
Aqueous immiscible layered double hydroxides – AIM-LDHs†
Kanittika
Ruengkajorn
 ,
Christopher M. R.
Wright
,
Nicholas H.
Rees
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
,
Christopher M. R.
Wright
,
Nicholas H.
Rees
,
Jean-Charles
Buffet
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA, UK. E-mail: Dermot.OHare@chem.ox.ac.uk
First published on 11th October 2018
Abstract
Post synthesis treatment of Layered Double Hydroxides (LDHs) with aqueous immiscible solvents (AIM solvents) yields highly dispersible, high surface area materials (up to 377 m2 g−1). The effect of solvent functional groups and structure, initial LDH particle morphology along with parameters such as dispersion time, and solvent recycling properties of AIM-LDHs are explored. The interactions between solvents and LDHs via hydrogen bonding are investigated using high field rapid magic angle spinning (MAS) solid state nuclear magnetic resonance spectroscopy (SSNMR). The strength of the hydrogen bonding interactions that a given solvent can offer appears a crucial factor for the effectiveness of the treatment.
Introduction
Layered Double Hydroxides (LDHs) have recently gained a lot of attention for use in catalysis and catalyst supports,1–3 adsorbents,1,4,5 flame retardants and polymer additives;6,7 owing to their highly flexible composition, morphology and properties.8,9 When used as heterogeneous catalysts or catalyst supports for organic transformations, reactions are often conducted in organic media, with reactivity often localised at the LDH surface. It is therefore beneficial to develop high surface area materials which can be well dispersed in common organic solvents. However, it is well known that the interlamellar hydrogen bonding networks and interlayer electrostatic interactions play a very important role in the structure of LDHs, leading to a tight stacking and heavily aggregated layers.10 As a result, synthesis of primary LDH particles intercalated with inorganic anions (nitrate, chloride, carbonate, and sulfate) usually yield low surface area materials (5–15 m2 g−1).11 It has previously been reported that such LDH materials can be delaminated in formamide, butanol, and acrylate which are thought to disrupt the hydrogen bonding networks and the electrostatic interactions of the layers; hence, facilitating the exfoliation and delamination of LDHs.12–16 However, isolation and recovery of the delaminated LDHs without aggregation is still a big challenge and not practical for commercial scale. Polar solvents have also been used to delaminate graphene oxide to monolayer graphene.16 This was attributed to the presence of hydroxyl, carbonyl and carboxyl groups.Recently, O’Hare and co-workers reported a LDH post-synthetic solvent treatment17–19 that dramatically increases their dispersion and organophilicity by using aqueous miscible organic solvents, so-called Aqueous Miscible Organic Layered Double Hydroxides (AMO-LDHs).17,20 The composition of AMO-LDHs is defined as [M1−xz+M′xy+(OH)2]a+(Xn−)a/n·bH2O·c(AMO-solvent), which instantly distinguishes them from the normal general formula of LDH, [M1−xz+M′xy+(OH)2]a+(Xn−)a/n·bH2O, wherein M and M′ are metal cations, z = 1 or 2; y = 3 or 4, 0 < x < 1, 0 < b < 1, 0 < c < 1, X is an anion, n is 1 to 3 and a = z(1 − x) + xy − 2.19 It has been observed that AMO-LDHs are highly dispersed in non-polar hydrocarbon solvents and exhibit significantly higher surface areas.19,20 AMO treatment can lead to dispersion into thin nanosheets, exfoliation, or even formation of single layers.17 Surface areas and total pore volumes as high as ∼460 m2 g−1 and 2.15 cc g−1 have been reported.17
These high surface area materials have been employed as catalyst supports for metallocene catalysed ethylene polymerisation in conjunction with methylaluminoxane. Activities recorded were up to ten times higher than when conventionally synthesised and commercial LDH supports were employed.21
Despite the improved properties gained using AMO-LDHs compared to conventional water washed materials, one of the main drawbacks is the large volume of solvents required to achieve well-dispersed, high surface area materials. It was proposed that if immiscible solvents could be used to exfoliate LDHs, this may improve solvent recyclability, as displaced water molecules could be easily separated. Furthermore, if good dispersions can be achieved in a variety of common organic solvents, this may improve the efficacy of these materials as slurry-phase catalysts.
Herein, a new post treatment method using a variety of different aqueous immiscible solvents bearing different functional groups and hydrogen bonding capabilities is described. Their effect on physicochemical properties such as surface area, morphology and solvent content of layered double hydroxides (LDHs) are determined and compared with both AMO and conventional water-washed LDHs.
Results and discussion
Both AMO and AIM solvents were compared in this study along with a conventional water-washed sample. The AMO solvents were acetone, ethanol, isopropyl alcohol, and 1-methyl-2-pyrrolidinone. Several AIM solvents with different functional groups and structures were employed; alcohols (1-butanol and 1-hexanol), esters (ethyl acetate), ethers (diethyl ether, diisopropyl ether, di-n-butyl ether, tert-amyl methyl ether, cyclopentyl methyl ether, methyl tert-butyl ether (MTBE) and anisole), amines (triethylamine), nitro groups (nitromethane), ketones (2-butanone, 3-pentanone, diisopropyl ketone, 4-heptanone, 2-pentanone, 3-methyl-2-butanone, 4-methyl-2-butanone or methyl ethyl ketone (MEK)), hydrocarbons (hexane, cyclohexane or toluene), and halogen (chloroform and dichloromethane).Mg4Al–CO3 LDH was synthesised either by co-precipitation at constant pH (pH = 10) or by the urea hydrothermal method. After washing with DI water several times to reach neutral pH, the LDH was rinsed and re-dispersed in a given solvent for a minimum of 4 hours. The shorthand term AMO-LDHs and AIM-LDHs are given for Aqueous Miscible Organic and Aqueous IMmiscible solvent treated LDHs, respectively.
AIM dispersing treatment
It has previously been reported that AMO treatment of Mg3Al–CO3 in acetone for 1 hour is enough to achieve complete dispersion and high surface area LDH (up to 363 m2 g−1).20 To ensure the maximum surface area is achieved, a dispersion time of 4 hours is commonly employed. Hence, in order to compare the AIM-LDHs with AMO and water washed analogues a dispersion time of 4 hours was chosen, after which the physico-chemical properties of the isolated materials was studied. Comparison of the powder XRD patterns for a selection of the solvent dispersed samples used in this study with the conventional water washed sample shows essentially no change in positions of the Bragg reflections with the (003) and (110) reflections unchanged (Fig. S1, ESI†). It can be inferred that solvent treatment has no effect on the crystal packing compared to the water washed sample, with no observed swelling of the layers after introduction of the solvent. There is, however, a change in the peak widths. By employing the Scherrer equation22 to determine the mean crystalline domain length (CDL) an estimate of the number of LDH layers can be made. The results illustrate a dramatic reduction in the stacking length of LDHs from about 260 to 30 Å. Moreover, the number of LDH layers decreases from ∼35 to 4–9 layers after treatment with all solvents, indicating that all solvents are capable of disrupting the LDH hydrogen bonding network to delaminate the layers.Analysing the LDH particles by Transmission Electron Microscopy (TEM) (Fig. 1), specific surface area and pores analyses (Fig. S2–S5, ESI†) and tap density measurements (Fig. S6, ESI†) show a stark contrast in the effectiveness of some of the AIM solvents after 4 hours of dispersion (Fig. 1 and Table 2). For AMO and AIM solvents containing conventional hydrogen bond donor or acceptor groups (N, O, F), high specific surface area materials are obtained (213–377 m2 g−1) with thin platelets that are much less aggregated than the water washed sample. An 80–90% reduction in powder density compared to water washed LDHs was also observed (from 1.21 to 0.20 g mL−1). In contrast, toluene, hexane and chloroform which do not contain such donor or acceptor groups were characterised as having low specific surface areas (1–24 m2 g−1) and high tap densities (1.15–1.33 g mL−1), with particle aggregation also being observed.
 | ||
| Fig. 1 TEM images of different solvent treated Mg4Al–CO3 LDHs. | ||
In all cases, the LDHs exhibit type IV BET isotherms with a H3 type hysteresis loop representing plate-like particles with slit-shape pores, according to the IUPAC classification.23–25 This behaviour reveals the mesoporous character of the samples, with the H3 hysteresis loop suggesting aggregation of plate-like particles with lots of non-uniform slit pore sizes and/or shape present in samples.26 Pore size distribution curves (Fig. S4 and S5, ESI†) determined by the Barrett–Joyner–Halenda (BJH) desorption method demonstrate the effect of the solvent washing process on the range of pore sizes for the LDH samples. According to IUPAC technical report, pores are classified into three types: macropores (>50 nm), mesopores (2–50 nm), and micropores (<2 nm).27–29 For the water washed sample a sharp peak at a maximum around 40 Å is apparent, indicating the presence of micropores and very small mesopores. Clearly defined multi-pore ranges are generated upon AMO or AIM treatment, with large mesopores and some macropores being formed.
The micropores contribute to the near edge zone of the interlayer region and the meso-/macropores are attributed to the external specific surface area of the LDH platelets.24
In order to determine the amount of solvent incorporated into the LDH structure, thermogravimetric analysis (TGA) in the low temperature region19,20 and elemental analysis were employed. Fig. S7 (ESI†) shows the TGA and the first derivative curves (DTG) of water washed and different solvent-treated LDHs. It can be seen from the region below 250 °C that the acetone treated LDHs produces two mass loss events; the first of which is believed to be due to loss of acetone. High boiling point solvents (such as hexanol, b.p. 155 °C) and hydrocarbon solvents (such as hexane and toluene) exhibit a broad peak in this region making solvent content determination by this method difficult. A summary of the determined contents is summarised in Table 1 and Table S4 (ESI†). In all cases a reduction in the water content of the material is observed with incorporation of the solvent which is most likely bound to the surface of the LDH via hydrogen bonding and/or intercalated in the galleries of the LDH. Noticeably, for toluene and hexane, the amount of solvent is 10–100 times lower than for other AMO and AIM solvents.
| Washing solvent | d 003 (Å) | d 110 (Å) | CDL003 (Å) | No. of layers | |
|---|---|---|---|---|---|
| Water | 7.53 | 1.52 | 262.8 | 35 | |
| AMO | Acetone | 7.81 | 1.53 | 34.1 | 4 |
| Ethanol | 7.80 | 1.53 | 34.3 | 4 | |
| Isopropyl alcohol | 7.80 | 1.53 | 31.1 | 4 | |
| AIM | Ethyl acetate | 7.79 | 1.53 | 32.9 | 4 |
| Diethyl ether | 7.75 | 1.53 | 40.8 | 5 | |
| 1-Hexanol | 7.76 | 1.52 | 34.8 | 4 | |
| Triethylamine | 7.74 | 1.52 | 34.7 | 4 | |
| Nitromethane | 7.76 | 1.53 | 60.2 | 8 | |
| Toluene | 7.67 | 1.52 | 60.1 | 8 | |
| Hexane | 7.68 | 1.52 | 59.0 | 8 | |
| Chloroform | 7.65 | 1.52 | 67.8 | 9 | |
| Solvent | BET specific surface area (m2 g−1) | Tap density (g mL−1) | b water contenta | c solvent contenta | |
|---|---|---|---|---|---|
| a Determined by elemental analysis using the formula [M1−xz+M′xy+(OH)2]a+(Xn−)a/n·bH2O·c(solvent) and Mg/Al = 4. | |||||
| Water | 9 | 1.21 | 0.634 | 0.000 | |
| AMO | Acetone | 354 | 0.24 | 0.225 | 0.113 |
| Ethanol | 332 | 0.27 | 0.512 | 0.092 | |
| Isopropyl alcohol | 302 | 0.22 | 0.245 | 0.215 | |
| 1-Methyl-2-pyrrolidinone | 297 | 0.30 | 0.033 | 0.109 | |
| AIM | Ethyl acetate | 377 | 0.20 | 0.041 | 0.099 |
| 1-Butanol | 369 | 0.24 | 0.110 | 0.087 | |
| 1-Hexanol | 364 | 0.23 | 0.370 | 0.021 | |
| Triethylamine | 363 | 0.22 | 0.051 | 0.082 | |
| Nitromethane | 331 | 0.27 | 0.247 | 0.040 | |
| Methyl ethyl ketone | 353 | 0.23 | 0.214 | 0.049 | |
| Diethyl ether | 327 | 0.20 | 0.124 | 0.036 | |
| tert-Butyl methyl ether | 213 | 0.47 | 0.402 | 0.001 | |
| Toluene | 1 | 1.33 | 0.548 | 0.002 | |
| Hexane | 7 | 1.15 | 0.593 | 0.042 | |
| Chloroform | 24 | 1.22 | 0.225 | 0.113 | |
Despite the low solvent contents and the low surface areas for toluene, hexane and chloroform, delamination is still observed. This suggests that such solvents can disrupt the hydrogen bonding network, albeit less effectively. In 1999, Desiraju–Steiner gave a definition for a new type of hydrogen bonding, so called ‘weak hydrogen bonding’, as an interaction X–H⋯A, wherein a hydrogen atom forms a bond between two structural moieties X and A, of which one or even both are of moderate to low electronegativity.30 It is now clearly recognised that X can be any element having higher electronegativity than H, and A could be any of these elements or π-electrons.31,32 Hence, hydrocarbon and halogenated solvents can be recognised as weak hydrogen bonding solvents.
On the other hand, weak hydrogen bonding solvents would have a weak interaction with the LDH surface, potentially requiring much longer dispersion times in order to interact with the surface hydroxyl groups of LDHs displacing surface bound water and generating high surface area LDHs.30,33
Dispersion time
To test whether weak hydrogen bond donor solvents could yield, well dispersed, high surface area LDH materials; a variety of alkyl (hexane, cyclohexane), aryl (toluene) and halogenated solvents (chloroform, dichloromethane) were employed in the washing process. The LDHs were contacted with a solvent and stirred until complete dispersions were obtained. For all solvents, except chloroform, aggregation of the LDH material on the sides of the reaction flask occurred during mixing and required regular redistribution into the solvent slurry.With increasing dispersion times, higher surface areas and lower densities were obtained (Fig. S8–S10, ESI†) (Table 3). For the halogenated and aryl solvents complete dispersions could be obtained after 72 hours; but with alkyl solvents, 120 hours was required. TEM imaging of the fully dispersible materials showed the formation of thin platelets with significantly reduced aggregation compared to shorter dispersion times (Fig. 2 and Fig. S11, S12, ESI†). Specific surface areas in the range 99–247 m2 g−1 could be obtained with reduced tap densities (0.33–0.97 g mL−1) and increased solvent content when compared to the 4 hour dispersions.
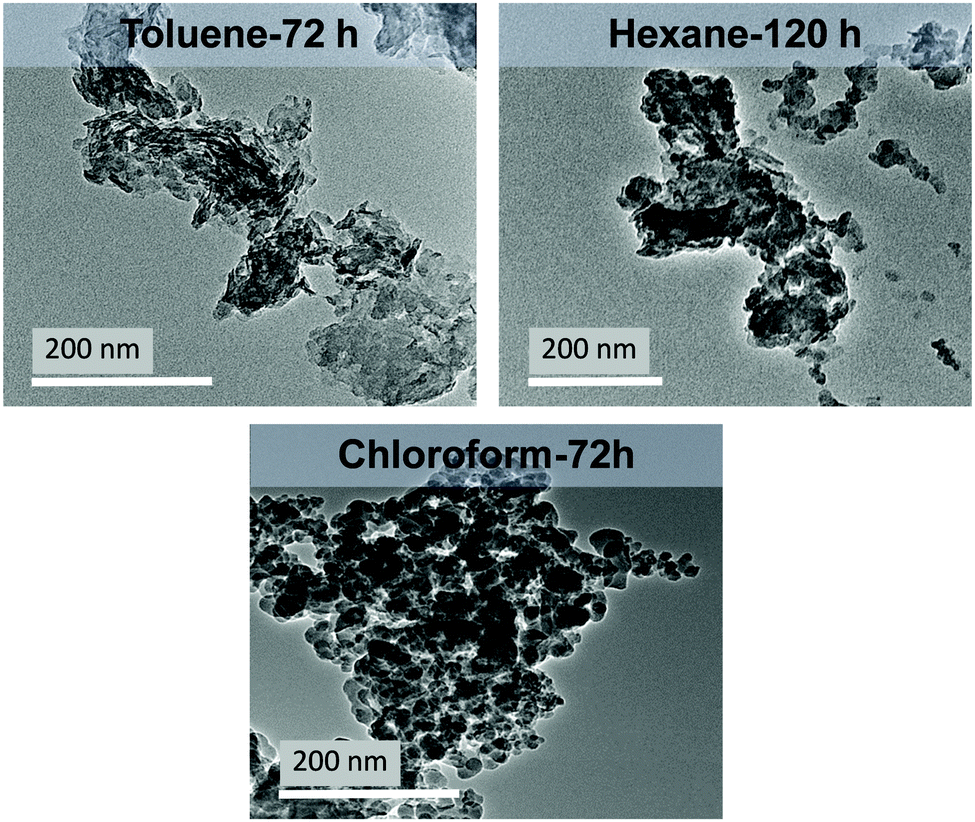 | ||
| Fig. 2 TEM images of different weak hydrogen-bond solvent-treated Mg4Al–CO3 LDHs at varied dispersion time. | ||
The results obtained can be rationalised based on the polarity and donor number of the solvents. For chloroform, three chlorine atoms are able to form weak hydrogen bonds with free hydroxyl groups (either in water or LDHs). These chlorine atoms induce the C–H bond to act as a weak acid (proton donor group) and interact with a free electron pair on the hydroxyl frameworks (i.e. a free proton acceptor centre).34 This could be a possible explanation for the higher surface area of the chloroform AIM-LDH (247 m2 g−1). In comparison, dichloromethane which has one fewer chlorine atom, possesses a weaker proton donor capacity, hence less acidity of the C–H group, resulting in a lower surface area product than in chloroform (141 m2 g−1). However, dichloromethane is quite polar35 making it more efficient than the alkyl hydrocarbon solvents (Table S11, ESI†). Toluene having a higher polarity than hexane or cylcohexane gives the second highest surface area (206 m2 g). The hydrocarbon solvents (hexane and cyclohexane) have low cohesion and minimal capacity for polar interactions (dipole-type and hydrogen-bond).
| Solvent | Dispersion time (h) | BET specific surface area (m2 g−1) | Tap density (g mL−1) | b water contenta | c solvent contenta |
|---|---|---|---|---|---|
| a Determined by elemental analysis using the formula [M1−xz+M′xy+(OH)2]a+(Xn−)a/n·bH2O·c(solvent) and Mg/Al = 4. | |||||
| Chloroform | 72 | 247 | 0.33 | 0.634 | 0.000 |
| Dichloromethane | 72 | 141 | 0.38 | 0.225 | 0.113 |
| Toluene | 72 | 206 | 0.37 | 0.512 | 0.092 |
| Hexane | 120 | 99 | 0.92 | 0.245 | 0.215 |
| Cyclohexane | 120 | 138 | 0.51 | 0.033 | 0.109 |
Although hexane and cyclohexane have similar characteristics, cyclohexane is slightly more effective than hexane (138 vs. 99 m2 g−1). An unifying explanation is still unclear. One possibility is that the lack of functionality and low electric polarisation of hexane results in a very weak interaction through dispersive force as well as the sterics of the long alkyl chain.36 Another option is that cyclohexane is preferentially adsorbed onto the LDH surface over linear alkanes;37 hence, it is more effective in the disruption of the hydrogen bonding network which may result in a higher surface area of LDHs than in hexane.38,39
Solvent structure
Erastova and co-workers have shown via molecular dynamics studies that bulkier molecules should be better at disrupting H bonding networks as very non-polar groups point away from the LDH surface towards the 2nd and 3rd coordination spheres of water/solvent/carbonate.39a Bulky substrates were also shown to struggle to penetrate into the first coordination sphere (closest to the LDH surface). However, tert-butanol (C4) was the bulkiest solvent studied. We recently showed that hydrogen bonding characteristics of the solvent play an important role for removing residual water present between the layers or on the surface of the LDHs.39bLDHs treated with various symmetrical and unsymmetrical etheral (Fig. S14–S17, ESI†) and ketone (Fig. S18–S21, ESI†) solvents were studied. In both cases increasing the chain length or the introduction of an aromatic moiety led to a reduction in the effectiveness of the treatment (lower specific surface areas, higher bulk densities). A closer look at the properties of the three aforementioned etheral solvent washed materials (EtOMe, Et2O and tBu2O) suggests that steric effects may also be important. Increasing the steric bulk around the oxygen atom leads to a decrease in both BET specific surface area (353–213 m2 g−1) and tap density (0.20–0.47). Whilst a complete dispersion could be obtained with all ketones, for longer chain ethers and anisole this was not the case after 4 hours (Fig. S22–S24, ESI†). Simulation studies of AMO solvents suggest that small solvent molecules are able to adsorb in close proximity to the LDH surface disrupting the hydrogen bonding network between water and LDHs.39 Hence, the steric effect of increasing the hydrocarbon chain length may interfere with the accessibility of the solvent to the LDHs surface.
As observed for the alkyl and aryl solvents, increasing the dispersion time resulted in complete dispersions with all the solvents.
An increase in the amount of solvent used (diethyl ether vs. ethyl acetate, both having C4) led to only slightly increased specific surface areas but also significant reduction in the water content of the LDHs, which agrees with a previous simulation study where Erastova and co-workers reported that the hydration shell of water would be more overlapped with solvent once large amount of solvent is used.39 The reduction in the amount of H-bonds between the two adjacent inner hydration layers becomes apparent only at high AMO concentrations.39a
However, it also can be seen than when comparing solvents with similar number of carbon atom, i.e. C4, ethyl acetate (377 m2 g−1), 1-butanol (369 m2 g−1), methyl ethyl ketone (353 m2 g−1) and diethyl ether (327 m2 g−1); the ether solvents seems to be less effective than carbonyl based solvents (attributed to a more polarised oxygen atom) and alcohol, since ether has less capability for H-bond forming than others.
Solvent recycling
One of the potential benefits of using AIM solvents over AMO is that the water removed from the LDH during the washing process may be easily separated from the solvent mix allowing the solvent to be reused. It has previously been reported that to obtain the highest surface areas using acetone, as the AMO solvent, 1 L of acetone is required for 3 g of LDH.19 To compare the recyclability of AMO and AIM solvents, consecutive batches of Mg4Al–CO3 were washed with either ethanol or 1-hexanol (representative of AMO and AIM solvents, respectively and chosen for their boiling point, similarity in structure but difference in miscibility with water). Considering the amount of solvent used in each step during solvent treatment, the rinsing step consumes the largest volume. Therefore, the solvent from this step was recycled and used to rinse LDH in the next batch. Analysis of the BET surface areas and tap densities of the isolated materials, shows the effectiveness of the AIM solvent treatment.With 1-hexanol the specific surface area and tap density remains essentially constant even after 4 recycling steps. In contrast, the specific surface area of the ethanol washed material begins to decrease even after the first recycling step with an increase in tap density observed after 3 cycles (Fig. 3 and Fig. S25, ESI†). The surface properties and composition of these LDHs are summarised in Tables S14 and S15 (ESI†).
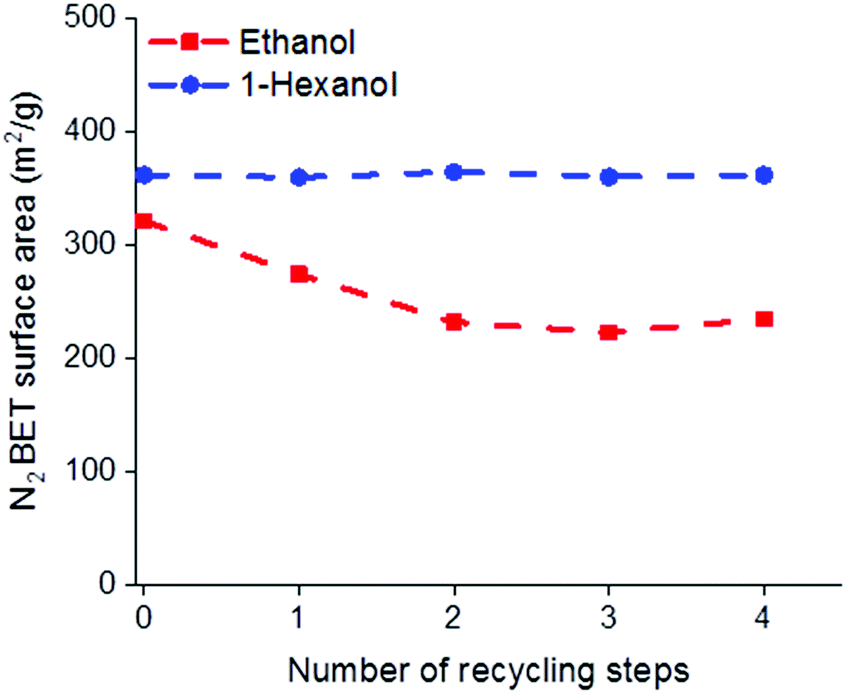 | ||
| Fig. 3 N2 BET specific surface areas of LDHs after solvent recycling. | ||
Effect of solvent washing on platelet like LDHs
The effect of AIM solvent washing was also explored on plate-like LDHs, prepared by urea hydrothermal method with an average particle size of about 3 μm.40 All AMO-LDHs and AIM-LDHs exhibit the same XRD patterns as the water washed LDHs (Fig. S31, ESI†), indicating that both AMO and AIM treatments do not affect the structure of the LDHs (Fig. S26, ESI†). The BET specific surface area of the LDHs slightly increased after treatment with AIM solvents from 24 to 64 m2 g−1 for water washed and 1-hexanol treated LDHs, respectively (Fig. 4). Surprisingly, the surface area slightly decreased after AMO treatment with acetone or ethanol. All LDHs exhibit type IV isotherm and H3 type hysteresis loop which represents the plate-like particles with slit-shape pores of LDHs (Fig. S27, ESI†). A pore size of 44 Å increased significantly with 1-hexanol dispersion and slightly increased in diethyl ether and ethyl acetate dispersion (Fig. S28, ESI†). Ethanol and ethyl acetate dispersion show a similar increase in pore size of 27 Å. For all solvents, tap density was seen to decrease after dispersion indicating displacement of water molecules by the dispersion solvent had occurred (Fig. S29, ESI†). The specific surface properties of these LDHs are summarised in Table S18 (ESI†). All solvent dispersed LDHs display a similar platelet morphology (∼3 μm diameter) as shown by TEM imaging (Fig. 5). Slightly thinner platelets can be distinguished after solvent treatment compared to the water dispersed sample. | ||
| Fig. 4 N2 BET surface areas of different solvent-treated platelet Mg4Al–CO3 LDHs. | ||
 | ||
| Fig. 5 SEM images of LDHs (a) water, (b) acetone, (c) ethanol, and (d) ethyl acetate dispersed LDHs. | ||
Solid-state NMR spectroscopy studies
Magic angle spinning solid state nuclear magnetic resonance (MAS SSNMR) studies have previously been used to probe site-specific structural information about LDHs, such as cation ordering and intercalated water, carbonate and nitrate anions.40–44 At MAS speeds below 25 kHz, the strong dipolar coupling between surface OH groups and bound water molecules result in ill-defined spectra preventing both ready identification and quantification of chemically distinct 1H environments.41,44–46 At spinning speeds above 40 kHz, the 1H homonuclear coupling can be suppressed, resolving the individual hydroxyl environments. There are four possible 1H environments in the case of MgAl LDHs due to different hydroxyl coordination modes (each either Mg and/or Al): Mg3–OH, Mg2Al–OH, MgAl2–OH and Al3–OH,44 while brucite (Mg3OH) contains a single proton environment at ∼0 ppm.40,47 The 1H chemical shifts and relative intensities are dominated by the presence of Al nuclei. Cadars et al. reported that only a maximum of three hydroxyl resonances (Mg3–OH, Mg2Al–OH and MgAl2–OH) were observed in the 1H MAS NMR spectra of MgAl–NO3 LDHs with various Mg/Al ratios which are summarised in Table S19 (ESI†).47In Mg rich LDHs (high Mg/Al ratios), only two hydroxyl environments corresponding to Mg3–OH and Mg2Al–OH should predominate.40,43,47 With increasing Al content the acidity of the hydroxyl group is increased resulting in a downfield shift,40,48 suggesting stronger hydrogen bonding to the interlayer anions and water with increasing charge on the hydroxide layers.47 In general, chemical shifts observed in the range 4.1–4.7 ppm can be ascribed to structural OH groups in the LDH and to the intercalated and/or adsorbed water.40,44,45,47 The MgAl2–OH moieties generally overlap with this water resonance but due to their low concentration, similar to Mg3–OH species, they can be difficult to identify.40,47
It was therefore proposed that multinuclear SSNMR spectroscopy could be used to probe the hydroxyl, aluminium cation and interlayer carbonate ion environments as well as probing the solvent interaction for both AMO and AIM LDHs, compared to conventional LDH samples. A previous report from O’Hare and co-workers on AMO-LDHs only probed the 27Al environments, concluding that no change in the octahedral Al sites of LDHs after AMO treatment was observed.20
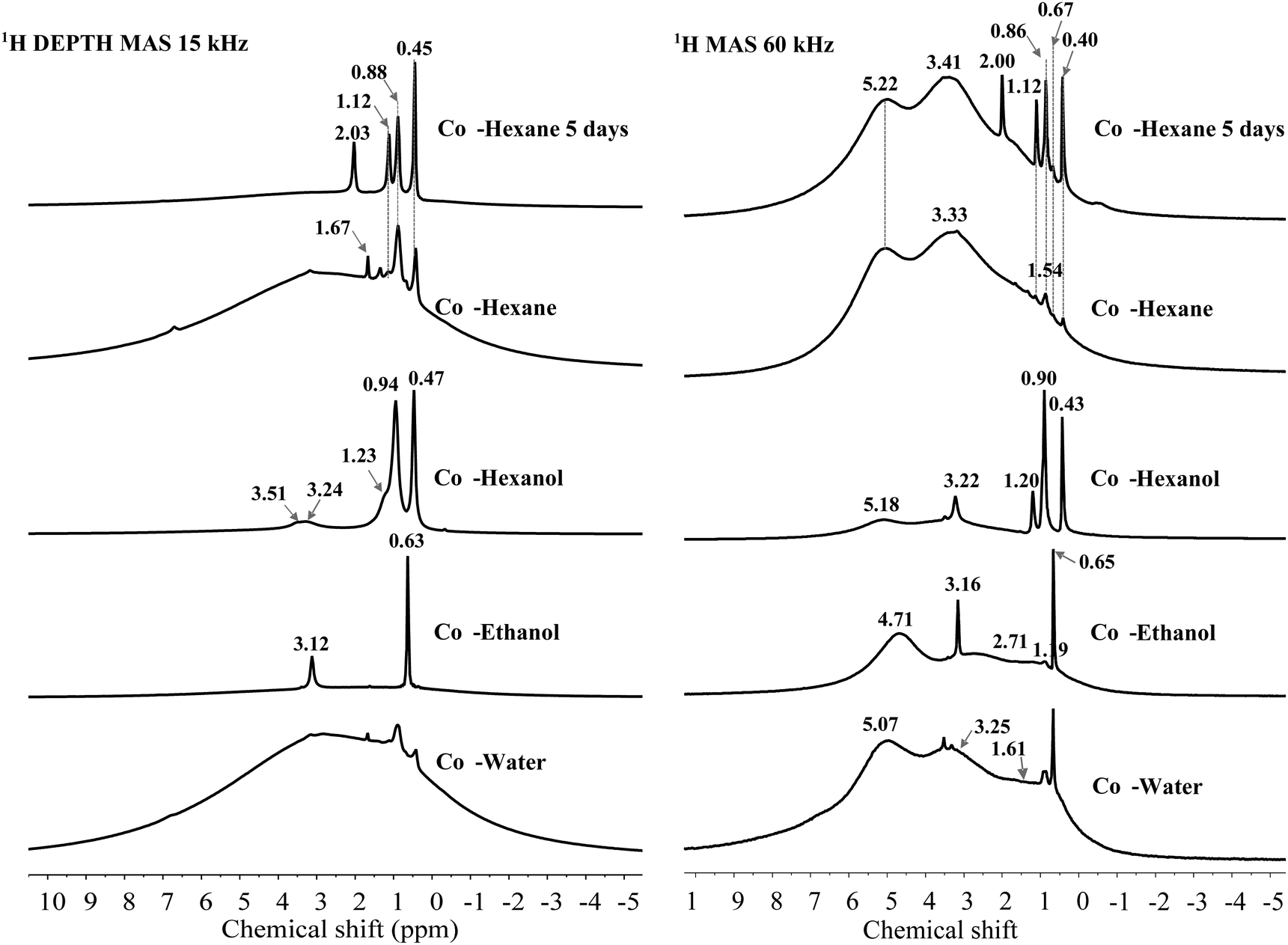
1H SSNMR spectroscopy was conducted at two spinning speeds (15 kHz, 9.4 T spectrometer and 60 kHz, 20 T spectrometer) comparing ethanol, 1-hexanol and hexane (4 and 120 h dispersion time) with a water dispersed sample (Fig. 6). Other solvent washed LDHs were only recorded at 15 kHz and their spectra are shown in Fig. S34 (ESI†). Comparison of the spectra at the two spinning speeds shows the vastly improved resolution of the hydroxyl environments at 60 kHz. Of note is that at both speeds the solvent resonances are well resolved with sharp signals, suggesting mobility of the solvent molecules on the LDH surface or within the LDH layers. Molecular dynamic simulations of the interaction of different concentrations of various AMO solvents within the interlayer region, suggested that smaller water-miscible solvents e.g. ethanol, are capable of penetrating the space between the first and second hydration layers. Such molecules close to the LDH surface are shown to preferentially orientate with the hydrogen bond donor/acceptor group pointing towards the surface.39 As such it may be anticipated that preferential orientation will cause anisotropy which may lead to broadening of the resonances for such molecules. Xu et al. utilised small organic molecules as probes to understand host–guest interactions inside the pores of mesoporous carbon materials by 1H SSNMR spectroscopy.49 At low concentrations of benzene or pyridine, interaction of the solvent with the surface either by π–π stacking or hydrogen bonding, lead to a proposed parallel alignment of the molecules with the surface. A broad resonance shifted upfield from bulk benzene due to ring currents from the surface was observed, which may be due to restricted motion leading to partial anisotropy, with similar results observed for pyridine which is a stronger hydrogen bond donor. At higher solvent loadings, sharper resonances corresponding to the bulk solvents were observed. In the case of AMO- and AIM-LDHs, major solvent resonances correspond in both number and chemical shift with what is expected for the bulk solvents. In all cases, weaker resonances can also be observed which appear to be shifted downfield from the bulk resonances and may correspond to molecules experiencing a stronger surface interaction. By fitting the 60 kHz data chemical shifts; intensities can be extracted allowing integration of resonances corresponding to surface hydroxyls as well as water and solvent molecules (Fig. S31–S35 and Table S20, ESI†). For the water dispersed sample (Cop-water LDH) at 60 kHz, broad 1H resonances at 0.60, 1.61, 3.25 and 5.07 ppm can be resolved.
 | ||
| Fig. 6 1H MAS NMR spectra of Cop-W LDH recorded at spinning speeds of 10 and 60 kHz. (Solvent impurities in the Cop-water sample (60 kHz) are consistent with cleaning solvent (ethanol) used to clean rotors between sample changing.) | ||
The broad resonance at 5.07 ppm corresponds to structural water molecules which have previously been observed at ∼4.9 ppm.43,47 A small shoulder at 6.90 ppm was also observed, which may be due to background signal, water or strongly hydrogen-bonded intercalated water or carbonate anions.43 The other broad resonances whose centres are at 3.25, 1.61 and 0.60 ppm, are consistent with the assignment of Mg2Al–OH and Mg3–OH environments.
For the ethanol dispersed sample, a downfield shift of the resonances attributed to structural water and hydroxyl environments is observed, which may be due to strong interaction of the AMO solvent with the surface. Very little change is observed for the hexane dispersed material. However, a slight downfield shift is observed when 1-hexanol had been employed, which could be indicative of a weaker interaction with the surface hydroxyl groups for these solvents possibly due to reduced polarity and increased sterics respectively.39a
Intense ethanol solvent resonances corresponding to ∼4% of the total 1H integral are observed at 0.66 and 3.15 ppm and assigned to protons from the methyl group and methylene species respectively. Exchange of the alcoholic proton with the surface hydroxyl groups means there is no clear resonance that can be inferred. Weaker resonances at 0.89 and 3.42 ppm can also be observed which may be due to solvent in close contact with the surface. For the hexane dispersed samples, the dispersion time clearly affects the amount of solvent incorporated within the structure, with a clear increase in the intensity of the hexane resonances at 0.4, 0.86, 1.10 ppm which increases from >1 to ∼3% of the total 1H integral. Due to the large number of protons in a hexane molecule this is still quite a low solvent incorporation matching the experimentally determined structures. For the 1-hexanol dispersed sample, rapid spinning was able to resolve the shoulder observed at 1.88 ppm in the 15 kHz spectrum with a resonance at 3.21 ppm also apparent and assigned to the two methylene groups closest to oxygen in the solvent. This may suggest that there is some interaction of this solvent with the surface leading to broadening of these resonances. Integration of the solvent resonances with respect to structural hydroxyl groups and water molecules shows it accounts for ∼27% of the total 1H integral, even accounting for the greater number of proton environments than for example ethanol, this is still a large quantity and may be due to incomplete removal of adsorbed and weakly bound solvent under vacuum due to the high boiling point of 1-hexanol (157 °C).
The 13C CP MAS NMR spectra of water washed and different solvent-treated LDHs are shown in Fig. S36 (ESI†). The isotropic chemical shift at 170 ppm, which is assigned to the intercalated carbonate anion was observed in all samples.9 There is a shoulder in this peak for most samples, which suggests defined orientation of the carbonate ions, the anisotropy is not removed fully by the MAS spinning. For all solvent-treated LDHs, the resonances corresponding to the carbon environments in the respective solvents were observed. In some cases weak resonances shifted downfield from the bulk solvent could also be seen which may again correspond to solvent experiencing a stronger surface interaction. All samples show the same 27Al MAS NMR spectra displaying a single resonance at 8.90 ppm (Fig. S37, ESI†), confirming that solvent treatment does not affect the distribution of octahedral Al environments of LDHs, as shown previously.20
Conclusions
A novel post-production organic solvent treatment step for LDHs using an aqueous immiscible organic solvent treatment (AIM-LDH) has been investigated. Results show that solvent incorporation into the structure leads to high dispersibility in the dispersing solvent and finer, free-flowing LDH powders with very high surface areas and low powder densities when compared to conventional LDHs. It is proposed that the hydrogen bonding characteristics of the solvent play an important role in displacing interstitial and surface water molecules in the LDH, which can be confirmed by elemental analysis and solid state MAS NMR studies. The solvent molecules appear to interact with the hydroxyl framework, structural water molecules and intercalated species via hydrogen bonding, as suggested by the 1H rapid MAS NMR studies.The resulting LDH properties are dependent upon the types of solvent used; AMO and AIM solvents containing strong hydrogen bond donor or acceptor groups generally yield high surface area LDHs after short dispersion times. Sterically hindered solvents or those with low hydrogen bond donor/acceptor capacity require longer dispersion times (up to 120 h) to achieve, well dispersed, high specific surface area LDHs.
This study allows flexibility of solvent selection for future applications. Furthermore, the use of AIM treatment could greatly enhance the efficiency of the manufacturing process, allowing the solvent to be easily separated from the displaced residual water and readily recycled for use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. R. would like to thank SCG Packaging PLC, LTD, and C. M. R. W. and J.-C. B. would like to thank SCG Chemicals Co., Ltd for funding. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). Collaborative assistance from the 850 MHz Facility Manager (Dinu Iuga, University of Warwick) is acknowledged.References
- C. Li, M. Wei, D. G. Evans and X. Duan, Small, 2014, 10, 4469–4486 CrossRef CAS PubMed.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- S. Omwoma, W. Chen, R. Tsunashima and Y. F. Song, Coord. Chem. Rev., 2014, 258–259, 58–71 CrossRef CAS.
- S. P. Newman and W. Jones, New J. Chem., 1998, 22, 105–115 RSC.
- K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- S. Guo, D. G. Evans and D. Li, J. Phys. Chem. Solids, 2006, 67, 1002–1006 CrossRef CAS.
- D. G. Evans and X. Duan, Chem. Commun., 2006, 485–496 RSC.
- X. Duan and D. G. Evans, Layered Double Hydroxides, Springer, 2006 Search PubMed.
- V. Rives, Layered Double Hydroxides: Present and Future, Nova Science Publishers, 2001 Search PubMed.
- R. Ma, Z. Liu, L. Li, N. Iyi and T. Sasaki, J. Mater. Chem., 2006, 16, 3809–3813 RSC.
- Q. Wang, H. H. Tay, Z. Guo, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Appl. Clay Sci., 2012, 55, 18–26 CrossRef CAS.
- T. Hibino and W. Jones, J. Mater. Chem., 2001, 11, 1321–1323 RSC.
- Q. Wang and D. O’Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- S. O'Leary, D. O'Hare and G. Seeley, Chem. Commun., 2002, 1506–1507 RSC.
- M. Adachi Pagano, C. Forano and J. P. Besse, Chem. Commun., 2000, 91–92 RSC.
- Q. Wu, A. Olafsen, Ø. B. Vistad, J. Roots and P. Norby, J. Mater. Chem., 2005, 15, 4695–4700 RSC.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- M. Yang, O. McDermott, J. C. Buffet and D. O'Hare, RSC Adv., 2014, 4, 51676–51682 RSC.
- C. Chen, M. Yang, Q. Wang, J. C. Buffet and D. O'Hare, J. Mater. Chem. A, 2014, 2, 15102–15110 RSC.
- C. Chen, A. Wangriya, J. C. Buffet and D. O'Hare, Dalton Trans., 2015, 44, 16392–16398 RSC.
- J.-C. Buffet, N. Wanna, T. A. Q. Arnold, E. K. Gibson, P. P. Wells, Q. Wang, J. Tantirungrotechai and D. O’Hare, Chem. Mater., 2015, 27, 1495–1501 CrossRef CAS.
- V. Drits, J. Srodon and D. D. Eberl, Clays Clay Miner., 1997, 45, 461–475 CrossRef CAS.
- G. Carja, R. Nakamura, T. Aida and H. Niiyama, Microporous Mesoporous Mater., 2001, 47, 275–284 CrossRef CAS.
- P. Gonzalez Rodriguez, M. de Ruiter, T. Wijnands and J. E. ten Elshof, Sci. Rep., 2017, 7, 481 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- B. D. Zdravkov, J. J. Čermák, M. Šefara and J. Janků, Cent. Eur. J. Chem., 2007, 5, 385–395 CAS.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CAS.
- J. Haber, Pure Appl. Chem., 1991, 63, 1227–1246 Search PubMed.
- G. R. Desiraju and T. Steiner, The weak hydrogen bond: in structural chemistry and biology, Oxford University Press, 2001 Search PubMed.
- L. C. Allen and P. A. Kollman, Chem. Rev., 1972, 72, 283 CrossRef.
- E. S. Kryachko, A. Karpfen and F. Remacle, J. Phys. Chem. A, 2005, 109, 7309–7318 CrossRef CAS PubMed.
- G. Marie-Pierre, S. Michiel and S. Marialore, J. Phys.: Condens. Matter, 2012, 24, 124106 CrossRef PubMed.
- K. Kulińska and M. Wiewiórowski, Can. J. Chem., 1988, 66, 2166–2171 CrossRef.
- C. F. Poole, The Essence of Chromatography, Elsevier Science, Amsterdam, 2003, ch. 4, p. 267 Search PubMed.
- T. M. Letcher, Thermodynamics, Solubility and Environmental Issues, Elsevier, 2007 Search PubMed.
- B. Bozbiyik, T. Duerinck, J. Lannoeye, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Microporous Mesoporous Mater., 2014, 183, 143–149 CrossRef CAS.
- I. Smallwood, Handbook of Organic Solvent Properties, Butterworth-Heinemann, 2012 Search PubMed.
- (a) V. Erastova, M. T. Degiacomi, D. O'Hare and H. C. Greenwell, RSC Adv., 2017, 7, 5076–5083 RSC; (b) K. Ruengkajorn, V. Erastova, J.-C. Buffet, H. C. Greenwell and D. O'Hare, Chem. Commun., 2018, 54, 4394–4397 RSC.
- P. J. Sideris, U. G. Nielsen, Z. Gan and C. P. Grey, Science, 2008, 321, 113 CrossRef CAS PubMed.
- A. van der Pol, B. L. Mojet, E. van de Ven and E. de Boer, J. Phys. Chem., 1994, 98, 4050–4054 CrossRef CAS.
- G. Marcelin, N. J. Stockhausen, J. F. M. Post and A. Schutz, J. Phys. Chem., 1989, 93, 4646–4650 CrossRef CAS.
- P. J. Sideris, F. Blanc, Z. Gan and C. P. Grey, Chem. Mater., 2012, 24, 2449–2461 CrossRef CAS.
- J. P. Yesinowski, H. Eckert and G. R. Rossman, J. Am. Chem. Soc., 1988, 110, 1367–1375 CrossRef CAS.
- M. A. Aramendia, V. Borau, C. Jiménez, J. M. Marinas, F. J. Romero and J. R. Ruiz, J. Solid State Chem., 1997, 131, 78–83 CrossRef CAS.
- F. Rey, V. Fornes and J. M. Rojo, J. Chem. Soc., Faraday Trans., 1992, 88, 2233–2238 RSC.
- S. Cadars, G. Layrac, C. Gérardin, M. Deschamps, J. R. Yates, D. Tichit and D. Massiot, Chem. Mater., 2011, 23, 2821–2831 CrossRef CAS.
- U. Sternberg and E. Brunner, J. Magn. Reson., 1994, 108, 142–150 CrossRef CAS.
- Y. Xu, T. Watermann, H.-H. Limbach, T. Gutmann, D. Sebastiani and G. Buntkowsky, Phys. Chem. Chem. Phys., 2014, 16, 9327–9336 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General details, PXRD, TEM, BET, BJH, TGA, dTGA, NMR, density measurements. See DOI: 10.1039/c8qm00407b |
| This journal is © the Partner Organisations 2018 |