
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyesters with bio-based ferulic acid units: crosslinking paves the way to property consolidation†
Doris
Pospiech
 *a,
Andreas
Korwitz
a,
Hartmut
Komber
a,
Dieter
Jehnichen
a,
Kerstin
Arnhold
a,
Harald
Brünig
a,
Holger
Scheibner
a,
Michael T.
Müller
a and
Brigitte
Voit
ab
*a,
Andreas
Korwitz
a,
Hartmut
Komber
a,
Dieter
Jehnichen
a,
Kerstin
Arnhold
a,
Harald
Brünig
a,
Holger
Scheibner
a,
Michael T.
Müller
a and
Brigitte
Voit
ab
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: pospiech@ipfdd.de
bOrganic Chemistry of Polymers, Technische Universität Dresden, 01069 Dresden, Germany
First published on 23rd August 2021
Abstract
Aromatic–aliphatic polyesters can be prepared as bio-based materials by incorporating monomers from biogenic sources. Here, 2-methyl (E)-3-(4-(2-hydroxyethoxy)-3-methoxyphenyl)acrylate, a derivative of ferulic acid, was synthesised and inserted into the structure of aliphatic and aromatic–aliphatic polyesters as a comonomer. Terpolymers with high relative molar masses Mw from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 100000 g mol−1 can be successfully prepared by transesterification polycondensation in the melt. Incorporation of ferulate units with intact double bonds is proven by NMR spectroscopy. Insertion of low molar amounts of ferulate units reduces the crystallinity of the base polyesters, but enhances the glass transition temperature. It is demonstrated that the materials after melt processing to a suitable shape (fibres, parts, films) can be crosslinked by electron beam irradiation, which enhances the stability of the materials.
000 to 100000 g mol−1 can be successfully prepared by transesterification polycondensation in the melt. Incorporation of ferulate units with intact double bonds is proven by NMR spectroscopy. Insertion of low molar amounts of ferulate units reduces the crystallinity of the base polyesters, but enhances the glass transition temperature. It is demonstrated that the materials after melt processing to a suitable shape (fibres, parts, films) can be crosslinked by electron beam irradiation, which enhances the stability of the materials.
1 Introduction
Over the years, the development and utilization of bio-based polymers have gained significant attention in an effort to increase the sustainability of the plastics industry.1 Bio-based polymers originate exclusively from natural biomass as the only source of renewable carbon.2 The main feedstocks are natural polymers that occur directly in nature (for instance cellulose and chitosan), plant oils, natural rubber, food feedstock (so-called sugar platform) and biomass that remains after industrial extraction of cellulose.2,3 Lignocellulose is the most abundant natural polymer.4 After extracting the cellulose, a lignin-rich liqueur remains to about 14–30 wt% forming the second most abundant source of natural polymers and the most important source for renewable aromatic compounds, among them furans, terpenes, and monolignols (p-coumaryl alcohol, sinapyl alcohol and coniferyl alcohol).3,5–8 So far, the only industrial process to generate pure low molar mass compounds out of lignin is the production of vanillin (4-hydroxy-3-methoxybenzaldehyd) from lignosulfonate via sulphite pulping7 or, more recently, by electrochemical transformation of Kraft lignin9 to vanillin. Vanillin can easily be converted into several monomers, thus forming a significant chemical platform for synthesis of bio-based polymers.1,7,10–13 Two of the most important derivatives are 4-hydroxy-3-methoxybenzoic acid and 2-methyl-hydroquinone which have frequently been used as comonomers for polyesters.11,12,14–18 Mialon et al.,12 for instance, converted vanillin into poly(dihydroferulic acid), a bio-based analogue of poly(ethylene terephthalate).Another interesting monomer that is either incorporated directly into the lignin structure (for instance in plants such as Pinus radiata and Popoulus tremula x Polpulus Alb), or is converted from coniferaldehyde (from lignin) via oxidation by hydroxyl-cinnamaldehyde dehydrogenase, is the bifunctional monomer ferulic acid (FA, (E)-3-(4-hydroxy-3-methoxy-phenyl)acrylic acid or 4-hydroxy-3-methoxy-cinnamic acid), an hydroxycarboxylic acid.19 FA acid has been reported to show antimicrobial,20 antioxidant21 and even anticancer22 properties that are ascribed to the phenolic structure of the compound.
A few attempts exist in which ferulic acid was subjected directly to polycondensation. Llevot et al.7 reported homopolyesters based on FA. The weight-averaged molar masses Mw were in most cases below 10000 g mol−1. Ouimet et al.23,24 protected the carboxylic group by a tert-butyl group and reacted the OH group with carboxylic acid dichlorides. Deprotection of the tert-butyl functionalities and reaction of the resulting diols with triphosgene in solution yielded poly(anhydride-ester)s with intermediate Mw (15000–18000 g mol−1) and intact double bonds. These polyesters were developed for applications as antioxidant requiring the preserved double bond. The procedure appears rather complicated to be transferred to industry. However, most of the studies used FA in hydrogenated form (typical hydrogenation with Pd/C catalyst).7,17 Llevot et al.7 reported homopolycondensation of hydrogenated and glycolised FA yielding polymers with molar masses in the range of 5000 g mol−1. Pion et al.17 reacted hydrogenated FA enzymatically with isosorbide resulting in new bio-based monomers. Upton et al.25 dimerised FA by UV irradiation for 20 h to yield a dicarboxylic acid as monomer for polyamides.
In this contribution, the incorporation of ferulate units into aromatic–aliphatic and aliphatic polyesters was studied to evaluate the possibility of preparation of random terpolymers (copolyesters with three basic units). The carboxylic group of FA was activated by conversion into the corresponding methyl ester, while the phenolic OH groups were transformed into alcoholic ones by reaction with ethylene carbonate. It was studied whether this modified monomer (abbreviated F) allows the use of a standard melt transesterification polycondensation as applied, e.g., for poly(butylene terephthalate) (PBT).26,27 Five series of FA-containing copolyesters were synthesised to modify poly(ethylene terephthalate) (PET), poly(trimethylene terephthalate (PTT), poly(isopropylene terephthalate) (PiPrT), poly(butylene terephthalate) (PBT), and the aliphatic polyester poly(butylene succinate) (PBS). All these polyesters can be (at least potentially) produced from bio-based monomers.1 The molar content of the bio-based ferulic acid monomer was systematically enhanced in all series to find out the concentration that allows suitable modification of the base polyesters. The chemical structure of the polyesters was analysed by NMR spectroscopy also to demonstrate that the double bonds survived the rather harsh polycondensation conditions. The conditions were carefully optimised to supress side reactions of the double bond which would, e.g., result in crosslinking by thermal polymerisation of the acrylic group. The influence of the FA content on the solid state structure (crystallinity) and the melting behaviour was analysed in order to pre-estimate the material properties. Furthermore, the ability of the double bonds to polymerise under different conditions (thermally with radical initiator and by treatment with electron beams) was examined. Post-polymerisation was expected to form crosslinked structures that stabilise the polymer material. Thus, a thermoplastic material would result that can be processed in the melt and crosslinked after melt processing to stabilise the outer surface of the material. This is of particular importance for parts with low dimensions such as fibres and for 3D printed parts with inherently occurring heterogeneities originating from the process.28,29 The final content of FA in the copolyesters was intended to be as low as possible to achieve an optimum between the material properties of the base polymer and the ability to crosslink.
2 Experimental
Materials
1,4-Butanediol (99+%, over molecular sieves, ACROS Organics, Fisher Scientific GmbH, Schwerte, Germany), 1,3-propanediol (99.7+%, over molecular sieves, VWR International GmbH, Darmstadt, Germany), 1,2-propanediol (99.5+%, over molecular sieve, Sigma-Aldrich Chemie GmbH, Munich, Germany), dimethyl succinate (DMS, 98%, Sigma-Aldrich Chemie GmbH, Munich, Germany), ethylene glycol (99.8% anhydrous, Sigma-Aldrich Chemie GmbH, Munich, Germany), ethylene carbonate (anhydrous, Sigma-Aldrich Chemie GmbH, Munich, Germany), methanol (99.9%, ACROS Organics, Fisher Scientific GmbH, Schwerte, Germany), (E)-3-(4-hydroxy-3-methoxyphenyl)acrylic acid (ferulic acid) (99%, abcr, Karlsruhe, Germany), dimethyl terephthalate (99+%, Sigma-Aldrich Chemie GmbH, Munich, Germany), dimethylacetamide (99.5+%, Riedel-De-Haen, Munich, Germany), potassium acetate (99+%, ACROS Organics, Fisher Scientific GmbH, Schwerte, Germany), titanium(IV) butoxide (97%, Sigma-Aldrich Chemie GmbH, Munich, Germany), antimony trioxide (99.9%, ACROS Organics, Fisher Scientific GmbH, Schwerte, Germany) were used as received.Methods
NMR spectroscopy was performed using a Bruker Avance III 500 spectrometer (Bruker, Germany) at 500.13 MHz for 1H and at 125.74 MHz for 13C. Dimethylsulfoxide-d6 (DMSO-d6) was used as solvent for the monomers (δ(1H) = 2.50 ppm, δ(13C) = 39.6 ppm). A mixture of CDCl3 and trifluoroacetic acid (TFA-d; 4:1 v/v) was the solvent for the polyesters. Polymer spectra were referenced on the CHCl3 (δ(1H) = 7.26 ppm) and CDCl3 signal (δ(13C) = 77.0 ppm), respectively.
The solution viscosities were measured at 25 °C in an Ubbelohde viscometer (Schott GmbH, Jena, Germany) equipped with capillary I. The polymers were dissolved in a mixture of pentafluorophenol (PFP)/chloroform (1:1 v/v) with a polymer concentration of 0.5 g dL−1.
The relative molar masses were determined by size exclusion chromatography (SEC) with a Knauer modular SEC (Knauer, Berlin, Germany) equipped with PL MiniMix-D separation column (Polymer Standards, Mainz, Germany) and refractive index detection. A mixture of PFP/CHCl3 (1:2 v/v) was employed as eluent with a flow rate of 0.3 mL min−1. The measurements were performed at 45 °C. The relative molar masses were calculated using polystyrene standards (Polymer Standards, Mainz, Germany).
The melting behaviour was investigated by differential scanning calorimetry (DSC) using a DSC Q2000 (TA Instruments, New Castle, DE, USA) in the temperature range of −80 to 300 °C with a scan rate of 10 K min−1 under nitrogen. The second heating run was employed to determine the glass transition temperature Tg by the half-step method, the melting peak maximum Tmax and the melting enthalpy ΔHm.
The degradation behaviour was investigated by thermogravimetric analysis (TGA) with a Q 5000 (TA Instruments, USA) in the temperature range 30 to 800 °C with a heating rate of 10 K min−1 under nitrogen.
Wide-angle X-ray scattering (WAXS) profiles were recorded in symmetric transmission using the 2-cycle diffractometer XRD 3003 T/T (GE Sensing & Inspection Technologies GmbH, Ahrensburg, Germany). The measurements were conducted at 40 kV and 30 mA, CuKα radiation (monochromatised with Goebel mirror, λ = 0.15418 nm) with the following parameters: 2θ-range: 0.5–45°; measuring time per measuring point: 30 s; step size Δ2θ: 0.05°, room temperature. Samples were wrapped in Al foil or filled in glass tubes (Ø = 1 mm). For calculation of the crystallinity α, based on the intensity parts scattered by crystalline as well as amorphous amounts peak-fitting with pseudo-Voigt functions was executed by using the software package Analyze (included in RayfleX, software of the X-ray device).
Raman measurements were performed on melt-pressed samples using RAMAN Imaging System WITec alpha300R (WITec GmbH, Ulm, Germany) equipped with 785 nm laser (10 mW). 200 accumulations with integration time of 0.5 s were recorded for one spectrum.
The shore hardness of melt-pressed samples (thickness 4 mm, diameter 25 mm) was analysed by means of a Zwick 3140 device (Zwick GmbH & Co. KG, Ulm, Germany) according to DIN EN ISO 868 Shore D (15 s). The tensile strength of fibres was measured by an UPM Zwick-2.5 kN instrument (Zwick GmbH & Co. KG, Ulm, Germany) with a measuring velocity of 1 mm min−1 until deformation.
Synthesis of methyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate
The synthesis was performed according to the procedure described by Kreye et al.30 Ferulic acid (10 g, 51.5 mmol) was dissolved in methanol (50 mL, 1.236 mol) in a one-neck flask equipped with condenser and magnetic stirrer. Concentrated sulphuric acid (0.5 mL, 9.38 mmol) was added under vigorous stirring and the reaction mixture was refluxed for four hours. After that time, the solution was cooled, neutralised with sodium hydrogen carbonate and filtered. The reaction solution was added to ethyl acetate and shaken several times in a separating funnel with saturated sodium hydrogen carbonate solution. Finally, it was treated with saturated sodium chloride solution and the organic phase was dried with sodium sulphate. Ethyl acetate was removed from the dried organic phase and the product was dried under reduced pressure at 50 °C overnight.Yield: 90%. The product was obtained as dark brown, highly viscous liquid, which crystallises upon standing at room temperature.
1H NMR (DMSO-d6): δ = 9.55 (br s, 1H; OH), 7.56 (d, 15.9 Hz, 1H; ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Ph), 7.31 (d, 2.0 Hz, 1H; HAr), 7.12 (dd, 8.3 Hz, 2.0 Hz, 1H; HAr), 6.79 (d, 8.3 Hz, 1H; HAr), 6.45 (d, 15.9 Hz, 1H; CH–C(O)), 3.82 (s, 3H; PhOCH3), 3.70 ppm (s, 3H; C(O)OCH3).
CH–Ph), 7.31 (d, 2.0 Hz, 1H; HAr), 7.12 (dd, 8.3 Hz, 2.0 Hz, 1H; HAr), 6.79 (d, 8.3 Hz, 1H; HAr), 6.45 (d, 15.9 Hz, 1H; CH–C(O)), 3.82 (s, 3H; PhOCH3), 3.70 ppm (s, 3H; C(O)OCH3).
Synthesis of methyl (E)-3-(4-(2-hydroxyethoxy)-3-methoxyphenyl)acrylate (F)
Ethylene carbonate (15.851 g, 180 mmol), methyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (24.985 g, 120 mmol), dimethylacetamide (125.5 mL, 1.349 mol) and potassium iodide (0.478 g, 2.88 mmol) were placed in a two-necked flask with condenser and magnetic stirrer. The mixture was heated to 150 °C under nitrogen flow and under vigorous stirring. The reaction proceeded at this temperature for three hours. After completion, the mixture was cooled down and a large excess of iced water was added under vigorous stirring. The precipitate was filtered off after one day of stirring, washed with water and dried at 50 °C under reduced pressure.
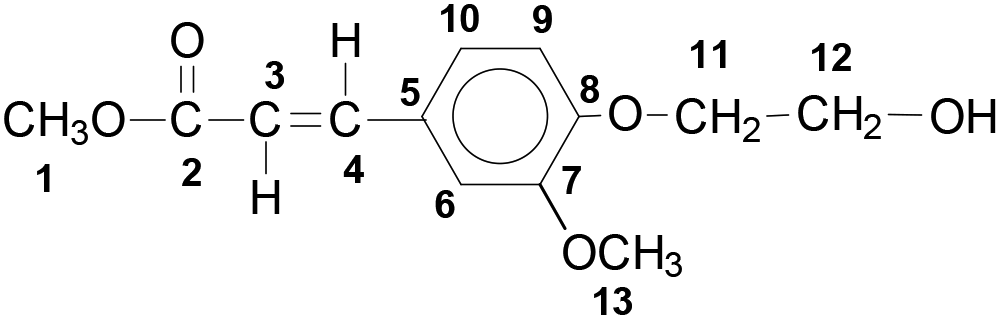
Yield: 85%. The product was obtained as beige solid. Mp.: 99 °C.
1H NMR (DMSO-d6): δ = 7.59 (d, 15.9 Hz, 1H; 4), 7.34 (d, 2.0 Hz, 1H; 6), 7.22 (dd, 8.4 Hz, 2.0 Hz, 1H; 10), 6.99 (d, 8.4 Hz, 1H; 9), 6.55 (d, 15.9 Hz, 1H; 3), 4.84 (t, 5.5 Hz, 1H; OH), 4.02 (t, 4.9 Hz, 2H; 11), 3.81 (s, 3H; 13), 3.72 (m, 2H; 12), 3.71 ppm (s, 3H; 1) (ESI Fig. 1†).
13C NMR (DMSO-d6): δ = 167.0 (2), 150.5 (8), 149.2 (7), 144.8 (4), 126.9 (5), 122.9 (9), 115.3 (3), 112.7 (10), 110.8 (6), 70.3 (11), 59.6 (12), 55.6 (13), 51.3 ppm (1) (ESI Fig. 2†).
Terpolyester synthesis
The melt polycondensations were carried out on laboratory scale in a three-necked 100 mL-glass flask equipped with mechanical stirrer, nitrogen inlet and distillation bridge. The monomers (dried overnight at 50 °C under reduced pressure) and catalysts (see Table 1, catalyst amount: 1 wt% based on the total weight of the monomers) were added to the flask in the appropriate composition (molar ratio dimethyl ester/diol/ferulic acid methyl ester according to Table 1). The flask was evacuated and flushed three times with nitrogen and was then placed in a metal bath pre-heated to 150 °C. Transesterification was carried out under stirring in nitrogen atmosphere with a continuous temperature raise within 60 min to 245 °C (series P(T-Pr-F), P(T-iPr-F), P(T-B-F) and P(S-B-F)) or 270 °C (series P(T-E-F)), respectively. The melt was stirred for 30 min under moderate nitrogen flow after the final reaction temperature was reached. Polycondensation was carried out with stirring under reduced pressure (10 Pa–5 Pa) for 30 min or, respectively, the time given in Table 1. At the end of the reaction, the flask was filled with nitrogen, taken out of the metal bath and the product was removed mechanically. Average yield: 90%.| Polymer | Molar ratiobT-diol-F (mol%/mol%/mol%) | Cat.c | t (h) |
M
ne (g mol−1) |
M
we (g mol−1) |
M w/Mn | η inh (dL g−1) | α |
T
gh (°C) |
T
mh (°C) |
ΔHmh (J g−1 K−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Conventional name of the homopolyesters.
b Determined by 1H NMR spectroscopy; estimated error ± 0.5 mol%.
c Catalyst, 1: potassium acetate; 2: mixture of titanium tetrabutylate and antimony trioxide (1:1 wt/wt); 3: titanium tetrabutylate, (1 wt% with respect to the total monomer weight).
d Polycondensation time under reduced pressure.
e Determined by SEC.
f Determined by viscometry.
g Determined by WAXS.
h Determined by DSC, 2nd heating. All experimental details are given in the Experimental section.
i Not completely soluble.
j Not measured.
k Synthesised at 150 °C.
|
|||||||||||
| P(T-E) [PET] | 50.8/49.2/0 | 1 | 7 | 37000 |
83000 |
2.24 | 0.72 | 0.69 | 84 | 222/253 | 53.4 |
| P(T-E-F)-2 | 47.4/48.3/4.3 | 1 | 3 | 28000 |
68000 |
2.43 | 0.63 | 0 | 78 | 225/236 | 34.5 |
| P(T-E-F)-4 | 45.6/46.1/8.3 | 1 | 3 | 15000 |
67000 |
4.47 | 0.60 | 0 | 79 | 169 | −12.0 |
| 211 | 13.3 | ||||||||||
| P(T-E-F)-6 | 32.7/32.7/34.6 | 1 | 3 | 13000 |
142000 |
10.92 | 0.42 | 81 | — | — | |
| P(T-Pr) [PTT] | 39.5/60.5/0 | 2 | 7 | 62000 |
138000 |
2.24 | 1.27 | 0.64 | 57 | 220/228 | 66.0 |
| P(T-Pr-F)-2 | 47.7/47.7/4.6 | 2 | 1 | 25000 |
73000 |
2.92 | 0.90 | 0 | 60 | 217 | 44.8 |
| P(T-Pr-F)-4 | 45.7/45.4/8.9 | 2 | 1 | 22000 |
95000 |
4.32 | 0.95 | 72 | 173 | 34.3 | |
| P(T-Pr-F)-7 | 32.8/33.8/33.4 | 2 | 1 | 21000 |
108000 |
5.14 | 0.84 | 0 | 74 | — | — |
| P(T-iPr) | 50.0/50.0/0 | 2 | 3 | 8,000 | 21000 |
2.62 | 0.26 | 0 | 80 | — | — |
| P(T-iPr-F)-1 | 48.2/47.3/4.5 | 2 | 1 | 19000 |
87000 |
4.58 | 0.76 | 0 | 81 | — | — |
| P(T-iPr-F)-2 | 45.5/45.5/9.0 | 2 | 1 | 21000 |
95000 |
4.52 | 0.78 | 0 | 90 | 209 | 0.8 |
| P(T-iPr-F)-6 | 33.0/33.7/33.3 | 2 | 0.5 | 18000 |
107000 |
5.94 | 0 | 92 | — | — | |
| P(T-B) [PBT] | 50.3/49.7/0 | 3 | 7 | 32000 |
78000 |
2.44 | 0.90 | 0.54 | 42 | ||
| P(T-B-F)-1 | 48.0/47.7/4.3 | 3 | 3 | 15000 |
53000 |
3.53 | 0.69 | 0.28 | 49 | ||
| P(T-B-F)-2 | 46.6/45.7/7.7 | 3 | 3 | 19000 |
79000 |
5.27 | 0.95 | 0 | 53 | ||
| P(T-B-F)-4 | 33.3/34.0/32.7 | 3 | 1 | 24000 |
91000 |
3.79 | 66 | — | — | ||
| S-diol/F | |||||||||||
| P(S-B) [PBS] | 50.0/50.0/0 | 2 | 3 | 41000 |
93000 |
2.27 | 0.90 | 0.37 | −35 | 111/114 | 71.8 |
| P(S-B-F)-2 | 45.5/48.2/6.3 | 1 | 3 | 10000 |
22000 |
2.20 | 0.36 | −27 | 37/76 97 | −41.2 | |
| 46.1 | |||||||||||
| P(S-B-F)-5 | 42.9/45.8/11.3 | 1 | 3 | 12400 |
19600 |
2.11 | 0.41 | 0 | −15 | — | — |
| P(S-B-F)-7 | 30.5/34.4/35.1 | 1 | 1 | 11000 |
53000 |
4.82 | 0.37 | 0 | 29 | — | — |
| P(F) | 0/0/100 | 3 | 3 | 1700 | 2800 | 1.65 | 0.10 | 0.71 | 80 | 223 | 40.3 |
Selected samples were upscaled by polycondensation in a 2.4 L stirring autoclave (Juchheim Laborgeräte GmbH, Bernkastel Kues, Germany). The dried monomers and catalysts were given in the appropriate proportions into the agitated autoclave pre-heated to 150 °C. The autoclave was closed, evacuated and flushed with nitrogen three times, and then the transesterification was started under stirring and moderate nitrogen flow. The autoclave was heated to 200 °C within 10 min, the temperature was maintained at 200 °C for 20 min before heating up to 245 °C (series P(T-Pr-F), P(T-iPr-F), P(T-B-F) and P(S-B-F)) or 280 °C (series P(T-E-F)), respectively, within 45 min or 60 min, respectively. The polycondensation reaction was continued under moderate nitrogen flow for four hours or seven hours, respectively. After that time, the autoclave was allowed to cool down. Then, the material was mechanically removed and ground.
Melt spinning
Melt spinning was performed with a self-constructed piston-type spinning device equipped with a 0.2 mm nozzle (capillary diameter) as described earlier.31 The polymer was dried before processing at 80 °C for 4 h under reduced pressure. The spinning temperature was set to 240 °C, the take-up speed (winding speed) was 100 m min−1, and the resulting fibres had a diameter of about 80 μm.Preparation of samples for electron beam treatment and mechanical tests
Before processing, the materials were dried at 50 °C for 12 hours under reduced pressure. The samples for e-beam treatment and mechanical tests (hardness) were melt pressed using a Specac press (Specac Ltd Science and Innovation Centre, Orpington, UK) with hydraulic Atlas foil pressing tool between two polyimide sheets separated by a spacer ring (100 μm) at 200 °C with a weight of 4000 kg for one minute. The films were cooled to room temperature in a water-cooled case. The final thicknesses of the transparent films were in the range of 50–70 μm and had a diameter of 2 cm.Electron beam (EB) treatment of terpolyester films
The samples were irradiated in a special irradiation chamber32 using an electron accelerator ELV-2 (Budker Institute of Nuclear Physics, Novosibirsk, Russia33). Before irradiation, the samples were dried at 50 °C for 8 hours under reduced pressure in a vacuum oven. After the drying process, the copolyester samples were placed into the irradiation chamber. Before irradiation, the chamber was heated up to 80 °C and kept for 5 minutes under reduced pressure to remove moisture and oxygen. Subsequently, the chamber was cooled down or heated up to the target irradiation temperatures (25 °C, 110 °C or 160 °C, respectively) in nitrogen atmosphere. The irradiation was carried out with constant electron energy (1.5 MeV) and electron current (4 mA), the dose was applied by several steps of 25 kGy until the target dose was reached. After the electron beam treatment, the samples were cooled down under nitrogen and subsequently taken out of the irradiation chamber.Determination of the gel content
The irradiated samples (120 mg) were dissolved in a mixture of PFP/CHCl3 (1:1 v/v, 15 mL) and stirred for 4 h. The solvent was removed and a new portion (15 mL) was added. The samples were again stirred for 48 h. Then, the gel content was separated by filtration, washed with chloroform and dried at 50 °C under reduced pressure for 60 h, and the mass was determined.
3 Results and discussion
Terpolyester synthesis and characterisation
FA was first converted into the methyl ester followed by reaction with ethylene carbonate to obtain methyl (E)-3-(4-(2-hydroxyethoxy)-3-methoxyphenyl)acrylate (denominated as F), see Fig. 1, eqn (1). The polycondensation of this activated monomer with dimethyl terephthalate or dimethyl succinate, respectively, and aliphatic diols was examined (Fig. 1, eqn (2)) and yielded five series of copolyesters (1: P(T-E-F) with ethylene glycol as the diol; 2: P(T-Pr-F) with 1,3-propanediol as the diol; 3: P(T-iPr-F) with 1,2-propanediol as the diol; 4: P(T-B-F) with 1,4-butanediol as the diol; and 5: P(S-B-F)) with dimethyl succinate and 1,4-butandiol as comonomers. The polycondensation was performed in melt (melt transesterification polycondensation) under the conditions briefly mentioned in Table 1.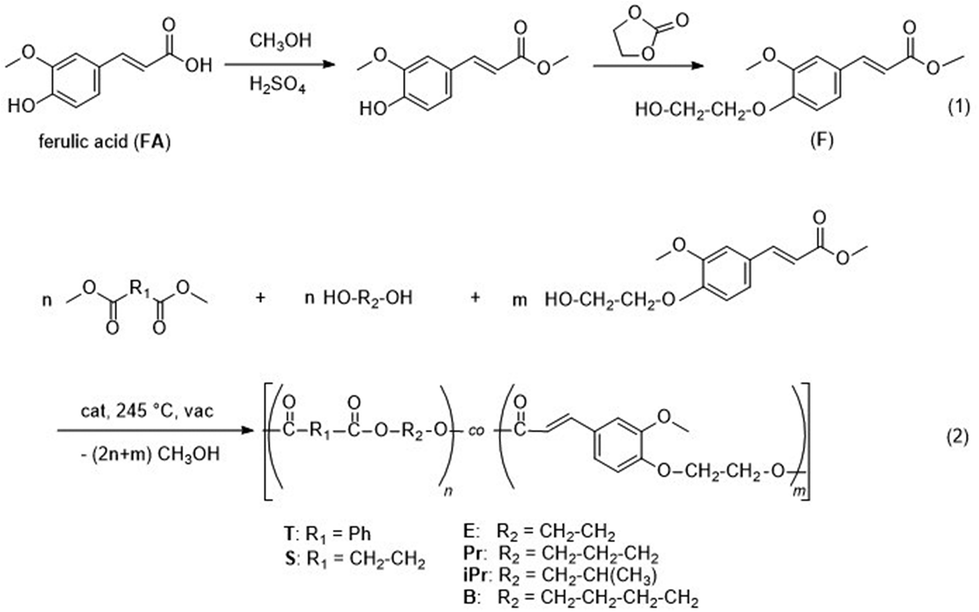 | ||
| Fig. 1 Synthesis of ferulic acid methyl (E)-3-(4-(2-hydroxyethoxy)-3-methoxyphenyl)acrylate (eqn (1)) used as monomer for the copolycondensation with dimethyl terephthalate or dimethyl succinate, respectively, and aliphatic diols (eqn (2)). | ||
Both, the final temperature and the time for polycondensation at this temperature depended on the structure of the comonomers. Series P(T-E-F) required 270 °C (in accordance to PET34), the other series were prepared by a final polycondensation temperature of 245 °C, as for PBT35 and PBS.36 The standard polycondensation time under reduced pressure was three hours (seven hours in the up-scaled experiments in a 2.4 L stirring autoclave). For the copolyesters with the highest content of F the polycondensation times had to be shorter to suppress crosslinking (1 h for P(T-Pr-F)-7, 0.5 h for P(T-iPr-F)-6, and 1 h for P(T-B-F)-4.
First, the homopolymerisation of methyl (E)-3-(4-(2-hydroxyethoxy)-3-methoxyphenyl)acrylate (F) was attempted. The NMR spectra (ESI Fig. 3–5†) proved condensation occurred for sample P(F), but intense methylester and hydroxyethyl signals revealed that only oligomers were formed.
In contrast, the copolymerisation of F with diacids (T, S) and diols (E, Pr, iPr and B) resulted in terpolymers with a composition in very good agreement with the composition expected from monomer feed ratio. The molar ratios of the three monomers of the terpolymers (Table 1) were determined from appropriate 1H NMR signals as given in the ESI (section 2.2†). Apparently, the incorporation of the F comonomer into the polyesters occurred without problems, at least up to contents of about 35 mol%. All polymers were characterised by 1H NMR spectroscopy and spectra and signal assignments are compiled in the ESI (Fig. 6, 9, 12, 15 and 18†). 13C NMR spectra were recorded only from one polymer of each series for more detailed structure confirmation (ESI Fig. 7, 10, 13, 16 and 19†). The 13C NMR signal assignments are based on the evaluation of 1D and of 2D NMR spectra (ESI Fig. 8, 11, 14, 17 and 20†). For example, Fig. 2 depicts the 1H spectrum and two regions of the 13C NMR spectrum of P(T-Pr-F)-7. The 1H NMR spectrum agrees with the expected structure, and side reactions of the acrylic moiety of F could not be detected, which is also true for the other polymers. In contrast to the copolymers from diacid and diol with alternating monomer units in the backbone, the incorporation of F with an acid and alcohol functionality results in a more complex microstructure (ESI, section 1†). As indicated in Fig. 2, several 1H and 13C NMR signals show splitting due to the different dyads or triads. For all terpolymers with highest F content, a quantitative evaluation of several diad and triad contents, respectively, revealed that a random distribution of the monomers occurred and no formation of longer F sequences could be stated.
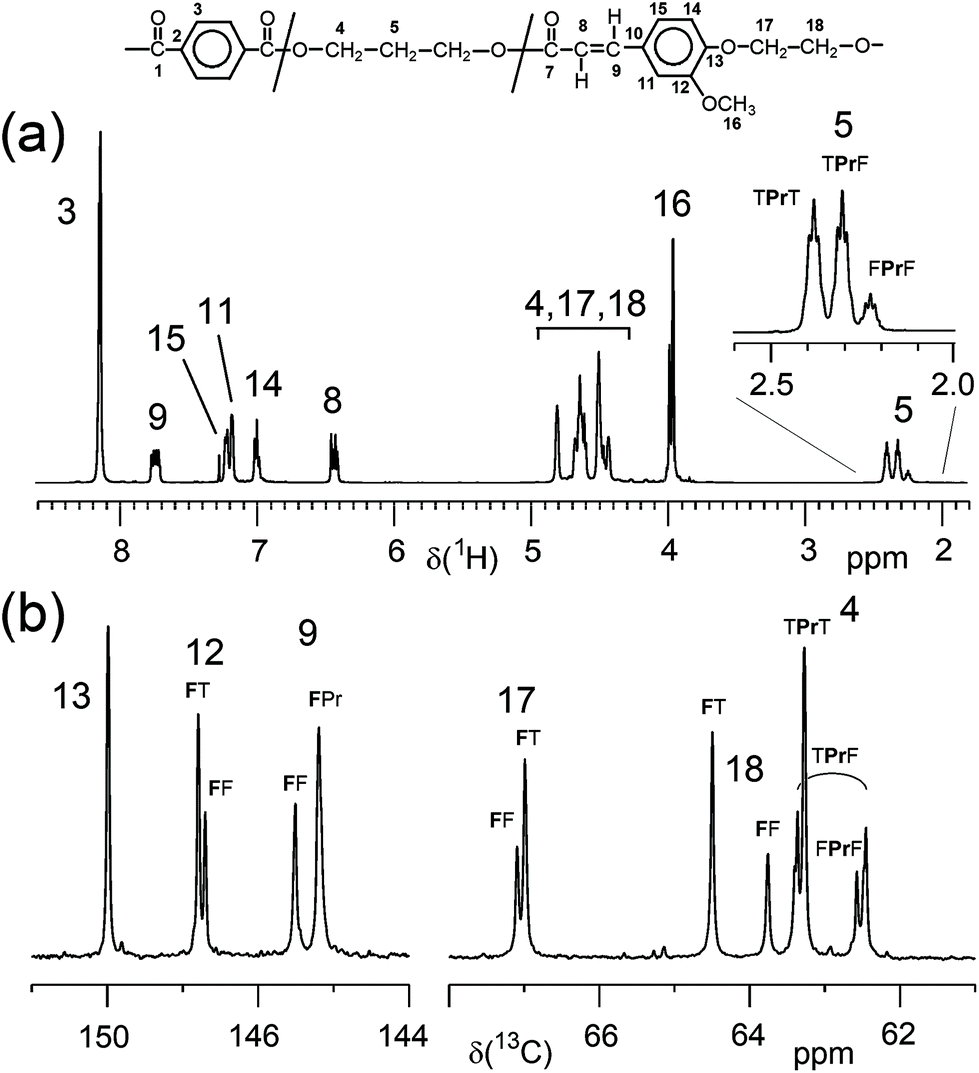 | ||
| Fig. 2 (a) 1H NMR spectrum and (b) two regions of the 13C NMR spectrum of P(T-Pr-F)-7 (33.4 mol% F; solvent: CDCl3:TFA-d 4:1 v/v). Assignments to diad and triad signals are given. | ||
The molar masses and dispersity of the copolyesters were measured by size exclusion chromatography (SEC). The solvent mixture used (pentafluorphenol/chloroform) only allowed the calculation of molar masses relative to polystyrene standards and not the use of a light scattering detector. The elution curves of all polyesters (ESI Fig. 21–25†) were unimodal.
The relative molar masses in the P(T-E-F) series and in the P(T-B-F) series were not significantly affected by the ferulate comonomer at molar F concentrations up to 8 mol%, yielding Mw values up to 95000 g mol−1. In contrast, the relative molar masses in the P(S-B-F) series were significantly lower than for the starting PBS. The samples of all series with the highest amount of ferulate units in the range of 30 mol% showed a higher dispersity Mw/Mn and higher Mw. It is assumed that this might be caused by side reactions of the double bond. The double bonds may have polymerised thermally to a very low extent and thus caused long-chain branching which alters the dispersity of the sample.
The general trend of the SEC results is also reflected by the solution viscosities (ηinh) that were measured in the same solvent mixture of pentafluorophenol/chloroform.
It can be summarised at this point that the polycondensations undertaken yielded copolyester samples with intact double bonds and molar masses in the typical range of standard polyesters.
Solid state structure
The solid state structure of the as-synthesised terpolyesters was analysed by WAXS to outline the influence of ferulate units. The main focus of the work was on samples with low molar content of F. The influence of the F content is discussed for the P(T-E-F) series as an example. The WAXS curves are displayed in Fig. 3. The curve for the homopolyester PET (curve 1) corresponds to those reported in the literature.34,37–39 The oligomeric ferulate homopolyester P(F) (curve 5) also exhibited a semicrystalline structure. A comparison from literature could not be found. The variety of reflections might be caused by the low molar mass of the sample presumably containing contributions of monomer crystallinity. However, this sample represents a necessary control for the terpolyesters P(T-E-F). The incorporation of low molar amounts of ferulate units into the PET structure (4.3 mol% in P(T-E-F)-2, curve 2; and 8.3 mol% in P(T-E-F)-4, curve 3) resulted in a drastic reduction of crystallinity α to zero (see Table 1). An enhanced molar content of 34.6 mol% in P(T-E-F)-6, curve 4, yielded a polymer with a new type of crystalline structure that is neither the one of PET, nor the one of P(F) (curve 5). This special crystalline structure has not been observed in the other terpolyester series. | ||
| Fig. 3 WAXS patterns of the copolyester series P(T-E-F), 1: P(T-E) (PET); 2: P(T-E-F)-2 (4.3 mol% F); 3: P(T-E-F)-4 (8.3 mol% F); 4: P(T-E-F)-6 (34.6 mol% F); 5: (P(F)). Curves shifted for better visualisation. | ||
The WAXS curves of all other polymer series are displayed in the ESI (ESI Fig. 26–28†). Incorporation of F in the P(T-Pr-F) series resulted in complete disappearance of crystallinity (ESI Fig. 26†). In contrast, the series P(T-iPr-F) was completely amorphous due to the amorphous nature of the homopolymer P(T-iPr) caused by the suppression of crystallinity by the methyl substituent (ESI Fig. 27†). Incorporation of low molar amounts of F into PBT (5.3 mol% in P(T-E-F)-1) preserved the PBT crystallinity, but at a somewhat lower degree (ESI Fig. 28†).
Melting behavior
DSC was employed to support the results of the X-ray measurements and to analyse the melting behaviour of the terpolyesters. The second heating run was used to exclude the influences of the thermal history of the samples. The parameters determined (Tg, Tm, ΔHm) are summarised in Table 1.Fig. 4 shows as example the DSC curves of the P(T-E-F) series.
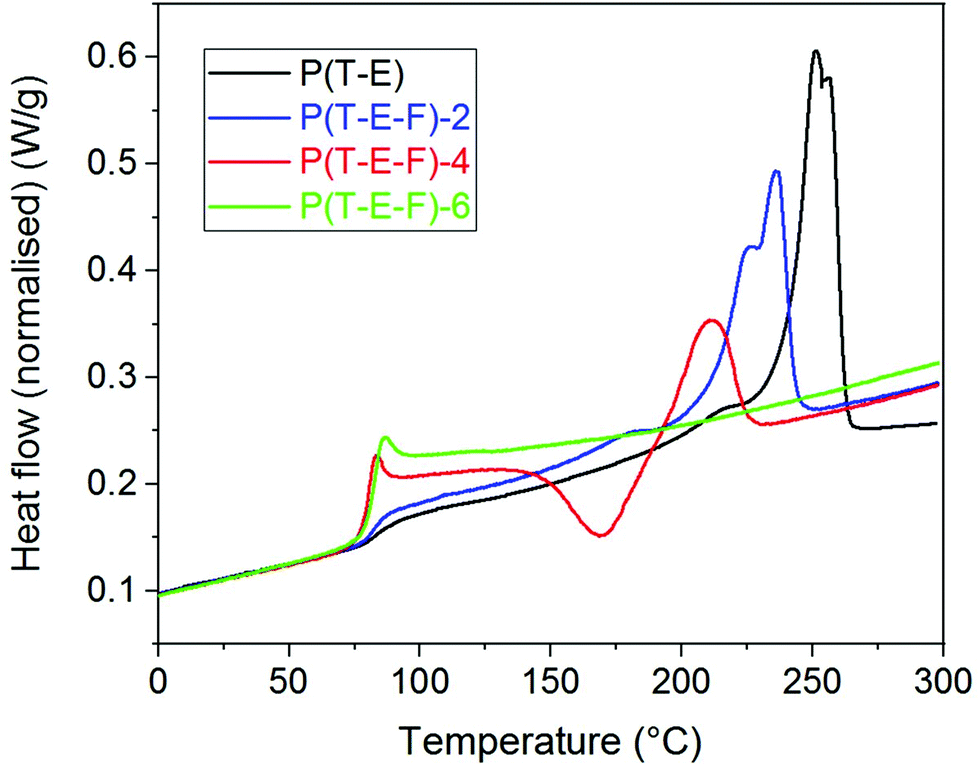 | ||
| Fig. 4 DSC curves (2nd heating) of the terpolyesters P(T-E-F) with different molar content of F. | ||
The DSC curve of the PET control (P(E-T)) reflects the thermal behaviour known from literature34,40 with a glass transition temperature Tg at 84 °C and a main melting peak Tm at 253 °C. Incorporation of ferulate units reduced both, the melting peak maximum Tm as well as the melting enthalpy ΔHm. This crystallinity was not present in the as-synthesised samples, in accordance with the WAXS results, but was generated in the recrystallisation during the first heating. The sample P(T-E-F)-6 only showed a glass transition within the measuring range until 300 °C. It is assumed that the melting of this special crystalline structure containing F units occurs far above 300 °C, similar to poly(hydroxybenzoic acid).41,42 A comparable behaviour was observed for the P(T-Pr-F) series (ESI Fig. 29†).
The series P(T-iPr-F) showed only glass transitions, in accordance with the WAXS findings. ESI Fig. 30† illustrates that both, glass transition temperature as well as Δcp increased with inceasing F content. This behaviour was observed in all series. PBT showed two melting peaks as described earlier.36 Incorporation of 4.3 mol% F shifted the maxima to lower temperature, but preserved the basic behaviour (ESI Fig. 31†). This is again following the WAXS results that proved the preserved PBT crystallinity in the sample. Higher amounts of F suppressed the crystallinity. The melting peak maxima Tm of the series show the following tendency: P(T-E-F) > P(T-Pr-F) > P(T-B-F) > P(S-B-F). The DSC curves of P(S-B-F) are displayed in ESI Fig. 32.† The incorporation of ferulate units enhanced in all series the glass transition temperature (except for PET, where the homopolyester PET had the highest Tg) as shown in Fig. 5. The Tg of the low molar mass homopolyester P(F) was found to be 80 °C, lower than reported by Nguyen et al.43 However, it should not be overestimated because of the low molar mass of the sample.
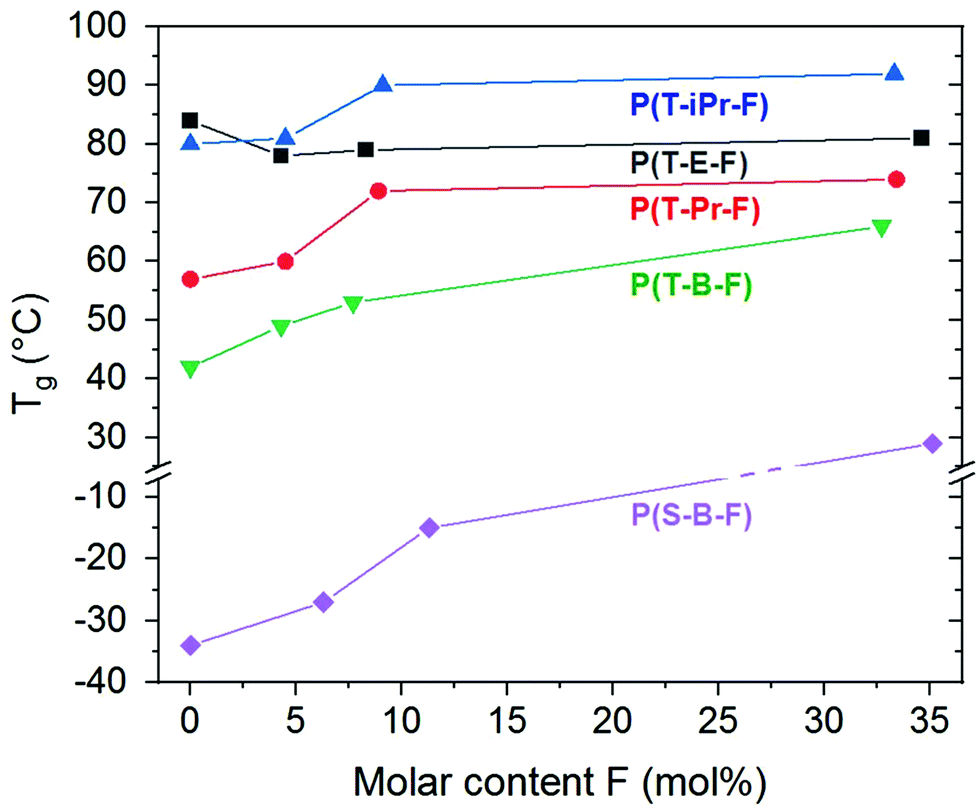 | ||
| Fig. 5 Influence of the molar content of ferulate units F on the Tg of the terpolyester series. | ||
Thermal stability of the terpolyesters
The thermal stability of the polyesters is of high importance for most of the applications and was further examined by TGA. The complete data are summarized in ESI Table 1.† The thermogravimetric curves of series P(T-Pr-F) are displayed in Fig. 6, the curves of the other series are given in the ESI (Fig. 33–36†).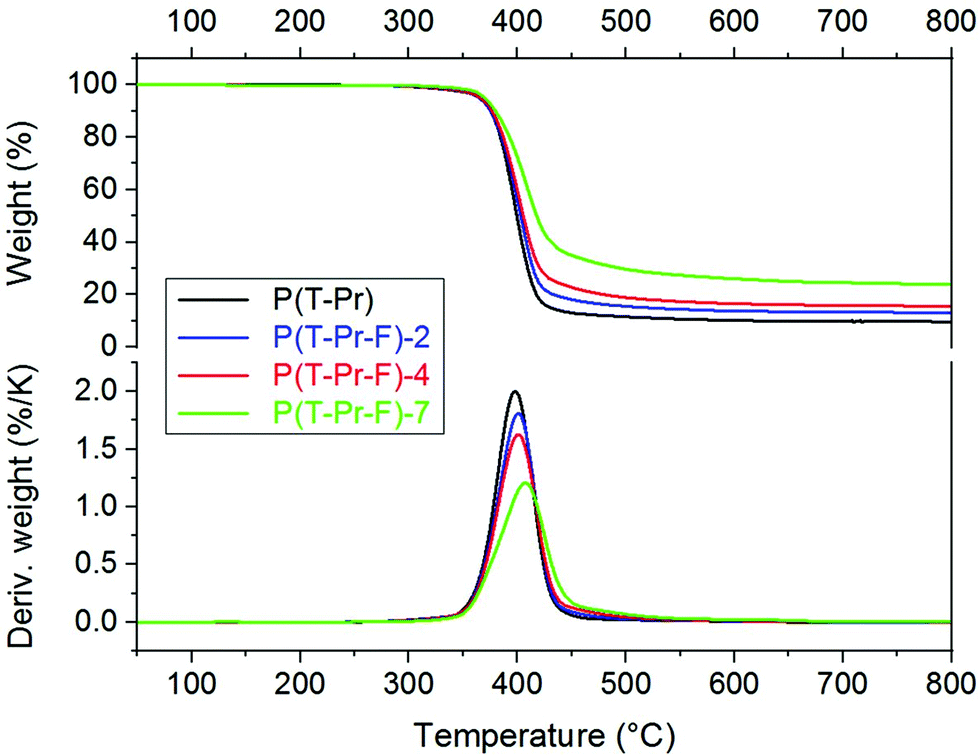 | ||
| Fig. 6 TGA curves of the terpolyesters of series P(T-Pr-F). | ||
The thermal decomposition maximum shifted to higher temperature with increasing F comonomer content. This is explained by the enhancement of stability caused by the incorporation of aromatic F units. The influence of the spacer on the decomposition maxima was clearly visible and raised in the following order: P(T-iPr-F) < P(T-Pr-F) and P(T-B-F) < P(T-E-F), in accordance to the tendency found for the homopolyesters.36 Additionally, the residue obtained after measurement at 800 °C (char) enhanced with increasing F content, see Fig. 7. The only exception was found in the P(T-E-F) series where the char of the homopolyester P(T-E) was the highest which is due to the very special decomposition mechanism of PET resulting in a significant amount of aromatic ring structures.44
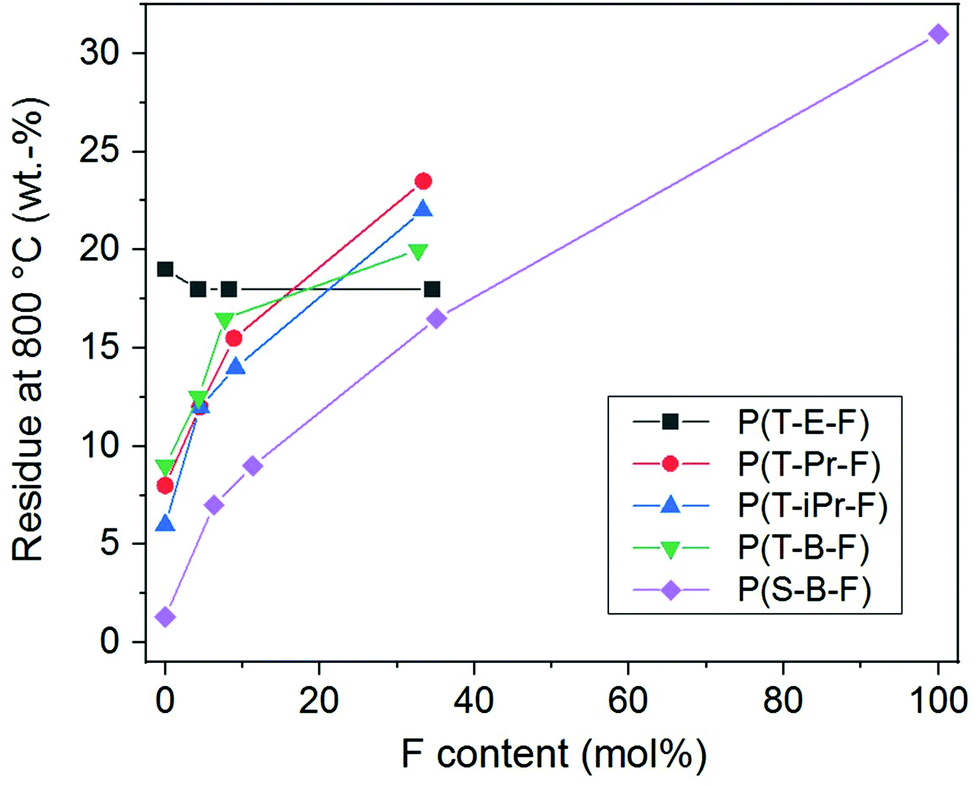 | ||
| Fig. 7 Influence of the molar content of F units on the residue after TGA at 800 °C. | ||
In summary, the TGA results indicated a higher thermostability of terpolyesters compared to homopolyesters by incorporation of ferulate units. This is taken as a first indication to a more favourable fire behaviour. All copolyesters are stable until 300 °C.
Study of crosslinkability by electron beam treatment
The ability to polymerise the double bond in the F units of the terpolyesters after polycondensation and melt processing was studied using the P(T-E-F) series and one sample of the P(T-B-F) series as representative samples. The monomeric FA is a sterically hindered 1,2-disubstituted acrylic monomer. Thus, it is not expected to easily undergo thermal polymerisation. Only a few reports described thermal copolymerisation with styrene initiated by the radical initiator azobis(isobutyronitrile) (AIBN), resulting in rather low molar mass oligomers.45Preliminary experiments with the sample P(T-E-F)-2 in solution and bulk at 250 °C did not yield any sign of crosslinking. The same was observed after UV irradiation. Thus, we choose for the study presented the method of e-beam irradiation (EB). Initial experiments with sample P(T-E-F)-2 at room temperature with doses between 100 and 600 kGy also did not result in crosslinking. Consequently, the melt-pressed copolyester samples as well as the melt-pressed non-modified homopolyesters (PET and PBT) were irradiated at a temperature above Tg (typically 110–120 °C) to ensure high molecular mobility of the polymer chains in the amorphous phase. Different doses up to 600 kGy were applied and the resulting samples were evaluated using solubility in the solvent mixture PFP/CHCl3 after 45 min. The photographic images of all irradiated samples are shown in ESI Fig. 37,† their appearance in PFP/CHCl3 (gel contents) are summarised in ESI Fig. 38.†
All non-irradiated samples independent of their F content were completely soluble in PFP/CHCl3. Increasing F content yielded melt-pressed films with a brownish colour (ESI Fig. 37†). PET and PBT films were transparent and remained colourless after EB treatment with all doses. They were completely soluble in PFP/CHCl3 after EB irradiation. That means, EB treatment did not cause significant changes in the chemical structure of PET and PBT and no crosslinking. Irradiated samples of the P(T-E-F) series showed partial crosslinking, visible by swollen, gelled parts in PFP/CHCl3. The gel content was enhanced with both increasing F content as well as increasing dose (ESI Fig. 38†), which is demonstrated in Fig. 8 for sample P(T-E-F)-2 with 4.3 mol% F.
 | ||
| Fig. 8 Influence of the applied EB dose on the gel content in sample P(T-E-F)-2. | ||
Samples P(T-B-F)-1 showed comparable behaviour (ESI Fig. 39†). Selected samples were analysed before and after EB irradiation by Raman spectroscopy. The double bond in the starting sample P(T-E-F)-2 was only weakly visible by broadening of the band at 1645 cm−1. EB irradiation resulted in a very weak change of this shoulder (ESI Fig. 40†), which cannot be taken as proof of the reaction of the double bonds.
The results discussed above demonstrated that melt-processed terpolyesters with F comonomers can be successfully crosslinked by EB treatment.
Mechanical tests
Selected copolyesters were melt-pressed to obtain samples for a preliminary evaluation of mechanical properties and the influence of EB treatment. Hardness tests (Shore D and compression tests) did not reveal significant differences between the samples. The shore D hardness of the F-containing sample P(T-E-F)-2 was comparable to PET (see ESI Table 2†). No influence of EB treatment was observed. This could be due to the thickness of the sample which exceeded the irradiation depth by several times. Therefore, melt-spun fibres of sample P(T-E-F)-2 were further analysed with and without EB treatment, to determine whether fibres as thinner samples would show a more pronounced effect of EB treatment. The fibre thickness of about 80 μm allowed complete irradiation of the sample volume. The values of the tensile tests of non-treated and EB treated fibres are summarised in Table 2, the stress–strain curves are given in ESI Fig. 40 and 41.†| Polymer P(T-E-F)-2 | E t (MPa) | σ m (MPa) | ε b (%) |
|---|---|---|---|
| As-spun | 4200 ± 137 | 28.7 ± 9.7 | 0.6 ± 0.2 |
| EB treated (600 kGy) | 4286 ± 69 | 57.6 ± 7.6 | 1.4 ± 0.2 |
The preliminary experiments showed an increase of E modulus Et, tensile strength σm and elongation at break εb after EB treatment. The stress–strain curves are given in ESI Fig. 41 and 42.† Detailed examinations will be performed in the future.
4 Conclusions
In this contribution, terpolyesters based on PET, PBT, PTT, PiPrT, PBS and containing ferulate units were reported. The terpolyesters could be prepared successfully by transesterification copolycondensation without thermal polymerisation of the double bonds and yielded soluble products. This is, to the best of our knowledge, the first report about such polyesters with significant molar content of ferulate units. The chemical structure of the terpolyesters were studied in detail by NMR spectroscopy and SEC. These methods demonstrated that high molar mass polyesters with unimodal molar mass distribution and random monomer sequences were formed.The incorporation of ferulate units had significant influence on the reduction of crystallinity, which can only be preserved in the PBT series at low molar contents of about 5 mol%. In contrast, the glass transition temperatures raised with increasing content of ferulate, indicating a better thermostability of the terpolyesters. Crosslinking by EB treatment occurred via the double bonds of the terpolyester, proven by the fact that the base homopolyesters could not be crosslinked by EB irradiation.
In summary, it can be stated that incorporation of bio-based ferulate units into conventional copolyesters enhances the sustainability of polyester materials and adds interesting aspects into the property profile, in particular, the opportunity to crosslink materials after thermoplastic processing, successfully demonstrated here by EB treatment. EB treatment resulted in crosslinking within the surface layers of the materials and is most effective in fibres, but also offers the opportunity to stabilise injection-moulded or 3D printed parts.
Author contributions
Conceptualization: D. P., A. K. and D. J.; Data curation: D. P., A. K., H. K. and D. J.; Funding acquisition: D. P.; Investigation: D. P., A. K., H. K., D. J., K. A., H. B., H. S. and M. T. M.; Methodology: D. P. and D. J.; Project administration: D. P.; Resources: D. P. and B. V.; Supervision: D. P. and B. V.; Validation: D. P., H. K. and D. J.; Visualization: D. P., D. J. and K. A.; Writing – original draft: D. P., A. K.; H. K., D. J., M. T. M. and B. V.; Writing – review & editing: D. P. and D. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (project number PO 575/13-1) is gratefully acknowledged. Furthermore, the authors wish to thank Mrs Petra Treppe (IPF) for SEC measurements, Mrs Julia Muche (IPF) for Raman measurements, and Norbert Smolka (IPF) for fibre spinning of sample P(T-E-F)-2.Notes and references
- H. Nakajima, P. Dijkstra and K. Loos, Polymers, 2017, 9, 1–26 CrossRef PubMed.
- E. Piorkowska, Adv. Polym. Sci., 2019, 1–35 CAS.
- R. M. O′dea, J. A. Willie and T. H. Epps, ACS Macro Lett., 2020, 9, 476–493 CrossRef.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- L. Costes, F. Laoutid, S. Brohez and P. Dubois, Mater. Sci. Eng., R, 2017, 117, 1–25 CrossRef.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- M. Firdaus and M. A. R. Meier, Eur. Polym. J., 2013, 49, 156–166 CrossRef CAS.
- L. Mialon, R. Vanderhenst, A. G. Pemba and S. A. Miller, Macromol. Rapid Commun., 2011, 32, 1386–1392 CrossRef CAS PubMed.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- L. H. Bock and J. K. Anderson, J. Polym. Sci., 1958, 28, 121–127 CrossRef CAS.
- W. Lange and O. Kordsachia, Holz Roh- Werkst., 1981, 39, 107–112 CrossRef CAS.
- H. R. Kricheldorf and G. Löhden, Polymer, 1995, 36, 1697–1705 CrossRef CAS.
- F. Pion, P. H. Ducrot and F. Allais, Macromol. Chem. Phys., 2014, 215, 431–439 CrossRef CAS.
- C. H. R. M. Wilsens, J. M. G. A. Verhoeven, B. A. J. Noordover, M. R. Hansen, D. Auhl and S. Rastogi, Macromolecules, 2014, 3306–3316 Search PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- R. Puupponen-Pimiä, L. Nohynek and C. Meier, J. Appl. Microbiol., 2001, 90, 494–507 CrossRef PubMed.
- H. R. El-Seedi, A. M. A. El-Said, S. A. M. Khalifa, U. Göransson, L. Bohlin, A. K. Borg-Karlson and R. Verpoorte, J. Agric. Food Chem., 2012, 60, 10877–10895 CrossRef CAS PubMed.
- S. Ou and K. C. Kwok, J. Sci. Food Agric., 2004, 84, 1261–1269 CrossRef CAS.
- M. A. Ouimet, J. J. Faig, W. Yu and K. E. Uhrich, Biomacromolecules, 2015, 16, 2911–2919 CrossRef CAS PubMed.
- M. A. Ouimet, J. Griffin, A. L. Carbone-Howell, W. H. Wu, N. D. Stebbins, R. Di and K. E. Uhrich, Biomacromolecules, 2013, 14, 854–861 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- M. Schwarzer, A. Korwitz, H. Komber, L. Häußler, B. Dittrich, B. Schartel and D. Pospiech, Macromol. Mater. Eng., 2018, 303, 1–16 Search PubMed.
- O. Fischer, D. Pospiech, A. Korwitz, K. Sahre, L. Häußler, P. Friedel, D. Fischer, C. Harnisch, Y. Bykov, M. Döring, L. Häußler, P. Friedel, D. Fischer, C. Harnisch, Y. Bykov and M. Döring, Polym. Degrad. Stab., 2011, 96, 2198–2208 CrossRef CAS.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- M. Vaezi, H. Seitz and S. Yang, Int. J. Adv. Des. Manuf. Technol., 2013, 67, 1721–1754 CrossRef.
- O. Kreye, S. Oelmann and M. A. R. Meier, Macromol. Chem. Phys., 2013, 214, 1452–1464 CrossRef CAS.
- D. Pospiech, A. Korwitz, K. Eckstein, H. Komber, D. Jehnichen, M. Suckow, A. Lederer, K. Arnhold, M. Göbel, M. Bremer, A. Hoffmann, S. Fischer, A. Werner, T. Walther, H. Brünig and B. Voit, J. Appl. Polym. Sci., 2019, 136, 48257 CrossRef.
- H. Körber, U. Lappan, U. Geißler, K. Lunkwitz and R. Hanke, Institute of Polymer Research Dresden, DE19930742A1, 1999.
- H. Dorschner, W. Jenschke and K. Lunkwitz, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 161, 1154–1158 CrossRef.
- D. Pospiech, A. Korwitz, H. Komber, D. Voigt, D. Jehnichen, J. Müller, A. Janke, T. Hoffmann and B. Kretzschmar, High Perform. Polym., 2007, 19, 565–580 CrossRef CAS.
- T. Köppl, S. Brehme, D. Pospiech, O. Fischer, F. Wolff-Fabris, V. Altstädt, B. Schartel and M. Döring, J. Appl. Polym. Sci., 2013, 128, 3315–3324 CrossRef.
- D. Pospiech, A. Korwitz, H. Komber, D. Jehnichen, L. Häussler, H. Scheibner, M. Liebmann, K. Jähnichen and B. Voit, Macromol. Chem. Phys., 2015, 216, 1447–1461 CrossRef CAS.
- N. S. Murthy, S. T. Correale and H. Minor, Macromolecules, 1991, 24, 1185–1189 CrossRef CAS.
- A. A. Gaonkar, V. V. Murudkar and V. D. Deshpande, Thermochim. Acta, 2020, 683, 178472 CrossRef CAS.
- D. Pospiech, D. Jehnichen, H. Komber, A. Korwitz, A. Janke, T. Hoffmann, B. Kretzschmar, L. Häussler, U. Reuter, M. Döring, S. Seibold, R. Perez-Graterol, J. K. W. Sandler, V. Altstädt, F. Bellucci and G. Camino, J. Nanostruct. Polym. Nanocompos., 2008, 4, 62–75 Search PubMed.
- A. Mehta, U. Gaur and B. Wunderlich, J. Polym. Sci., Part B: Polym. Phys., 1978, 16, 289–296 CAS.
- H. Yoon, L. F. Charbonneau and G. W. Calundann, Adv. Mater., 1992, 4, 206–214 CrossRef CAS.
- H. R. Kricheldorf and G. Schwarz, Polymer, 1984, 25, 520–528 CrossRef CAS.
- H. T. H. Nguyen, M. H. Reis, P. Qi and S. A. Miller, Green Chem., 2015, 17, 4512–4517 RSC.
- B. J. Holland and J. N. Hay, Polymer, 2002, 43, 1835–1847 CrossRef CAS.
- H. Sun, Y. D. Young, S. Kanehashi, K. Tsuchiya, K. Ogino and J. Sim, J. Fiber Sci. Technol., 2016, 72, 74–79 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00851j |
| This journal is © The Royal Society of Chemistry 2021 |