
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnthraquinone-2,6-disulfamidic acid: an anolyte with low decomposition rates at elevated temperatures†
Philip Rohlandab,
Kristin Schreyerab,
Martin D. Hager ab and
Ulrich S. Schubert*ab
ab and
Ulrich S. Schubert*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstraße 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Friedrich Schiller University Jena, Philosophenweg 7a, 07743 Jena, Germany
First published on 3rd December 2021
Abstract
A new sulfamidic acid anthraquinone derivative was synthesized from 2,6-diaminoanthraquinone with high yields, designed for utilization in redox flow batteries. The active material was investigated with cyclic voltammetry, revealing a reversible redox reaction at approximately −0.65 V vs. Ag/AgCl at pH-values above 12. A stress test in a redox flow battery was applied with hold times at critical states of charge and at 32 °C as well as at 60 °C. Furthermore, the stability was investigated at the maximum concentration of the anolyte. All in all, the material showed the lowest decomposition rates at 60 °C reported so far for an organic anolyte in a redox flow battery.
Introduction
In the course of ongoing climate change, energy production has to switch from fossil fuels to renewable resources to dampen the effects of global warming.1 But for the breakthrough of clean energy conversion, the introduction of green, environmentally friendly storage technologies is necessary to compensate for the inherent discontinuous power output of the main, solar- and wind-based, technologies. Nowadays, many possible approaches have been evaluated for this mammoth task.2 Rechargeable batteries provide a broad range of applicable dimensions, which would enable the installation of a decentralized energy storage grid. However, market-ready products, like lithium batteries or vanadium redox flow batteries (RFB), suffer from expensive and dangerous electrolytes or raw materials that are mined under environmentally and socially harmful conditions.3–5 The recently emerging research field of RFB based on organic materials is capable of targeting these drawbacks.Numerous redox-active molecules, like ferrocenes,6,7 quinones,8,9 viologens,9–11 stable radicals,11–13 alloxazine,14,15 and flavine,16 have been subject to research showing the broad range of possible scaffolds of organic RFB (ORFB).
These molecules comprise earth-abundant elements targeting lower costs for battery electrolytes. Moreover, some materials, like TEMPO derivatives, do not require drastic electrolyte pH-values or are even bioinspired, like flavines, which are based on vitamin B2.16
Among these possible structures, quinones, in particular anthraquinones (AQ), are the best investigated anolyte materials.17 They combine a reversible redox chemistry involving two electrons per molecule (see Scheme 1) to result in a high specific capacity. Furthermore, their redox potential is highly depending on the pH-value of the electrolyte and the substitution pattern at the anthraquinone core resulting in a great potential for tailoring the molecule characteristic to the targeted application.18 They also show extraordinarily high stabilities during cycling and against side reactions,19 but this is also one of their major drawbacks. The high stability hinders derivatization reactions at the anthraquinone core and necessitates drastic or complex reaction conditions20 resulting in the need of often cost-intensive starting materials. Nevertheless, these reactions are necessary to install linkers and solubility-promoting groups at the core to overcome the intrinsically low solubility of anthraquinone in water.
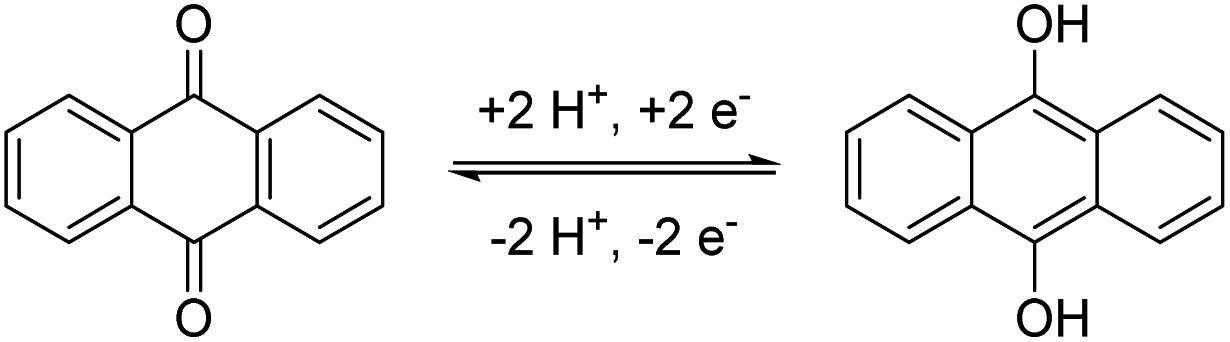 | ||
| Scheme 1 Schematic representation of the electrochemistry of anthraquinone. | ||
In this contribution, we report a fast, straight-forward one-step synthesis with high yields towards a highly water-soluble anthraquinone derivative. By utilizing cheap starting materials, reactants, solvents and simple reaction conditions, we report a new, economic anthraquinone active material based on 2,6-diaminoanthraquinone. The current literature lacks examples where newly developed active materials are tested under more realistic conditions, like dwell times at high states of charge (SOC) as well as elevated temperatures. Nevertheless, these parameters are crucial, in particular for a future commercialization of the active material. Thus, we tested a 0.1 M solution of our new active material and included dwell times in combination with electrolyte temperatures of up to 60 °C in the cycling protocol. The molecules revealed a high stability during the hold times even when the temperature was increased. These findings where then confirmed by a long-term cycling test at maximum concentration.
Experimental
Chemicals and materials
All chemicals were bought with a purity of at least 95% and were used as received. Detailed information is provided in the ESI.† The ion-selective membrane (FKS-30, Fumatec GmbH, Germany) was cut into appropriate pieces and was prewetted in 1.5 M lithium chloride solution for at least 24 h prior to installation. GFA-6 (SGL SE, Germany) felts are used as pours electrodes.Electrochemical analytics
Battery experiments
All experiments were performed with the VMP-3 (Bio-Logic, France) potentiostat/galvanostat under an argon atmosphere in a glovebox (UNIlab Plus Eco, MBraun, Germany, O2 < 1 ppm, H2O < 1 ppm, argon as protective gas). The electrolyte was pumped in the RFB through Norprene® A-60-G (øinn 1.6 mm, øout 4.8 mm, Saint-Gobain, France) with a pumping speed of approximately 25 mL min−1 by a Hei-Flow Value 01 equipped with a C8 multi-channel pumping head (Heidolph, Germany). The RFB was designed by JenaBatteries GmbH (Germany) as a flat type cell with a membrane active area of 5 cm2. A detailed overview of the components is provided in earlier publications of our group.11 The standard temperature in the glovebox was 32 °C. For experiments at elevated temperatures, the capacity-limiting side was tempered in a sand bath (Hei-Tec, Heidolph, Germany), equipped with an external temperature sensor (PT 1000, Heidolph, Germany), at 60 °C. All tests were performed with an unbalanced, compositionally symmetric cell setup.Results and discussion
Synthesis
The concept of using sulfamidic acids or sulfonamides as solubility enhancer in water represents a common strategy in the design process of new drugs.21–23 We used this approach to enhance the solubility of anthraquinones with amino side groups. On the one hand, the often applied 2,6-dihydroxy anthraquinone24,25 can be substituted by the one order of magnitude cheaper 2,6-diamino anthraquinone and, on the other hand, sulfonamides are highly stable and cleavage or rearrangement reactions need drastic conditions and long reaction times.26 To further reduce the costs of the active material, we optimized the synthesis conditions by the usage of cheap reagents while maintaining high yields.The first synthesis of sulfonamides from anthraquinones dates back to 1924, when different amino-AQ were transformed into their water-soluble derivatives by using chlorosulfuric acid, inert solvents like nitrobenzene, and organic nitrogen bases.27 We adapted the conditions but changed to less dangerous solvents, and we receive high yields of above 87% (see Scheme 2). To increase the solubility of the active material, we applied a lithium chloride-based electrolyte to impose polarization effects similar to the lithium chloride/N,N-dimethylacetamide system for cellulose.28 To prevent the formation of salts with different metal ions, lithium hydroxide was used for sulphate salt formation, despite the fact that, e.g., sodium or potassium hydroxide would be much more cost effective.
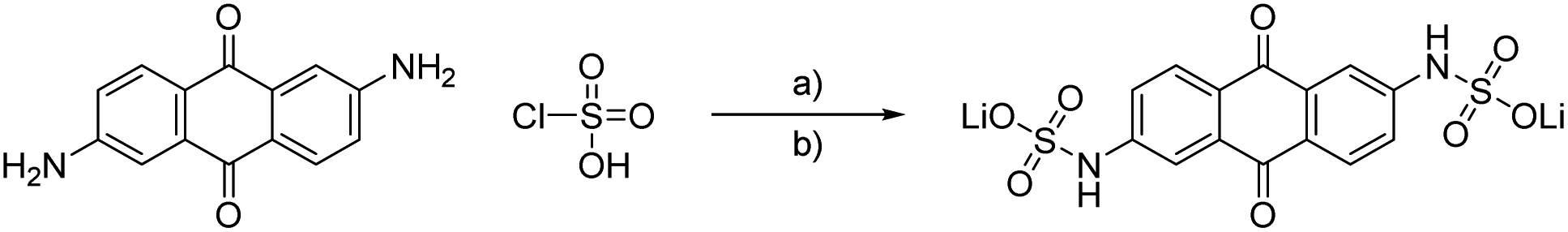 | ||
| Scheme 2 Schematic representation of the synthesis of LiAQS. (a) Dichloromethane, pyridine, 0 °C to room temperature, 16 h. (b) Water, lithium hydroxide, room temperature, 30 min, 87%. | ||
Electrochemical analysis
The redox properties of the synthesized lithium anthraquinone 2,6-disulfamidic acid (LiAQS) were investigated via CV investigation. Since the redox chemistry of anthraquinones is known to be highly pH-dependent,18 firstly the redox reversibility of the material was studied. As expected, the reoxidation of the hydroquinone to the quinone is hindered at pH values between 7 and 11 (see Fig. 2). At pH values of 13 and above, the AQ-typical redox pattern in the CV is visible.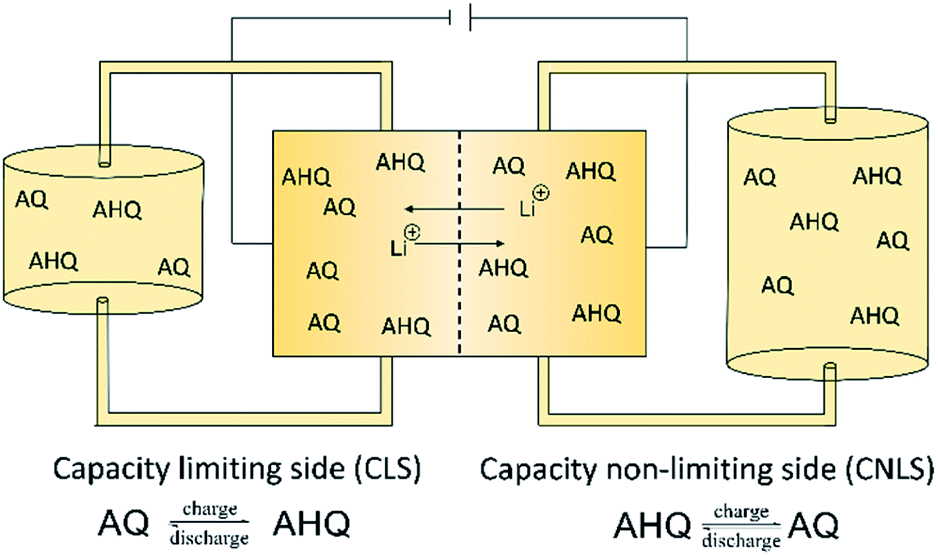 | ||
| Fig. 1 Schematic representation of an unbalanced compositionally symmetric redox flow cell setup utilizing anthraquinone (AQ) as well as anthrahydroquinone (AHQ) and the representative half-cell reactions in the capacity limiting side (CLS) and non-limiting side (CLNS). | ||
The parameters of the redox reaction of the LiAQS was investigated by applying different scan speeds during the CV experiments at a pH-value of 14. While the reduction to the hydroquinone produces a sharp negative peak even at very fast scan speeds of 2000 mV s−1, the peak of the reoxidation is broadened (see Fig. 2).
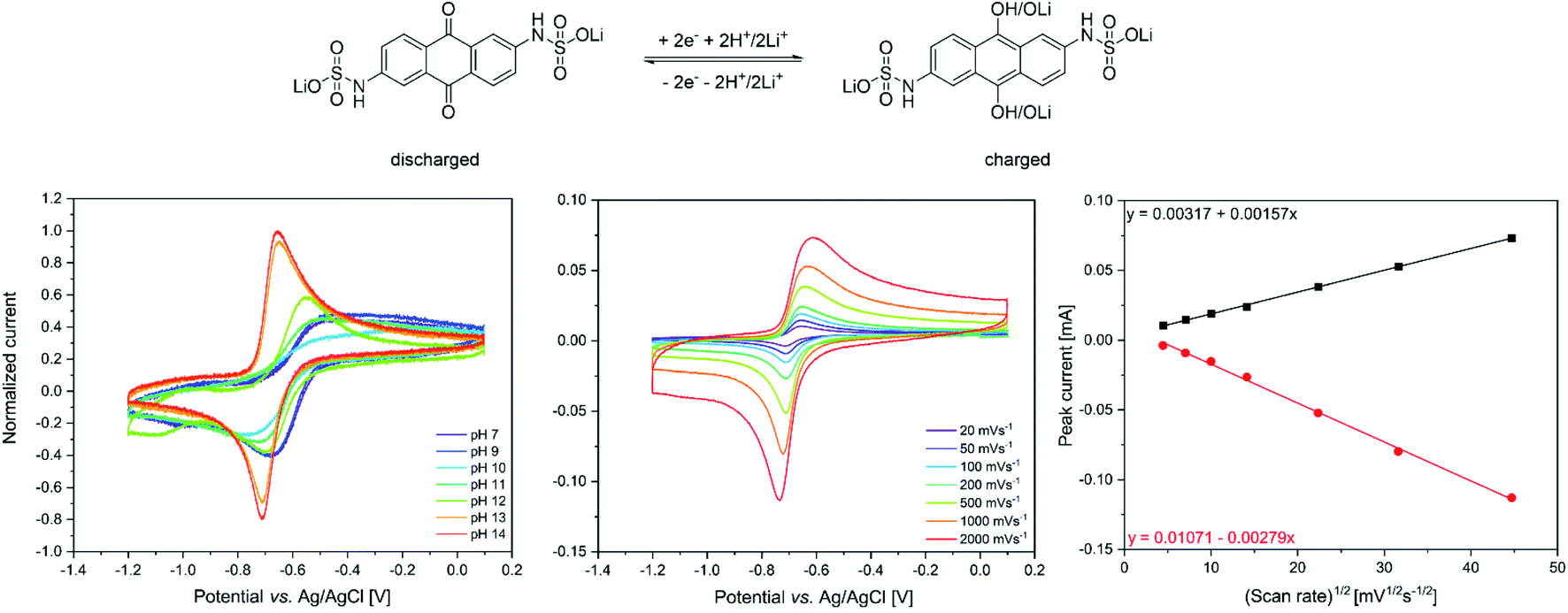 | ||
| Fig. 2 Top: schematic representation of the redox chemistry of the LiAQS. Bottom: cyclovoltammetry of a 50 mM LiAQS in 0.1 M lithium chloride solution. Scan direction: 0 V to −1.2 V and back to 0 V. For each condition five scans were carried out and the third is shown. (left) pH-dependent investigation utilizing lithium hydroxide for adjusting the pH-value with a scan speed of 100 mVs−1, (middle) CV at different scan speeds at a pH-value of 14, (right) plot of the peak current for the oxidation (black) and reduction process (red) vs. the square root of the scan speed at a pH-value of 14. | ||
The reversibility of the redox reaction was investigated by plotting the peak current vs. the square root of the scan speed. The data points for the reduction and reoxidation can then be fitted linearly. The curves fit nicely to the datapoints, indicating reversible redox reactions of the LiAQS and a diffusion-controlled process.29 Based on these findings we decided to use electrolytes with a pH-value between 13 and 14 for the following battery experiments. We thus used a lithium hydroxide concentration of 0.25 M for the battery electrolyte.
Battery experiments
In the recent years, anthraquinones have shown to be one of the most promising anolytes for redox flow batteries.17 Nevertheless, the need for high pH-values is one of their main drawbacks, limiting the selection of catholytes. The ferri–ferro cyanate has been established over the last years as the catholyte material of choice when using anthraquinones.18,19,24,25 However, we renounced full battery tests and focused our experiments on the investigation of the new LiAQS and its performance under unfavorable but realistic conditions. For this, we utilized the concept of an unbalanced, compositionally symmetric redox flow cell. The setup was firstly introduced by the group of Brushett30 and further developed by Goulet and Aziz.31 It uses the same electrolyte in both half cells of the RFB, largely avoiding crossover artefacts (Fig. 1). Furthermore, we exposed our material to hold times in vulnerable SOCs and at elevated temperatures to mimic more realistic conditions. The protocol includes 50 consecutive cycles with potentiostatic and galvanostatic parts (see Experimental details). This ensures that the cell reaches its maximum SOC. After 50 cycles, a hold time of 24 h at maximum SOC is applied. After 250 cycles, the protocol is repeated for 200 additional cycles at 60 °C.The relatively low concentration of 0.1 M was used to keep the experiment's time in a manageable range and to highlight possible decomposition reactions.
The long-term test at low concentrations revealed an average decomposition rate of 0.4% d−1 (see Fig. 3 upper left) during the cycling phases, which is low in comparison to other anthraquinone derivatives.17 Moreover, the decay rates during the hold times are barely visible, indicating a stable charged state, which is favorable regarding the integration of the battery in an electrical grid for long-term storage of energy. The high stability in high SOCs reduces the overall decay to 0.18% d−1 (including hold times). Notably, the elevated temperatures in the glove box must be considered at this point since most of the published data are measured at room temperature, i.e. between 20 and 25 °C. The increase in temperature, although not extensive, might already trigger decomposition reactions. Nevertheless, in comparison to other quinone derivatives that feature the solubility-promoting group (SPG) directly attached to the quinoidic core, the synthesized LiAQS is remarkably stable. The decay window ranging from 35% d−1 for the 2,5-dimethoxy-3,6-dihydroxy-1,4-benzoquinone32 to 0.14% d−1 for the 2,6-dihydroxyanthraquinone33 in full battery setup and 70% d−1 for tetramorpholinohydroquinone34 and 0.08% d−1 2,7-anthraquinone disulfonic acid33 in a symmetric cell set up. The topic of electrolyte lifetime in aqueous RFB is excellently summed up in the review of Kwabi et al.17
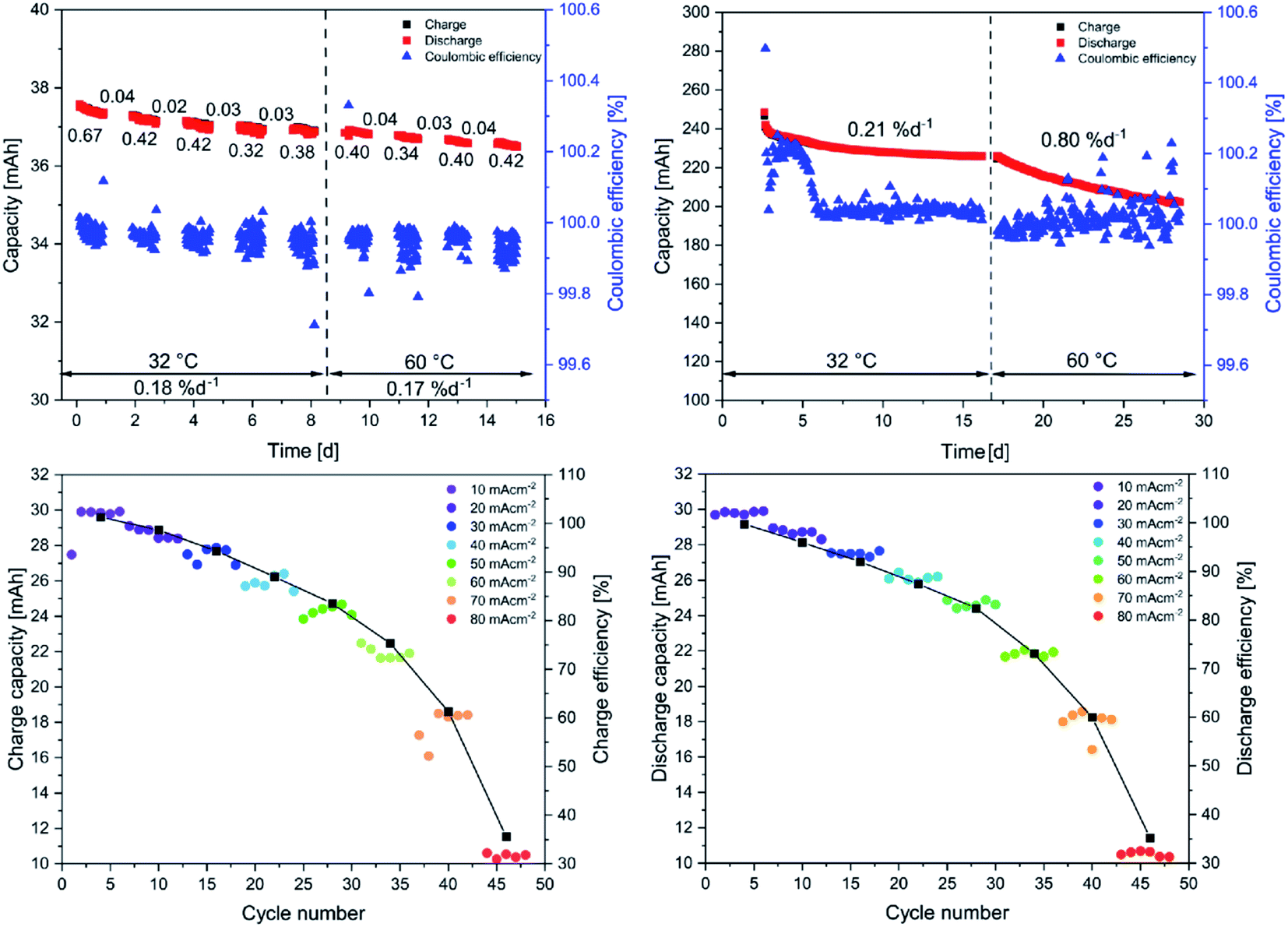 | ||
| Fig. 3 (Top left) Long-term stability test using 15 mL of an aqueous 0.1 M LiAQS, 1 M LiCl and 0.25 M LiOH solution vs. 35 mL of the same solution. The values below the red and black squares indicate the decay rate during the representative 50 cyles in % d−1. After every 50th cycle, the electrolyte is kept at maximum SOC for 24 h. The values above the squares represent the deacy rates during this hold times. After 250 cycles, the temperature of the electrolyte is raised to 60 °C (indicated by the dashed line). The values below the temperature represent the decay rates over the whole temperature phases. (Top right) long-term test using 10 mL of a 0.75 M LiAQS, 1 M LiCl and 0.25 M LiOH solution vs. 25 mL of the same solution. Bottom: current capacity tests using 11.5 mL of a 0.1 M LiAQS, 1 M LiCl and 0.25 M LiOH solution vs. 35 mL of the same solution. Either the charge (left) or the discharge (right) current density is varied. The current density of the counter process is kept at 10 mA cm−2. | ||
The Aziz group demonstrated that the introduction of a linker between AQ core and SPG can further increase the stability by one magnitude.8,19 However, for our system, this approach will be subject of future investigations.
Noteworthy, the 0.1 M LiAQS electrolyte reveals the same stability at 60 °C as at 32 °C, which is remarkable since increased temperatures usually promote decomposition reactions. The average decomposition rates during cycling phases and during hold times are nearly the same, resulting in an overall decay rate of 0.17% d−1 (0.18% d−1 at 32 °C). This is, to the best of our knowledge, the lowest ever measured value for an organic anolyte material at 60 °C. Furthermore, the coulombic efficiencies are not affected by the temperature change and is, with 99.95%, high. For an overview of all data of the low concentration long-term stability test see Table 1 in the ESI.† In the next step, the current capacity of the active material was investigated. Two experimental series were conducted, either with a changing charge or discharge current. The data reveals that a charge as well as a discharge current of 60 mA cm−2 can be applied until the achievable capacity drops below 80%, which is remarkably high for a 0.1 M solution (see Fig. 3).
In the last step, the behavior of the electrolyte at the maximal material concentration of 0.75 M was investigated in a long-term stability test. The electrolyte was charged potentiostatically and galvanostatically for 200 cycles at 32 °C and 200 cycles at 60 °C, like described for the low concentration test but without hold times. For an overview of all data of the high concentration long-term stability test see Table 2 in the ESI.† After an equilibration over ten cycles, during which the capacity strongly fades, the decay rate stabilizes over the next 190 cycles. Meanwhile, the coulombic efficiency (CE) rises until the 25th cycle and then slowly decreases until the 50th cycle. This might be caused by a longer equilibration time due to the maximal electrolyte concentration. In the 0.1 M LiAQS solution for comparison the CE is stable starting from the first cycle. After the equilibration, the CE of the 0.75 M solutions stabilizes above 99.95% and the capacity decay remains at around 0.2% d−1, confirming the findings for the 0.1 M solution. However, the subsequent 200 cycles at 60 °C show a more pronounced fade of 0.8% d−1, indicating a temperature- and concentration-dependent decay mechanism. Nevertheless, the measured fade rates are the lowest obtained so far for anolytes at 60 °C. In comparison to the experiments at 32 °C, the CE fluctuates a bit more but is on average also above 99.95%.
Nevertheless, only approximately 50% of the theoretical capacity is addressable. We ascribe this behavior to the formation of quinhydrone complexes, formed from a fully charged, doubly reduced anthrahydroquinone and an uncharged anthraquinone, formally consisting of two half charged, singly reduced semiquinones.35–38 This process would be enhanced by an initial dimer formation, as shown for other 2,6-disubstituted anthraquinones,39,40 and hydrogen bonds formed by the sulfamic acid. The usage of lithium-based electrolytes might even enhance this behavior due to its oxophilicity and the possibility of complex formation based on lithium enolates.41–44
Conclusion
A new active material was synthesized starting from an inexpensive 2,6-diamino anthraquinone. While the reaction conditions are mild and the reactants readily available, the yields are high, enabling the possibility for a further commercialization.The RFB performance of the active material was investigated at low concentrations but under realistic stress conditions, like hold times in vulnerable SOCs or elevated temperatures. The LiAQS showed low decompositions rates of 0.2% d−1 at 32 °C, which places it among the best performing AQ derivates with the SPG directly linked to the AQ core. Furthermore, the stability at 60 °C is comparable to that at 32 °C. Based on this data, the LiAQS revealed, to the best of our knowledge, the highest stability of an organic anolyte at 60 °C so far. With regard to a possible commercialization, we also determined the stability of the anolyte at the maximum concentration of 0.75 M. The LiAQS showed similar decomposition rates at 32 °C as at low concentration. However, the stability at 60 °C is slightly decreased. Nevertheless, it is the best performing anolyte material under the used conditions so far.
A further increase in stability might be achieved by the introduction of a linker between the SPG and the anthraquinone core as the data by the group of Aziz suggests;8,19 this is the topic of ongoing investigations.
Author contributions
P. R. designed this project and wrote the manuscript. P. R. and K. S. carried out the experiments. M. D. H. and U. S. S. supervised the project. All authors provided critical feedback, discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Thuringian Ministry of Economic Affairs, Science and Digital Society (TMWWdG), the Thüringer Aufbaubank (TAB) and the European Regional Development Fund (ERDF), for financial support (2016 IZN 0009 and CEEC-01/2020). We further want to thank Dr Christian Friebe for his help and suggestions and Oliver Nolte for inspirations.Notes and references
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable Sustainable Energy Rev., 2009, 13, 1513–1522 CrossRef.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 CrossRef CAS.
- C. Banza Lubaba Nkulu, L. Casas, V. Haufroid, T. De Putter, N. D. Saenen, T. Kayembe-Kitenge, P. Musa Obadia, D. Kyanika Wa Mukoma, J. M. Lunda Ilunga, T. S. Nawrot, O. Luboya Numbi, E. Smolders and B. Nemery, Nat. Sustain., 2018, 1, 495–504 CrossRef PubMed.
- P. Navalpotro, E. Castillo-Martínez and J. Carretero-González, Sustainable Energy Fuels, 2021, 5, 310–331 RSC.
- P. S. Borchers, M. Strumpf, C. Friebe, I. Nischang, M. D. Hager, J. Elbert and U. S. Schubert, Adv. Energy Mater., 2020, 10, 2001825–2001833 CrossRef CAS.
- Y. L. Ji, M. A. Goulet, D. A. Pollack, D. G. Kwabi, S. Y. Jin, D. De Porcellinis, E. F. Kerr, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900039–1900046 CrossRef.
- S. J. Jin, Y. Jing, D. G. Kwabi, Y. L. Ji, L. C. Tong, D. De Porcellinis, M. A. Goulet, D. A. Pollack, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2019, 4, 1342–1348 CrossRef CAS.
- C. DeBruler, B. Hu, J. Moss, J. Luo and T. L. Liu, ACS Energy Lett., 2018, 3, 663–668 CrossRef CAS.
- T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2016, 55, 14427–14430 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J. F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS.
- K. X. Lin, R. Gomez-Bombarelli, E. S. Beh, L. C. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- W. Lee, B. W. Kwon and Y. Kwon, ACS Appl. Mater. Interfaces, 2018, 10, 36882–36891 CrossRef CAS.
- A. Orita, M. G. Verde, M. Sakai and Y. S. Meng, Nat. Commun., 2016, 7, 13230–13238 CrossRef CAS.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS.
- D. P. Tabor, R. Gomez-Bombarelli, L. C. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, J. Mater. Chem. A, 2019, 7, 12833–12841 RSC.
- M. Wu, Y. Jing, A. A. Wong, E. M. Fell, S. J. Jin, Z. J. Tang, R. G. Gordon and M. J. Aziz, Chem-Us, 2020, 6, 1432–1442 CrossRef CAS.
- Y. Jing, M. Wu, A. A. Wong, E. M. Fell, S. Jin, D. A. Pollack, E. F. Kerr, R. G. Gordon and M. J. Aziz, Green Chem., 2020, 22, 6084–6092 RSC.
- G. L. Perlovich, V. P. Kazachenko, N. N. Strakhova and O. A. Raevsky, J. Chem. Eng. Data, 2014, 59, 4217–4226 CrossRef CAS.
- F. J. Bandelin and W. Malesh, J. Am. Pharm. Assoc., 1959, 48, 177–181 CrossRef CAS.
- F. MartÍnez and A. GÓmez, Phys. Chem. Liq., 2010, 40, 411–420 CrossRef.
- Z. J. Yang, L. C. Tong, D. P. Tabor, E. S. Beh, M. A. Goulet, D. De Porcellinis, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2018, 8, 1702056–1702065 CrossRef.
- D. G. Kwabi, K. Lin, Y. Ji, E. F. Kerr, M.-A. Goulet, D. De Porcellinis, D. P. Tabor, D. A. Pollack, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Joule, 2018, 2, 1894–1906 CrossRef CAS.
- S. Searles and S. Nukina, Chem. Rev., 1959, 59, 1077–1103 CrossRef CAS.
- F. Baumann, H. Walter, W. Hentrich and L. Zeh, IG Farbenindustrie AG, Germany Pat., DE469564C, 1924 Search PubMed.
- C. L. Mccormick, P. A. Callais and B. H. Hutchinson, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- J. Winsberg, C. Stolze, A. Schwenke, S. Muench, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2017, 2, 411–416 CrossRef CAS.
- J. D. Milshtein, J. L. Barton, R. M. Darling and F. R. Brushett, J. Power Sources, 2016, 327, 151–159 CrossRef CAS.
- M. A. Goulet and M. J. Aziz, J. Electrochem. Soc., 2018, 165, A1466–A1477 CrossRef CAS.
- P. Sun, Y. Liu, Y. Li, M. A. Shehzad, Y. Liu, P. Zuo, Q. Chen, Z. Yang and T. Xu, Ind. Eng. Chem. Res., 2019, 58, 3994–3999 CrossRef CAS.
- M. A. Goulet, L. Tong, D. A. Pollack, D. P. Tabor, S. A. Odom, A. Aspuru-Guzik, E. E. Kwan, R. G. Gordon and M. J. Aziz, J. Am. Chem. Soc., 2019, 141, 8014–8019 CrossRef CAS PubMed.
- E. Drazevic, C. Szabo, D. Konya, T. Lund, K. Wedege and A. Bentien, ACS Appl. Energy Mater., 2019, 2, 4745–4754 CrossRef CAS.
- F. D'Souza, J. Am. Chem. Soc., 1996, 118, 923–924 CrossRef.
- N. Hayashi, T. Yoshikawa, M. Kurakawa, T. Ohnuma, Y. Sugiyama and H. Higuchi, Mol. Cryst. Liq. Cryst., 2006, 456, 133–137 CrossRef CAS.
- K. Molčanov, C. Jelsch, B. Landeros, J. Hernández-Trujillo, E. Wenger, V. Stilinović, B. Kojić-Prodić and E. C. Escudero-Adán, Cryst. Growth Des., 2018, 19, 391–402 CrossRef.
- S. Patai and Z. Rappoport, The Quinonoid Compounds, John Wiley & Sons Ltd., Hoboken, 1988, vol. 1 Search PubMed.
- T. J. Carney, S. J. Collins, J. S. Moore and F. R. Brushett, Chem. Mater., 2017, 29, 4801–4810 CrossRef CAS.
- C. Wiberg, T. J. Carney, F. Brushett, E. Ahlberg and E. G. Wang, Electrochim. Acta, 2019, 317, 478–485 CrossRef CAS.
- D. Seebach, Angew. Chem., Int. Ed., 1988, 27, 1624–1654 CrossRef.
- M. P. Sibi, J. Am. Chem. Soc., 1998, 120, 2991 CrossRef CAS.
- A. Streitwieser, J. Mol. Model., 2006, 12, 673–680 CrossRef CAS.
- A. Streitwieser, J. Org. Chem., 2009, 74, 4433–4446 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05545c |
| This journal is © The Royal Society of Chemistry 2021 |