
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV-assisted synthesis of hydroxyapatite from eggshells at ambient temperature: cytotoxicity, drug delivery and bioactivity
Sazia Sultana a,
Md. Sahadat Hossaina,
Monika Mahmuda,
Mashrafi Bin Mobaraka,
Md Humayun Kabira,
Nahid Sharmina and
Samina Ahmed*ab
a,
Md. Sahadat Hossaina,
Monika Mahmuda,
Mashrafi Bin Mobaraka,
Md Humayun Kabira,
Nahid Sharmina and
Samina Ahmed*ab
aInstitute of Glass and Ceramic Research and Testing (IGCRT), Bangladesh Council of Scientific and Industrial Research (BCSIR), Dr Qudrat-i-Khuda Road, Dhanmondi, Dhaka-1205, Bangladesh
bBCSIR Laboratories, Dhaka, Bangladesh Council of Scientific and Industrial Research (BCSIR), Dr Qudrat-i-Khuda Road, Dhanmondi, Dhaka-1205, Bangladesh. E-mail: shanta_samina@yahoo.com; Tel: +880-1817549816
First published on 19th January 2021
Abstract
Hydroxyapatite [Ca10(PO4)6(OH)2], an important biomaterial, retains a chemical structure that is similar to the mineral phase of bone. Consequently, the ability of hydroxyapatite (Hap) to augment bone growth within bone tissue has made it a potential candidate for use as a hard tissue-implant material. In this work, adopting a UV-mediated solid-state method for the first time, hydroxyapatite was synthesized from eggshells and no thermal treatment was used but ambient temperature was maintained. This simple synthesis process involved a combination of ball milling of the starting materials followed by UV-irradiation. UV-excitation of the Ca and P precursors resulted in the desired Hap and X-ray diffraction (XRD), Fourier-transform infrared (FT-IR) and Raman spectroscopic techniques were used for characterization. The potency of UV-Hap as a biomaterial was examined via the bioactivity, cytotoxicity and the drug (ciprofloxacin) loading–releasing response, which was encouraging. The results of the cell viability assays complied an insignificant cytotoxicity and the simulated body fluid immersion test indicated the bioactivity was within the acceptable range. On the other hand, to better understanding the drug ejection and associated transport phenomenon, two kinetic models (Higuchi and Ritger–Peppas models) were used and a diffusion controlled ciprofloxacin release mechanism was observed using the Higuchi model. However, the experimental outcomes of a drug delivery response exposed UV-Hap as a favorable vehicle for drug loading and release. Hence, this research highlights the prospects of a UV-assisted synthesis method as a green route for the synthesis of Hap to be applied in biomedical fields.
1. Introduction
In recent years, the application of synthesized biomaterials in bone and dentistry fields has received significant attention and notable efforts have been reported for the synthesis of biomaterials via different routes using various sources.1–5 In particular, hydroxyapatite [Ca10(PO4)6(OH)2], which retains a close chemical similarity to the mineral phase of bone, is ranked highly as one of the most promising biomaterials.5–7 It is well-known that this biomaterial possesses first-rate biocompatibility, biodegradability, bioactivity, nontoxicity and non-inflammatory characteristics which demonstrate its extensive use in various biomedical fields, for example, as a bone cavity filling material, for drug delivery applications, bone substitutes, implant components, coatings on implants, the treatment of hyperthermia in cancer patients, dental materials and so on.8,9 Indeed, the ability of hydroxyapatite (Hap) to augment bone growth within the bone tissue has made it a potential candidate for use as a hard tissue-implant material. Furthermore, apart from biomedical applications, many other remarkable and diverse uses of Hap have also been reported.10,11With regard to these particular applications, the development of novel approaches to synthesize Hap using various Ca and P sources is now a vital task for researchers. Nevertheless, to expand the biomaterial research field, a significant number of procedures have been developed by scientists to synthesize Hap, such as wet chemical precipitation,1,12,13 electrochemical methods,14,15 solid-state reactions,16 hydrothermal,17,18 microemulsion,19 sol–gel20 and microwave irradiation21 methods and so on. It must be mentioned here that the synthesis protocols strictly regulate the morphology and structural characteristics of Hap, which eventually control its biomedical applications. For instance, Hap in hollow microparticle format has a high surface area and an extended drug loading ability, which makes it a promising candidate vehicle for drug release.22 Consequently, during the last couple of years, a significant volume of research studies have explored numerous schemes using Hap-based drug carriers.8,9,11,22–24 However, researchers have drawn a generalized consensus about the perfect bone substitution material, in which together with the biocompatibility, biodegradability and bioactivity, the material should have adequate drug delivery and bactericidal properties, but no systematic toxicity.24
Keeping the above discussed viewpoints in mind, in the present study we aimed to develop a novel protocol to synthesize hydroxyapatite at room temperature. Indeed, in recent years, instead of using conventional methods, researchers have focused on the development of eco-friendly methods, in which room temperature based protocols, rather than high temperatures, have been used to facilitate the synthesis of Hap.11 Herein, our approach was confined to the development of a UV-assisted solid-state method to synthesize hydroxyapatite (Hap) from eggshells for the first time, in which no thermal treatment was applied. The potency of the UV-assisted Hap as a biomaterial was examined via the bioactivity, cytotoxicity and drug loading-releasing response. Ciprofloxacin is a well-known broad-spectrum second generation fluoroquinolone antibiotic used to treat osteomyelitis, a bacterium causing bone infections,25 we examined the suitability of the UV-mediated Hap as a drug carrier by monitoring the loading–releasing profile of ciprofloxacin as the targeted drug.
2. Materials and methods
2.1 Synthesis of UV-sensitized Hap
Prior to UV-excitation, Ca and P precursors (eggshells and (NH4)2HPO4 respectively) were treated together for 8 h with a high energy planetary ball mill, which resulted in a homogeneous mixture of the starting materials in the form of fine particles. Focused UV-illumination ensured that the maximum output level of intensity was applied to this mixture through a halogen lamp of 500 W for a period of 8 h. The entire operation triggered the formation of the desired Hap. The schematic diagram shown in Fig. 1 illustrates the developed procedure.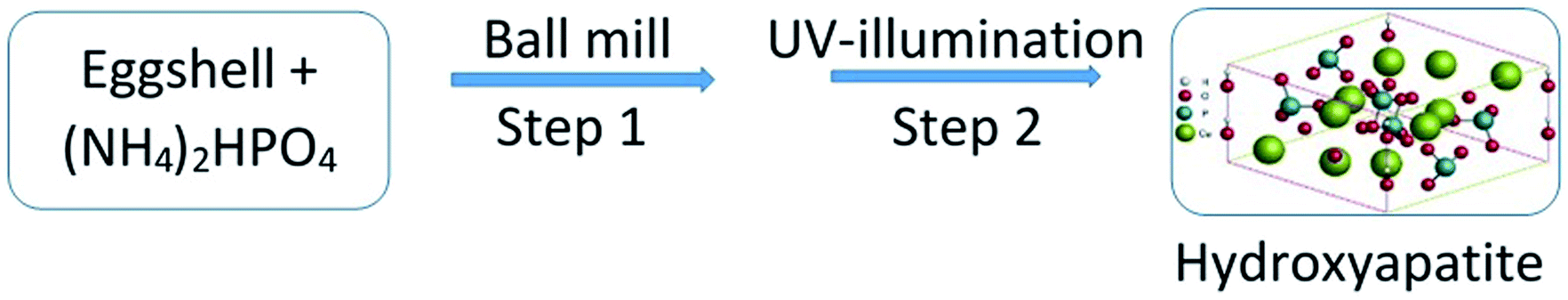 | ||
| Fig. 1 Schematic representation of the UV-mediated solid-state synthesis of Hap. | ||
2.2 Characterization of UV-sensitized Hap
Customary characterization of this UV-assisted Hap was performed using X-ray diffraction (XRD), Fourier-transform infrared (FT-IR) and Raman spectroscopy techniques, and to validate our observations, Hap formation was confirmed by comparing the observed data with the respective characteristic values.The XRD analysis (model: PANalytical X'pert PRO XRD PW 3040) facilitated the phase categorization of the UV-mediated Hap. To collect the data, continuous scanning with 0.01 steps and the 2θ = 5–75° using CuKα (λ = 1.54060) radiation operating at a combination of 40 kV and 30 Ma was used, and the cooling temperature was fixed at 19–20 °C. The observed phases were confirmed by comparison with the standard JCPDS files representative of pure Hap. Additionally, in the case of the starting materials, analysis conducted prior to the UV-illumination phase was also taken into account.
Next, to clarify the functional groups present in Hap, we applied FT-IR and Raman spectroscopic techniques. Using the FT-IR-Prestige 21 (SHIMADZU) equipped with an attenuated total reflection (ATR) set-up, the FT-IR band positions were noted according to previous studies.19 Conversely, Raman mapping was performed with a high-resolution Raman spectrometer (HORIBA MacroRAM™) in the 3400 cm−1 to 100 cm−1 region using the 785 nm wavelength excitation from a diode pumped solid-state laser. The adjustable output power was set at 5 mW.
2.3 Evaluation of the biomedical performance
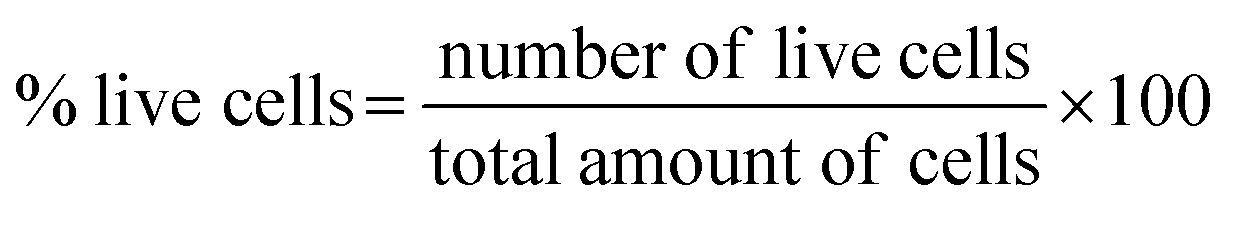 | (1) |
To compare the observed results, the entire approach was repeated using a control.
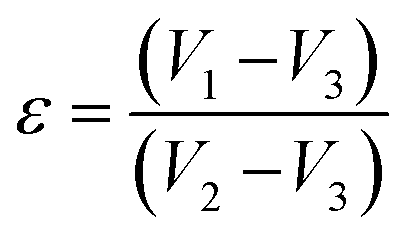 | (2) |
The fabricated scaffolds were then submerged in 50 mL aqueous ciprofloxacin solution (conc. = 20 μg mL−1) using an air-tight reaction vessel. After 24 h the scaffolds were withdrawn from the vessel and subjected to various examinations: (i) a few scaffolds were oven dried at 100 °C for phase analysis and molecular characterization using XRD and FT-IR, respectively; and (ii) using the rest of the Hap tablets impregnated with ciprofloxacin, the in vitro drug release was examined in synthesized body fluids (SBF) following the protocol reported elsewhere.28 The percentage of ciprofloxacin liberated from the scaffold was evaluated using the following equation:
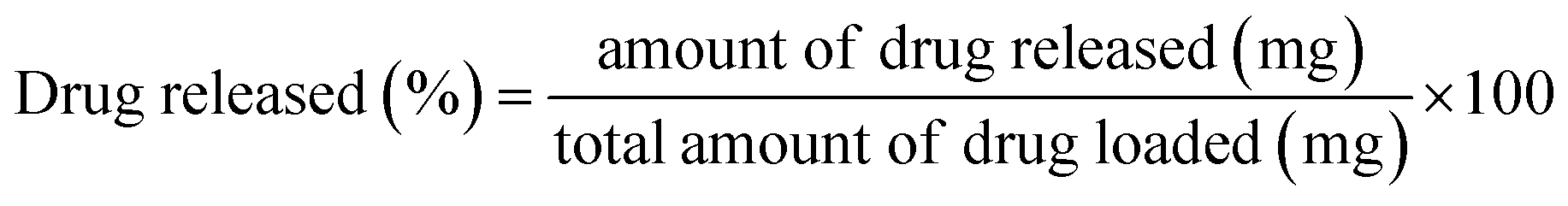 | (3) |
In contrast, the amount of ciprofloxacin loaded into the scaffolds (Fig. 5a) was computed by analyzing the concentration of the residual ciprofloxacin solution according to the Beer–Lambert law using a UV-spectrophotometer. The wavelength maximum, otherwise known as the λmax, of Cipro is 278 nm and this wavelength was chosen for the absorbance measurement.
3. Results and discussion
3.1 Characterization of the UV-sensitized Hap
The XRD patterns of the starting materials are depicted in Fig. 2a–d which provides an impression of how the associated phases are presented in the respective raw materials. Noticeably, in Fig. 2a and b, distinctive peaks for calcite and diammonium hydrogen phosphate (ICDD codes # 01-072-4582 and 00-008-0033 respectively)29 can be observed. In contrast, the XRD spectrum (Fig. 2c) of these two materials in the mixed form only shows their characteristic peaks, with no peaks observed for Hap or β-TCP. This observation proves that no phase transformation occurs at this stage. However, the diffractogram obtained for the ball milled and oven dried mixture (without applying UV-illumination) of the raw materials, as shown in Fig. 2d, clearly reveals a change. A wide hump covering the 2θ positions around 31–35° appeared along with a strong diffraction peak at the 29.41° position (104 plane) responsible for CaCO3. These peaks confirm that this dual mode operation (i.e., ball milling and oven drying only) was not sufficient to complete the reaction for the full-fledged formation of hydroxyapatite.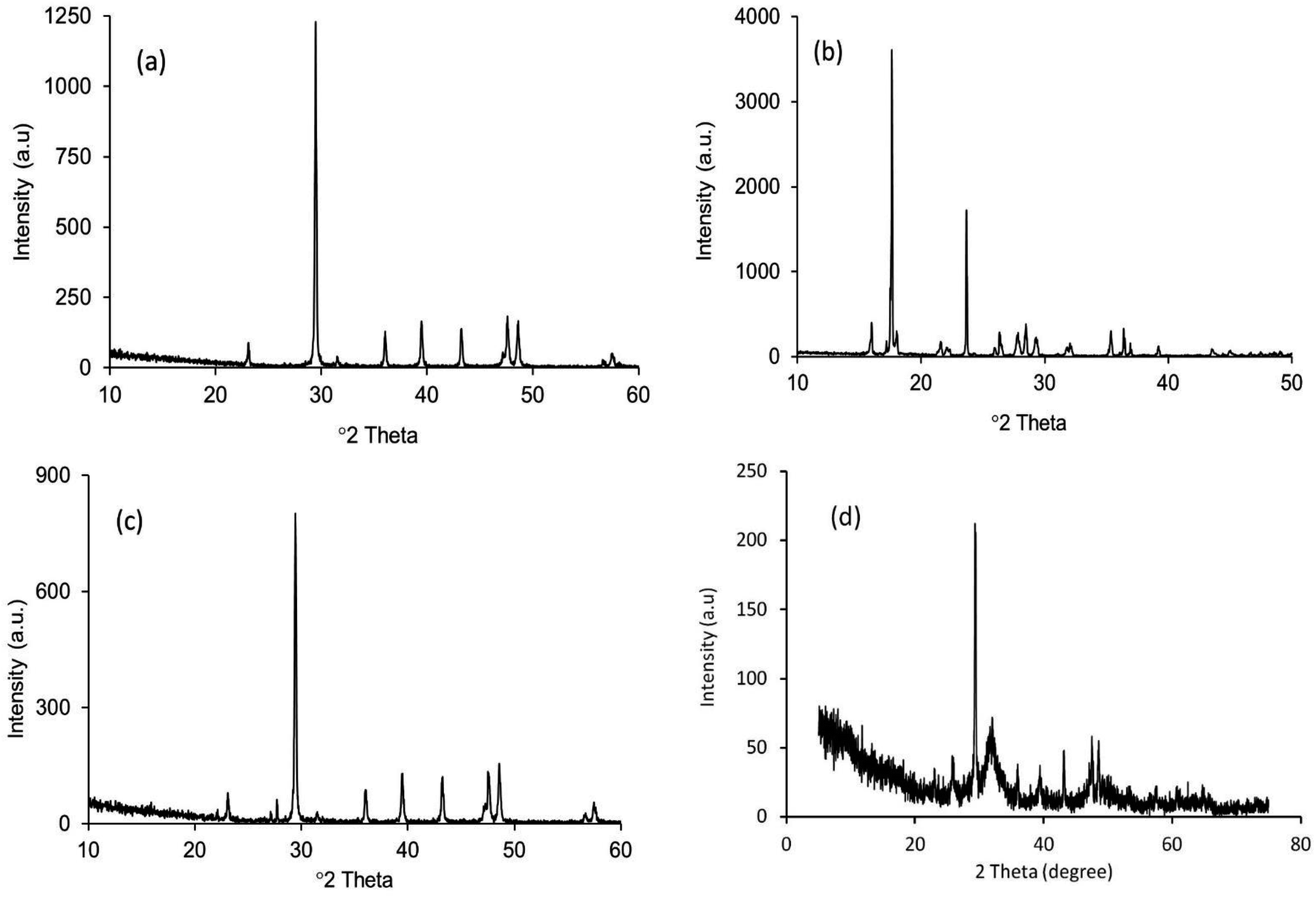 | ||
| Fig. 2 XRD patterns of the starting materials: (a) eggshell; (b) (NH4)2HPO4; (c) mixture of eggshell and (NH4)2HPO4 without any treatment; and (d) the eggshell and (NH4)2HPO4 mixture after ball milling followed by oven drying at 100 °C. | ||
The XRD diffractogram (Fig. 3) displays well-matched typical characteristic d-spacing values (Å) of 3.44 (002), 3.08 (210), 2.81 (211), 2.77 (112) and 2.71 (300) supporting the formation of crystalline Hap with a hexagonal phase structure.19,30 Further validation of this observation was accomplished by deducing the crystallographic parameters using the well-established equations7 and a very good match was observed between the calculated crystallographic values of the UV-assisted Hap and the JCPDS (file # 09-0432)19 data. This proved that irradiation of the eggshell with (NH4)2HPO4 resulted in the successful formation of the desired Hap. Nevertheless, in addition to Hap, peaks for β-TCP and CaCO3 were also indexed in the recorded XRD (Fig. 3). Having d-spacing values (Å) of 2.87 and 2.60, β-TCP appeared as the second phase and the presence of CaCO3 was proven by the peak noted at the 2θ position 29.36°.
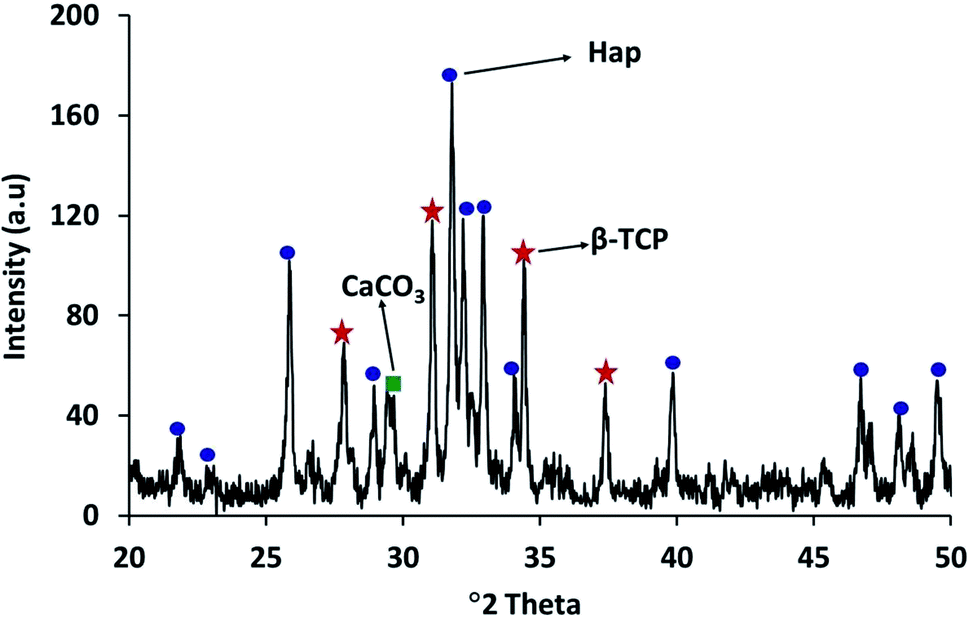 | ||
| Fig. 3 XRD pattern of Hap synthesized by UV-excitation of eggshells. | ||
Next, to clarify the functional groups present in Hap, we used the FT-IR and Raman spectroscopic techniques. Both the FT-IR and Raman spectroscopic data, as tabulated in Tables 1 and 2tbl2, respectively, reveal that all the characteristic band positions, as well as the Raman shifts, are in good agreement with those observed in previous studies.19,31 Moreover, the presence of CaCO3 in the UV-synthesized Hap is also supported by the peaks, as evidenced for the CO32− group at band positions 877 and 1418 cm−1 in the FT-IR spectrum. Further confirmation of the CO32− group was provided by the Raman spectroscopic data, in which the Raman shift for the CO32− group was observed at 1070 cm−1.
| Band position, cm−1 | Corresponding assignment |
|---|---|
| 471 | PO43− bending (ν2) |
| 562 | Asymmetric mode (ν4) of the PO43− group |
| 603 | Symmetric mode (ν4) of the PO43− group |
| 632 | OH liberation |
| 958 | PO43− stretching (ν1) |
| 1028 | Asymmetric stretching mode (ν3) of the PO43− group |
| 1651 | Bending mode of H–O–H |
| Raman shift, cm−1 | Corresponding assignment |
|---|---|
| 427 | Bending mode (ν2) representative of the PO43− group |
| 585 | Bending mode (ν4) representative of the PO43− group |
| 963 | Symmetric stretching mode (ν1) ascribed to the tetrahedral PO43− group |
The likely reason for the formation of Hap as a result of the UV-illumination of the blended starting materials at ambient temperature can be justified by the following explanation provided by Verwilghen et al.32 primarily, upon UV-illumination of the mixture, (NH4)2HPO4 releases NH3 and the hydrogen phosphate groups are adsorbed onto the carbonate surface, deprotonation then occurs through discharging of the carbonate anions into the solution, forming calcium orthophosphate. Conversely, UV-excitation of the moistened CaCO3 triggers the slow evolution of CO2, buffering the environment as basic conditions are favorable for the formation of Hap.
3.2 Cytotoxic assessment
Fig. 4a–d shows microscopic images of the Vero cells treated with different concentration of Hap under controlled conditions, and Fig. 4e shows the corresponding cell viability. Clearly, although the survival rate of the Vero cells was found to decrease slowly as a function of the Hap concentration, the cell viability is still within a fairly significant limit. More specifically, the calculated percentages of live cells (Fig. 4e) were 88.41%, 86.68% and 82.30% in the presence of Hap concentrations of 30, 50 and 100 mg mL−1 respectively. This indicates that in each case, after a 24 h incubation period, the cell viability was more than 50%, which indicates that the UV-sensitized Hap exhibited a negligible cytotoxicity, complying with the concentration limit allowed by ISO standards.33 This observation indicates that the Hap synthesized using UV-sensitization of eggshells is certainly safe enough to be used as a biomaterial.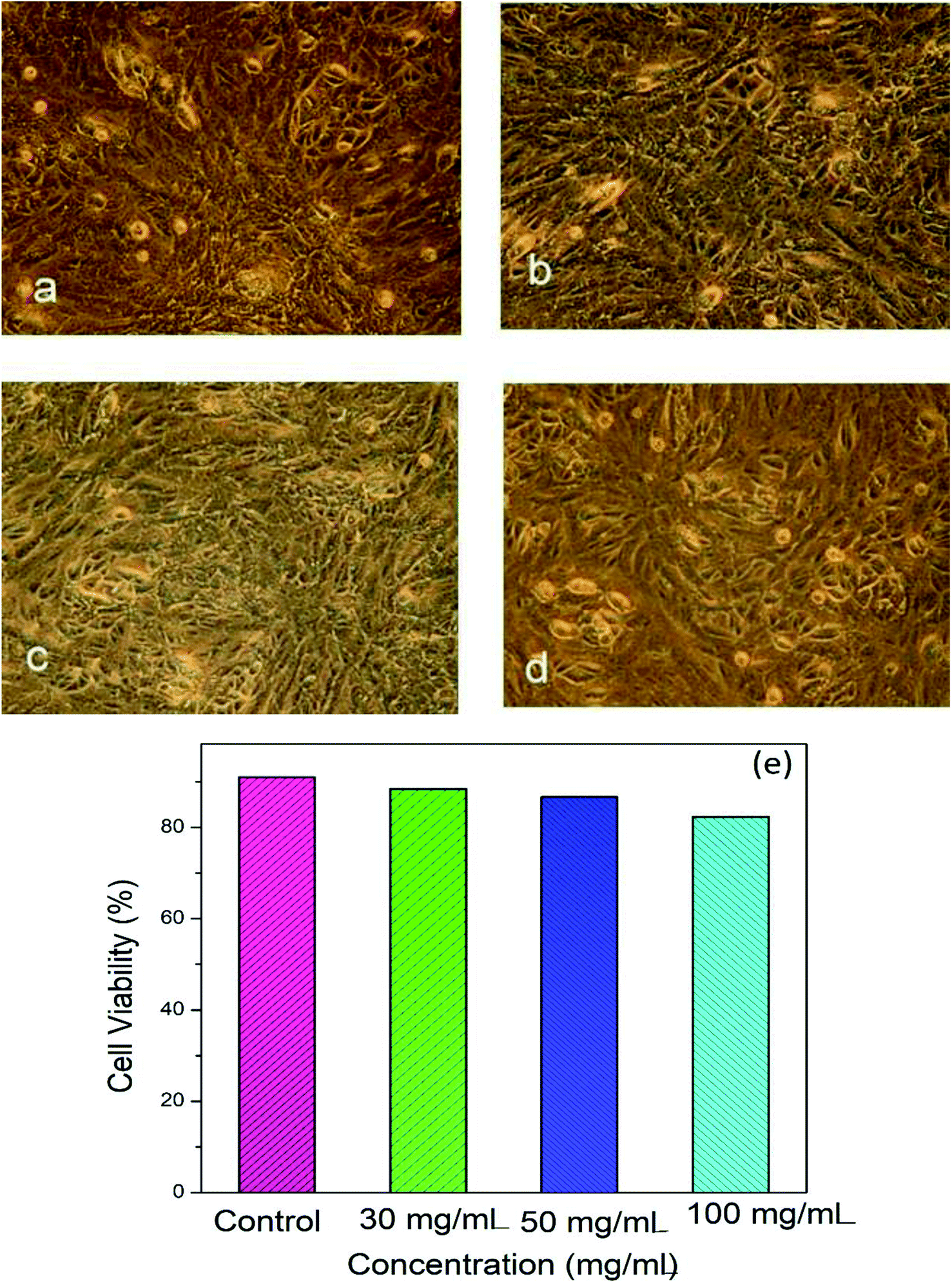 | ||
| Fig. 4 Cytotoxicity response of UV-mediated Hap: (a–d) microscopic images of the Vero cells (controlled conditions (a); Hap = 30 mg mL−1 (b); Hap = 50 mg mL−1 (c); and Hap = 100 mg mL−1 (d)). (e) The cytotoxic effects observed for the Vero cell line after incubation for 24 h. Results are compared with the control (DMSO). | ||
3.3 Evidence of ciprofloxacin loading and releasing
The characteristic d-spacing values (Å) 2.81, 2.77 and 2.71 accountable for Hap,19,30 and the XRD pattern of the Cipro loaded Hap were coupled with the peaks for Cipro (2θ positions at 25.81°, 26.51° and 29.35°)25 as expected. It should be noted that the 2θ position 29.35° possibly overlapped with calcite. However, this data obviously supports the assimilation of ciprofloxacin within the scaffold. Further investigation to provide a better understanding about the ciprofloxacin loading was performed using the FT-IR approach. The FT-IR spectrum (Fig. 5b) of the Cipro-incorporated Hap clearly attests to the successful loading of the drug by showing a broad peak at 1652 cm−1, which is a combination of the two individual peaks observed (in the case of Cipro; Fig. 5c) at 1707 and 1625 cm−1 for ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)c (c is carboxylic) and the ν(CO)p (p is pyridone) vibrations, respectively,28 whereas these two peaks were absent in Hap (Fig. 5a).
O)c (c is carboxylic) and the ν(CO)p (p is pyridone) vibrations, respectively,28 whereas these two peaks were absent in Hap (Fig. 5a).
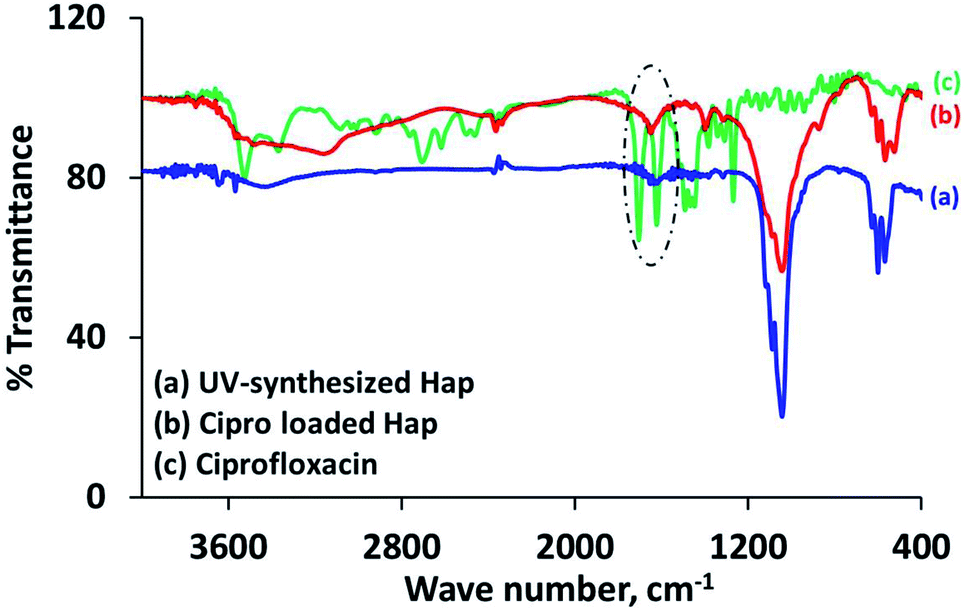 | ||
| Fig. 5 FT-IR spectra: (a) UV-Hap; (b) Cipro loaded Hap; and (c) Cipro. | ||
The profile picture of the loaded ciprofloxacin (as a percentage) versus the amount of porogen added is shown in Fig. 6a, in which the clear contribution from the pore forming agent is visible; without any accumulation of glucose, the percentage of drug loading was at the minimum and this percentage tends to increase as a function of the porogen addition and maximum loading (78.58%, i.e., = 15.72 μg mL−1) was achieved when 10% (w/w) glucose was added with Hap. This is because upon sintering, the higher percentage of glucose forms more pores within the Hap scaffold. Accordingly, the porosity of the porogen incorporated scaffolds was calculated using eqn (2) and a higher percentage (21–23%) of porosity was witnessed when 10% (w/w) glucose was added to the scaffold, which facilitates the incorporation of more of the drug. This observation is in agreement with a previous study34 in which the permeability of the Hap tablets was found to be reliant on the porosity, as well as on the connectivity of the porous network.
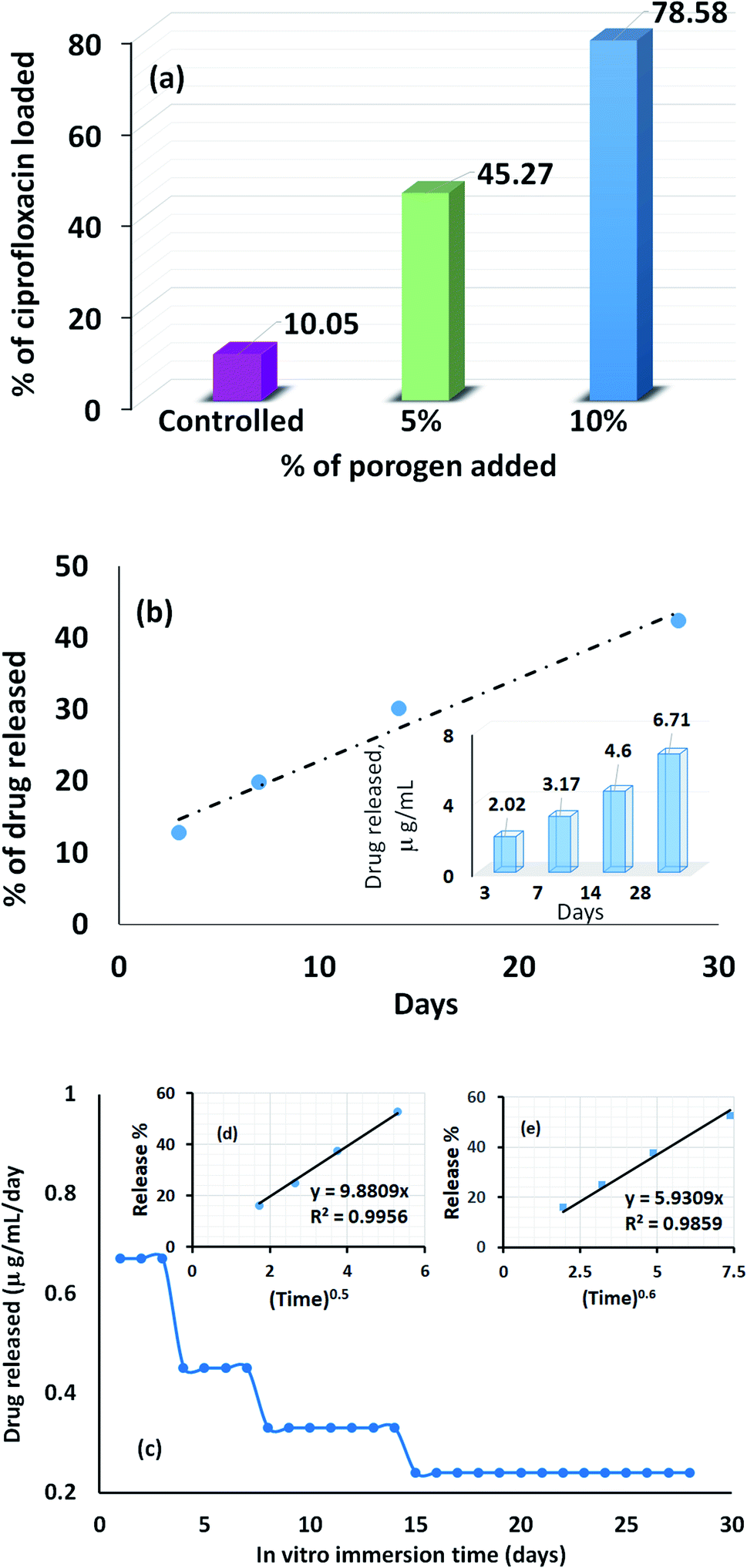 | ||
| Fig. 6 Drug loading characteristics of the UV-synthesized Hap scaffold: (a) ciprofloxacin loading efficiency; (b) time dependent drug release efficiency in SBF; and (c) time dependent average release of ciprofloxacin per day (inset (d) the Higuchi model, and inset (e) the Ritger–Peppas model). | ||
The recorded percentage of drug released at different time intervals (3, 7, 14 and 28 days) from the 15.72 μg mL−1 ciprofloxacin loaded scaffold is given in Fig. 6b. A gradual increase in the drug discharge profile is noticed as the time progresses and after 28 days, 42.37% of the loaded ciprofloxacin was liberated in the non-stop mode from the scaffold. A similar trend was observed in previous investigations.28,35 Considering the sustained release behavior, we presume that the drug delivery carriers we developed may have a mesoporous structure; but to come up with a concrete conclusion more investigations are needed and further research in this direction is being performed in our group. However, interestingly, the collective release profile of ciprofloxacin from the Hap scaffold at each time interval showed (inset in Fig. 6b) that the ciprofloxacin release pattern followed a therapeutic level of ciprofloxacin for osteomyelitis (2–50 μg mL−1).36,37 Consequently, we anticipate that the UV-sensitized Hap could have potential for use as a local antibiotic therapy by controlling ciprofloxacin release in the treatment of osteomyelitis.
The average amount released per day (Fig. 6c) was deduced by dividing the measured concentration by the soaking period (i.e., the number of days between changing the SBF solution). Fig. 6c demonstrates that for the first 3 days, the maximum per diem drug discharge was observed. This rapid initial release is due to the significant difference in the drug concentration between the scaffolds and the release medium. A sharp decrease was monitored on the 4th day and remained fairly stable for the next 3 days. Again, each time the drug releasing medium was replenished, a noticeable change was visualized on the 8th and 14th day, respectively, but the release profile remained almost constant throughout the last 14 days. Nevertheless, to gain a better understanding of the ciprofloxacin ejection and associated transport phenomenon, two release kinetic models: (i) the Higuchi model (Q = Kt0.5); and (ii) the Ritger–Peppas model (Q = Kt0.6) were used to interpret the collective release percentage data.38,39 In both models, Q represents the cumulative amount of drug released during the in vitro immersion time, t, per unit area and K is the dissolution constant characterizing the rate of drug release. The cumulative release percentage was plotted against t0.5 and t0.6 for the Higuchi model (inset (d) in Fig. 6c) and the Ritger–Peppas model (inset (e) in Fig. 6c). A higher correlation coefficient (0.9956) was obtained in case of the Higuchi model, which showed that the ciprofloxacin release mechanism is strictly diffusion controlled.
3.4 Bioactive response
The recorded apatite formation ability, otherwise known as the bioactive response of the scaffold, is depicted in Fig. 7a, which shows a time dependent increment in the measured weight of the scaffold. This ensures the bone in-growth ability of the prepared scaffold while incubated in SBF. The apatite formation/precipitation on the scaffold surface can be elucidated with the assistance of a previously published report.40 Briefly, Hap typically comprises two types of negative ions, (e.g., OH− and PO43−) coupled with the positive ion, Ca2+. The negatively charged ions of Hap usually stay on the surface and attract the positively charged calcium ions in the SBF solution. This phenomenon results in an extra positive charge on the surface of the SBF nursed Hap which will then react again with more OH− and PO43− ions that are present in the SBF. This mechanism continues to form Hap nuclei on the surface of the scaffold and thus provide the bioactive nature of the developed UV-sensitized Hap. Fig. 7b shows this bioactive mechanism.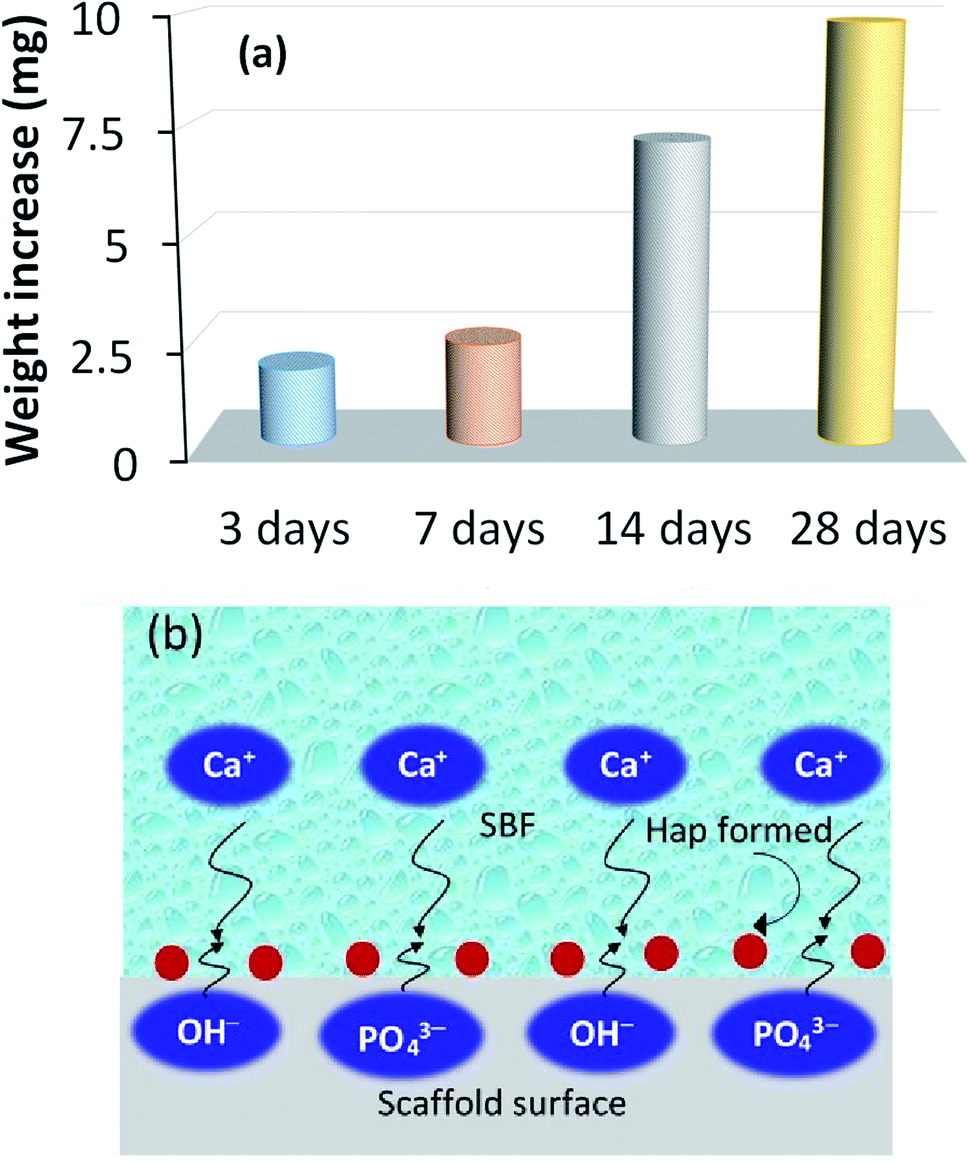 | ||
| Fig. 7 Bioactive response of the UV-synthesized Hap scaffold: (a) time dependent increment of the scaffold weight while incubated in SBF; and (b) pictorial representation of the bioactive process. | ||
Images of this apatite formation phenomenon at different time intervals were captured to show the changes in the surface morphology of the UV-excited Hap and the corresponding pictures are shown in Fig. 8a–d. The image of the blank scaffold clearly shows the pores which gradually started to fill during the course of SBF immersion, as expected.
 | ||
| Fig. 8 SBF treated UV-synthesized Hap scaffold at various time intervals: (a) blank scaffold; (b) after 7 days; (c) after 14 days; and (d) after 28 days. | ||
4. Conclusions
In conclusion, Hap has been developed from eggshells for the first time via UV-irradiation of the source materials. A negligible cytotoxicity, together with the efficient delivery of a therapeutic level of ciprofloxacin for the treatment of osteomyelitis, and the bioactive behavior of this newly developed Hap support its' application in the biomedical field. Hence, UV-radiation could be an effective alternative approach for the synthesis of Hap at room temperature.Conflicts of interest
There is no conflict to declare.Acknowledgements
Sazia Sultana gratefully acknowledges BCSIR for the AAMS Postgraduate Fellowship to accomplish this research work. We thank the BCSIR authority for financial support (R&D ref. no. 39.02.0000.011.14.012.2017.1386, date 18.04.2018 and 39.02.0000.011.14.111.2019.224, date 06.11.2019). Assistance from Pharmaceutical Sciences Research Division, BCSIR Laboratories Dhaka and CARF, BCSIR is also appreciated.Notes and references
- G. T. El-Bassyouni, S. S. Eldera, S. H. Kenawy and E. M. A. Hamzawy, Heliyon, 2020, 6, e04085 CrossRef.
- C. Suresh Kumar, K. Dhanaraj, R. M. Vimalathithan, P. Ilaiyaraja and G. Suresh, J. Asian Ceram. Soc., 2020, 8(2), 416–429 CrossRef.
- S. M. Naga, A. M. Hassan, M. Awaad, A. Killinger, R. Gadow, A. Bernstein and M. Sayed, J. Asian Ceram. Soc., 2020, 8(2), 373–386 CrossRef.
- M. Saleem, S. Rasheed and C. Yougen, Sci. Technol. Adv. Mater., 2020, 21, 242–266 CrossRef CAS.
- A. Haider, S. Haider, S. S. Han and I. Kang, RSC Adv., 2017, 7, 7442–7458 RSC.
- N. A. S. M. Puad, P. Koshy, H. Z. Abdullah, M. I. Idris and T. C. Lee, Heliyon, 2019, 5, e01588 CrossRef.
- S. Ahmed, F. Nigar, A. I. Mustafa and M. Ahsan, Trans. Ind. Ceram. Soc., 2017, 76(4), 215–221 CrossRef CAS.
- D. Li, J. He, X. Huang, J. Li, H. Tian, X. i Chen and Y. Huang, RSC Adv., 2015, 5, 30920–30928 RSC.
- Q. Wu, C. Liu, L. Fan, J. Shi, H. Jia, Q. Qi, L. Sun and F. Chen, RSC Adv., 2013, 5, 7486–7494 RSC.
- K. Alorku, M. Manoj and A. Yuan, RSC Adv., 2020, 10, 40923–40939 RSC.
- L. Sheikh, S. Tripathy and S. Nayar, RSC Adv., 2016, 6, 62556–62571 RSC.
- S. Mondal, A. Dey and U. Pal, Advances in Nano Research, 2016, 4(4), 295–307 CrossRef.
- A. Ibrahim, X. Li, Y. Zhou, Y. W. Chen, H. Wang and J. Li, Int. J. Mol. Sci., 2015, 16, 7960–7975 CrossRef CAS.
- A. Nur and H. Setyawan, J. Chem. Eng, Japan, 2016, 49(2), 144–151 CrossRef CAS.
- T. Salsabila, W. Widiyastuti and H. Setyawan, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 858, 012021, DOI:10.1088/1757-899X/858/1/012021.
- S. Wu, H. Hsu, S. Hsu, Y. Chang and W. Ho, J. Asian Ceram. Soc., 2016, 4, 85–90 CrossRef.
- A. R. Noviyanti, N. Akbar, Y. Deawati, E. E. Ernawati, Y. T. Malik, R. P. Fauzia and Risdiana, Heliyon, 2020, 6, e03655 CrossRef.
- A. A. Chaudhry, S. Haque, S. Kellici, P. Boldrin, I. Rehman, F. A. Khalid and J. A. Darr, Chem. Commun., 2006, 2286–2288 RSC.
- S. A. Jahan, M. Y. A. Mollah, S. Ahmed and M. A. B. H. Susan, Mater. Today: Proc., 2017, 4, 5497–5506 Search PubMed.
- K. Ishikawa, E. Garskaite and A. Kareiva, J. Sol-Gel Sci. Technol., 2020, 94(3), 551–572 CrossRef CAS.
- M. N. Hassan, M. M. Mahmoud, A. A. El-Fattah and S. Kandil, Ceram. Int., 2016, 42, 3725–3744 CrossRef CAS.
- D. Feng, J. Shi, X. Wang, L. Zhang and S. Cao, RSC Adv., 2013, 3, 24975–24982 RSC.
- T. Long, Y. Guo, Y. Liu and Z. Zhu, RSC Adv., 2013, 3, 24169–24176 RSC.
- S. Tang, B. Tian, Q. Ke, Z. Zhu and Y. Guo, RSC Adv., 2013, 3, 41500–41509 Search PubMed.
- S. Thangadurai, S. K. Shukla, A. K. Srivastava and Y. Anjaneyulu, Acta Pharm., 2003, 53, 295–303 CAS.
- N. Khan, F. Afroz, M. N. Begum, S. R. Rony, S. Sharmin, F. Moni, C. M. Hasan, K. Shaha and M. H. Sohrab, Toxicol. Rep., 2018, 5, 970–976 CrossRef CAS.
- M. S. Rahman, M. M. Rana, L. Spitzhorn, N. Akhtar, M. Z. Hasan, N. Choudhury, T. Fehm, J. T. Czernuszka, J. Adjaye and S. M. Asaduzzaman, Prog. Biomater., 2019, 8, 137–154 CrossRef CAS.
- S. Sasikumar, Front. Mater. Sci., 2013, 7(3), 261–268 CrossRef.
- S. Ahmed, M. H. Kabir, F. Nigar, S. F. Kabir, A. I. Mustafa and M. Ahsan, Science Vision, 2011, 16 & 17, 81–92 Search PubMed.
- Q. Wang, B. Zhou, X. Weng, S. Lv, F. Schüthb and A. Lu, Chem. Commun., 2019, 55, 10420–10423 RSC.
- P. E. Timchenko, E. V. Timchenko, E. V. Pisareva, M. Y. Vlasov, L. T. Volova, O. O. Frolov and A. R. Kalimullina, J. Opt. Technol., 2018, 85(3), 130–135 CrossRef CAS.
- C. Verwilghen, M. Chkir, S. Rio, A. Nzihou, P. Sharrock and G. Depelsenaire, Mater. Sci. Eng., C, 2009, 29, 771–773 CrossRef CAS.
- ISO10993-5, Biological evaluation of medical devices, test for in vitro cytotoxicity, International organization or standardization, Geneva, Switzerland, 3rd edn, Publication date Committee, ISO/TC: 2009-06, pp. 1–34, Technical 194 Biological and clinical evaluation of medical devices, http://www.iso.org/standard/36406.html Search PubMed.
- A. Cosijns, C. Vervaet, J. Luyten, S. Mullens, F. Siepmann, L. Van Hoorebeke, B. Masschaele, V. Cnudde and J. P. Remon, Eur. J. Pharm. Biopharm., 2007, 67, 498–506 CrossRef CAS.
- T. J. Mäkinen, M. Veiranto, P. Lankinen, N. Moritz, J. Jalava, P. Törmälä and H. T. Aro, J. Antimicrob. Chemother., 2005, 56, 1063–1068 CrossRef.
- K. C. Rani, R. Primaharinastiti and E. Hendradi, Int J Pharm Pharm Sci, 2016, 8(1), 45–51 CAS.
- J. K. Koort, T. J. Mäkinen, E. Suokas, M. Veiranto, J. Jalava, P. Törmälä and H. T. Aro, Acta Orthop., 2008, 79(2), 295–301 CrossRef.
- M. Ciocilteu, A. G. Mocanu, A. Mocanu, C. Ducu, O. E. Nicolaescu, V. C. Manda, A. Turcu-Stiolica, C. Nicolicescu, R. Melinte, M. Balasoiu, O. Croitoru and J. Neamtu, Acta Pharm., 2018, 68, 129–144 CAS.
- R. Gouda, H. Baishya and Z. Qing, J. Dev. Drugs, 2017, 6, 171, DOI:10.4172/2329-6631.1000171.
- S. C. S. Song, S. W. Phang and W. H. Yap, AIP Conf. Proc., 2019, 2137, 020015, DOI:10.1063/1.5120991.
| This journal is © The Royal Society of Chemistry 2021 |