
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic fermentation of β-peptide macrocycles by thiadiazole-forming ring-closing reactions†
Jonathan G.
Hubert
a,
Iain A.
Stepek
a,
Hidetoshi
Noda
 b and
Jeffrey W.
Bode
*a
b and
Jeffrey W.
Bode
*a
aLaboratorium für Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zürich, Zürich, Switzerland 8093. E-mail: bode@org.chem.ethz.ch; Web: http://www.bode.ethz.ch
bInstitute of Microbial Chemistry (Bikaken), 3-14-23 Kamiosaki, Shinagawa-ku, Tokyo 141-0021, Japan
First published on 8th January 2018
Abstract
Macrocyclic β-peptides were efficiently prepared using a thiadiazole-forming cyclization reaction between an α-ketoacid and a thiohydrazide. The linear β-peptide precursors were assembled from isoxazolidine monomers by α-ketoacid-hydroxylamine (KAHA) ligations with a bifunctional initiator – a process we have termed ‘synthetic fermentation’ due to the analogy of producing natural product-like molecules from simpler building blocks. The linear synthetic fermentation products underwent Boc-deprotection/thiadiazole-forming macrocyclization under aqueous, acidic conditions to provide the cyclic products in a one-pot process. This reaction sequence proceeds from easily accessed initiator and monomer building blocks without the need for additional catalysts or reagents, enabling facile production of macrocyclic β-peptides, a relatively underexplored structural class.
Introduction
Macrocyclic compounds are an increasingly valuable platform for biologically active molecules owing to their unique structural properties. Macrocycles have emerged as ideal candidates for the modulation of protein–protein interactions (PPIs), an important area of modern drug discovery.1–3 In comparison to their linear counterparts, macrocycles frequently exhibit higher affinity and selectivity for a given target, and often display enhanced in vivo stability.4–8These desirable traits have led to the emergence of innovative methodologies for the generation of libraries of macrocyclic peptides.9–11 Contemporary methods include mRNA/ribosome/phage display technology,12–15 selection-based techniques,16,17 DNA-encoded/programming chemistry18 and diversity-oriented synthesis.19,20 The generation of diverse arrays of macrocyclic compounds using multicomponent reactions, often in a combinatorial setting, has also gained significant attention.21–29
While amide-forming cyclizations remain the most common approach to peptide macrocycles, considerable attention has been paid to alternative ring-closing strategies. Leading methods include ring-closing metathesis,30–33 alkyne–azide cycloaddition,34–38 cross-coupling reactions39–44 and Ugi-type reactions.45–47 Yudin has recently reported an oxadiazole-forming cyclization, leading to peptides with superior physicochemical properties.26 These are all promising technologies, but require construction of the linear peptides one at a time or cyclization of partially protected precursors.
Our group recently introduced “synthetic fermentation” – the production of natural product-like molecules by controlled oligomerization under aqueous conditions without additional reagents, protecting groups, or additives.48 The resulting mixtures can be used in biological assays without isolation, and the mixtures are easily reproduced and optimized. In our first implementation, linear, bioactive β-peptides were produced by combining small building blocks under aqueous conditions (Fig. 1). This report employed three types of building blocks: α-ketoacid initiators (I), elongation monomers (M) and terminators (T) and was applied to the discovery of an HCV protease inhibitor. The further optimization of these compounds; however, is limited by the poor properties of small linear peptides as drugs.
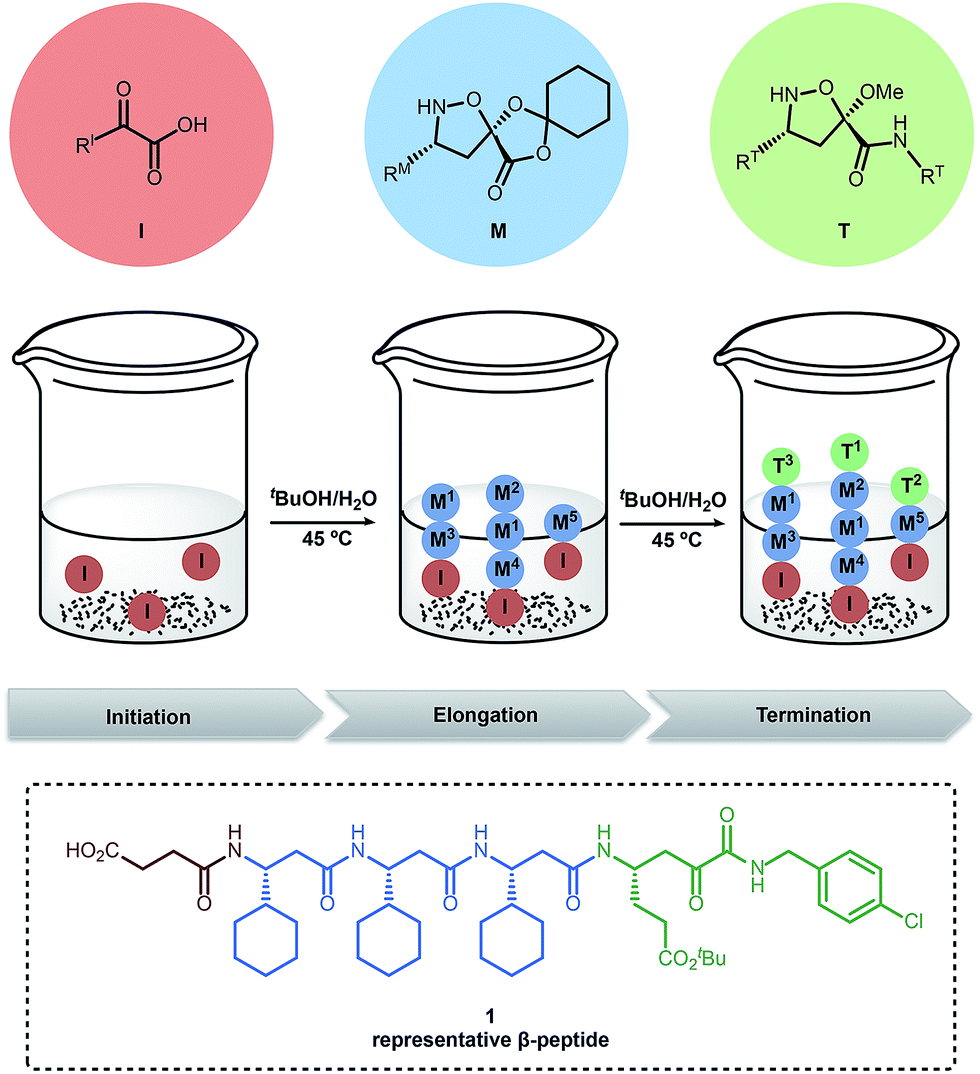 | ||
| Fig. 1 Preparation of β-peptide mixtures using synthetic fermentation.48 | ||
Motivated by both the increasing interest in macrocyclic compounds for biological screening and the limitations of linear β-peptides, we aimed to identify approaches to form macrocyclic peptides under the conditions of synthetic fermentation. In this report, we document a first-generation approach to the synthetic fermentation of β-peptide macrocycles using a unique thiadiazole-forming ring-closing reaction, including optimization of this novel transformation and its application to the one-pot preparation of β-peptide macrocycles from simple building blocks (Scheme 1).
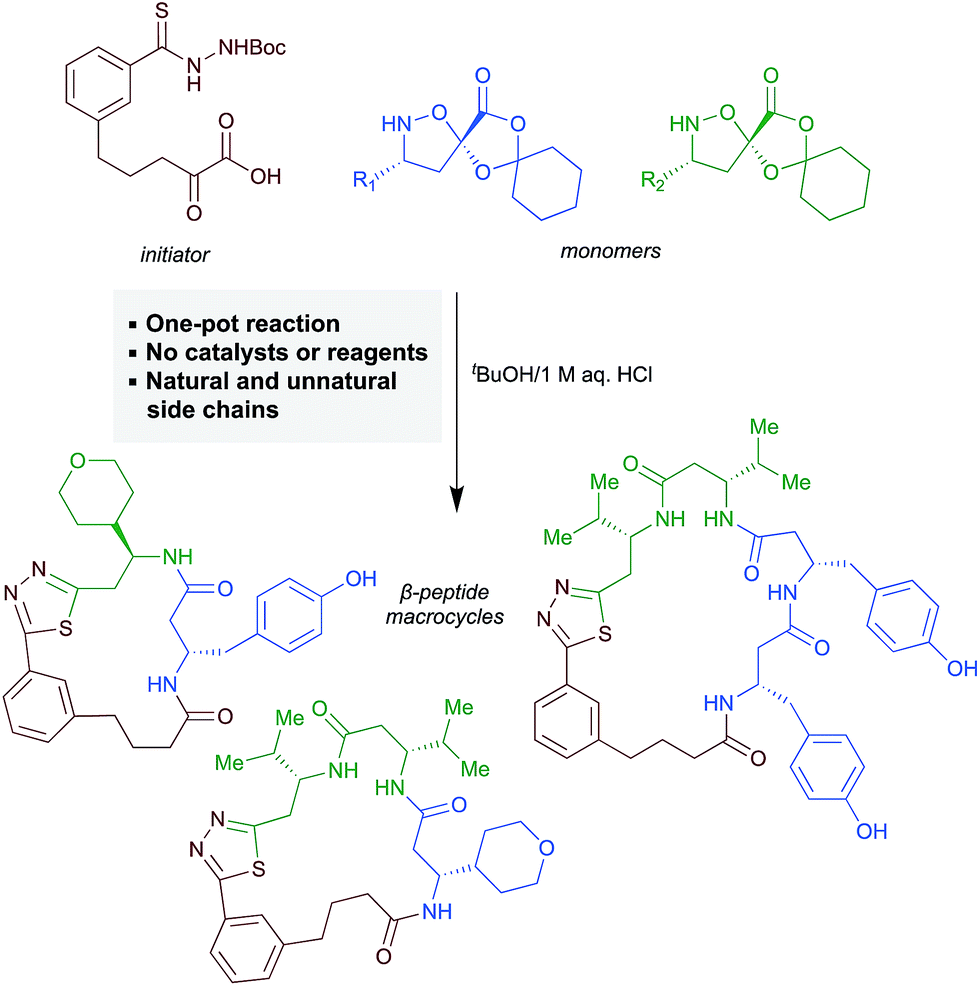 | ||
| Scheme 1 Synthesis of β-peptide macrocycles by synthetic fermentation/thiadiazole-forming macrocyclization. | ||
Results and discussion
Cyclization strategy
Two general strategies for macrocyclization were initially considered (Scheme 2). In the first, cyclization would involve a “lynchpin” terminator that would react with both the C-terminal α-ketoacid and an orthogonal functional group on the initiator. Although such approaches are possible, we quickly learned that this process leads to mixtures of cyclic products and various linear precursors. In the second approach, we sought to directly engage the α-ketoacid in a macrocyclization reaction, as this would simplify the overall process by obviating the need for specially functionalized terminators; only an initiator and elongation monomers would be required.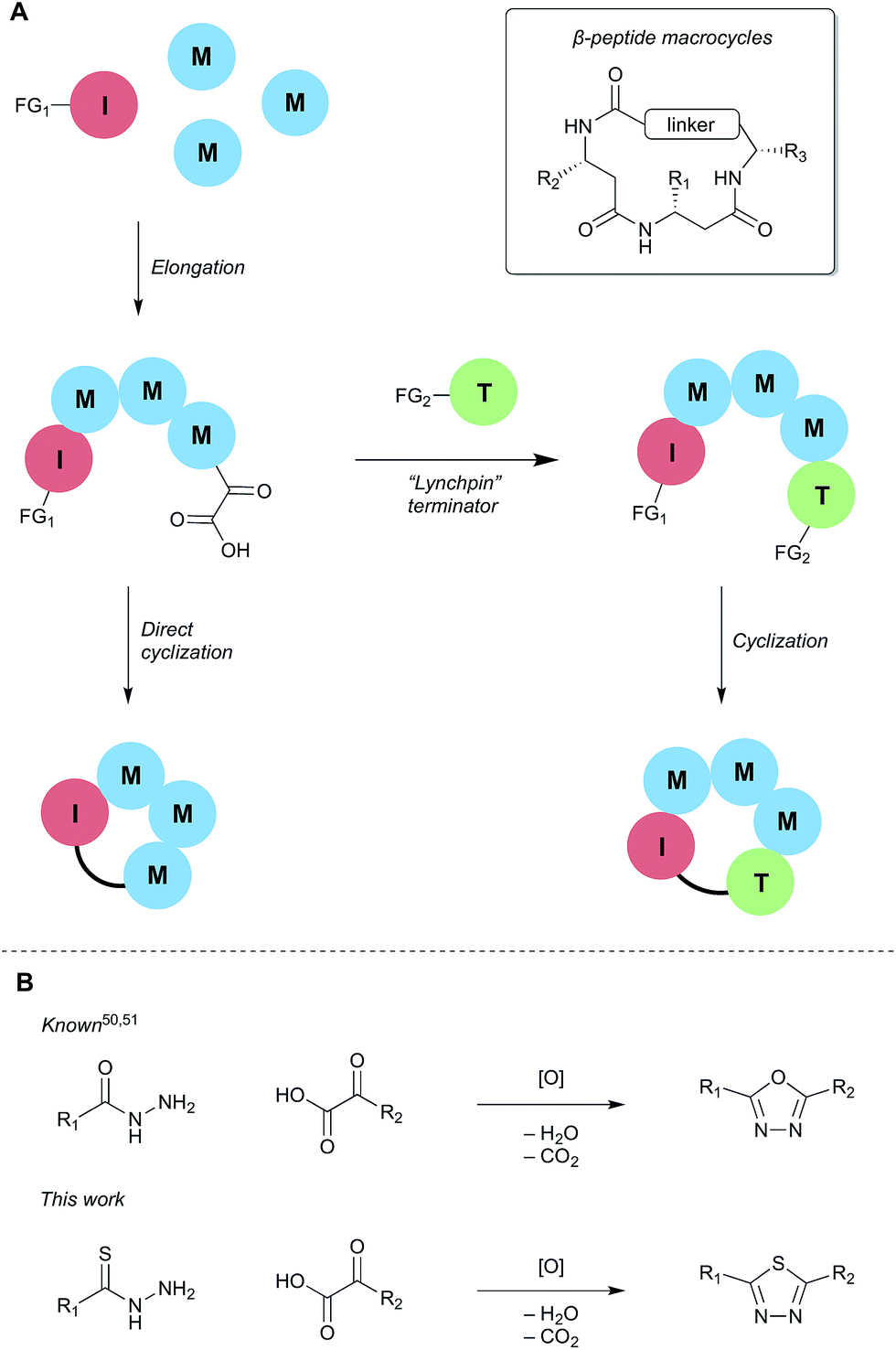 | ||
| Scheme 2 Strategies to adapt synthetic fermentation methodology for the preparation of β-peptide macrocycles. (A) General cyclization strategies; (B) proposed 1,3,4-thiadiazole-forming reaction for macrocyclization. | ||
Although ring closure using the KAHA (α-ketoacid-hydroxylamine) ligation – an outstanding reaction for peptide macrocycle formation49 – is possible, we wished to identify a process that would form something other than an amide bond. We were attracted by recent reports of oxidative coupling of α-ketoacids and hydrazides to form 1,3,4-oxadiazoles,50,51 the same moiety used in Yudin's exciting work.26 We envisaged that using a thiohydrazide in place of the hydrazide would allow for milder oxidation conditions and provide the equally attractive 1,3,4-thiadiazoles.
Intermolecular thiadiazole formation
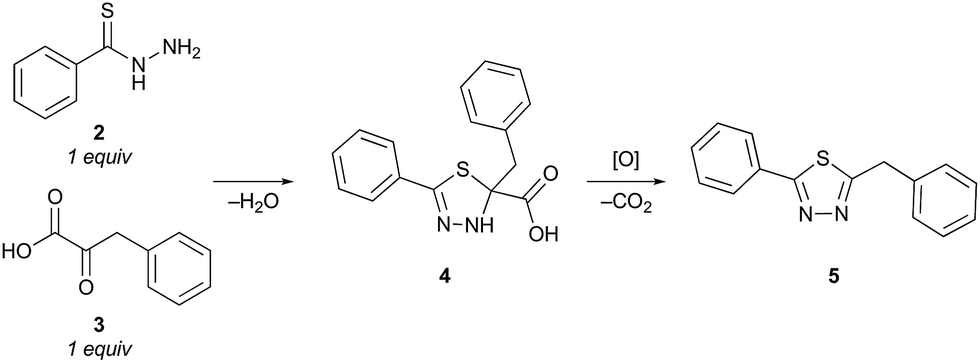
To evaluate the proposed thiadiazole-forming reaction, we investigated the intermolecular reaction of benzothiohydrazide (2) and phenylpyruvic acid (3) (Table 1). Under standard synthetic fermentation conditions, condensation of thiohydrazide 2 with 3 and subsequent cyclization provided adduct 4, while the formation of thiadiazole 5 was not observed (entry 1). Adduct 4 was also the major product formed under basic reaction conditions (entry 2). However, the addition of aqueous HCl to the reaction media resulted in considerable thiadiazole formation and full conversion to 5 was achieved when the reaction was performed at 70 °C (entries 3–4). The observation that the decarboxylation/oxidation process required for the conversion of 4 to 5 could proceed without adding an additional oxidant was particularly pleasing, as it would allow the planned macrocyclization reaction to be carried out under simpler conditions than initially anticipated. In the absence of oxygen (degassed solvent under an inert atmosphere) conversion of adduct 4 to thiadiazole 5 was suppressed (entry 5). When oxygen was reintroduced to the previously degassed solvent, full conversion to thiadiazole 5 was observed (entry 6). Combined, these observations suggest that oxygen is acting as the oxidant in this transformation.|
|
|||
|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Thiadiazole 5: intermediate 4b |
a Reactions performed by combining 0.1 M stock solutions of 2 and 3 (15 μL each) and heating at the given temperature for 16 h.
b Ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 determined by LC-MS.
c Solvent degassed and reaction performed under nitrogen atmosphere.
d Degassed solvent exposed to air prior to reaction set up.
e Precipitation observed during the reaction. :4 determined by LC-MS.
c Solvent degassed and reaction performed under nitrogen atmosphere.
d Degassed solvent exposed to air prior to reaction set up.
e Precipitation observed during the reaction.
|
|||
| 1 |
t
BuOH/H2O (5:1) |
45 | <1:20 |
| 2 |
t
BuOH/1 M NaOH (5:1) |
45 | <1:20 |
| 3 |
t
BuOH/1 M HCl (5:1) |
45 | 1:1 |
| 4 |
t
BuOH/1 M HCl (5:1) |
70 | >20:1 |
| 5 |
t
BuOH/1 M HCl (5:1)c |
70 | 3:2 |
| 6 |
t
BuOH/1 M HCl (5:1)d |
70 | >20:1 |
| 7 |
t
BuOH/1 M HCl (1:1) |
70 | >20:1e |
| 8 | MeOH/1 M HCl (5:1) |
70 | >20:1 |
| 9 | DMSO/1 M HCl (5:1) |
70 | Complex mixture |
| 10 | Dioxane/1 M HCl (5:1) |
70 | 1:1 |
| 11 | DMF/1 M HCl (5:1) |
70 | 3:2 |
| 12 |
t
BuOH/1 M AcOH (5:1) |
70 | 1:2 |
| 13 |
t
BuOH/1 M citric acid (5:1) |
70 | 1:3 |
| 14 |
t
BuOH/1 M TFA (5:1) |
70 | >20:1 |
The effect of varying the reaction media was investigated. While thiadiazole formation proceeded cleanly when a greater proportion of water was used, precipitation of the product was observed under these conditions (entry 7). The reaction was equally effective when methanol was used instead of t-butanol, but the use of other water-miscible solvents either slowed conversion or promoted the formation of side products (entries 8–11). A brief screen of alternative acid sources revealed that, while any acid is sufficient to promote formation of thiadiazole 5, full conversion is only observed when relatively strong acids (i.e. HCl or TFA) are used (entries 12–14).
Although not the main focus of this work, we briefly investigated the scope of the oxygen-promoted thiadiazole formation (Table 2).
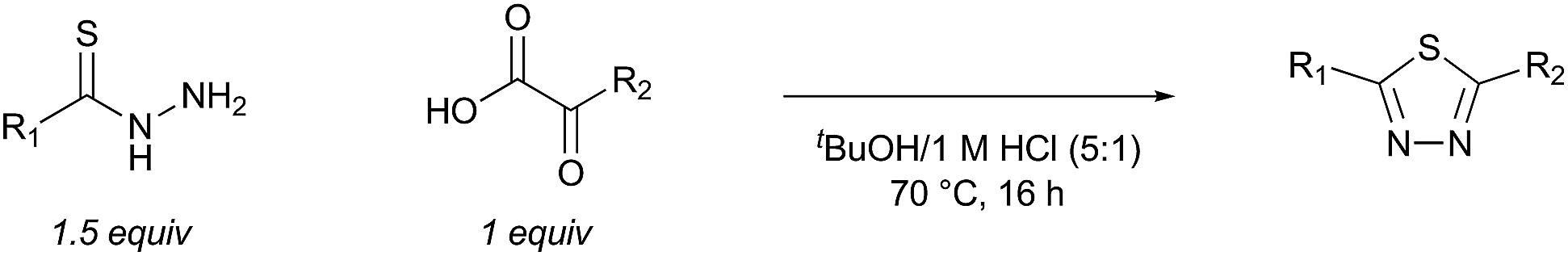
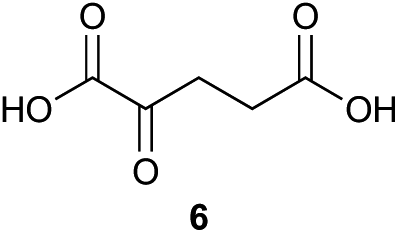
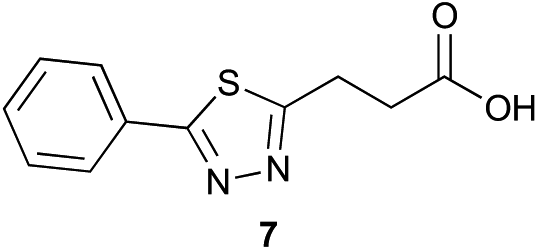
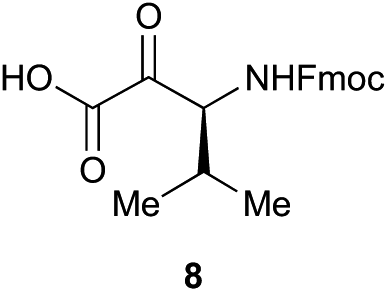
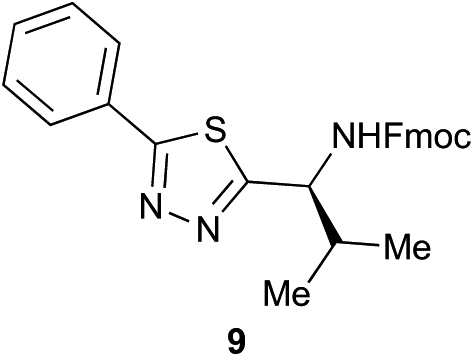
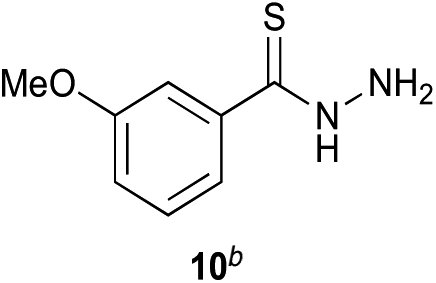
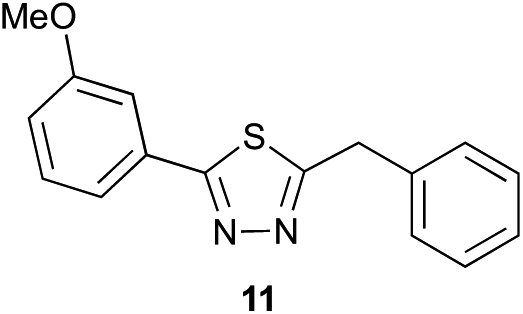
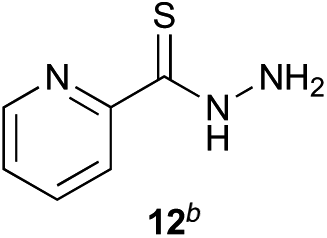
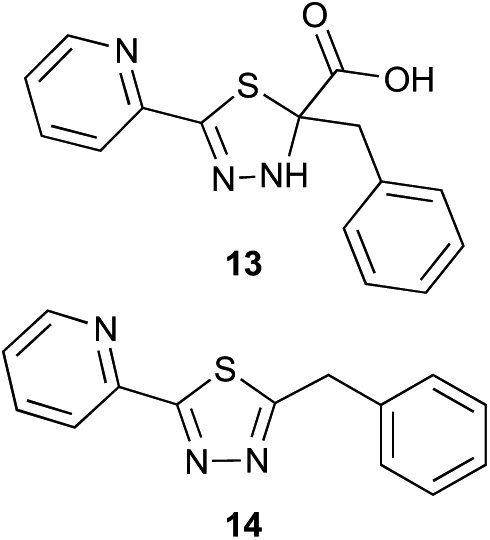
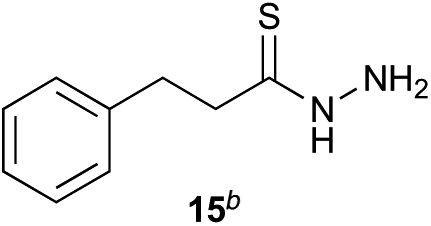
Preliminary studies revealed that competing dimerization of the thiohydrazide led to the formation of a symmetric thiadiazole side product. Accordingly, a slight excess of the thiohydrazide was used to ensure full conversion of the α-ketoacid. Under optimized conditions the model reaction between 2 and 3 afforded thiadiazole 5 in good isolated yield (entry 1). α-Ketoacids bearing carboxylic acids or protected amines were also tolerated (entries 2–3). The reaction of methoxyphenyl thiohydrazide 10 and α-ketoacid 3 also proceeded cleanly to give thiadiazole 11 in good yield (entry 4). However, pyridyl and alkyl thiohydrazides failed to provide the corresponding thiadiazoles in desirable quantities, due to either slow conversion or the formation of unidentified side products (entries 5–6). The relative success of thiohydrazides 2 and 10 as coupling partners, when compared with 12 and 15, suggests that a reasonably electron-rich, aromatic thiohydrazide is optimal for this reaction, presumably facilitating the decarboxylation/oxidation process required for thiadiazole formation.
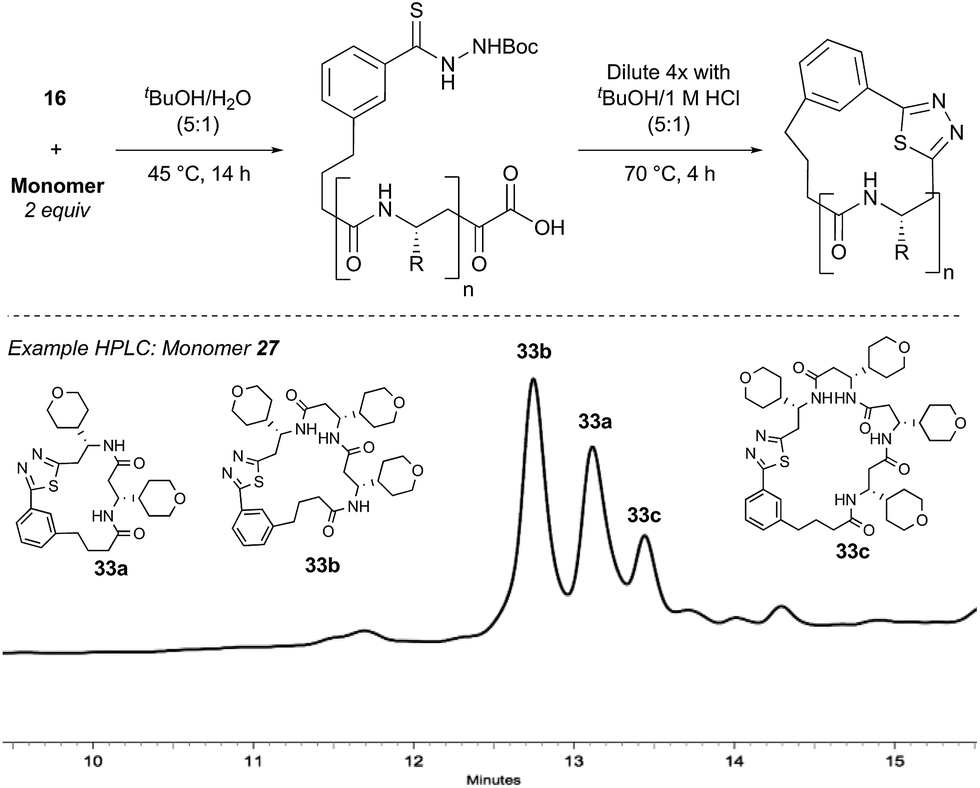
Synthesis of bifunctional initiator 16
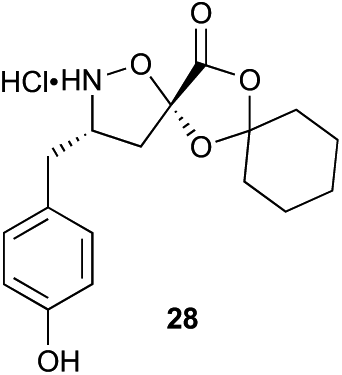
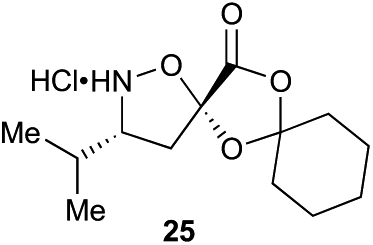
The proposed synthetic fermentation/macrocyclization process required a bifunctional initiator, bearing both an α-ketoacid moiety to elicit elongation and a thiohydrazide for subsequent cyclization (Scheme 3). To prevent interference in the elongation process, we elected to protect the thiohydrazide moiety with a Boc group. We postulated that a Boc-thiohydrazide would be stable under the neutral elongation conditions and subsequently cleaved when the reaction mixture was acidified to promote the thiadiazole-forming macrocyclization. As such, α-ketoacid 16 was proposed as a suitable initiator to investigate the elongation/macrocyclization process.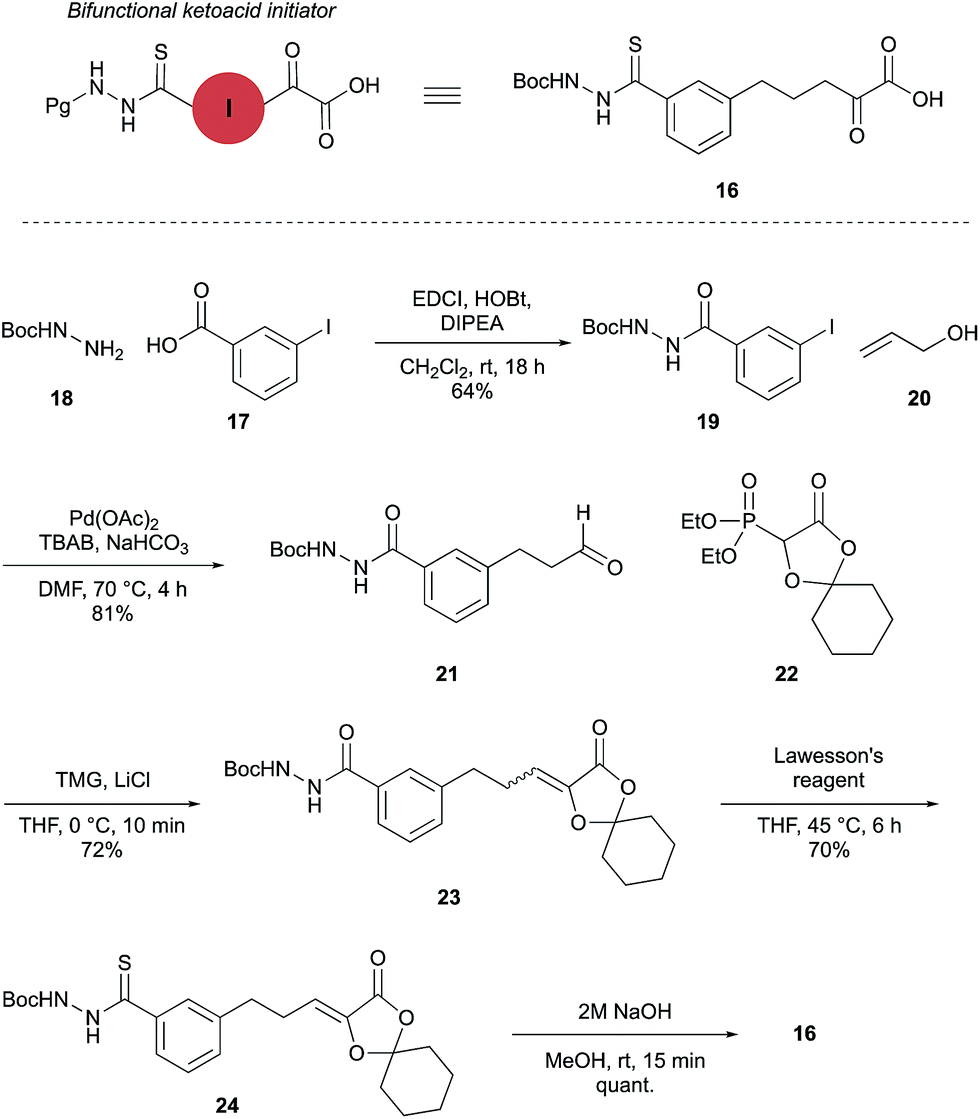 | ||
| Scheme 3 Synthesis of thiohydrazide-functionalized α-ketoacid initiator 16. | ||
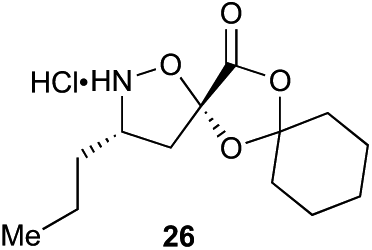
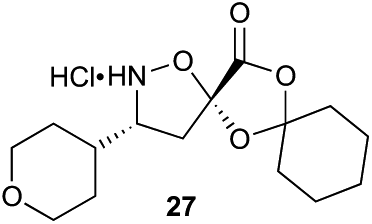
α-Ketoacid initiator 16 was readily synthesized in five steps from commercially available 3-iodobenzoic acid (17). Following EDCI/HOBt-mediated coupling with Boc-hydrazine 18, a Heck reaction between iodide 19 and allyl alcohol (20) provided aldehyde 21.52 An HWE reaction with phosphonate 22 allowed installation of the acetal-protected α-ketoacid framework.53,54 Subsequent conversion to thiohydrazide 24 using Lawesson's reagent, followed by unveiling of the free α-ketoacid via ester hydrolysis afforded initiator 16.
Synthesis of β-peptide macrocycles
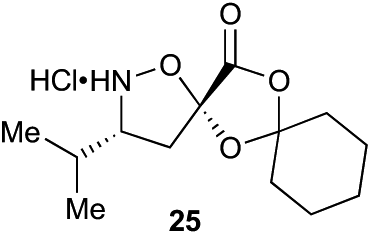
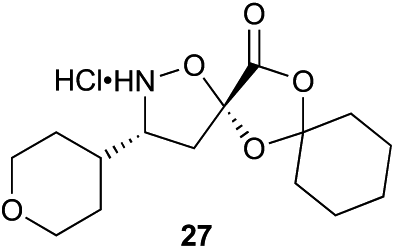
With bifunctional initiator 16 in hand, we investigated the β-peptide elongation/macrocyclization reaction (Table 3). The elongation monomers used in this process were synthesized according to previous reports.48,54 Initially, oligomerization using only one elongation monomer was performed, reducing the complexity of the resulting peptide mixtures and simplifying initial analysis. Elongation was carried out as previously described: two equivalents of the monomer were employed to favor formation of peptides containing 2–3 monomer units.48 To promote Boc-deprotection and thiadiazole-forming cyclization, the mixtures of peptides were diluted with tBuOH/1 M HCl (5:1) and heated to 70 °C.55 The reactions were monitored by LC-MS and HPLC and the major components of the reaction mixtures are shown in Table 3 (see ESI† for HPLC/MS traces).
|
|
||
|---|---|---|
| Entry | Monomer | Major products observed |
|
a Reactions performed by combining 0.1 M stock solutions of 16 (10 μL) and monomer (20 μL), heating at 45 °C for 14 h, diluting with tBuOH/1 M HCl (5:1, 90 μL) and heating at 70 °C for 4 h. The resulting mixtures were analyzed by HPLC and LC-MS with the major products shown in the table above.
|
||
| 1 |
|

|
| 2 |
|
|
| 3 |
|
|
| 4 |
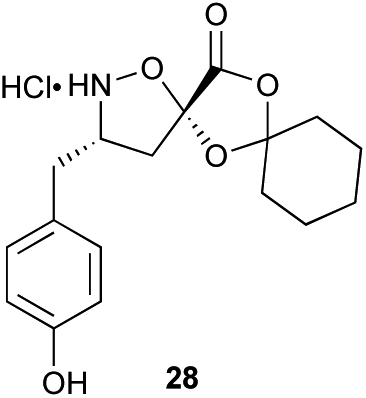
|
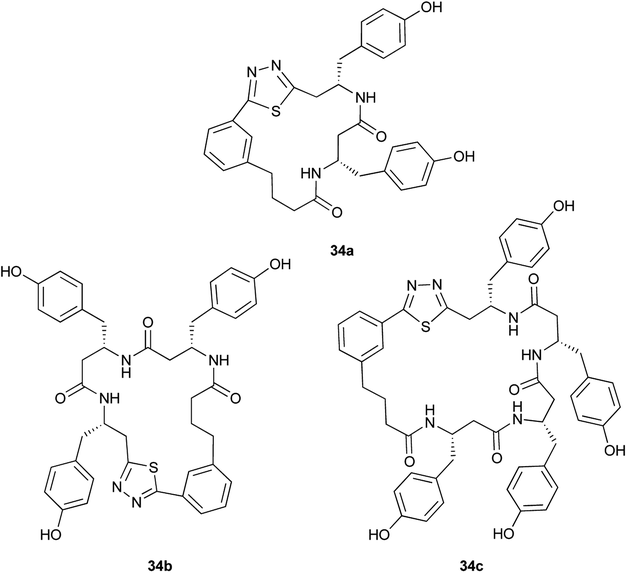
|
| 5 |
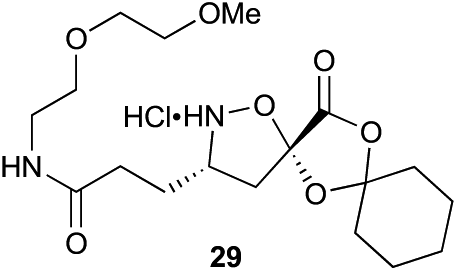
|
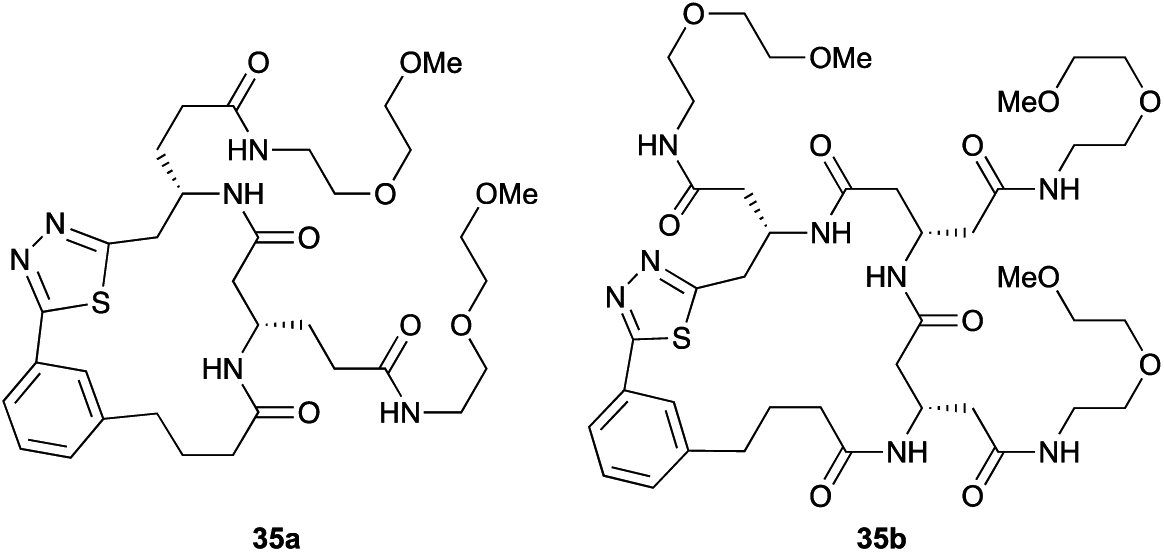
|
| 6 |

|
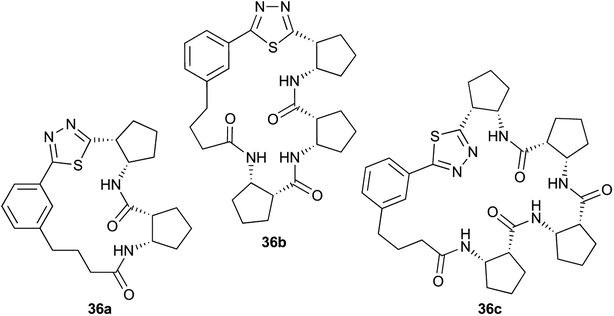
|
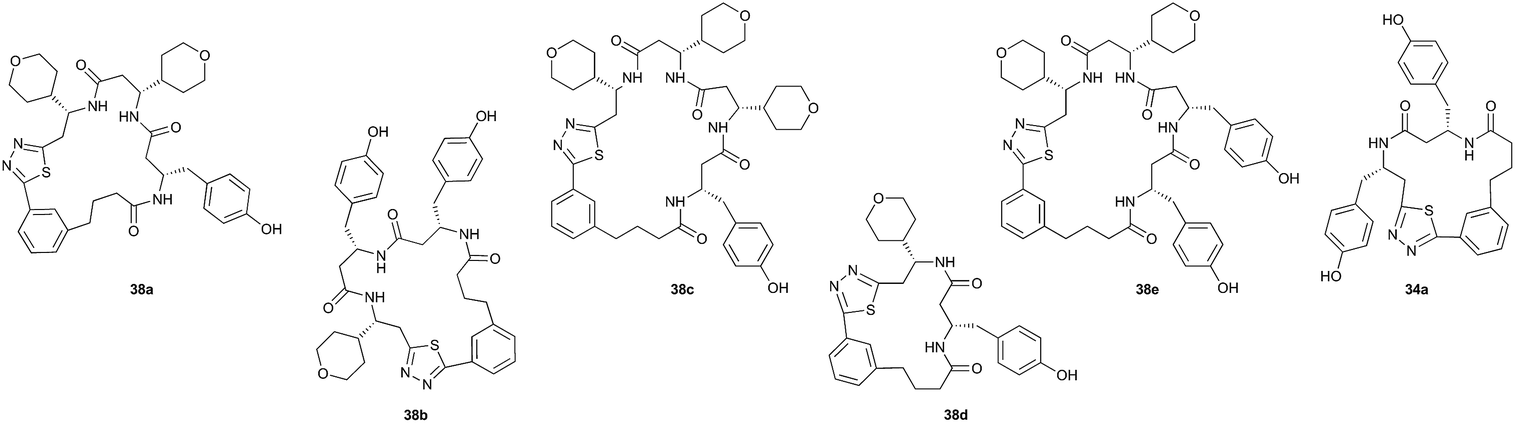
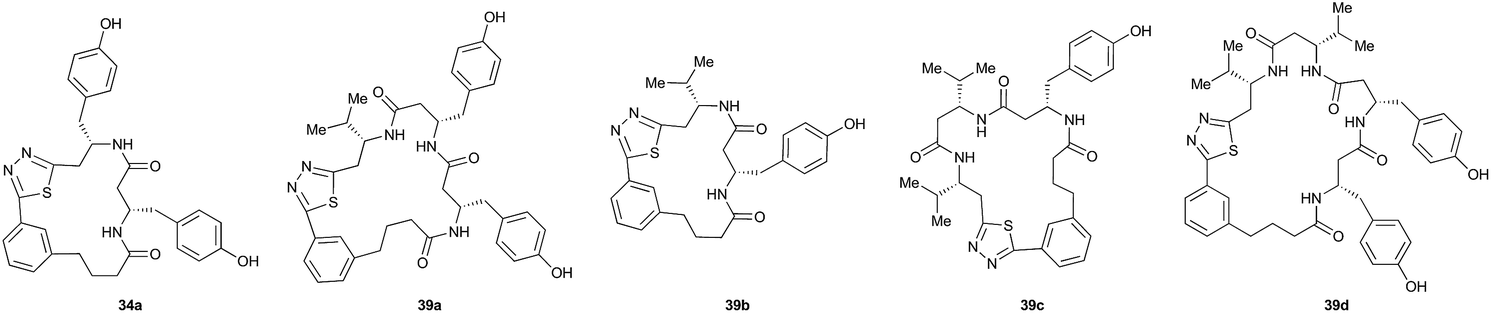
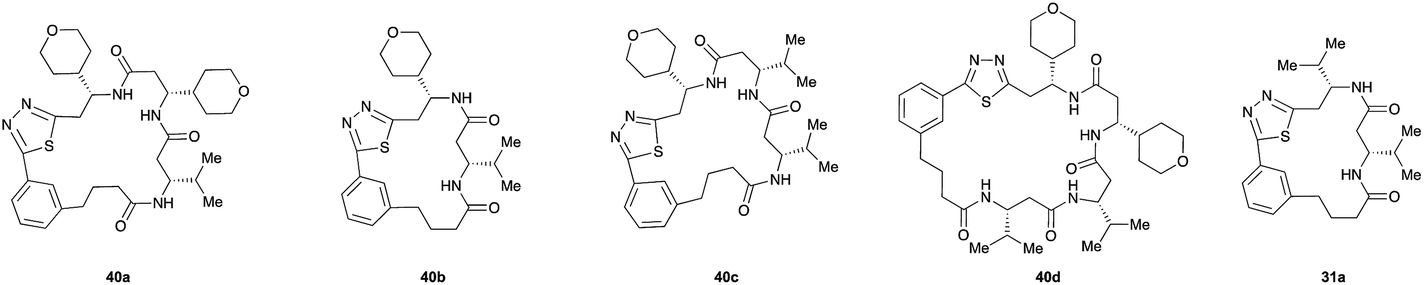
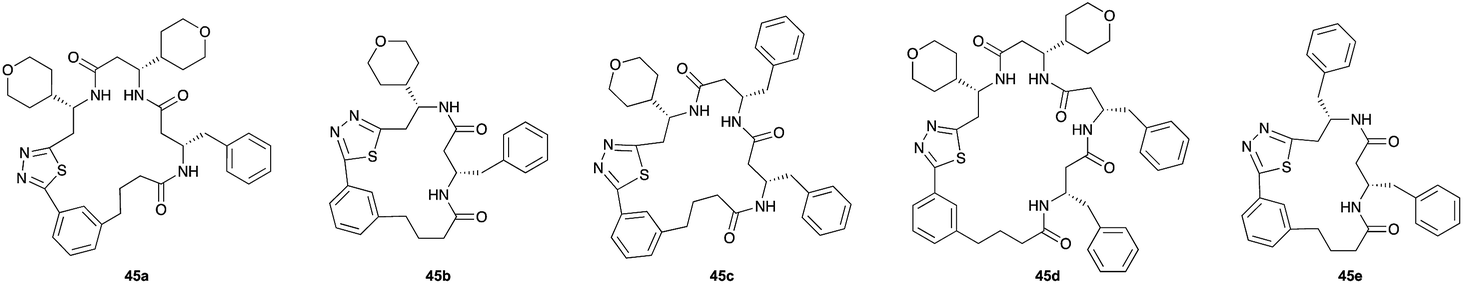
Pleasingly, the desired macrocyclic β-peptides bearing a 1,3,4-thiadiazole were the major products observed in the reaction mixtures. The predominant products were 17- and 21-membered macrocycles – containing 2 or 3 monomer units respectively – however, larger macrocycles were occasionally seen as minor components of the mixtures.
Interestingly, despite the relative complexity of the overall process (multiple KAHA ligations, Boc-deprotection, intramolecular condensation and subsequent oxidation/decarboxylation) the formation of the thiadiazole-linked cyclic products is strongly favored. The reactions were carried out with a linear β-peptide concentration of approximately 8 mM and higher molecular weight compounds resulting from oligomerization of these linear precursors were generally not observed.
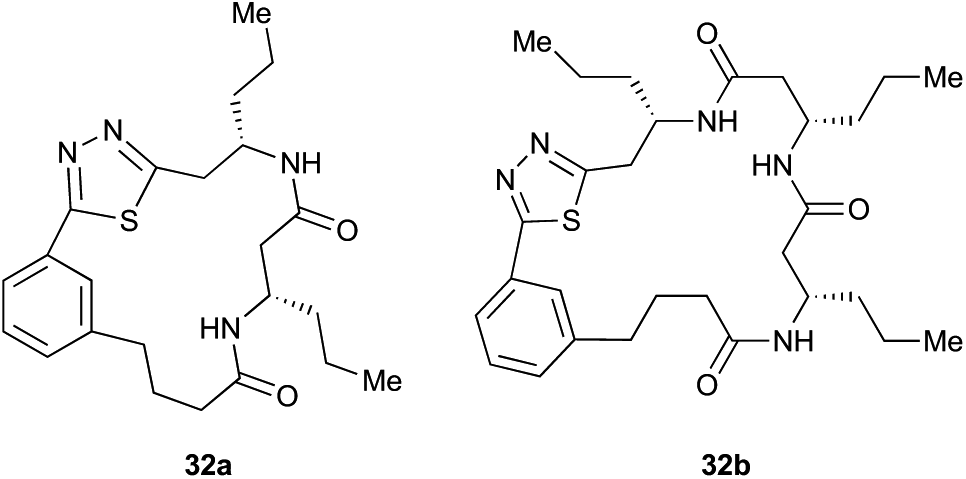
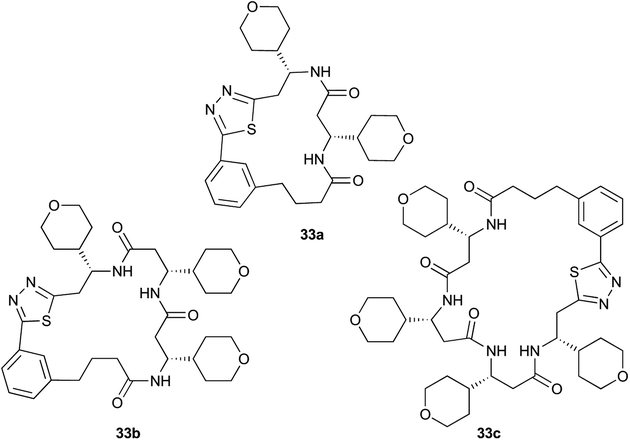
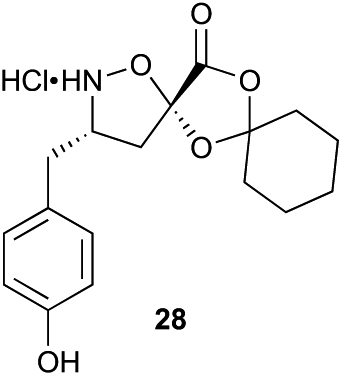
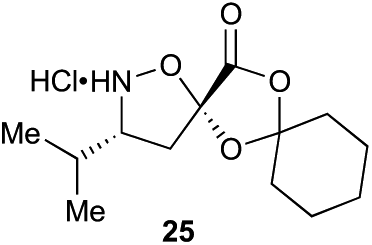
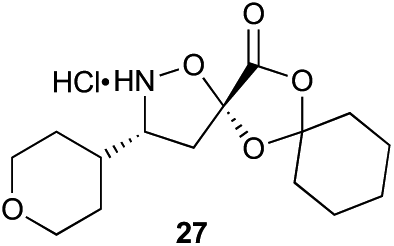
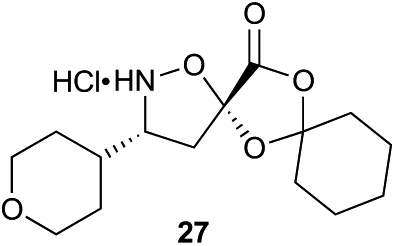
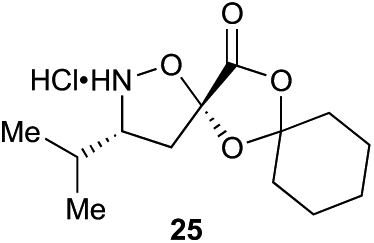
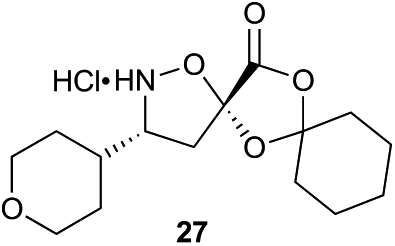
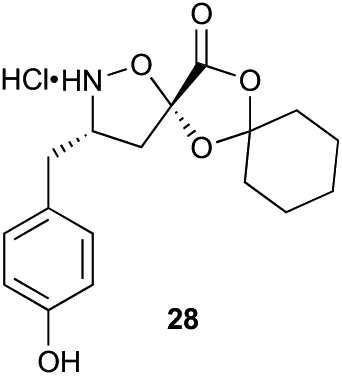
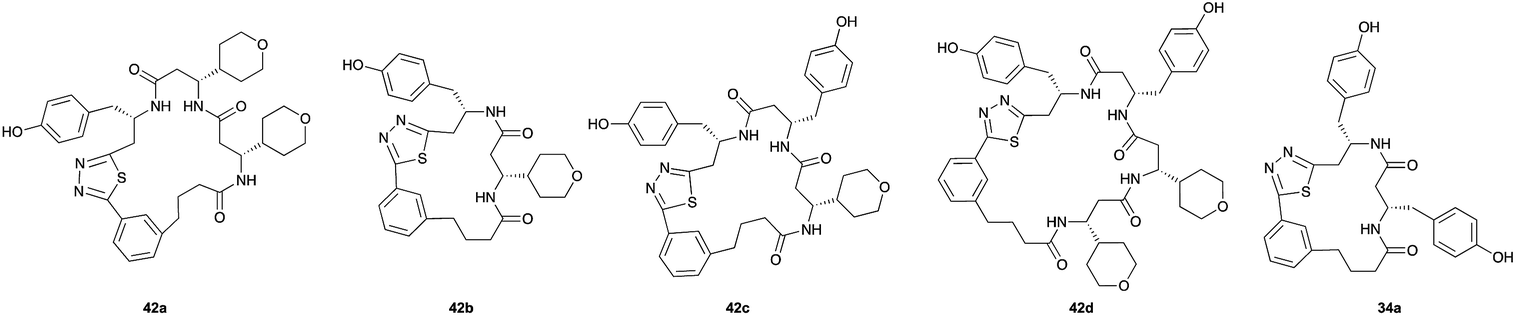
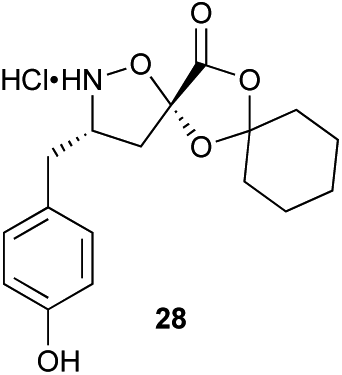
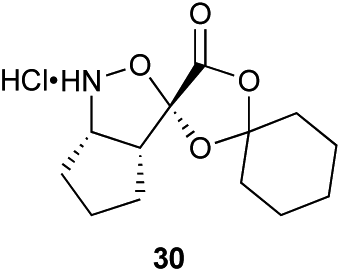
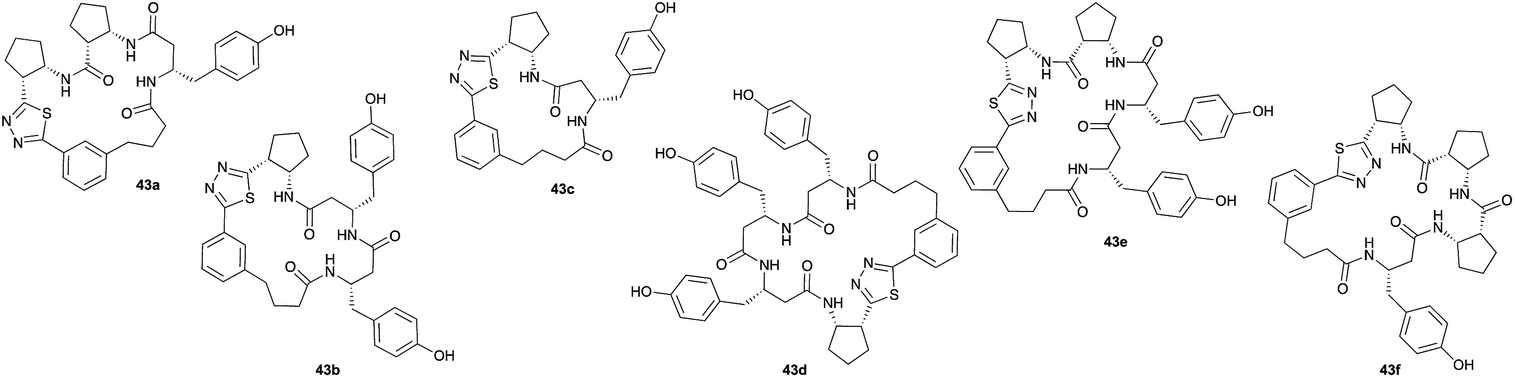
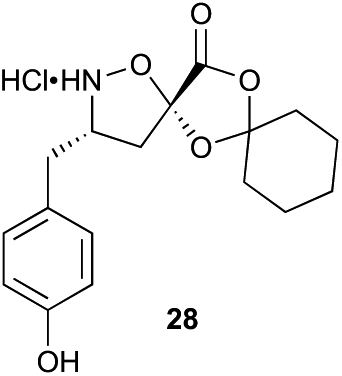
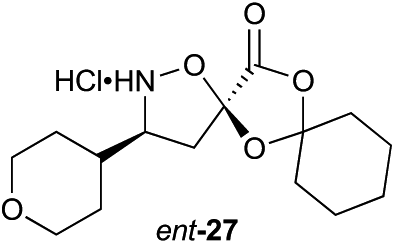
Aliphatic monomers 25 and 26 were employed without incident, as was pyran monomer 27 (entry 1–3). The reaction was tolerant to phenol groups, with macrocycles 34a–c observed as the major products when monomer 28 was used (entry 4). Amide-bearing monomer 29 was also compatible (entry 5). Finally, monomer 30 could also be employed to generate a range of unique peptide macrocycles containing cyclic β-amino acids (entry 6).
The reactions were generally run on small scale (1.0 μmol) to mirror conditions commonly employed for the preparation of synthetic fermentation libraries. However, the reaction scale could be increased (100 μmol) without significantly altering product distribution. The major products were isolated by preparative HPLC and fully characterized, confirming successful macrocyclic β-peptide formation (see ESI† for details).
During our investigations some limitations of the types of monomers that are compatible with this process were identified. Specifically, non-polar aromatic monomers were unsuitable, as the (assumed) products were poorly soluble in solvents required for LC-MS or HPLC analysis. Additionally, monomers bearing acid-sensitive functionality such as tert-butyl esters or Boc-protected amines gave complex reaction mixtures containing only trace quantities of the desired cyclic compounds.
After completion of the reaction, the media could be readily modified to provide solutions suitable for biological screening. The addition of 1 M NaOH (1/6 of reaction volume) followed by a 10-fold or greater dilution with a suitable buffer provided solutions of macrocyclic products in the micromolar concentration range containing <10% tBuOH and a pH equivalent to the chosen buffer.
The key feature of synthetic fermentation is the ability to rapidly prepare mixtures of natural-product like peptides in a controlled fashion. To extend this concept to the formation of macrocyclic peptides, we investigated the synthetic fermentation/macrocyclization process using mixtures containing two different elongation monomers (Table 4). The monomers were added sequentially, with separate elongation periods, in order to control the amino acid sequence of the resulting β-peptides and limit mixture complexity.
|
|
|||
|---|---|---|---|
| Entry | Monomer 1 | Monomer 2 | Major products observed |
|
a Reactions performed by combining 0.1 M stock solutions of 16 (10 μL) and monomer 1 (10 μL), heating at 45 °C for 6 h, adding monomer 2 (10 μL), heating at 45 °C for 14 h, diluting with tBuOH/1 M HCl (5:1, 90 μL) and heating at 70 °C for 4 h. The resulting mixtures were analyzed by HPLC and LC-MS with the major products shown in the table above.
|
|||
| 1 |
|
|
|
| 2 |
|
|
|
| 3 |
|
|
|
| 4 |
|
|
|
| 5 |
|
|
|
| 6 |
|
|
|
| 7 |
|
|
|
| 8 |
|

|
|
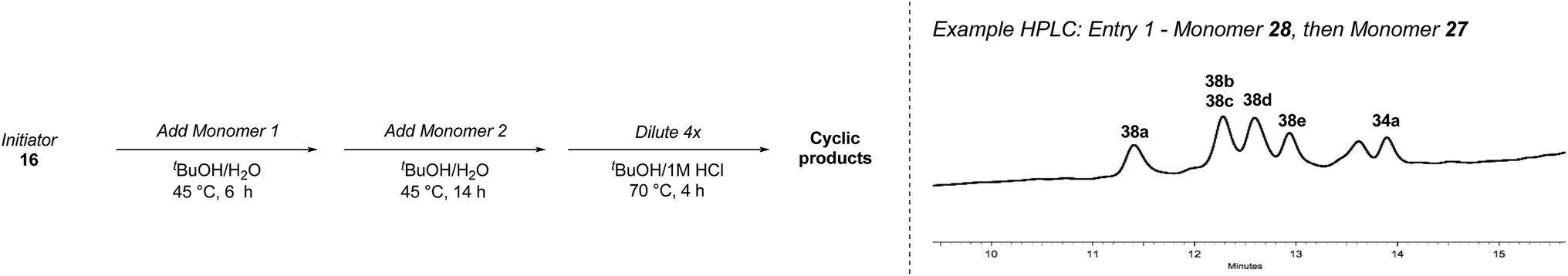
Following the second elongation the reaction mixtures were diluted with tBuOH/1 M HCl (5:1) and heated to 70 °C to effect cyclization. Pleasingly, the enhanced complexity of the β-peptide mixtures had no noticeable effect on the Boc-deprotection/macrocyclization process and the macrocyclic β-peptides were again the major products.
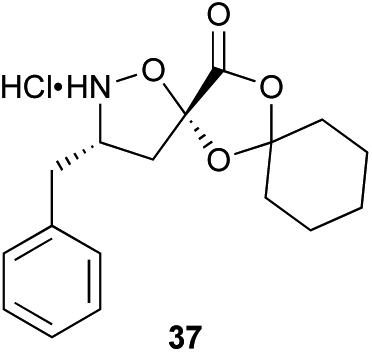
Monomers 25, 27 and 28, having proven effective in single monomer cyclizations, were used in various combinations to provide a range of macrocyclic compounds (entries 1–5). Including two monomers in each reaction allowed for the combination of acyclic and cyclic monomers (entry 6) as well as monomers with differing absolute stereochemistry (entry 7). Monomers that gave poorly soluble products when used alone (e.g. benzyl monomer 37) could be used in combination with more polar monomers, providing soluble mixtures that were readily analyzed by HPLC (entry 8).
Unsurprisingly, given the relative complexity of the overall process, the mixtures sometimes contained minor products that could not be easily identified. While not ideal for the application to biological screening, as these minor side products can increase the chances of false positives, this limitation is balanced by the efficiency in which the macrocyclic products are obtained. The synthesis of a library of such complex, chiral β-peptide macrocycles by conventional means would require lengthy multistep syntheses and several purification steps for each compound.
For future applications to biological screening, we anticipate that this methodology could be extended to provide diverse mixtures, generated using more monomers per reaction and a single elongation process. Subsequent identification of bioactive compounds from these mixtures would be analogous to isolation of active cyclic peptides from natural sources56–60 or semi-synthetic combinatorial libraries.10
Conclusions
In summary, we have established methodology for the rapid generation of macrocyclic β-peptide libraries from a selection of simple building blocks, enabled by the development of a 1,3,4-thiadiazole-forming cyclization reaction between an α-ketoacid and a thiohydrazide. The process is carried out in aqueous acidic conditions and does not require additional catalysts or reagents, which will allow for direct biological evaluation of β-peptide macrocycles, a relatively underexplored structural class.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Commission for Technology and Innovation (CTI). We are grateful to Michael Bavand, Philipp Anstätt and Paula Nichols for helpful discussions and support. We thank the LOC MS Service and the LOC NMR Service for analyses.Notes and references
- B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS PubMed.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- J. Mallinson and I. Collins, Future Med. Chem., 2012, 4, 1409–1438 CrossRef CAS PubMed.
- L. Di, AAPS J., 2015, 17, 134–143 CrossRef CAS PubMed.
- N. K. Terrett, Drug Discovery Today: Technol., 2010, 7, e97–e104 CrossRef CAS PubMed.
- A. D. Foster, J. D. Ingram, E. K. Leitch, K. R. Lennard, E. L. Osher and A. Tavassoli, J. Biomol. Screening, 2015, 20, 563–576 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- K. Josephson, A. Ricardo and J. W. Szostak, Drug Discovery Today, 2014, 19, 388–399 CrossRef CAS PubMed.
- N. K. Bashiruddin and H. Suga, Curr. Opin. Chem. Biol., 2015, 24, 131–138 CrossRef CAS PubMed.
- K. Deyle, X.-D. Kong and C. Heinis, Acc. Chem. Res., 2017, 50, 1866–1874 CrossRef CAS PubMed.
- K. Ito, T. Passioura and H. Suga, Molecules, 2013, 18, 3502–3528 CrossRef CAS PubMed.
- T. Passioura, T. Katoh, Y. Goto and H. Suga, Annu. Rev. Biochem., 2014, 83, 727–752 CrossRef CAS PubMed.
- T. Morioka, N. D. Loik, C. J. Hipolito, Y. Goto and H. Suga, Curr. Opin. Chem. Biol., 2015, 26, 34–41 CrossRef CAS PubMed.
- W. H. Connors, S. P. Hale and N. K. Terrett, Curr. Opin. Chem. Biol., 2015, 26, 42–47 CrossRef CAS PubMed.
- S. Collins, S. Bartlett, F. Nie, H. F. Sore and D. R. Spring, Synthesis, 2016, 48, 1457–1473 CrossRef CAS.
- C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719–737 RSC.
- R. Liu, X. Li and K. S. Lam, Curr. Opin. Chem. Biol., 2017, 38, 117–126 CrossRef CAS PubMed.
- M. G. Ricardo, F. E. Morales, H. Garay, O. Reyes, D. Vasilev, L. A. Wessjohann and D. G. Rivera, Org. Biomol. Chem., 2015, 13, 438–446 CAS.
- L. A. Wessjohann, O. Kreye and D. G. Rivera, Angew. Chem., Int. Ed., 2017, 56, 3501–3505 CrossRef CAS PubMed.
- T. M. Vishwanatha, E. Bergamaschi and A. Dömling, Org. Lett., 2017, 19, 3195–3198 CrossRef CAS PubMed.
- E. M. M. Abdelraheem, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, J. Org. Chem., 2016, 81, 8789–8795 CrossRef CAS PubMed.
- J. R. Frost, C. C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef CAS PubMed.
- M. Dow, F. Marchetti, K. A. Abrahams, L. Vaz, G. S. Besra, S. Warriner and A. Nelson, Chem.–Eur. J., 2017, 23, 7207–7211 CrossRef CAS PubMed.
- G. Masson, L. Neuville, C. Bughin, A. Fayol and J. Zhu, in Synthesis of Heterocycles via Multicomponent Reactions II, Springer, Berlin, Heidelberg, 2010, pp. 1–24 Search PubMed.
- R. Madhavachary, E. M. M. Abdelraheem, A. Rossetti, A. Twarda-Clapa, B. Musielak, K. Kurpiewska, J. Kalinowska-Tłuścik, T. A. Holak and A. Dömling, Angew. Chem., Int. Ed., 2017, 56, 10725–10729 CrossRef CAS PubMed.
- S. J. Miller and R. H. Grubbs, J. Am. Chem. Soc., 1995, 117, 5855–5856 CrossRef CAS.
- S. J. Miller, H. E. Blackwell and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 9606–9614 CrossRef CAS.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- S. Cantel, A. Le Chevalier Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D'Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- G. Chouhan and K. James, Org. Lett., 2013, 15, 1206–1209 CrossRef CAS PubMed.
- V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra and J. H. van Maarseveen, Org. Lett., 2006, 8, 919–922 CrossRef CAS PubMed.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS PubMed.
- M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski, S. Voigt, G. Buntkowsky and H. Kolmar, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS PubMed.
- K. V. Lawson, T. E. Rose and P. G. Harran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3753–E3760 CrossRef PubMed.
- B. A. Hopkins, G. F. Smith and N. Sciammetta, Org. Lett., 2016, 18, 4072–4075 CrossRef CAS PubMed.
- F.-M. Meyer, J. C. Collins, B. Borin, J. Bradow, S. Liras, C. Limberakis, A. M. Mathiowetz, L. Philippe, D. Price, K. Song and K. James, J. Org. Chem., 2012, 77, 3099–3114 CrossRef CAS PubMed.
- V. Balraju, D. S. Reddy, M. Periasamy and J. Iqbal, J. Org. Chem., 2005, 70, 9626–9628 CrossRef CAS PubMed.
- L. Mendive-Tapia, S. Preciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- T. Willemse, W. Schepens, H. W. T. van Vlijmen, B. U. W. Maes and S. Ballet, Catalysts, 2017, 7, 74 CrossRef.
- L. A. Wessjohann, C. R. B. Rhoden, D. G. Rivera and O. E. Vercillo, in Synthesis of Heterocycles via Multicomponent Reactions I, Springer, Berlin, Heidelberg, 2010, pp. 199–226 Search PubMed.
- M. C. Morejón, A. Laub, B. Westermann, D. G. Rivera and L. A. Wessjohann, Org. Lett., 2016, 18, 4096–4099 CrossRef PubMed.
- E. M. M. Abdelraheem, M. P. de Haan, P. Patil, K. Kurpiewska, J. Kalinowska-Tłuścik, S. Shaabani and A. Dömling, Org. Lett., 2017, 19, 5078–5081 CrossRef CAS PubMed.
- Y.-L. Huang and J. W. Bode, Nat. Chem., 2014, 6, 877–884 CrossRef CAS PubMed.
- F. Rohrbacher, G. Deniau, A. Luther and J. W. Bode, Chem. Sci., 2015, 6, 4889–4896 RSC.
- C. Xu, F.-C. Jia, Q. Cai, D.-K. Li, Z.-W. Zhou and A.-X. Wu, Chem. Commun., 2015, 51, 6629–6632 RSC.
- P. Gao, J. Wang, Z. Bai, H. Cheng, J. Xiao, M. Lai, D. Yang and M. Fan, Tetrahedron Lett., 2016, 57, 4616–4619 CrossRef CAS.
- M. van Gemmeren, M. Börjesson, A. Tortajada, S.-Z. Sun, K. Okura and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 6558–6562 CrossRef CAS PubMed.
- N. Kaczybura and R. Brückner, Synthesis, 2007, 2007, 118–130 CrossRef.
- T. Gerfaud, Y.-L. Chiang, I. Kreituss, J. A. Russak and J. W. Bode, Org. Process Res. Dev., 2012, 16, 687–696 CrossRef CAS.
- The thiadiazole-forming macrocyclization reaction was also tested on a purified linear tri-β-peptide. See ESI† for details.
- M. Dreyfuss, E. Härri, H. Hofmann, H. Kobel, W. Pache and H. Tscherter, Eur. J. Appl. Microbiol. Biotechnol., 1976, 3, 125–133 CrossRef CAS.
- H. Ueda, H. Nakajima, Y. Hori, T. Fujita, M. Nishimura, T. Goto and M. Okuhara, J. Antibiot., 1994, 47, 301–310 CrossRef CAS PubMed.
- O. Ohno, Y. Ikeda, R. Sawa, M. Igarashi, N. Kinoshita, Y. Suzuki, K. Miyake and K. Umezawa, Chem. Biol., 2004, 11, 1059–1070 CrossRef CAS PubMed.
- K. M. Witherup, M. J. Bogusky, P. S. Anderson, H. Ramjit, R. W. Ransom, T. Wood and M. Sardana, J. Nat. Prod., 1994, 57, 1619–1625 CrossRef CAS.
- L. T. Tan, X. C. Cheng, P. R. Jensen and W. Fenical, J. Org. Chem., 2003, 68, 8767–8773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data of all new compounds. See DOI: 10.1039/c7sc05057g |
| This journal is © The Royal Society of Chemistry 2018 |