
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous carbon–carbon composite electrodes for vanadium redox flow batteries synthesized by twin polymerization†
Maike Schnucklake *a,
Lysann Kaßnerb,
Michael Mehringb and
Christina Rothc
*a,
Lysann Kaßnerb,
Michael Mehringb and
Christina Rothc
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, D-14195 Berlin, Germany. E-mail: maike.schnucklake@fu-berlin.de
bTechnische Universität Chemnitz, Fakultät für Naturwissenschaften, Institut für Chemie, Professur Koordinationschemie, D-09107 Chemnitz, Germany
cElectrochemical Process Engineering, Universität Bayreuth, Universitätsstraße 30, D-95447 Bayreuth, Germany
First published on 18th November 2020
Abstract
Highly porous carbon–carbon composite electrodes have been synthesized by surface twin polymerization on a macroporous polyacrylonitrile (PAN)-based substrate. For this purpose the compound 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (Spiro), being a molecular precursor for phenolic resin and silica, was polymerized onto PAN-based felts with subsequent thermal transformation of the hybrid material-coated felt into silica-containing carbon. The following etching step led to high surface carbon–carbon composite materials, where each carbon component served a different function in the battery electrode: the carbon fiber substrate possesses a high electron conductivity, while the amorphous carbon coating provides the catalytic function. For characterization of the composite materials with respect to structure, porosity and pore size distribution scanning electron microscopy (SEM) as well as nitrogen sorption measurements (BET) were performed. The electrochemical performance of the carbon felts (CF) for application in all-vanadium redox flow batteries was evaluated by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). Compared to the pristine PAN-based felt the composite electrodes show significantly enhanced surface areas (up to 35 times higher), which increases the amount of vanadium ions that could be adsorbed onto the surface and thus contributes to an increased performance.
Introduction
For the integration of only intermittently available renewable power sources into the grid, efficient and reliable energy storage devices are imperative. Besides lithium-ion batteries1–3 and supercapacitors,4–6 redox flow batteries (RFBs) play an important role in energy storage.7–10 One major advantage of RFBs is the scalability of storage capacity due to the applied electrolyte volume independent from the size of the electrode area, which makes them favorable compared to other technologies.11 A further advantage of this type of battery is their less pronounced degradation (e.g. no solid-state phase transformations as in lithium-ion batteries) and therefore longer cycle life. Contamination of the electrolyte solutions due to diffusion of redox species across the membrane can be widely avoided by employing the same element in both half cells.12,13Vanadium redox flow batteries (VRFBs) are a specific type of these batteries, which are already commercialized and readily available. Here, the energy is stored in the form of vanadium-ion containing electrolytes, which consist of V(II)/V(III) at the negative and V(IV)/V(V) at the positive side. The redox reaction takes place at porous, stable, cheap and inert carbon electrodes, which commonly are commercial carbon felts. In general, porous carbon materials are widely used for energy applications, like conversion and storage.14–19 They also recently attracted interest in other fields, such as aeronautics,20,21 hydrogen storage19,22 and biomedical applications,23 where they can be used e.g. for tissue engineering.24 Porous carbon fiber materials satisfy all the requirements for energy applications, including mechanical and thermal strength, as well as relatively low cost.25 However, the poor catalytic activity of carbon felts for the respective redox reactions of the flow batteries is one significant drawback.26 Therefore, one focus of current research is the development of suitable methods for tailoring the felts' properties to enhance their electrochemical performance.27–29 For example Cheng et al. report about the preparation of porous nitrogen doped carbon electrodes derived from biomass.29 Ideal electrodes for use in redox flow batteries should provide high surface area combined with good electrical conductivity and electrochemical stability to facilitate the redox reaction as well as the electron transfer.30
Basically, there exist different strategies to incorporate porosity into carbon materials.31 Due to pyrolysis and subsequent physical or chemical activation of organic precursors, such as wood or coal, porous carbons could be conventionally synthesized. Nowadays, however, the utilization of templates is the most common strategy, as they exert more control on the pore characteristics in the final carbonized product.32 Here, two approaches are equally popular: the hard-templating and the soft-templating method, where in the latter case various surfactants or polymeric detergents are used as structure directing agents.33 During the hard-templating process nanostructured inorganic SiO2 particles are often deployed as porogens, and as a consequence the precursor has to be removed after the carbonization step.31 Both methods generate highly ordered nanostructures with large surface areas.
Spange et al. developed a sophisticated synthesis route called twin polymerization, to produce simultaneously two interpenetrating polymers forming an organic–inorganic hybrid material with only one monomer and one catalyst required.34–39 Moreover, the twin polymerization approach offers several major advantages. As a well-established example the twin monomer 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] forms phenolic resin and silica without producing any side products, which reduces the shrinking commonly experienced during the stabilization step.36–38,40–42 Ideally, the twin polymerization facilitates the synthesis of nanocomposite domains of 0.5 nm to 3 nm, which is beneficial for the envisaged application.37 It is possible to transform the hybrid materials into highly porous carbon materials by pyrolysis under inert conditions. The silicon dioxide of the hybrid material can serve as an intrinsic template for the porous carbon and can be easily removed by leaching with hydrofluoric acid or sodium hydroxide solution.36–38,42–45 Depending on the choice of the monomer, it is also possible to design the structure of the emerging carbon material. Materials based on the twin monomer Spiro possess micropores which facilitate the adsorption of the redox species, while mesopores are preferentially formed when the precursor tetrafurfuryloxysilane is used. Quantities of monomer, catalyst and substrate can be varied, which allows to develop a huge variety of different porous carbon materials, varying in their properties like porosity and morphology. Furthermore, twin polymerization provides the opportunity to modify the surface of various solid substrates. To obtain a successful coating, the polymerization parameters have to be adapted by considering characteristic features of the used substrate. For example, carbon black as well as Stöber particles were coated by Windberg et al. in this way.45
In 2018, Choudhury et al. used the twin polymerization approach to synthesize a promising carbon cathode for lithium–sulfur batteries.46 Depending on the monomer, which was applied, hollow carbon spheres with microporous or mesoporous shell were synthesized by Böttger-Hiller et al.43 In this study different silicon dioxide particles were used as spherical hard templates to form the porous carbon material. They figured out, that the morphology of the resulting spheres can be influenced by varying the ratio of monomer/substrate and not only by the utilization of different templates. Ebert et al. coated graphite sheets with different layers of amorphous carbon by utilizing the twin polymerization procedure.44 Inspired by these works, we report a synthesis of porous electrode materials for use in redox flow batteries with a significantly increased surface area compared to the state-of-the-art carbon electrodes and with particularly ordered structure. In our preliminary work, highly porous, co-doped composite electrodes were synthesized.47 We demonstrated that the introduction of a greater number of heteroatom-sites enhances the kinetics of the positive half-cell reaction of the redox flow battery. However, it was not possible to differentiate between the effects of the implementation of heteroatoms or the higher surface on the enhanced performance observed for the electrodes.47 Therefore, in this work we focus on the enlargement of the specific surface area without doping the electrode with additional heteroatoms in order to investigate the effect of the increased surface in more detail. With the twin polymerization approach it was possible to produce electrodes, which are suitable for both positive and negative half-cell reactions. The carbon felt structure is used as scaffold with high electron conductivity, whereas the amorphous carbon deposit on its surface should provide the catalytic functionality.
The influence of the catalyst as well as the reproducibility of the twin polymerisation method were investigated and showed that using an acid as catalyst provided a longer lasting stability of the increased electrochemical activity of the electrode. The twin polymerization approach shows a good reproducibility.
Experimental
Materials
All chemicals were received from commercial sources: toluene (99.8%, Sigma-Aldrich), 2-hydroxybenzyl alcohol (99%, Sigma-Aldrich), tetramethoxysilane (≥99.0%, Sigma-Aldrich), tetrabutylammonium fluoride solution (1.0 M in THF, Sigma-Aldrich), hexane (anhydrous, 95%, Sigma-Aldrich), dichloromethane, sodium hydroxide. The chemicals were used as obtained if not otherwise stated. Toluene and dichloromethane were dried with standard methods under inert atmosphere and freshly distilled before use. Deionized water was used for the syntheses and ultrapure Milli-Q® water (18.2 MΩ cm) for electrochemical measurements.Polyacrylonitrile (PAN)-based felts (only stabilized, not yet carbonized) were received from Freudenberg Performance Materials SE & Co. KG (Weinheim, Germany) and utilized as substrate. It should be mentioned here, that the untreated carbon felt is called ‘pristine’. The composite electrodes are named according to the following general scheme – containing the synthesis conditions: material type (catalyst, carbonization time, repeat sequences of etching). The following abbreviations are used for the different catalysts: MSA for acid-catalyzed polymerization and T for thermally induced reaction.
Methods
Synthesis of 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (Spiro)
2,2′-Spirobi[benzo-4H-1,3,2-dioxasiline] was synthesized according to the literature.37 In brief, it is prepared by a transesterification of salicyl alcohol and tetramethoxysilane under argon atmosphere. The synthesis is catalyzed by tetra-n-butylammonium fluoride.Coating of PAN-based felts
PAN-based felts were coated on the basis of coating procedures of silica materials by acid-catalyzed twin polymerization as described earlier.43 The felt (2.5 × 2.5 cm) was weighed and placed in a baked out round-bottom flask under inert atmosphere with dichloromethane (DCM, 200 mL per 1 g substrate) and methanesulfonic acid (MSA, 240 mg per 1 g substrate). The mixture was stirred for 30 minutes and afterwards the solvent was evaporated under reduced pressure. The substrate loaded with methanesulfonic acid was transferred into a second baked out round-bottom flask under inert atmosphere and then toluene was added (200 mL per 1 g substrate). Furthermore, 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (0.5 g per 1 g substrate) dissolved in toluene (100 mL per 1 g 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline]) was added and the mixture was stirred slightly. After some minutes the solution became turbid and flocculation began besides staining of the felt due to polymerization of the twin monomer on the substrate surface. After 22 hours the coated substrate was removed from the flask, washed with toluene, dried under air and weighed.In case of the thermally induced polymerization the felt was given to a solution containing dried DCM (6 mL per 1 g substrate) and 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (0.56 g per 1 g substrate). The solution was completely absorbed. After evaporation of the solvent by purging with argon, the felt was heated to 230 °C at a heating rate of 4.1 K min−1 and held at this temperature for one hour in order to carry out the thermally induced polymerization.
Carbonization of coated PAN-based felts
The dried felts were transferred into an appropriate glass tube and were carbonized under continuous argon flow. The samples were heated to 1000 °C with a heating rate of 4.1 K min−1 and held at this temperature for one hour unless otherwise stated. The samples were cooled under argon flow and weighed.Etching of silica with sodium hydroxide solution
The weighed sample was transferred into a Teflon round-bottom flask equipped with a reflux condenser and a magnetic stir bar. 5 M sodium hydroxide solution was added (300 mL per 1 g sample) and the mixture was refluxed with slight stirring for 3 h. After cooling to room temperature the etched sample was separated by suction filtration, washed with water till neutralization of washing water and then dried under air. A detailed description of all weights of PAN-based samples and used materials is given in the ESI (Table S1†).Structural characterization
Electron microscopy images were taken by an instrument from type Nova NanoSEM 200 of FEI Company (TU Chemnitz, Laboratory of Solid Surfaces Analysis), as well as with a SEM 840 J A from Jeol with an acceleration voltage of 10 kV (University of Bayreuth).For transmission electron microscopy (TEM) analysis, ultra-thin sections of the embedded sample were prepared and investigated with a CM20 FEI instrument.
The specific surface area was measured with an instrument of Quantachrome (Autosorb iQ2) by means of nitrogen adsorption according to Brunauer, Emmett and Teller (BET) with determination of pore size distribution. Before measurement, samples were baked out for 24–48 hours at 80 °C under vacuum (10−4 to 10−5 mbar). In case of the carbon felt electrodes the samples were cut into pieces without grinding and the carbon material was measured as prepared. The specific surface area was calculated by the DFT method and the pore size distributions were determined by applying the nonlocal density functional theory (NLDFT) equilibrium mode method based on slit pores.
Elemental analyses were obtained utilizing the VarioEL Organic Elemental Analyzer from Elementar Analysesysteme GmbH to identify the carbon, hydrogen, nitrogen and sulfur content. The average of the measured values was determined and listed in Table 2.
Thermogravimetric measurements were realized on a Thermogravimetric Analyzer 7 (TGA 7), of PerkinElmer Company (TU Chemnitz, Laboratory of Physical Chemistry). The samples were heated from 30–900 °C with a heating rate of 20 K min−1 under constant air flow. This temperature was held for another 10 to 30 min.
Electrochemical characterization
All electrochemical measurements were carried out with a Reference 600 potentiostat from Gamry Instruments operating in a three electrode setup. A saturated calomel electrode (SCE) and a platinum electrode made out of a one millimeter thick platinum piece (0.6 cm × 0.7 cm) were used as reference (243 ± 2 mV vs. SHE) and counter electrodes, respectively. The respective composite electrodes with a size of 0.5 × 0.5 mm served as working electrodes. For contacting they were pierced with a 1 mm thick glassy carbon rod, taking care to hit the center. Electrochemical measurements were performed in a solution containing 0.2 mol L−1 vanadyl sulfate (VOSO4, Sigma Aldrich) in 2 M sulfuric acid (H2SO4, Sigma Aldrich) for the positive half-cell reaction, while for the negative half-cell reaction the electrolyte consists of 0.2 mol L−1 vanadium (50% V2+ and 50% V3+) in 2 mol L−1 sulfuric acid, which was obtained by dilution of previously charged commercial vanadium electrolyte. In all cases the cyclic voltammograms were recorded with a scan rate of 2 mV s−1 and the electrolyte was purged with nitrogen before every measurement.The potentiostatic EIS measurements were implemented in a frequency range from 105 to 10−1 Hz with an AC amplitude of 10 mV and a DC potential of 0.75 V vs. SCE. The EIS data were fitted in the frequency range between 31.25–0.999 Hz and the working electrode was conditioned five minutes at the DC potential before the measurement started.
Results and discussion
Porous C–C composite electrodes with tuned porosity were obtained by coating a pristine PAN-based felt with hybrid material using twin polymerization (Fig. 1) and subsequent pyrolysis and etching. To initiate the polymerization reaction, methanesulfonic acid was utilized as a catalyst and in a second synthesis procedure the polymerization was induced thermally39 to study the effect of catalysis. After carbonization under inert atmosphere a subsequent etching step, in which the silicon dioxide is removed, was performed to obtain the final porous electrode. The overall synthetic procedure is illustrated in Fig. 1.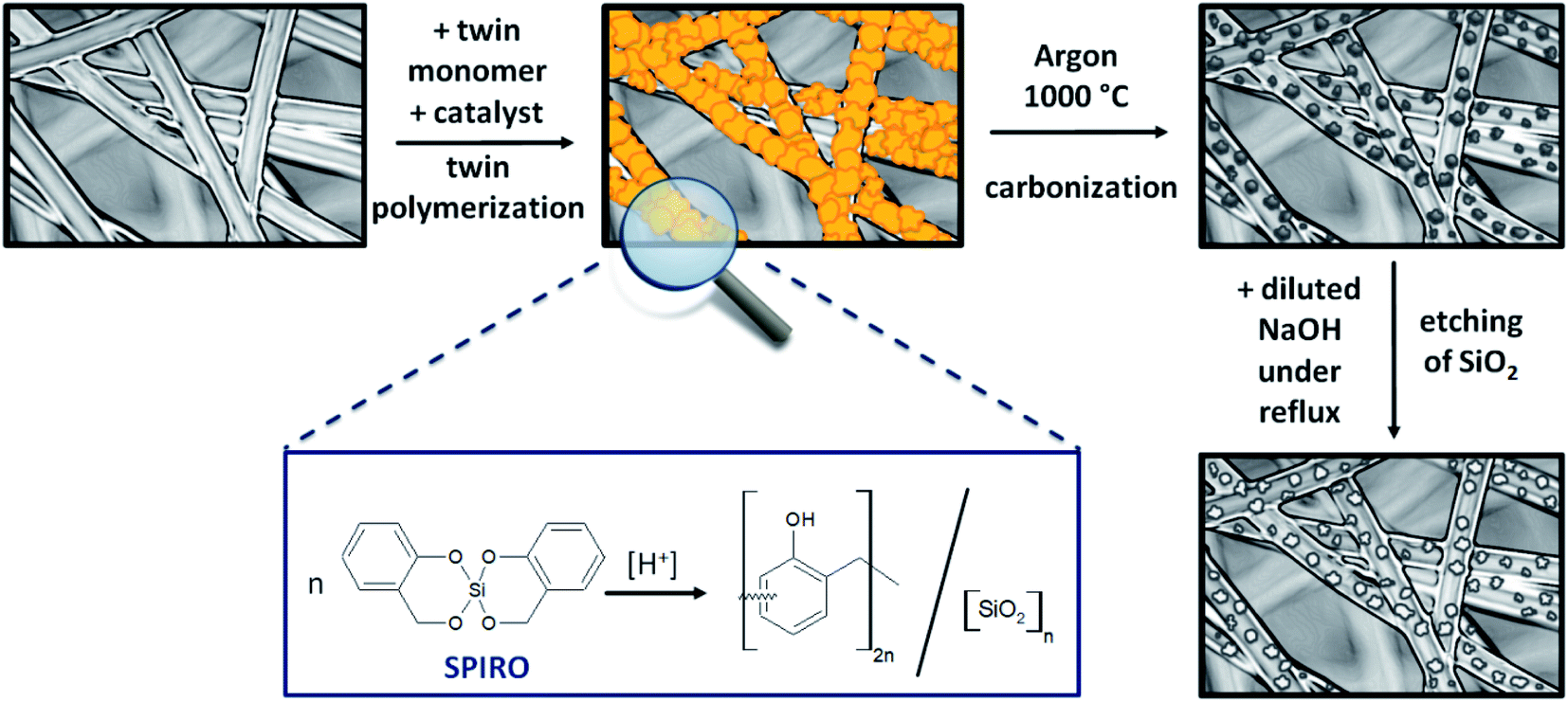 | ||
| Fig. 1 Schematic illustration of the synthesis of porous C–C composite electrode material by coating a PAN-based felt substrate with hybrid material using the twin polymerization and subsequent pyrolysis and etching. | ||
The carbon material obtained from phenolic resin–silica hybrid material by thermally induced substrate-free twin polymerization is referred to as “carbon material” in the following text, while the synthesized amorphous carbon-coated electrodes are called “composites”.
Structural characterization
For a detailed insight into the coating process of the PAN-based fiber material, SEM images of the felt were captured after certain steps of the treatment (Fig. 2). The pristine felt is comprised of carbon fibers with an average thickness of ten micrometers, showing a smooth surface. The single fiber strands appear to have only random orientation. During the polymerization step, the fibers become covered with a thick layer of polymer (Fig. 2b and e). Also some amount of the polymer is present in the form of isolated particles next to the fibers. In contrast to that finding, after the carbonization step the coating appears incomplete and the fibers are no longer homogeneously covered (Fig. 2c and f). | ||
| Fig. 2 High resolution images of the carbonized felt (left: a and d), PAN-based felt with twin polymer directly after the process of coating catalyzed by acid (middle: b and e) and composite electrode after carbonization and etching (right: c and f) at different magnifications. | ||
Additional TEM images of the carbonized, acid-catalyzed sample are given in the ESI (Fig. S1†).
To get rid of the silica phase in the last step of the synthesis procedure, the composite electrodes were etched with an aqueous solution of diluted sodium hydroxide (see Fig. 1). The alkaline treatment was chosen instead of hydrofluoric acid.43,45,46 Choosing this more cautious and environmentally benign approach, it was possible to prevent damage to the composite electrode. Thermogravimetric analysis was selected to test, whether the chosen method was suitable to remove the entire silica template (Fig. 3). During analysis under air, the residue of the TGA analysis gives information about the silicon dioxide content of the evaluated composite material. The composite electrodes show a negligibly small residue of maximum 4.8 wt%, which demonstrates that the template can be removed in an adequate way.
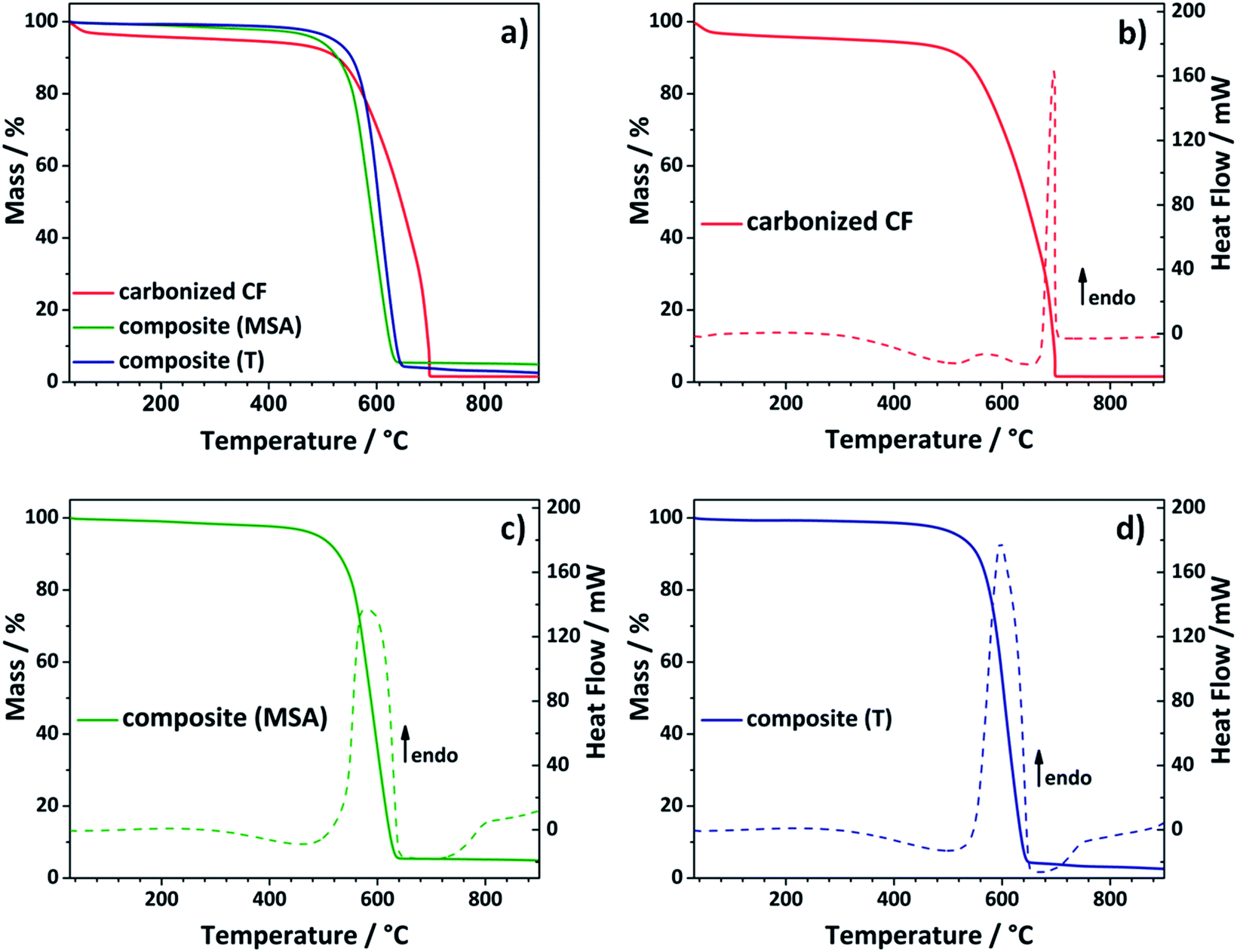 | ||
| Fig. 3 Thermogravimetric analysis examined under air shows the complete burning of the carbon composite electrode to demonstrate the effective removal of the silicon dioxide phase (a). DSC curves of carbonized felt (b, red) and composite material obtained by acidic catalysis (c, green) and composite material obtained by thermally induced polymerization (d, blue). | ||
Comparing the curve shapes of the carbonized felt and the composites, it is noticed that the carbonized material loses some water at the beginning, but then appears to be relatively stable. In the differential scanning calorimetry (DSC) curve a sharp endothermic peak is observed at about 700 °C (Fig. 3b). In contrast to that, the curves of the two composite electrodes appear distinctively different. In this case, the first thermolytic degradation already passes through a maximum at around 590 °C, which comes along with a significant mass loss. The two felts have already lost 50% of their mass at approximately 600 °C, while the carbonized sample shows a residual weight of 71 wt% at the respective temperature. Both composite materials show a similar stability. The material obtained by the acidic catalyzed twin polymerization starts to decompose at lower temperatures, while the curve of the composite received by thermally induced polymerization drops slightly faster. Comparison with the DSC curves (Fig. 3b–d) confirms this observation. Here, the endothermic peak of the acidic catalyzed sample seems to be more broadened. In the following it was evaluated whether the composites are stable enough under the conditions of the electrochemical measurements (see Fig. 5).
Nitrogen sorption measurements were conducted to analyze the porosity of the electrode materials and the carbon material. The different curves are shown in Fig. 4a and the respective DFT surface areas are listed in Table 1. The pristine felt shows the typical low surface area of 7 m2 g−1. Compared to the untreated felt, the composite materials show significantly enhanced surface areas (up to 35 times higher), which increases the amount of vanadium ions that could be adsorbed onto the surface. The isotherms of the composite electrodes and the carbon material show furthermore a gas uptake at lower relative pressures indicating a significantly increased contribution from microporosity. For a more detailed insight into the porosity, pore size distributions of the respective materials were calculated and are given in Fig. 4b–d. The corresponding total pore volumes (Vtotal) are listed in Table 1. The composite material, for which the polymerization was thermally induced, as well as its carbon material counterpart present a bimodal distribution with two maxima at a diameter of around 1.5 nm and 4–5 nm. In contrast to that, the reference material exhibits a unimodal distribution with a single maximum in the pore size range higher than 2 nm. It is worth mentioning here, that commercial pristine CFs show predominantly macropores with a literature value of <0.5 m2 g−1.48 Consequently, it is concluded that the micropores are introduced due to the amorphous carbon coating. These assumptions are confirmed by the contributions of the micro- and mesopore volumes to the total volume shown in Table 1. It is evident that the proportion of micropores increases in the order of pristine < carbonized CF < composite (MSA, 1h, E1) < composite (T, 1h, E1) < carbon material (T, 1h, E1) < carbon material (T, 1h, E2) as previously assumed.
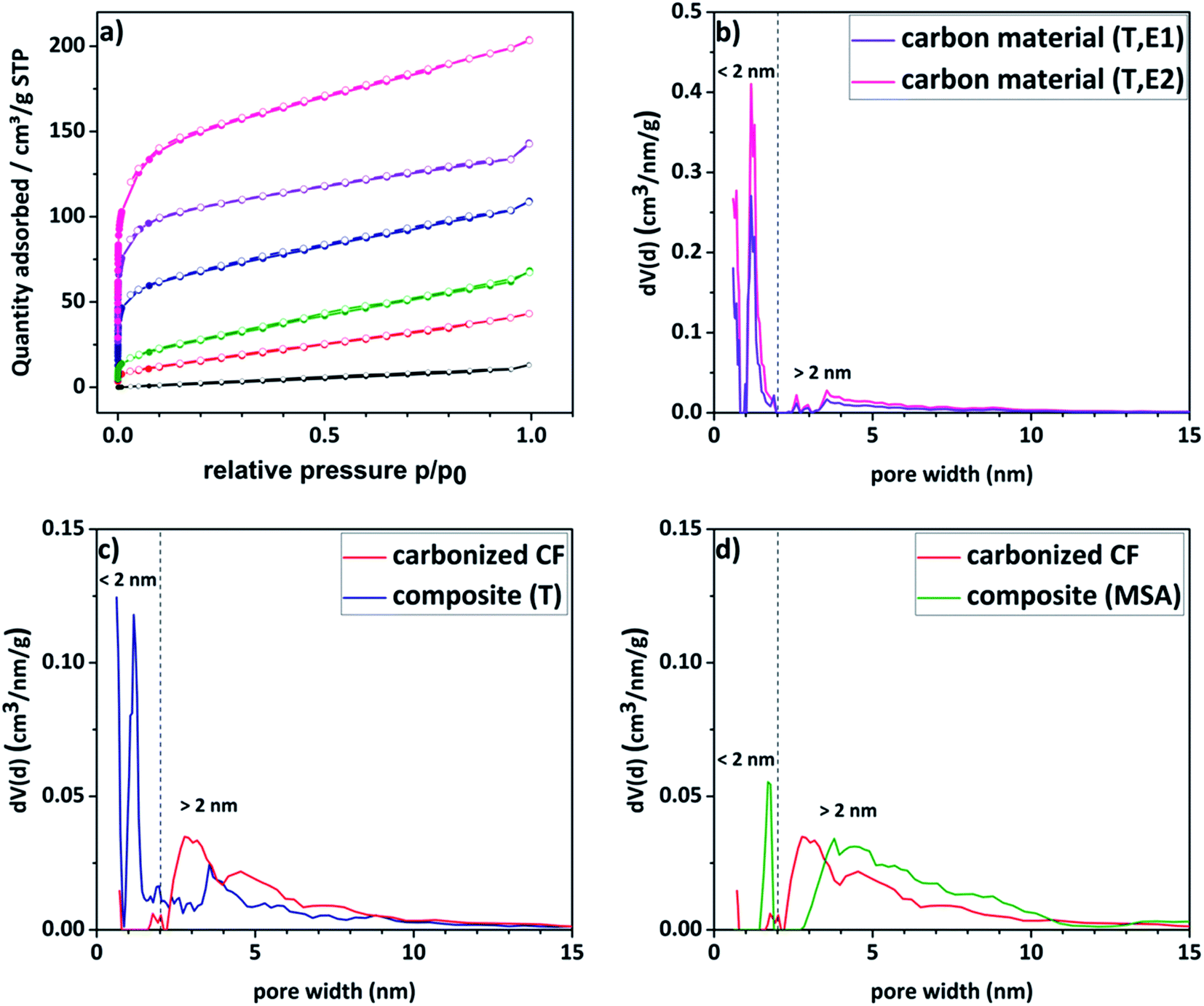 | ||
| Fig. 4 (a) Nitrogen sorption isotherms of the synthesized composite electrodes (blue, green), carbon material (etched for one or two times respectively, purple and pink) and the carbonized felt (red) compared to pristine felt (black). (b–d) Pore size distributions of the reference material (red), the composite electrodes (blue, green) and the carbon material etched for one or two times, respectively (purple, pink), obtained by using the nonlocal density functional theory (NLDFT) equilibrium model. | ||
| DFT surface area/m2 g−1 | Vtotal/cm3 g−1 | (Vmicro) (<2 nm)/cm3 g−1 | (Vmeso) (2–36 nm)/cm3 g−1 | |
|---|---|---|---|---|
| Pristine | 7 | 0.0156 | 0 | 0.0156 |
| Carbonized CF | 43 | 0.06069 | 0.00854 | 0.0521 |
| Composite (MSA, 1h, E1) | 123 | 0.1854 | 0.0127 | 0.1727 |
| Composite (T, 1h, E1) | 256 | 0.1569 | 0.0764 | 0.0805 |
| Carbon material (T, 1h, E1) | 444 | 0.2022 | 0.1495 | 0.0527 |
| Carbon material (T, 1h, E2) | 579 | 0.2978 | 0.2081 | 0.0897 |
Depending on the chosen twin monomer, it is possible to tailor the porosity of the resulting carbon. In this case, Spiro was utilized, since the introduction of micropores into the material appears to be beneficial for the intended application.49 It is frequently reported in the literature that a bimodal pore size distribution, in which the micropores facilitate the adsorption and larger mesopores provide fast electrolyte transport, exerts a positive impact on the performance of carbon-based electrodes.50,51
Due to a high amount of white residue in the TGA analysis, which is presumably remaining silicon dioxide, a second etching step of the carbon material became necessary (denoted with suffix E2). By this procedure, the specific surface area was enhanced even further from 444 m2 g−1 to 579 m2 g−1 as a result of further removal of silicon dioxide creating new pores (see Table 1). When looking at Fig. 4b it is striking to see, that an associated increase in microporosity is observed.
Table 2 summarizes the composition of the different carbon electrode materials. All examined samples are mainly composed of carbon, while the pristine felt has a high nitrogen content of 19 wt%, which decreases due to the carbonization step. Note that those values are typical for carbon materials obtained from PAN-based felts, while depending on the starting material the remaining nitrogen content could vary slightly. As intended, both composite materials do not have significantly higher nitrogen contents. Consequently the modification by twin polymerization only produces a coating of porous amorphous carbon material without doping the felts with additional nitrogen. We focused on the positive impact of the enhanced surface area, so that we were able to distinguish between the effect of the doping and the effect of the enhanced surface. Consistent with the aforementioned assumption, the microporous carbon material as received from the substrate-free polymerized monomer has only a negligibly small amount of 0.3 wt% nitrogen. This could be assigned to an impurity from the etching process, since all the nitrogen is contained within the PAN-derived felts.
| C/wt% | N/wt% | S/wt% | H/wt% | |
|---|---|---|---|---|
| a It is worth mentioning here that the residual mass can be attributed to oxygen content or incomplete combustion of the samples.b In accordance with the TGA findings, where we observed a white residue, the residual mass could be attributed to silicon dioxide and additionally from remaining oxygen from the phenolic resin. Oxygen cannot be measured with the chosen approach. | ||||
| Pristinea | 58.8 | 19.2 | 0.0 | 4.2 |
| Carbonized CF | 91.8 | 5.9 | 0.0 | 0.04 |
| Composite (MSA, 1h, E1)b | 81.3 | 6.7 | 0.2 | 0.6 |
| Composite (T, 1h, E1)b | 76.5 | 6.7 | 0.0 | 0.3 |
| Carbon material (T, 1h, E1)b | 65.7 | 0.3 | 0.0 | 0.3 |
It should be also noted here, that the composite material, for which the polymerization was catalyzed by acid, also contains a small amount of 0.2 wt% of sulfur. This can be attributed to negligible residues of the catalyst material i.e. methanesulfonic acid.
Electrochemical characterization: the positive and negative redox reaction
The modified composite electrodes have been investigated regarding their electrochemical performance for the positive (V(IV)/V(V)) and the negative (V(II)/V(III)) redox reaction. For this purpose, cyclic voltammetry measurements have been performed. The CV curves of the respective electrode materials are shown in Fig. 5. Here, the onset potential is defined as the value, when ten percent of the maximum current is reached. A rapid rise in the cyclovoltammetric curve and thus a low onset potential indicates fast kinetics of the reaction. The material shows good activity, in case the vanadium redox reaction starts early. When the system is fully reversible, the ratio between the anodic peak current and the cathodic peak current (ip) should be one and the separation of both peaks (ΔEp) should be small. All of those parameters taken together can be seen as an indication of an improved electrochemical activity and a very good reversibility.52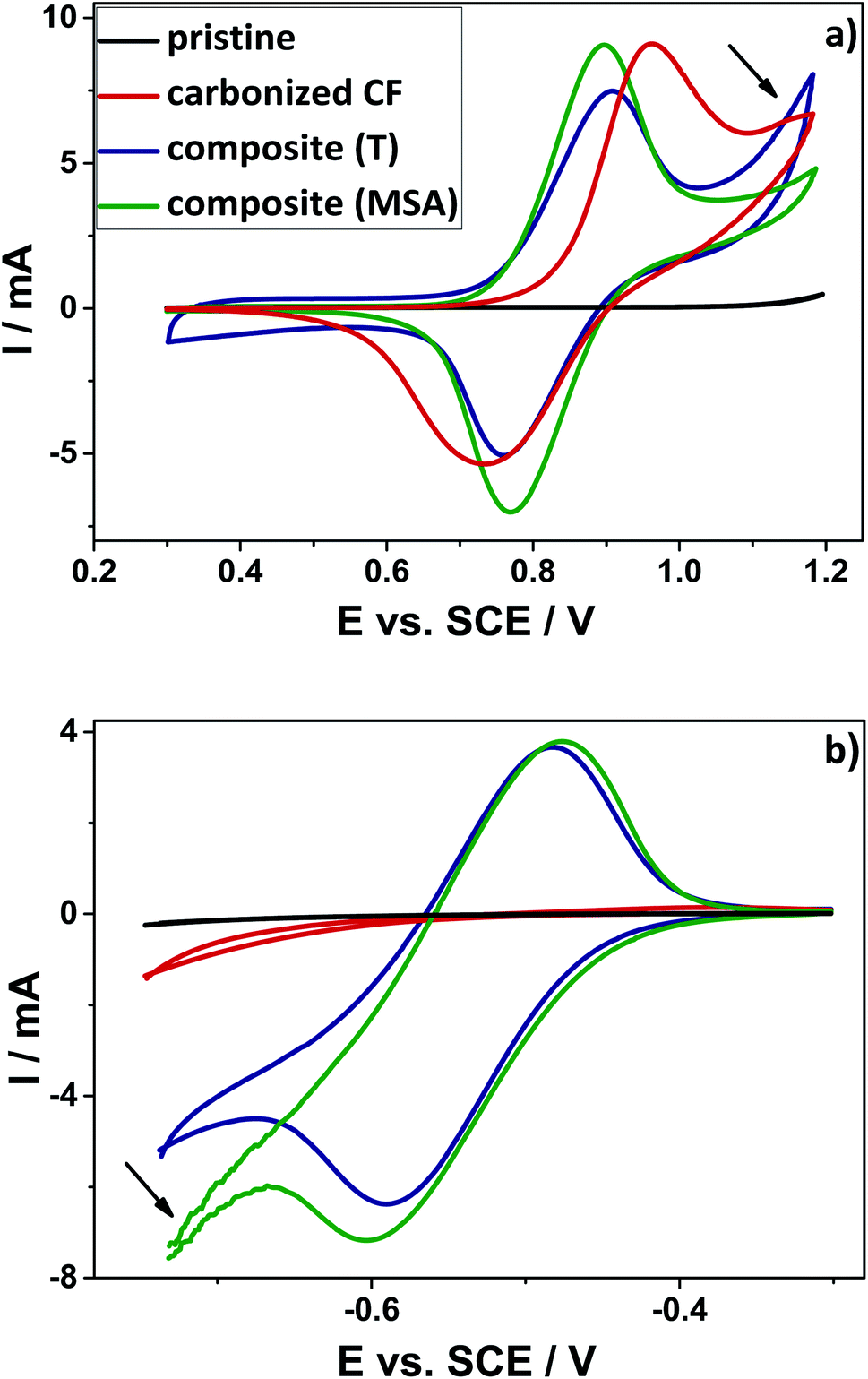 | ||
| Fig. 5 Comparison between the cyclic voltammetry curves of the pristine felt (black) and the carbonized CF (red) as well as the thermally catalyzed composite (blue) and the composite, for which the polymerization was catalyzed by acid (green) for the positive (a) and the negative (b) half-cell reaction. The electrolyte consists of 2 mol L−1 sulfuric acid and additional 0.2 mol L−1 vanadyl sulfate in case of the positive electrolyte. For the negative side an electrolyte was used, which consists of 0.2 mol L−1 vanadium (50% V2+ and 50% V3+) in 2 mol L−1 sulfuric acid, which was obtained by dilution of previously charged commercial vanadium electrolyte. | ||
Considering the positive half-cell reaction, it is apparent that both composite materials show a better electrochemical performance as compared to the pristine and the carbonized material. The reversibility of the reaction is significantly improved and a lower peak separation was observed. The maximum currents appear highly symmetric and the onset potentials are shifted to lower potentials for the oxidation and they are shifted to higher potentials for the reduction. In accordance with the nitrogen sorption measurements, the higher surface areas led to an increased double layer capacitance (DLC) in the region between 0.3 V and 0.6 V vs. SCE. The enhanced surface area of the composite is favorable, because due to the additional highly porous carbon material on top of the fibers' surface a higher amount of vanadium ions could be adsorbed. As a result of enhanced surface area, higher maximum currents could be noticed for the composite materials compared to the pristine and the carbonized sample. The composite obtained by acidic catalysis shows the highest maximum currents (see ESI, Table S2†).
Especially, the sample obtained by thermally induced polymerization is characterized by an additional oxidative current at a potential of about 1.0 V (marked with an arrow). Probably, this peak originates from the early onset of corrosion of carbon to carbon dioxide, which was demonstrated in earlier studies with differential electrochemical mass spectrometry (DEMS) measurements.53,54 With higher surface area and higher amount of additional amorphous carbon on the fibers' surface, this oxidative current increases. This observation suggests that the amorphous carbon obtained by the twin polymerization approach is more prone to oxidation. Typically, carbon corrosion in aqueous solution is detected at positive potentials higher than 1.2 V vs. SCE, which gives reason to assume that our amorphous carbon is less stable as compared to commercially routinely-applied carbon material.55 Based on the TGA-analysis, the carbon material also showed lower stability against combustion under air in comparison with the composite electrodes. Here the carbon material already starts to degrade at lower temperatures around 400 °C (see ESI, Fig. S3†). It should be determined, whether this unwanted parasitic carbon corrosion has an impact on the performance of the electrode after several cycles.
The reference material does not show any activity towards the negative redox couple (V(II)/V(III)). Neither an oxidation peak, nor a reduction peak could be observed for the pristine and the carbonized felt. However, the electrodes modified by means of twin polymerization show a good activity for the negative half-cell reaction. This can be seen from two very distinct oxidation- and reduction peaks. The composite, which was polymerized by thermal activation, demonstrates slightly lower maximum currents, while the sample achieved by acidic catalysis shows noticeably stronger evolution of hydrogen. This hydrogen evolution reaction (HER) is known as an undesirable parasitic side reaction,56 which may also be enhanced by an increased specific surface area (marked with an arrow).
Additional electrochemical impedance spectroscopy measurements were performed to study the charge transfer of the V(IV)/V(V) redox reaction. By fitting the EIS data, it was possible to determine the electrical charge transfer resistance and the double-layer capacitance. These are summarized in Table 3 and the corresponding Nyquist plots with utilized equivalent circuit model are shown in the ESI in Fig. S2.†
| RCT/Ω | CDL/mF | |
|---|---|---|
| Carbonized CF | 87.15 | 0.188 |
| Composite (MSA, 1h, E1) | 6.363 | 14.34 |
| Composite (T, 1h, E1) | 4.369 | 56.06 |
The results for these two parameters fit with our previous assumptions. The carbon coating leads to a larger double-layer capacitance of the electrode/electrolyte interface in agreement with the cyclic voltammetry curves. The charge transfer resistance decreases significantly, which correlates with an accelerated electron transfer. Additionally smaller charge transfer resistances are associated with better kinetics.
Electrochemical characterization: accelerated stress test
Twin polymerization is a suitable method to increase the electrochemical activity of the CFs. Nevertheless, it remains to be examined, whether the increase in electrochemical performance for the positive side is permanent, or if the corrosion of carbon will lead to significant degradation of the electrodes' performance. For this purpose, the samples were subjected to 150 cycles between 0.3 V and 1.2 V with a scan rate of 20 mV s−1, serving as an accelerated stress test (AST) treatment (see ESI, Fig. S4†).The CVs decisively change during the AST test. In case of the composite obtained by acidic catalysis only the maximum current decreases. Here, the peak separation remains unchanged, which means that the reversibility is still good. In contrast to that, the composite synthesized by thermally activated polymerization degrades much faster, which could be seen by a higher peak separation and subsequent DLC decrease after 150 cycles (blue, dashed line; marked by an arrow). In agreement with the recent literature a correlation between the corrosion of carbon and the decrease of the materials surface area could be drawn, which results in a less broadened DLC region.57
It is generally known, that the materials graphitization degree has a significant impact on the corrosion stability, which gives reason to think, that the porous carbon material on top of the fibers is less strongly graphitized.57 For further information about the amount of carbon dioxide produced due the electrochemical corrosion of carbon, differential electrochemical mass spectrometry (DEMS) measurements will be performed in the future. Furthermore, the graphitization degree can be investigated using Raman spectroscopy and the ID:IG ratio as an indication, although one has to consider that Raman spectroscopy is a bulk method, which may have a limited sensitivity to unravel the coating's properties.
It remained to be investigated, if the synthesis is reproducible and provides electrodes with similar activity. For that purpose the synthesis procedure was carried out a second time (see ESI, Fig. S5†). Both obtained material batches show the same activity, whereby the material obtained by the repeated synthesis exhibits slightly increased maximum currents. It is concluded that in this case apparently more porous material could be deposited on the fibers. A slightly increased oxidative current could be also observed, which is in line with the previous results. With increased amount of deposited material on the fibers, the oxidative current increases. This phenomenon will be examined in more detail in the future, to find a way to stabilize the amorphous coating against electrochemical corrosion at higher potentials.
Conclusions
Twin polymerization of 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] on PAN-based fiber substrates has been reported as a novel and promising synthetic route to obtain high-surface carbon–carbon composite electrodes for application in redox flow batteries. These highly porous electrode materials demonstrated excellent activities for the positive and, in particular, the negative redox reaction in all-vanadium redox flow batteries compared to the carbonized felts. It was found that doping the surface with heteroatoms is not necessary for increasing the electrochemical activity of the electrodes, although it is often described to the contrary in the literature.With the twin polymerization approach a porous composite electrode with a significantly increased surface area compared to the state-of-the-art electrodes was obtained, where the porosity of the material can be easily tailored depending on the used twin monomer and the chosen synthesis conditions, such as type of catalysis or maximum temperature. Moreover, a bimodal pore size distribution has been obtained and turned out beneficial for the performance of the electrode facilitating electrolyte transport and accessibility of the active surface. The electrochemical studies showed a significantly increased electrochemical activity and furthermore higher double-layer capacitance for the received material indicating a higher surface area in contact with the electrolyte. In future work, the synthesis will be scaled-up to investigate the composites as electrode materials in full cell battery cycling tests.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support by Freudenberg Performance Materials SE & Co. KG (Weinheim, Germany) providing pristine PAN-based felts is gratefully acknowledged. We would also like to acknowledge the assistance through elemental analyses of Core Facility BioSupraMol supported by DFG. Furthermore, the authors would like to thank Ingrid Otto (University of Bayreuth) for her support in recording the high resolution images, Benjamin Mielke and Benjamin Hentschel (TU Chemnitz) for excecuting nitrogen adsorption measurements, Benjamin Mielke (TU Chemnitz) for taking scanning electron microscopy measurements and Prof. Christoph Tegenkamp (TU Chemnitz) for access to the instrument. We also thank Sebastian Scholz (TU Chemnitz) for thermogravimetric measurements, Prof. Werner A. Goedel (TU Chemnitz) for access to the instrument and Ulrike Kunz (TU Darmstadt) for providing the TEM images.References
- B. Diouf and R. Pode, Renewable Energy, 2015, 76, 375–380 CrossRef.
- D. P. Opra, S. V. Gnedenkov and S. L. Sinebryukhov, J. Power Sources, 2019, 442, 227225 CrossRef CAS.
- A. Barré, B. Deguilhem, S. Grolleau, M. Gérard, F. Suard and D. Riu, J. Power Sources, 2013, 241, 680–689 CrossRef.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC.
- J. Libich, J. Máca, J. Vondrák, O. Čech and M. Sedlaříková, Journal of Energy Storage, 2018, 17, 224–227 CrossRef.
- X. Deng, B. Zhao, L. Zhu and Z. Shao, Carbon, 2015, 93, 48–58 CrossRef CAS.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 CrossRef CAS.
- C. Fabjan, J. Garche, B. Harrer, L. Jörissen, C. Kolbeck, F. Philippi, G. Tomazic and F. Wagner, Electrochim. Acta, 2001, 47, 825–831 CrossRef CAS.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 CrossRef CAS.
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776–9809 CrossRef CAS.
- M. Skyllas-Kazacos, J. Electrochem. Soc., 1986, 133, 1057 CrossRef CAS.
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef CAS.
- W. Xin and Y. Song, RSC Adv., 2015, 5, 83239–83285 RSC.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- J. Deng, M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- M. H. Chakrabarti, N. P. Brandon, S. A. Hajimolana, F. Tariq, V. Yufit, M. A. Hashim, M. A. Hussain, C. T. J. Low and P. V. Aravind, J. Power Sources, 2014, 253, 150–166 CrossRef CAS.
- Y. Tao, Z.-Y. Sui and B.-H. Han, J. Mater. Chem. A, 2020, 8, 6125–6143 RSC.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- M. F. Hassan, M. A. Sabri, H. Fazal, A. Hafeez, N. Shezad and M. Hussain, J. Anal. Appl. Pyrolysis, 2020, 145, 104715 CrossRef CAS.
- S. Chand, J. Mater. Sci., 2000, 35, 1303–1313 CrossRef CAS.
- Y. Xia, Z. Yang and Y. Zhu, J. Mater. Chem. A, 2013, 1, 9365–9381 RSC.
- N. Saito, K. Aoki, Y. Usui, M. Shimizu, K. Hara, N. Narita, N. Ogihara, K. Nakamura, N. Ishigaki, H. Kato, H. Haniu, S. Taruta, Y. Ahm Kim and M. Endo, Chem. Soc. Rev., 2011, 40, 3824–3834 RSC.
- S. K. Nataraj, K. S. Yang and T. M. Aminabhavi, Prog. Polym. Sci., 2012, 37, 487–513 CrossRef CAS.
- J. González-García, P. Bonete, E. Expósito, V. Montiel, A. Aldaz and R. Torregrosa-Maciá, J. Mater. Chem., 1999, 9, 419–426 RSC.
- O. Nibel, S. M. Taylor, A. Pătru, E. Fabbri, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, A1608–A1615 CrossRef CAS.
- D. Dixon, D. J. Babu, J. Langner, M. Bruns, L. Pfaffmann, A. Bhaskar, J. J. Schneider, F. Scheiba and H. Ehrenberg, J. Power Sources, 2016, 332, 240–248 CrossRef CAS.
- C. Flox, M. Skoumal, J. Rubio-Garcia, T. Andreu and J. R. Morante, Appl. Energy, 2013, 109, 344–351 CrossRef CAS.
- D. Cheng, M. Tian, B. Wang, J. Zhang, J. Chen, X. Feng, Z. He, L. Dai and L. Wang, J. Colloid Interface Sci., 2020, 572, 216–226 CrossRef CAS.
- R. Schweiss, C. Meiser and F. W. T. Goh, ChemElectroChem, 2017, 4, 1969–1974 CrossRef CAS.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111–2116 CrossRef CAS.
- L. Kaßner, A. Knoblauch, A. Seifert, R.-E. Grützner, G. Cox, A. Lange, S. Csihony, F. Simon, S. Anders, L. Kroll, M. Rahaman, D. Zahn, L. Mertens, M. Weber, M. Mehring and S. Spange, Macromol. Chem. Phys., 2016, 217, 2462–2472 CrossRef.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015, 36, 1623–1639 CrossRef CAS.
- S. Spange, P. Kempe, A. Seifert, A. A. Auer, P. Ecorchard, H. Lang, M. Falke, M. Hietschold, A. Pohlers, W. Hoyer, G. Cox, E. Kockrick and S. Kaskel, Angew. Chem., Int. Ed., 2009, 48, 8254–8258 CrossRef CAS.
- S. Spange, M. Mehring, A. Auer, M. Birkner, G. Bistoni, T. Ebert, B. Fiedler, J. Friedrich, M. Göring, S. Höhne, L. Kaßner, P. Kitschke, H. Lang, K. Nagel, J. Prehl, P. Kitschke, F. Roschke, K. Schreiter, A. Seifert and D. Uhlig, Twin Polymerization, De Gruyter, 2018 Search PubMed.
- P. Kempe, T. Löschner, A. A. Auer, A. Seifert, G. Cox and S. Spange, Chem.–Eur. J., 2014, 20, 8040–8053 CrossRef CAS.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef CAS.
- C. Huster, K. Nagel, S. Spange and J. Prehl, Chem. Phys. Lett., 2018, 713, 145–148 CrossRef CAS.
- P. Kitschke, A. A. Auer, T. Löschner, A. Seifert, S. Spange, T. Rüffer, H. Lang and M. Mehring, ChemPlusChem, 2014, 79, 1009–1023 CrossRef CAS.
- F. Böttger-Hiller, P. Kempe, G. Cox, A. Panchenko, N. Janssen, A. Petzold, T. Thurn-Albrecht, L. Borchardt, M. Rose, S. Kaskel, C. Georgi, H. Lang and S. Spange, Angew. Chem., Int. Ed., 2013, 52, 6088–6091 CrossRef.
- T. Ebert, G. Cox, E. Sheremet, O. Gordan, D. R. T. Zahn, F. Simon and S. Spange, Chem. Commun., 2012, 48, 9867–9869 RSC.
- T. Windberg, T. Ebert, D. Uhlig, S. Schulze and S. Spange, Microporous Mesoporous Mater., 2017, 246, 62–71 CrossRef CAS.
- S. Choudhury, T. Ebert, T. Windberg, A. Seifert, M. Göbel, F. Simon, P. Formanek, M. Stamm, S. Spange and L. Ionov, Part. Part. Syst. Charact., 2018, 35, 1800364 CrossRef.
- M. Schnucklake, L. Eifert, J. Schneider, R. Zeis and C. Roth, Beilstein J. Nanotechnol., 2019, 10, 1131–1139 CrossRef CAS.
- A. Fetyan, I. Derr, M. K. Kayarkatte, J. Langner, D. Bernsmeier, R. Kraehnert and C. Roth, ChemElectroChem, 2015, 2, 2055–2060 CrossRef CAS.
- T. X. Huong Le, M. Bechelany and M. Cretin, Carbon, 2017, 122, 564–591 CrossRef.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- M. Schnucklake, S. Kuecken, A. Fetyan, J. Schmidt, A. Thomas and C. Roth, J. Mater. Chem. A, 2017, 5, 25193–25199 RSC.
- L. Eifert, Z. Jusys, R. Banerjee, R. J. Behm and R. Zeis, ACS Appl. Energy Mater., 2018, 1, 6714–6718 CrossRef CAS.
- L. Eifert, Z. Jusys, R. J. Behm and R. Zeis, Carbon, 2020, 158, 580–587 CrossRef CAS.
- J.-G. Oh, W. H. Lee and H. Kim, Int. J. Hydrogen Energy, 2012, 37, 2455–2461 CrossRef CAS.
- A. Fetyan, G. A. El-Nagar, I. Lauermann, M. Schnucklake, J. Schneider and C. Roth, J. Energy Chem., 2018, 32, 57–62 CrossRef.
- J. Melke, R. Schuster, S. Möbus, T. Jurzinsky, P. Elsässer, A. Heilemann and A. Fischer, Carbon, 2019, 146, 44–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07741k |
| This journal is © The Royal Society of Chemistry 2020 |