simple electrostatics, the samples with the most linear arrangement of the CDyC motif and the shortest DyC bonds would be expected to have the highest Ueff value. Therefore, we would expect the 2Dy–cation and 4Dy to have larger Ueff than the 2Dy–anion and 3Dy, and this is found to be exactly the case experimentally (Fig. 7). Furthermore, by replacing the hard, equatorial N-donors with softer S-donors, the Ueff values for all of 2Dy–4Dy are larger than 5Dy by approximately 40%. This effect is most clearly demonstrated by comparison of 4Dy to 5Dy, which both have CDyC angles of approximately 176° and are free of alkali-metal coordination, with Ueff barriers of 1109(70) and 757(39) K respectively.
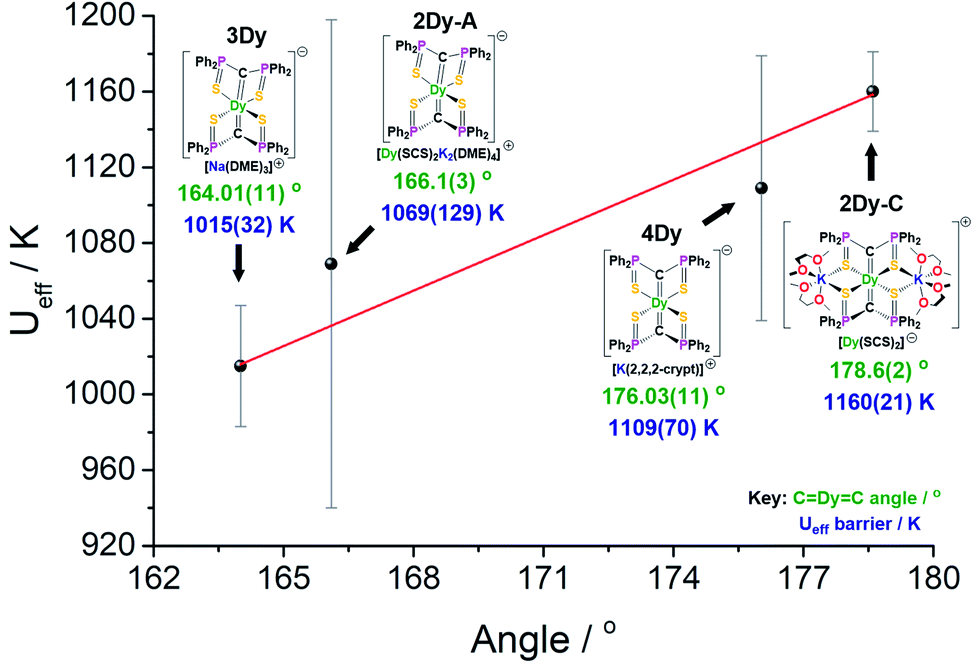 | ||
| Fig. 7
U
eff barrier as a function of CDyC angle for 2Dy–4Dy. | ||
We observe an interesting situation for the orientation of the principal gz axis of the ground |±15/2〉 state for the present compounds. This axis lies along the average DyC vector for 5Dy (Table S19†), as expected for two strong trans-methanediide donors, and yet despite similar variations between the pairs of DyC bond lengths in any given complex (ca. 0.04 Å), this is not repeated in the SCS variants. For each of the SCS complexes, the principal axis surprisingly aligns with the longer DyC bond (Table S19†). This orientation is not reconcilable with simple electrostatic interactions and reveals more complex interactions are at play. Additionally, the 2Dy–cation, which has the largest Ueff value and CDyC angle, also has the smallest average ∠gz-DyC angles at 0.84 and 0.55°. This is due to the coordination of potassium ions locking the molecule in place and increasing the rigidity of the system, as observed in the Cp-based systems.3d Furthermore, despite 4Dy having the shortest average and most symmetrical DyC distances, its Ueff value is less than that of the 2Dy–cation (1109 vs. 1160 K, respectively). We attribute this to the presence of the potassium ions bound to the sulphur groups in the 2Dy–cation, whose role could be two-fold: whilst enabling the CDyC bond angle to be more linear (176° vs. 179°), they also likely polarise negative charge away from the S-donors, weakening their donor strength to Dy. To examine this latter effect, we have performed a CASSCF-SO calculation on the [Dy(SCS)2K2(DME)4]+ structure where the {K(DME)2}+ moieties were removed (Table S20†). The energy spectrum reveals a slight decrease in CF splitting, reducing the energy of the highly mixed 3rd excited state by about 100 K. It would be interesting to isolate the 2Dy–cation to measure its properties without the neighbouring anion, however, thus far all experimental attempts have been unsuccessful.
An interesting result of our measurements on 5Dy reveals a single Orbach relaxation process with Ueff = 757(39) K, in contrast to the parent isomer a-Dy which shows two relaxation pathways with Ueff = 721(1) and 813(1) K. Comparison of the CASSCF-SO energy spectra of these two species shows only minor differences between the CF states, with the 3rd and 4th excited states for a-Dy calculated at 742 and 810 K. There is no clear reason why the relaxation appears as a single mechanism for 5Dy (α < 0.05) and the calculated barrier could be masking multiple pathways as it is unclear which CF states are involved in the Orbach relaxation mechanism. This work clearly shows how the Ueff energy barrier can be affected based on minor changes to the geometry and electrostatics of the coordination environment.
The comparison of the ZFC/FC measurements performed on these analogues highlights an important issue with the definition of TB1. As a number of high performing SMMs are temperature and/or moisture sensitive, they require similar preparation to the sealed NMR tubes used for the SCS samples presented here. Therefore, since there is no standard sweep rate for the assignment of TB1, it is difficult to be sure that reported relaxation behaviour has origins from SMM blocking, or additionally is influenced by temperature equilibration issues at the sample.
As shown in Fig. 7, there is a positive correlation between the Ueff barrier and CDyC angle. A similar trend can be shown for average ∠gz-DyC angle against Ueff barrier which, as expected, demonstrates a negative correlation (Fig. S32†). However, it is important to note some caveats. Firstly, the slight structural deviations between each bis-SCS structure must be addressed. As mentioned, the CDy bonds of 4Dy are marginally shorter than most other CDy bond lengths, although upon considering the magnitude of this difference, it is unlikely to have a major effect. Additionally, the coordinated potassium ions in the 2Dy–cation are expected to remove electron density from the S-atoms and consequently weaken their equatorial presence. Although computational models suggest that without this coordination the Ueff barrier would decrease, the magnitude of this effect is not great enough to disrupt the trend in linearity. Secondly, there is a gap in the middle region of the graph that is devoid of data points. As there is no clear method for producing bis-methanediide SCS compounds with specific CDyC angles, targeting this gap would present a major challenge. Thirdly, the uncertainty involved in calculation of the Ueff value is different for each point, and indeed has been correlated to atomic displacement parameters in crystal structures.19 The trend line passes directly through the two data points with smallest error. Given the experimental limitations, this data, along with the supported computational validation of Ueff, experimentally demonstrate a magneto-structural trend with respect to linearity with minimal deviations in structural facets, particularly compared to the existing literature.
Conclusions
We have prepared a series of SMMs with high energy barriers though deliberate tailoring of the coordination environment of an existing SMM in order to weaken the equatorial donors. Replacement of equatorial N-donors for softer S-donors results in a ca. 40% increase in Ueff. The longer Dy–S bonds and more defuse electron density reduces the effect of the equatorial CF and increases the stabilisation of the highly magnetic states of DyIII, as predicted. Additionally, this results in an increase in the blocking temperature, up to 15 K. The ability to isolate a number of [Dy(SCS)2]+/− variants has allowed the observation of a trend in Ueff across an analogous series, relating to the linearity of the bis-methanediide coordination.Author contributions
L. R. T.-H., M. G., E. Z., and F. O’D. prepared and characterised the complexes. L. R. T.-H. and M. J. G. obtained and analysed the magnetic data. A. J. W. collected, solved, and refined the crystal structures. N.F.C. and S.T.L. originated the central idea, directed the research, and analysed all the data. L. R. T.-H., M. J. G., N. F. C., and S. T. L. wrote the manuscript with input from all the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (EP/P002560/1, EP/P001386/1, EP/M027015/1), ERC (CoG612724, StG851504), Royal Society (University Research Fellowship to NFC) and The University of Manchester for support. We thank the Computational Shared Facility at The University of Manchester for access to computational resources and the National EPSRC UK EPR Facility.Notes and references
- (a) A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 1–4 CrossRef PubMed; (b) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC; (c) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (d) S. T. Liddle and J. van Slageren, in Lanthanides and Actinides in Molecular Magnetism, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 315–340 Search PubMed; (e) F. D. Natterer, K. Yang, W. Paul, P. Willke, T. Choi, T. Greber, A. J. Heinrich and C. P. Lutz, Nature, 2017, 543, 226–228 CrossRef CAS PubMed; (f) S. M. Aldoshin, D. V. Korchagin, A. V. Palii and B. S. Tsukerblatt, Pure Appl. Chem., 2017, 89, 1119–1143 CAS; (g) M. Feng and M. L. Tong, Chem.–Eur. J., 2018, 7574–7594 CrossRef CAS PubMed; (h) J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC; (i) K. L. M. Harriman, D. Errulat and M. Murugesu, Trends Chem., 2019, 1, 425–439 CrossRef CAS; (j) T. Komeda, K. Katoh and M. Yamashita, in Molecular Technology, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019, vol. 3, pp. 263–304 Search PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- (a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed; (b) J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed; (c) Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed; (d) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed; (e) D. S. Krylov, F. Liu, S. M. Avdoshenko, L. Spree, B. Weise, A. Waske, A. U. B. Wolter, B. Büchner and A. A. Popov, Chem. Commun., 2017, 53, 7901–7904 RSC; (f) F. Liu, D. S. Krylov, L. Spree, S. M. Avdoshenko, N. A. Samoylova, M. Rosenkranz, A. Kostanyan, T. Greber, A. U. B. Wolter, B. Büchner and A. A. Popov, Nat. Commun., 2017, 8, 16098–16106 CrossRef CAS PubMed; (g) E. Rousset, M. Piccardo, M.-E. Boulon, R. W. Gable, A. Soncini, L. Sorace and C. Boskovic, Chem.–Eur. J., 2018, 24, 14768–14785 CrossRef CAS PubMed; (h) G. Lu, Y. Liu, W. Deng, G. Huang, Y. Chen, J. Liu, Z.-P. Ni, M. Giansiracusa, N. F. Chilton and M.-L. Tong, Inorg. Chem. Front., 2020, 7, 2941–2948 RSC; (i) C. A. P. Goodwin, Dalton Trans., 2020, 49, 14320–14337 RSC; (j) A. Chiesa, F. Cugini, R. Hussain, E. Macaluso, G. Allodi, E. Garlatti, M. Giansiracusa, C. A. P. Goodwin, F. Ortu, D. Reta, J. M. Skelton, T. Guidi, P. Santini, M. Solzi, R. De Renzi, D. P. Mills, N. F. Chilton and S. Carretta, Phys. Rev. B, 2020, 101, 174402–174410 CrossRef CAS; (k) D. Shao and X. Y. Wang, Chin. J. Chem., 2020, 38, 1005–1018 CrossRef CAS; (l) V. S. Parmar, F. Ortu, X. Ma, N. F. Chilton, R. Clérac, D. P. Mills and R. E. P. Winpenny, Chem.–Eur. J., 2020, 26, 7774–7778 CrossRef CAS PubMed; (m) Z. Zhu, Y.-Q. Zhang, X.-L. Li, M. Guo, J. Gu, S. Liu, R. A. Layfield and J. Tang, CCS Chem., 2021, 3, 388–398 CrossRef; (n) R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728–1746 CrossRef CAS PubMed; (o) D. Shao and X.-Y. Wang, Chin. J. Chem., 2020, 38, 1005–1018 CrossRef CAS.
- (a) J. Sievers, Z. Phys. B: Condens. Matter., 1982, 45, 289–296 CrossRef CAS; (b) R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS; (c) C. R. Ganivet, B. Ballesteros, G. De La Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem.–Eur. J., 2013, 19, 1457–1465 CrossRef CAS PubMed; (d) N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551–2557 CrossRef PubMed; (e) N. F. Chilton, Inorg. Chem., 2015, 54, 2097–2099 CrossRef CAS PubMed; (f) J. Wu, J. Jung, P. Zhang, H. Zhang, J. Tang and B. Le Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC; (g) L. Escalera-Moreno, J. L. Baldovi, A. Gaita-Arino and E. Cronado, Chem. Sci., 2018, 9, 3265–3275 RSC; (h) A. K. Bar, P. Kalita, M. K. Singh, G. Rajaraman and V. Chandrasekhar, Coord. Chem. Rev., 2018, 367, 163–216 CrossRef CAS; (i) F.-S. Guo, A. K. Bar and R. A. Layfield, Chem. Rev., 2019, 119, 8479–8505 CrossRef CAS PubMed; (j) M. A. Dunstan, R. A. Mole and C. Boskovic, Eur. J. Inorg. Chem., 2019, 2019, 1090–1105 CrossRef CAS.
- (a) K. S. Pederson, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe, O. Waldmann, L. G. Chibotaru, J. Bendix and J. Dreiser, Chem. Sci., 2014, 5, 1650 RSC; (b) J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003–8010 CrossRef CAS PubMed; (c) M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and S. T. Liddle, Chem. Sci., 2016, 7, 155–165 RSC; (d) Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed; (e) S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181–5191 RSC; (f) J. Long, B. G. Shestakov, D. Liu, L. F. Chibotaru, Y. Guari, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov and J. Larionovaa, Chem. Commun., 2017, 53, 4706–4709 RSC; (g) A. B. Canaj, M. K. Singh, C. Wilson, G. Rajaraman and M. Murrie, Chem. Commun., 2018, 54, 8273–8276 RSC; (h) V. Dubrovin, A. A. Popov and S. Avdoshenko, Chem. Commun., 2019, 55, 13963–13966 RSC; (i) C. Gao, A. Genoni, S. Gao, S. Jiang, A. Soncini and J. Overgaard, Nat. Chem., 2020, 12, 213–219 CrossRef CAS PubMed.
- (a) T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. L. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6(7492), 1–8 Search PubMed; (b) T. Pugh, V. Vieru, L. F. Chibotaru and R. A. Layfield, Chem. Sci., 2016, 7, 2128–2137 RSC; (c) T. Pugh, N. F. Chilton and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 55, 11082–11085 CrossRef CAS PubMed; (d) B. M. Day, F.-S. Guo and R. A. Layfield, Acc. Chem. Res., 2018, 51, 1880–1889 CrossRef CAS PubMed.
- (a) G.-J. Chen, Y.-N. Guo, J.-L. Tian, J. Tang, W. Gu, X. Liu, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem.–Eur. J., 2012, 18, 2484–2487 CrossRef CAS PubMed; (b) M. R. Silva, P. Martín-Ramos, J. T. Coutinho, L. C. J. Pereira and J. Martín-Gil, Dalton Trans., 2014, 43, 6752–6761 RSC; (c) W. Chu, Q. Sun, X. Yao, P. Yan, G. An and G. Li, RSC Adv., 2015, 5, 94802–94808 RSC; (d) P.-P. Cen, S. Zhang, X.-Y. Liu, W.-M. Song, Y.-Q. Zhang, G. Xie and S. Chen, Inorg. Chem., 2017, 56, 3644–3656 CrossRef CAS PubMed; (e) X. Yao, P. Yan, G. An, C. Shi, Y. Li and G. Li, New J. Chem., 2018, 42, 8438–8444 RSC; (f) M. Guo, J. Wu, O. Cador, J. Lu, B. Yin, B. Le Guennic and J. Tang, Inorg. Chem., 2018, 57, 4534–4542 CrossRef CAS PubMed; (g) S. M. Chen, J. Xiong, Y. Q. Zhang, Q. Yuan, B. W. Wang and S. Gao, Chem. Sci., 2018, 9, 7540–7545 RSC; (h) S. Zhang, W. Mo, J. Zhang, Z. Zhang, B. Yin, D. Hu and S. Chen, Inorg. Chem., 2019, 58, 15330–15343 CrossRef CAS PubMed; (i) H. Wu, M. Li, B. Yin, Z. Xia, H. Ke, Q. Wei, G. Xie, S. Chen and S. Gao, Dalton Trans., 2019, 48, 16384–16394 RSC; (j) P. Cen, X. Liu, J. Ferrando-Soria, Y.-Q. Zhang, G. Xie, S. Chen and E. Pardo, Chem.–Eur. J., 2019, 25, 3884–3892 CrossRef CAS PubMed; (k) H.-H. Zou, T. Meng, Q. Chen, Y.-Q. Zhang, H.-L. Wang, B. Li, K. Wang, Z.-L. Chen and F. Liang, Inorg. Chem., 2019, 58, 2286–2298 CrossRef CAS PubMed; (l) Y. Gil, P. Fuentealba, A. Vega, E. Spodine and D. Aravena, Dalton Trans., 2020, 49, 17709–17718 RSC.
- K. Randall McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC.
- Y.-S. Ding, T. Han, Y.-Q. Zhai, D. Reta, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Chem.–Eur. J., 2020, 26, 5893–5902 CrossRef CAS PubMed.
- A. B. Canaj, S. Dey, O. Céspedes, C. Wilson, G. Rajaraman and M. Murrie, Chem. Commun., 2020, 56, 1533–1536 RSC.
- (a) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed; (b) C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed; (c) P. Evans, D. Reta, C. A. P. Goodwin, F. Ortu, N. F. Chilton and D. P. Mills, Chem. Commun., 2020, 56, 5677–5680 RSC; (d) P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2020, 141, 19935–19940 CrossRef PubMed; (e) A. Canaj, S. Dey, C. Wilson, O. Cespedes, G. Rajaraman and M. Murrie, Chem. Commun., 2020, 56, 12037–12040 RSC; (f) L. Zhu, B. Yin, P. Ma and D. Li, Inorg. Chem., 2020, 59, 16117–16121 CrossRef CAS PubMed; (g) X.-L. Ding, Y.-Q. Zhai, T. Han, W.-P. Chen, Y.-S. Ding and Y.-Z. Zheng, Chem.–Eur. J., 2020, 202003931 Search PubMed; (h) L. R. Thomas-Hargreaves, D. Hunger, M. Kern, A. J. Wooles, J. van Slageren, N. F. Chilton and S. T. Liddle, Chem. Commun., 2021, 57, 733–736 RSC.
- (a) C. A. P. Goodwin, D. Reta, F. Ortu, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2017, 139, 18714–18724 CrossRef CAS PubMed; (b) M. J. Giansiracusa, A. K. Kostopoulos, D. Collison, R. E. P. Winpenny and N. F. Chilton, Chem. Commun., 2019, 55, 7025–7028 RSC; (c) J. Liu, D. Reta, J. A. Cleghorn, Y. X. Yeoh, F. Ortu, C. A. P. Goodwin, N. F. Chilton and D. P. Mills, Chem.–Eur. J., 2019, 25, 7749–7758 CrossRef CAS PubMed; (d) A. Chakraborty, B. M. Day, J. P. Durrant, M. He, J. Tang and R. A. Layfield, Organometallics, 2020, 39, 8–12 CrossRef CAS; (e) M. He, F.-S. Guo, J. Tang, A. Mansikkamaki and R. A. Layfield, Chem. Sci., 2020, 11, 5745–5752 RSC.
- (a) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed; (b) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 CrossRef; (c) P.-B. Jin, Y.-Q. Zhai, K.-X. Yu, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2020, 59, 9350–9354 CrossRef CAS PubMed.
- (a) Y.-S. Meng, L. Xu, J. Xiong, Q. Yuan, T. Liu, B.-W. Wang and S. Gao, Angew. Chem., Int. Ed., 2018, 57, 4673–4676 CrossRef CAS PubMed; (b) Z. Jiang, L. Sin, Q. Yang, B. Yin, H. Ke, J. Han, Q. Wei, G. Xie and S. Chen, J. Mater. Chem. C, 2018, 6, 4273–4280 RSC; (c) S. Bala, G.-Z. Huang, Z.-Y. Ruan, S.-G. Wu, Y. Li, L.-F. Wang, J.-L. Liu and M.-L. Tong, Chem. Commun., 2019, 55, 9939–9942 RSC; (d) A. B. Canaj, S. Dey, E. R. Martí, C. Wilson, G. Rajaraman and M. Murrie, Angew. Chem., Int. Ed., 2019, 58, 14146–14151 CrossRef CAS PubMed; (e) M. J. Giansiracusa, S. Al-Badran, A. K. Kostopoulos, G. F. S. Whitehead, D. Collison, F. Tuna, R. E. P. Winpenny and N. F. Chilton, Dalton Trans., 2019, 48, 10795–10798 RSC; (f) T. Han, M. J. Giansiracusa, Z. H. Li, Y. S. Ding, N. F. Chilton, R. E. P. Winpenny and Y. Z. Zheng, Chem.–Eur. J., 2020, 26, 6773–6777 CrossRef CAS PubMed; (g) K. Yu, J. G. C. Kragskow, Y. Ding, Y. Zhai, D. Reta and N. F. Chilton, Chem, 2020, 6, 1777–1793 CrossRef CAS; (h) M. Li, H. Wu, Z. Xia, L. Ungur, D. Liu, L. F. Chibotaru, H. Ke, S. Chen and S. Gao, Inorg. Chem., 2020, 59(10), 7158–7166 CrossRef CAS PubMed; (i) J. Long, A. O. Tolpygin, E. Mamontova, K. A. Lyssenko, D. Liu, M. D. Albagami, L. F. Chibotaru, J. Larinova, Y. Guari and A. A. Trifonov, Inorg. Chem. Front., 2021 10.1039/D0qI01267J.
- (a) T. Cantat, F. Jaroschik, F. Nief, L. Ricard, N. Mézailles and P. Le Floch, Chem. Commun., 2005, 5178–5180 RSC; (b) T. Cantat, L. Ricard, P. Le Floch and N. Mézailles, Organometallics, 2006, 25, 4965–4976 CrossRef CAS; (c) T. Cantat, F. Jaroschik, L. Ricard, P. Le Floch, F. Nief and N. Mézailles, Organometallics, 2006, 25, 1329–1332 CrossRef CAS.
- (a) N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS; (b) C. V. Topping and S. J. Blundell, J. Phys.: Condens. Matter, 2019, 31, 013001–013027 CrossRef CAS PubMed.
- N. F. Chilton, C. A. P. Goodwin, D. P. Mills and R. E. P. Winpenny, Chem. Commun., 2015, 51, 101–103 RSC.
- (a) Y.-N. Guo, G.-F. Xu, P. Games, L. Zhao, S.-Y. Lin, R. Deng, J. Tang and H.-J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538–8539 CrossRef CAS PubMed; (b) Y.-N. Guo, G.-F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H.-J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948–11951 CrossRef CAS.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. CCDC 2051321–2051330. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00238d |
| This journal is © The Royal Society of Chemistry 2021 |