
Clean protocol for deoxygenation of epoxides to alkenes via catalytic hydrogenation using gold†
Jhonatan L.
Fiorio
 and
Liane M.
Rossi
*
and
Liane M.
Rossi
*
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000, São Paulo, SP, Brazil. E-mail: lrossi@iq.usp.br
First published on 5th November 2020
Abstract
The epoxidation of olefin as a strategy to protect carbon–carbon double bonds is a well-known procedure in organic synthesis, however the reverse reaction, deprotection/deoxygenation of epoxides is much less developed, despite its potential utility for the synthesis of substituted olefins. Here, we disclose a clean protocol for the selective deprotection of epoxides, by combining commercially available organophosphorus ligands and gold nanoparticles (Au NP). Besides being successfully applied in the deoxygenation of epoxides, the discovered catalytic system also enables the selective reduction N-oxides and sulfoxides using molecular hydrogen as reductant. The Au NP catalyst combined with triethylphosphite P(OEt)3 is remarkably more reactive than solely Au NPs. The method is not only a complementary Au-catalyzed reductive reaction under mild conditions, but also an effective procedure for selective reductions of a wide range of valuable molecules that would be either synthetically inconvenient or even difficult to access by alternative synthetic protocols or by using classical transition metal catalysts.
Introduction
The carbon–carbon double bond of alkenes is not often described as a central functional group of organic synthesis,1,2 but it can be useful to obtain alcohols, aldehydes, amines, diols, and epoxides, and more complex molecules via reactions including olefin metathesis, hydroformylation, oxidative cleavage, Diels–Alder reaction, and Heck coupling reaction among others.3 Due to the usually high number of steps in synthetic organic routes, reliable protection–deprotection strategies for the carbon–carbon double bond may find wide applications. Most methodologies suffer the disadvantages of harsh reagents or reaction conditions, or low chemoselectivity.4–6 Protecting group strategies for alkenes include the epoxidation of alkenes, followed by its regeneration using a range of reducing condition, but the deoxygenation of epoxides in the presence of other more reactive and reducible functional groups remains a major challenge owing to a lack of chemoselectivity.7–11 Efficient reduction of epoxides can be done by using stoichiometric amounts of Li,12 metal halides/Amberlyst15,13 SmI2,14,15 Me2PhSiLi,16 dimethyl diazomalonate/Rh2OAc4,17 ZrCl4/NaI,18 high valent oxorhenium complexes including MeReO3 and ReIO2(PPh3)2,19–21 and an oxoruthenium complex together with PPh3, H2, or Na2SO3 as reductants.22 These methods lack versatility due to the intolerance towards other reducible functional groups, reaction are mainly conducted using stoichiometric amounts of reagents, and the catalysts suffer from low activity, low atom efficiency, and tedious work-ups with moisture-sensitive reaction conditions.21,23–25 Therefore, the development of efficient catalytic systems for the chemo- and stereoselective deoxygenation of epoxides with readily available catalysts and practical reductants such as H2 under mild reaction conditions to the corresponding alkenes, is notably lacking in the arsenal of techniques available to synthetic organic chemists.Gold-based catalysts are plausible candidates aiming high selectivity in the reductive deoxygenation of epoxides into alkenes, due to their low propensity to reduce carbon–carbon double bonds. Pure gold demonstrates very low or no activity in hydrogenations, but the combination of gold nanoparticles (Au NPs) with ligands has disclosed the successful application of Au NPs as selective catalysts for hydrogenation reactions.26–33 The activating effect associated to added ligands is a result of the cooperative character that the ligand plays upon adsorption on metal's surface decreasing the unfavorable energetic barrier to H2 homolytic dissociation on Au surface,34 by triggering a new channel to H2 heterolytic dissociation on the ligand/Au interface.31,33,35–37 So far, the catalytic systems formed by Au/ligand have already been employed in the selective hydrogenation of alkynes to alkenes,31,38–40 aldehydes to alcohols,28,30,41 including allylic alcohols.26,27
To date, several heterogeneous catalysts have been reported for deoxygenation reactions using H2, CO/H2O or alcohols as reductants. These catalysts, however, fall shortly due to the lack of selectivity for alkenes and limited scope of substrates, especially when it comes to stereospecific deoxygenation of epoxides, usually a mixture of cis and trans alkenes are obtained.9,24,42–47 Metals, such as Pd and Pt, are well-known to catalyze the ring-opening (hydrogenolysis) of epoxides into the corresponding alcohols,48–56 or when the alkene is formed, the over hydrogenation of the product is difficult to prevent.8,57
Herein, we demonstrate that the Au NP catalyst combined with triethylphosphite, P(OEt)3, is remarkably more reactive than solely Au NPs for the deoxygenation of epoxides to alkenes. We achieved excellent catalytic activity for a broad scope of substrates while keeping high selectivity, e.g., intact carbon–carbon double bonds due to the lack of reactivity of gold to reduce the alkene.
Experimental section
General information
All the reagents used for the support and catalyst preparation were of analytical grade, purchased from Sigma-Aldrich and used as received. Tetrachloroauric(III) acid was purchased as a 30 wt% aqueous solution in dilute HCl (Sigma-Aldrich). Commercial reagents were purchased from Sigma Aldrich, and used as received. The glass reactor was thoroughly cleaned with aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1 v/v), rinsed with copious pure water, and then dried in an oven prior to use. Gold content in the catalyst was measured by FAAS analysis, on a Shimadzu AA-6300 spectrophotometer using an Au hollow cathode lamp (Photron). Metal leaching into the supernatant solution was measured by inductively coupled plasma measurements, performed on a Spectro Arcos ICP-OES. TEM analyses were performed with a JEOL 2100. Catalyst samples for TEM were prepared by sonicating the catalyst powder in propan-2-ol. A drop of the resulting dispersion was placed on a carbon-coated copper grid (Ted Pella, Inc.). The histogram of nanoparticle size distribution was obtained from the measurement of about 200 particles.
:HNO3 = 3:1 v/v), rinsed with copious pure water, and then dried in an oven prior to use. Gold content in the catalyst was measured by FAAS analysis, on a Shimadzu AA-6300 spectrophotometer using an Au hollow cathode lamp (Photron). Metal leaching into the supernatant solution was measured by inductively coupled plasma measurements, performed on a Spectro Arcos ICP-OES. TEM analyses were performed with a JEOL 2100. Catalyst samples for TEM were prepared by sonicating the catalyst powder in propan-2-ol. A drop of the resulting dispersion was placed on a carbon-coated copper grid (Ted Pella, Inc.). The histogram of nanoparticle size distribution was obtained from the measurement of about 200 particles.
Experimental procedures
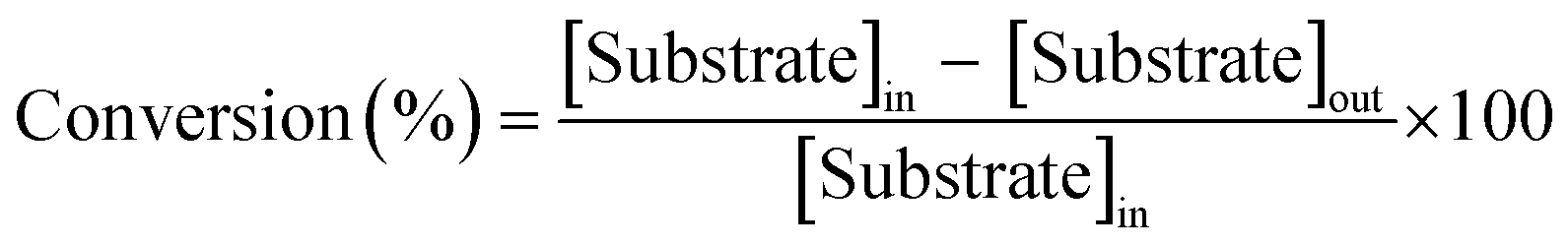 | (1) |
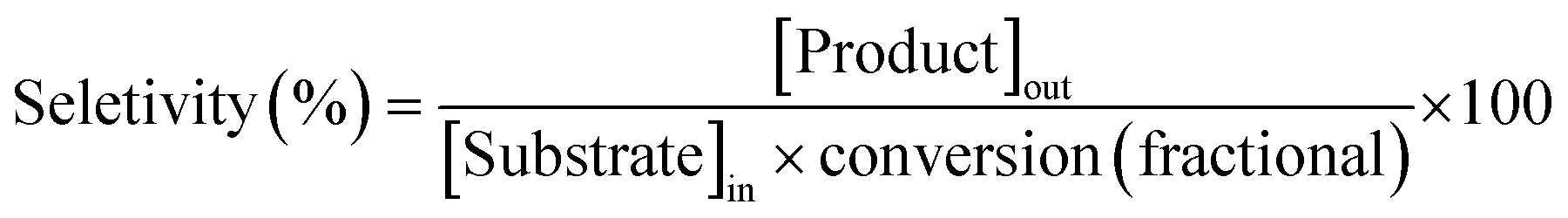 | (2) |
Results and discussion
The selective deoxygenation of styrene epoxide 1a was chosen as benchmark reaction to investigate the effect of the organophosphorus ligand on the catalytic activity of gold nanoparticles supported on silica. The Au/SiO2 catalyst without ligand reached 23% of conversion (Table 1, entry 1) after 6 h of reaction (1 mmol of epoxide, 1 mol% of Au, 2 mL of i-PrOH, 100 °C, 6 bar of H2). Under similar conditions, triethylphosphite (P(OEt)3), a commercially available, cheap and quite stable to air-phosphite, was added as an auxiliary ligand to gold, promoting full conversion of styrene epoxide 1a (Table 1, entry 2). The desired deoxygenate product was delivered with 90% isolated yield. In fact, the catalytic activity of the Au/SiO2 catalyst varied significantly depending on the identity of the organophosphorus ligand added to the reaction mixture (Table 1). The addition of alkyl-phosphine ligands to gold, such as PCy3 and PnBu3 (Table 1, entries 7 and 9), increased the conversion to 80–90%. In contrast, the addition of aryl-phosphine PPh3 (Table 1, entry 6) resulted in the same conversion as pure gold (23%). Additionally, phosphites were also tested as ligand, since the oxygen atom insulates the phosphorus (and therefore the metal to which it is attached) from steric and electronic effects arising from substituent modification.59 While the triphenylphosphite (Table 1, entry 10) was detrimental to reactivity, alkyl phosphites (Table 1, entries 10 and 11) afforded an improved reactivity of the gold catalyst. In contrast, tertiary phosphine oxide or organophosphate ligands did not show the same activating effect of the parent ligands (Table 1, entries 12–14).|
|
|||
|---|---|---|---|
| Entry | Ligand | Conversion of 1a (%) | Selectivity to 2a (%) |
| a Reaction conditions: 1 mmol of epoxide, 1 mol% of Au, 2 mol% of ligand (when added), 2 mL of i-PrOH, 100 °C, 6 bar of H2, 6 h. Conversion and selectivity were determined by GC analysis using biphenyl as an internal standard. Isolated yield is shown in parenthesis. b Au catalyst was not added. c Reaction was performed under N2 atmosphere. | |||
| 1 | No ligand | 23 | >99 |
| 2 | P(OEt)3 | 100 | >99 (90%) |
| 3b | P(OEt)3 | 0 | — |
| 4c | P(OEt)3 | 0 | — |
| 5 | PPh3 instead of P(OEt)3 | 23 | >99 |
| 6 | PCy3 instead of P(OEt)3 | 86 | >99 |
| 7 | Dppe instead of P(OEt)3 | 89 | >99 |
| 8 | PBu3 instead of P(OEt)3 | 94 | >99 |
| 9 | P(OPh)3 instead of P(OEt)3 | 12 | >99 |
| 10 | P(OMe)3 instead of P(OEt)3 | 81 | >99 |
| 11 | P(OBu)3 instead of P(OEt)3 | 86 | >99 |
| 12 | OP(OEt)3 instead of P(OEt)3 | 21 | >99 |
| 13 | OPCy3 instead of P(OEt)3 | 24 | >99 |
| 14 | PPh3O instead of P(OEt)3 | 25 | >99 |
A systematic survey of parameters such as the effect of the amount of added ligand, solvent, temperature and H2 pressure led to the optimized reaction conditions (Table S1–S4†). Lowering the amount of added phosphite from 2 to 1 mol% molar equivalent did not decrease the catalytic activity (>99% conversion) and selectivity to the corresponding alkene, but a decrease in the reaction conversion to 75% was noticed after further reducing the amount of ligand to 0.5 mol% (Table S1†). A series of blank experiments (Table 1, entries 3–4) indicate that the ligand is not acting as a reductant as previously reported for rhenium21 and molybdenum11 catalysts. Even the stoichiometric reaction of 1a with P(OEt)3 (1 mmol of ligand and 1 mmol of epoxide) showed no conversion after 24 h. In summary, the Au NP-catalyzed reaction can be conducted with only 1 mol% of P(OEt)3 in i-PrOH. Most of the catalytic tests were conducted at 100 °C and 6 bar of H2, but the hydrogenation reaction can be conducted at lower temperature and pressure.
The timepoint studies illustrate that the catalyst system Au NP and P(OEt)3 affords higher reaction rates when compared to the reaction without ligand (Fig. 1a). Moreover, the catalyst was easily separated from the reaction mixture, washed, and reused after adding new portions of substrate and P(OEt)3. By running kinetic curves, no loss of catalytic efficiency was noticed in five successive reactions (Fig. 1a). To gain insight into the catalytic active species, a hot filtration test was performed (Fig. 1b). No conversion took place after the Au/SiO2 catalyst was filtered off, indicating that the catalysis was heterogeneous in nature, and the catalytic activity and selectivity is not related to the formation of homogenous species. To corroborate with this, no gold content was detected by ICP-AES analysis of the reaction product. A homogeneous gold catalyst, such as Au(PPh3)Cl, was tested under similar conditions and was not able to catalyze the hydrogenation either. Comparing the morphology and size of as-prepared catalyst (Fig. 1c) with the recycled catalyst (Fig. 1d) by TEM images revealed only a slight increase of the mean diameter size from 2.3 ± 0.6 nm to 3.2 ± 1 nm after five recycles.
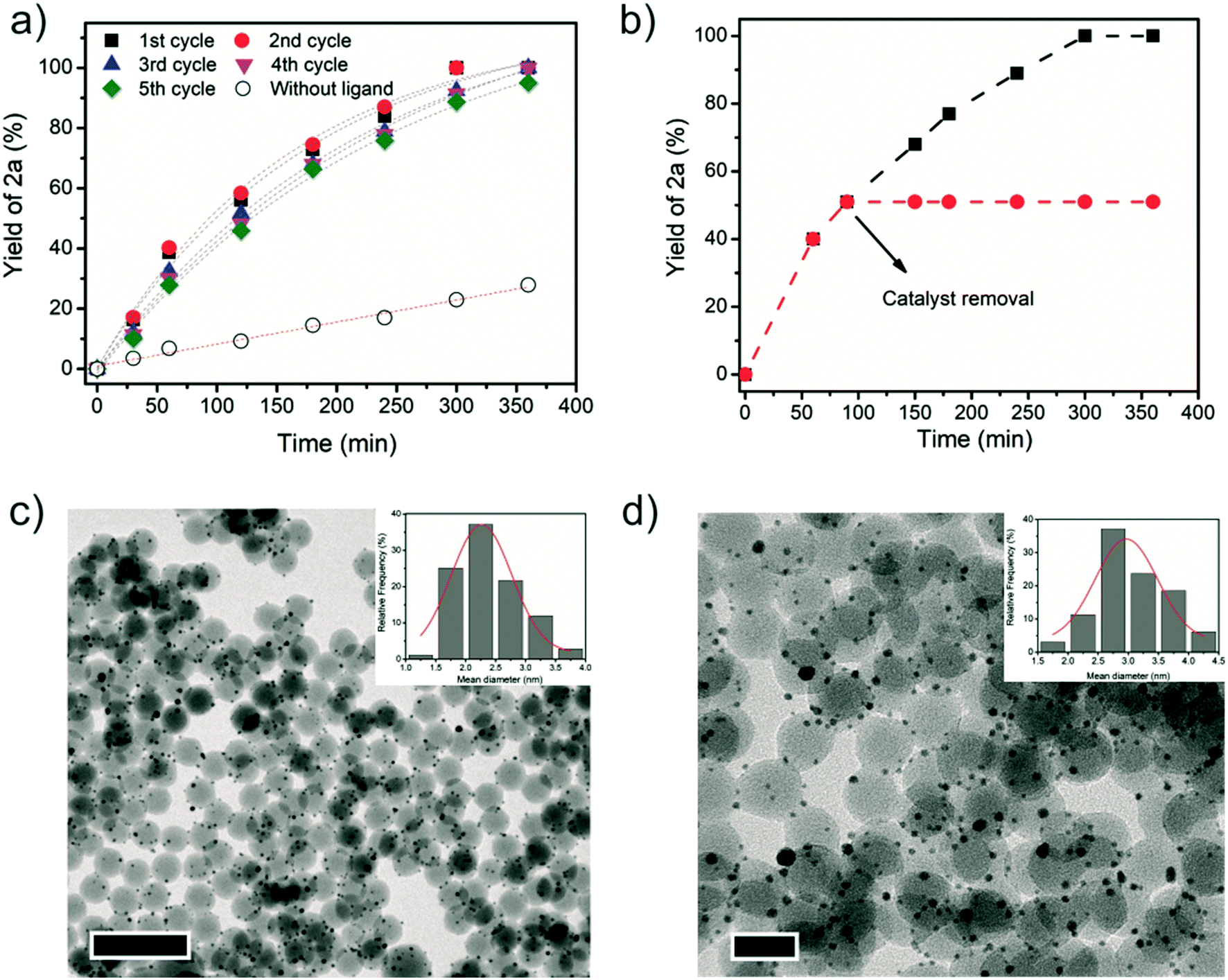 | ||
| Fig. 1 a) Timepoint study and recycles of Au/SiO2 magnetic catalyst with and without P(OEt)3 as ligand for the hydrogenation of styrene epoxide 1a; b) hot filtration test (black dots/traces represent the reaction conducted in the presence of Au/SiO2 catalyst and red dots/traces after catalyst removal); c) TEM image and size distribution of fresh Au/SiO2 catalyst (scale bar, 100 nm) and d) after the fifth reuse (scale bar, 25 nm). Reaction conditions: 1 mmol of epoxide, 1 mol% of Au, 1 mol% of P(OEt)3, 2 mL of i-PrOH, 100 °C, 6 bar of H2, 6 h. | ||
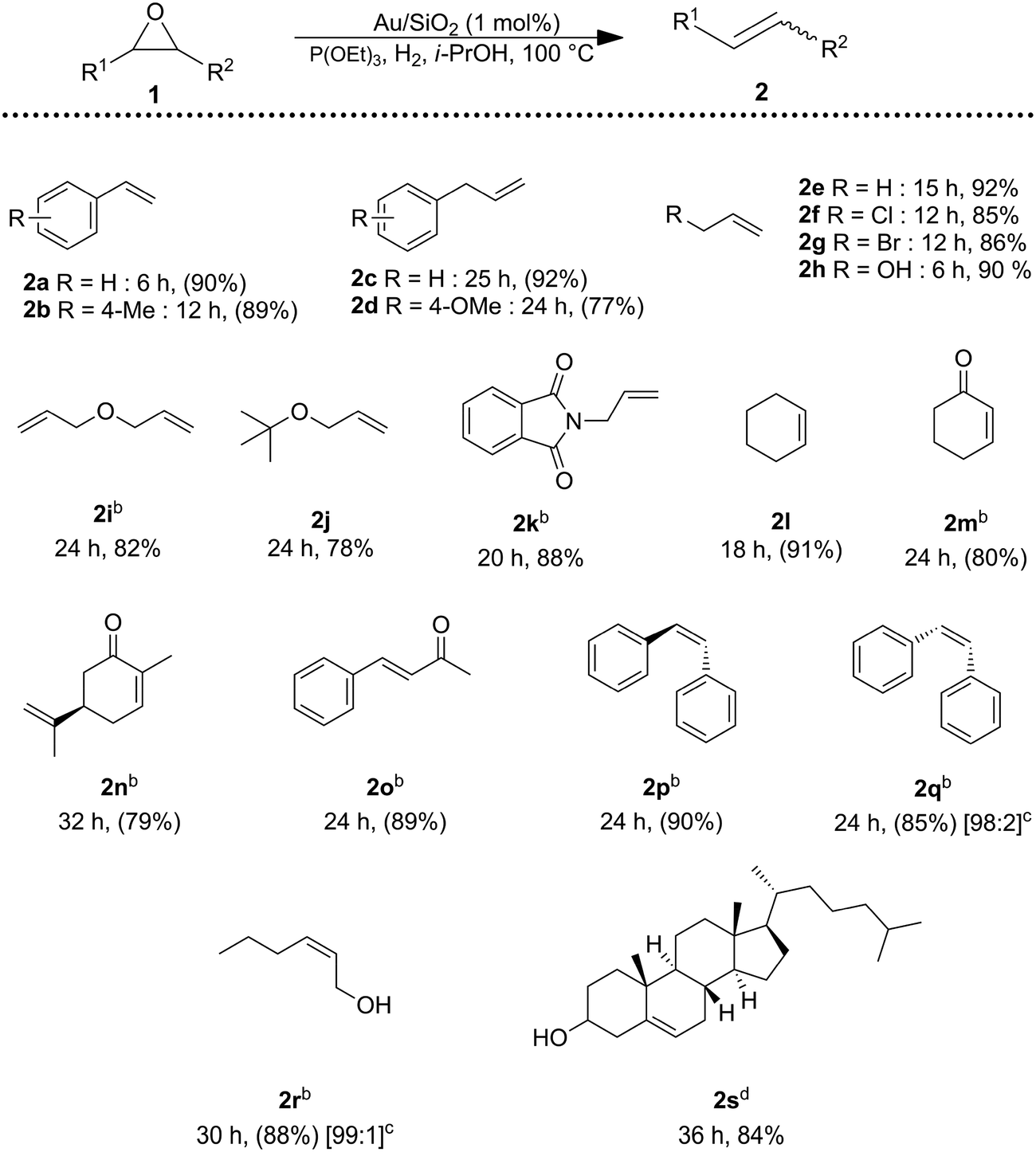
The catalytic system (Au/SiO2 + P(OEt)3) was able to catalyze the deoxygenation of a wide range of epoxides (Table 2), affording the corresponding alkenes in good to excellent yields, while hydrogenation of the alkene products did not occur. Styrene oxide substituted with methoxy group 2b was completely converted into the desired alkene. Benzylic epoxides could also be deoxygenated with yield over 75% of alkene (2c and 2d). Aliphatic epoxides were converted into the corresponding alkenes in high yields (2e–2j). Importantly, halides, hydroxy, ketone and alkene moieties remained intact during the deoxygenation of epoxides (2f–h, 2m, 2n, and 2i). Deoxygenation of α,β-epoxy carbonyl compounds occurred smoothly to the unsaturated α,β-carbonyl compound 2o. The deoxygenation proceeded in a stereospecific fashion: trans-stilbene oxide to trans-stilbene (2p) and cis-epoxides to cis-alkenes (2q and 2r). Moreover, 25-hydroxy-4,5-epoxy-cholestan-3-one, undergo the reaction in good yield, furnishing to 25-hydroxycholestenone (2s), the biologically active compound responsible for the regulation of calcium and phosphate ion concentration in cells.
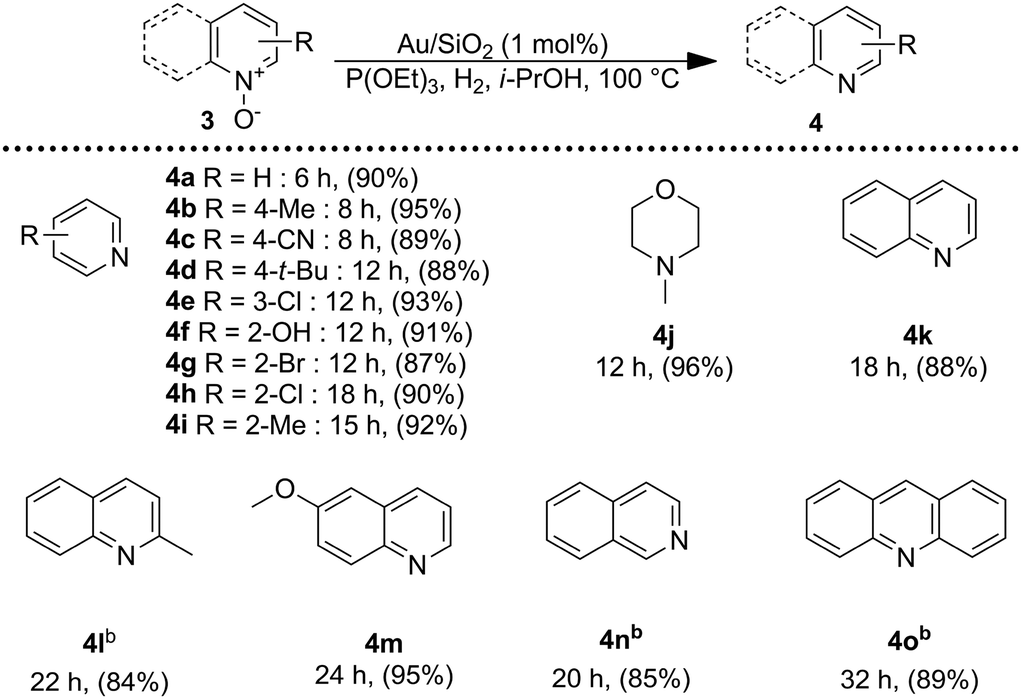
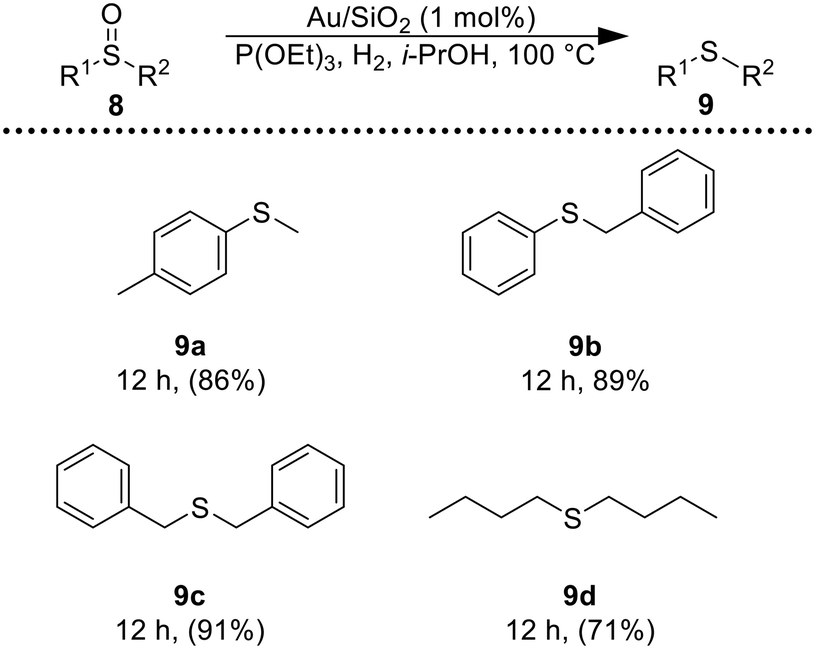
Encouraged by these results, we became interested in the deoxygenation of a series of heterocyclic N-oxides to N-heteroarenes, an important class of heterocycles key structures in a wide range of pharmacologically and biologically relevant natural products.60 Deoxygenation of a representative selection of heterocyclic N-oxides using H2 as reductant revealed to be compatible with the catalytic system affording the corresponding reduced products in good to excellent yields. Notably, substrates possessing reducible functionalities such as halides, cyano, and hydroxyl were also chemoselectively reduced to the corresponding N-heterocycles (Table 3). Furthermore, to investigate the scope of the deoxygenation ability of the present catalytic system, we next applied this system to deoxygenation of sulfoxides (Table 4). Phenyl, benzylic, and aliphatic sulfoxides were converted into the corresponding sulfides in yields over 70%.
| a Reaction conditions: 1 mmol of sulfoxide, 0.01 mmol of Au, 1 mol% of P(OEt)3, 2 mL of i-PrOH, 100 °C, 10 bar of H2. |
|---|
|
The utility of this protocol was also demonstrated under scaled-up conditions (eqn (1)); 1o (20 mmol; 3.92 g) was successfully converted to the alkene, 2o (90% isolated yield; 3.21 g), importantly, no side product of isomerization was detected. A turnover number (TON) of 1800 was reached with a productivity of approximately 75 mol mol−1 h−1, and the catalyst can be recycled as demonstrated above.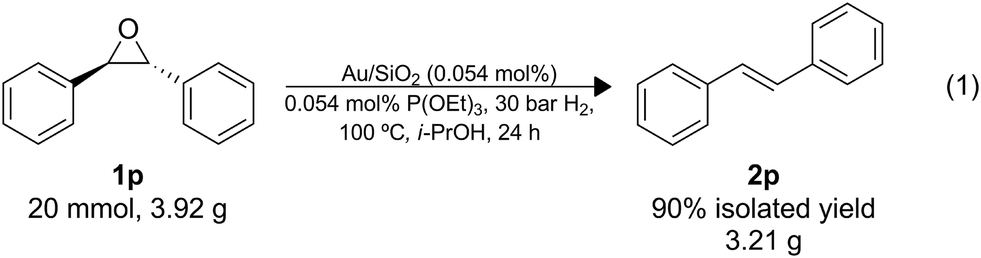
Although the mechanistic aspects of our gold catalyzed hydrogenation reactions remain somewhat speculative at this time, hydrogen activation might be occurring via a heterolytic pathway that happens due to a cooperative effect among the gold surface, the ligand and, possibly, the substrate. The concerted mechanism leading to heterolytic activation of H2 on gold surface has already been proposed to be effective in the hydrogenation of carbonyl groups using gold catalysts.26,27,35 The hydrogenation resulted from a combined effect of the ligand employed, usually secondary phosphine oxides, the gold surface catalyst, and also the substrate (α,β-unsaturated aldehydes), which was hydrogenated via the heterolytic cleavage of H2 and its concerted addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond to form the corresponding alcohol. The activity was governed by the basicity between ligand and reactant and the sites available on the nanoparticle surface. Here, we have expanded the practical applicability of gold catalysts possibly via a catalytic cycle where H2 is activated at the ligand–gold NP surfaces interface leading to the heterolytic splitting of H2 molecules at mild conditions; most important for epoxide to alkene conversion, using a metal that is inactive for the successive alkene reduction.
O bond to form the corresponding alcohol. The activity was governed by the basicity between ligand and reactant and the sites available on the nanoparticle surface. Here, we have expanded the practical applicability of gold catalysts possibly via a catalytic cycle where H2 is activated at the ligand–gold NP surfaces interface leading to the heterolytic splitting of H2 molecules at mild conditions; most important for epoxide to alkene conversion, using a metal that is inactive for the successive alkene reduction.
Conclusions
In summary, a general approach to selective deoxygenation of epoxides, N-oxides, and sulfoxides was developed by cooperative effect of Au NPs and P-containing ligands, through the formation of a cooperative interface Au/ligand. Notably, the deoxygenation reactions furnished the desired products in high yields for a broad scope of substrates, with excellent functional-group tolerance providing access to an array of diversely functionalized alkenes, sulfides and N-heteroarenes compounds, which are valuable building blocks in organic synthesis. We believe that the current work will be of broad synthetic and mechanistic interest for chemists, disclosing a promising and efficient way to improve the catalytic activity of Au NPs in hydrogenation reaction while showing new applications for the well-known phosphine chemistry in homogeneous catalysis. Further efforts on the application of this catalytic system as a synthetic organic chemistry tool are needed to consolidate this useful protocol, as well as continue with more detailed mechanism studies.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors are grateful to INCT-Catálise/FAPESC/CNPq/CAPES (CNPq 465454/2014-3; 444061/2018-5) and FAPESP (2016/16738-7, 2018/26253-6) for financial support, and CNPQ and CAPES for fellowships.References
- E. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474–1485 CrossRef CAS.
- P. E. Sonnet, Tetrahedron, 1980, 36, 557–604 CrossRef CAS.
- R. Bruckner, Organic Mechanisms, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010 Search PubMed.
- J. R. Lamb, A. K. Hubbell, S. N. MacMillan and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 8029–8035 CrossRef CAS.
- G. K. Pathe and N. Ahmed, Tetrahedron Lett., 2015, 56, 6202–6206 CrossRef CAS.
- J. Robertson and R. S. Srivastava, Mol. Catal., 2017, 443, 175–178 CrossRef CAS.
- F. L. Wu, B. P. Ross and R. P. McGeary, Eur. J. Org. Chem., 2010, 1989–1998 CrossRef CAS.
- S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS.
- A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2011, 50, 2986–2989 CrossRef CAS.
- S. C. A. Sousa and A. C. Fernandes, Coord. Chem. Rev., 2015, 284, 67–92 CrossRef CAS.
- S. Asako, T. Sakae, M. Murai and K. Takai, Adv. Synth. Catal., 2016, 358, 3966–3970 CrossRef CAS.
- K. N. Gurudutt and B. Ravindranath, Tetrahedron Lett., 1980, 21, 1173–1174 CrossRef CAS.
- G. Righi, P. Bovicelli and A. Sperandio, Tetrahedron, 2000, 56, 1733–1737 CrossRef CAS.
- P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- J. M. Concellón and E. Bardales, Org. Lett., 2002, 4, 189–191 CrossRef.
- M. T. Reetz and M. Plachky, Synthesis, 1976, 1976, 199–200 CrossRef.
- M. G. Martin and B. Ganem, Tetrahedron Lett., 1984, 25, 251–254 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and M. Jafarpour, Tetrahedron Lett., 2005, 46, 4107–4110 CrossRef CAS.
- Z. Zhu and J. H. Espenson, J. Mol. Catal. A: Chem., 1995, 103, 87–94 CrossRef CAS.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- T. Nakagiri, M. Murai and K. Takai, Org. Lett., 2015, 17, 3346–3349 CrossRef CAS.
- K. P. Gable and E. C. Brown, Organometallics, 2000, 19, 944–946 CrossRef CAS.
- S. Florio, F. Perna, A. Salomone and P. Vitale, in Comprehensive Organic Synthesis II, Elsevier, 2014, vol. 8, pp. 1086–1122 Search PubMed.
- T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545–5548 CrossRef CAS.
- J. Yu, Y. Zhou, Z. Lin and R. Tong, Org. Lett., 2016, 18, 4734–4737 CrossRef CAS.
- I. Cano, A. M. Chapman, A. Urakawa and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2014, 136, 2520–2528 CrossRef CAS.
- I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS.
- C. Liu, H. Abroshan, C. Yan, G. Li and M. Haruta, ACS Catal., 2016, 6, 92–99 CrossRef CAS.
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS.
- G. Li, H. Abroshan, Y. Chen, R. Jin and H. J. Kim, J. Am. Chem. Soc., 2015, 137, 14295–14304 CrossRef CAS.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- J. L. Fiorio, R. V. Gonçalves, E. Teixeira-Neto, M. A. Ortuño, N. López and L. M. Rossi, ACS Catal., 2018, 8, 3516–3524 CrossRef CAS.
- T. Whittaker, K. B. S. Kumar, C. Peterson, M. N. Pollock, L. C. Grabow and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 16469–16487 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- N. Almora-Barrios, I. Cano, P. W. N. M. van Leeuwen and N. López, ACS Catal., 2017, 7, 3949–3954 CrossRef CAS.
- R. Ouyang and D. Jiang, ACS Catal., 2015, 5, 6624–6629 CrossRef CAS.
- G. Lu, P. Zhang, D. Sun, L. Wang, K. Zhou, Z.-X. Wang and G.-C. Guo, Chem. Sci., 2014, 5, 1082–1090 RSC.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS.
- M. Yan, T. Jin, Y. Ishikawa, T. Minato, T. Fujita, L.-Y. Chen, M. Bao, N. Asao, M.-W. Chen and Y. Yamamoto, J. Am. Chem. Soc., 2012, 134, 17536–17542 CrossRef CAS.
- S.-S. Li, L. Tao, F.-Z.-R. Wang, Y.-M. Liu and Y. Cao, Adv. Synth. Catal., 2016, 358, 1410–1416 CrossRef CAS.
- R. Silva, J. Fiorio, P. Vidinha and L. Rossi, J. Braz. Chem. Soc., 2019, 00, 1–8 Search PubMed.
- A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Molecules, 2011, 16, 8209–8227 CrossRef CAS.
- Y. Shiraishi, H. Hirakawa, Y. Togawa and T. Hirai, ACS Catal., 2014, 4, 1642–1649 CrossRef CAS.
- Y. Li, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2013, 52, 12636–12640 CrossRef CAS.
- J. Ni, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem. Commun., 2011, 47, 812–814 RSC.
- J.-M. Huang, Z.-Q. Lin and D.-S. Chen, Org. Lett., 2012, 14, 22–25 CrossRef CAS.
- T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. – Eur. J., 2010, 16, 11818–11821 CrossRef CAS.
- H. Hirakawa, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Chem. Commun., 2015, 51, 2294–2297 RSC.
- H. Sajiki, K. Hattori and K. Hirota, Chem. Commun., 1999, 1041–1042 RSC.
- S. Nandi, P. Patel, A. Jakhar, N. H. Khan, A. V. Biradar, R. I. Kureshy and H. C. Bajaj, ChemistrySelect, 2017, 2, 9911–9919 CrossRef CAS.
- E. Thiery, J. Le Bras and J. Muzart, Green Chem., 2007, 9, 326–332 RSC.
- M. S. Kwon, I. S. Park, J. S. Jang, J. S. Lee and J. Park, Org. Lett., 2007, 9, 3417–3419 CrossRef CAS.
- P. S. Dragovich, T. J. Prins and R. Zhou, J. Org. Chem., 1995, 60, 4922–4924 CrossRef CAS.
- C. Yao, T. Dahmen, A. Gansäuer and J. Norton, Science, 2019, 364, 764–767 CrossRef CAS.
- S. Thiyagarajan and C. Gunanathan, Org. Lett., 2019, 21, 9774–9778 CrossRef CAS.
- W. Liu, W. Li, A. Spannenberg, K. Junge and M. Beller, Nat. Catal., 2019, 2, 523–528 CrossRef CAS.
- S. H. Pang and J. W. Medlin, J. Phys. Chem. Lett., 2015, 6, 1348–1356 CrossRef CAS.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
- K. D. Cooney, T. R. Cundari, N. W. Hoffman, K. A. Pittard, M. D. Temple and Y. Zhao, J. Am. Chem. Soc., 2003, 125, 4318–4324 CrossRef CAS.
- Y.-P. Han, X.-S. Li, X.-Y. Zhu, M. Li, L. Zhou, X.-R. Song and Y.-M. Liang, J. Org. Chem., 2017, 82, 1697–1704 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01695k |
| This journal is © The Royal Society of Chemistry 2021 |