
A single palladium site catalyst as a bridge for converting homogeneous to heterogeneous in dimerization of terminal aryl acetylenes†
Chao
Zhao‡
a,
Haizhu
Yu‡
d,
Jing
Wang
a,
Wei
Che
c,
Zhijun
Li
a,
Tao
Yao
 c,
Wensheng
Yan
c,
Min
Chen
a,
Jian
Yang
a,
Shiqiang
Wei
c,
Yuen
Wu
*a and
Yadong
Li
b
c,
Wensheng
Yan
c,
Min
Chen
a,
Jian
Yang
a,
Shiqiang
Wei
c,
Yuen
Wu
*a and
Yadong
Li
b
aDepartment of Chemistry, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, Hefei 230026, China. E-mail: yuenwu@ustc.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China
cNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029, China
dDepartment of Chemistry and Center for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, 230601, China
First published on 27th March 2018
Abstract
Herein, we utilize the surface dangling bond of MOFs as anchoring sites to access single Pd sites embedded on a hollow MOF nanobox. The stabilization of isolated Pd1 species is based on the strong coordination of the surface dangling bond of MOFs, followed by sequential reduction and phase transfer processes. The supported isolated single Pd sites can effect the highly active and selective process to produce conjugated dienes towards the dimerization of terminal aryl acetylenes, which has been previously only catalyzed by homogeneous catalysts. Unlike the commercial Pd/C and nanoparticles (NPs), the heterolytic cleavage of H2 and C–H bond efficient cleavage of terminal alkynes on atomically dispersed Pd1 sites ensure the high selectivity process and prevent the generation of styrene.
 Yuen Wu | Yuen Wu received his BS and PhD degrees from the Department of Chemistry, Tsinghua University in 2009 and 2014, respectively. He is currently a professor in the Department of Chemistry, University of Science and Technology of China. His research interests are focused on the synthesis, assembly, characterization and application exploration of functional nanomaterials. |
Introduction
Heterogeneous catalysts with satisfactory recyclability and easy separation have been widely used in the modern chemical industry. In the past decades, metal NP catalysts have achieved overwhelming successes in organocatalysis and electrocatalysis by realizing the activation of small molecules such as hydrogen, oxygen and carbon dioxides.1–12 Unlike the homogeneous catalysts commonly adopting well-defined metal centers and ligands, the diverse distribution of reactive sites and the coexistence of multiple surface configurations usually cause the reactions catalyzed by NP catalysts to occur via multifarious pathways. To date, the catalytic reactions realized by NP catalysts are mostly limited to oxidation/reduction and cross-coupling reactions.1 Towards the synthesis of complex molecules and chemicals involving more restructuring of the chemical bonds and intricate mechanisms, the homogeneous catalysts still remain the most powerful tools for the synthetic chemists. Still, it is highly desirable to exploit new catalytic systems that combine the versatility and selectivity of homogeneous catalysts and the fascinating features of heterogeneous catalysis.13–15Single site catalysts (SSCs) with well-defined single metal centers on a defect-rich support surface based on the strong Lewis acid–base interplay have become one of the new frontiers and hot spots in heterogeneous catalysis.16–21 Apart from the advantages attributed to heterogeneous catalysts including stability and reusability, the single site catalysts can serve as a promising alternative to heterogenize the homogeneous catalysts due to their unique quantum size effect, low-coordination environment and metal–support interactions.22,23 Towards the important transformations in homogeneous catalysis and biocatalysis, the unique interplay between the ligands and metal centres often guarantees the excellent selectivity of the homogeneous catalysts due to the well-defined geometric and electronic structures.24–26 Recently, the single site catalysts have been confirmed to be very selective for some important reactions including hydroformylation of olefins,23 electroreduction of carbon dioxide,27 electroreduction of oxygen to hydrogen peroxide,28 and so on.29 To some extent, the defects on the support such as oxygen vacancies or carbon vacancies can serve as organic modifiers (ligands) to make the SSCs behave like their homogeneous analogues and offer high selectivity towards a specific product.18,30 Therefore, it is highly believed that SSCs could bridge the gap between homogeneous and heterogeneous catalysis.18,23
To date, conjugated dienes are mainly produced by the Suzuki–Miyaura cross-coupling reaction, which has been exclusively achieved with expensive and environmentally unfriendly homogeneous catalysts (for example, copper salts, and palladium complexes).31–36 It remains a bottleneck to effectively generate conjugated dienes using recoverable and environmentally benign heterogeneous catalysts. Herein, we demonstrate that single Pd site catalysts can achieve a heterogenized process of homogeneous catalysts in the dimerization of terminal aryl acetylenes. Such an isolated Pd1 species exhibits high selectivity towards producing conjugated dienes, a very important class of intermediates in the synthesis of pharmaceuticals, pesticides, rubber and other chemical products.28,31 Density functional theory (DFT) calculations reveal that the heterolytic cleavage of H2 and the C–H bond efficient activation of terminal alkynes on Pd1 sites ensure the highly selectivity process.
In this work, we utilize the surface coordinatively unsaturated dangling N bond of ZIF-67 to synthesize a novel isolated single Pd site on a hollow MOF nanobox catalyst (denoted as Pd1/H-ZIF). The synthesis is based on the strong coordination between the lone pair electrons from the free amine groups and Pd d-orbital, enabling the atomic dispersion of surface Pd sites (Fig. 1a). During the catalytic reaction, the reactants could easily interact with the active sites without the limitation of mass diffusion due to the accessible surface.
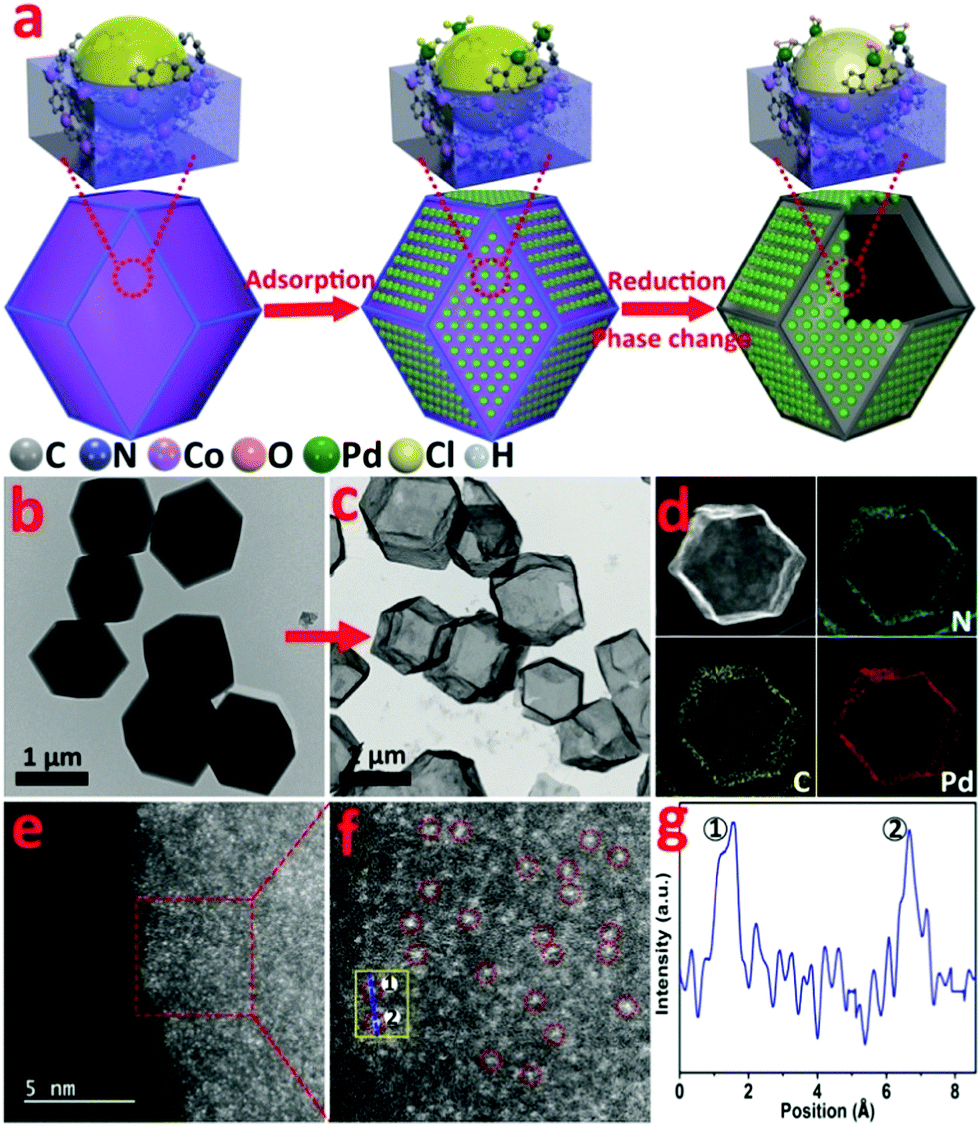 | ||
| Fig. 1 (a) Schematic illustration of the preparation of Pd1/H-ZIF, (b) TEM image of ZIF-67, (c) TEM image of Pd1/H-ZIF, (d) corresponding EDS mapping images of N, C and Pd, (e and f) magnified HAADF-STEM images of Pd1/H-ZIF, showing that only Pd atoms are present in Pd1/H-ZIF, and (g) the corresponding intensity profiles obtained on the yellow area in (f). | ||
Results
Synthesis and structure characterization of Pd1/H-ZIF
Zeolite imidazolate frameworks (ZIF-67) with uniform rhombic dodecahedron and excellent monodispersity were synthesized via a hydrothermal method, as evidenced by transmission electron microscopy (TEM) (Fig. 1b and Fig. S1, ESI†). As illustrated in Fig. 1a, the as-prepared ZIF-67, comprising similar sodalite coordination of Co2+ nodes and 2-methylimidazole, was used as a support to anchor Pd species with the surface unsaturated dangling N groups. Firstly, Na2PdCl4 was introduced into a turbid liquid of ZIF-67 to allow the full absorption of Pd due to the strong coordination interaction between Pd2+ and N. After being collected and washed with methanol, the resulting PdCl2/ZIF-67 was annealled at 200 °C under an H2/Ar atmosphere to reduce the isolated ionic Pd atoms and remove the coordinative Cl−. The as-obtained powders were re-dispersed in deionized water and heated at 100 °C for 2 h, during which a mild phase transformation occurred under hydrothermal conditions. As verified by the evolution from Fig. 1b to c, the internal ZIF-67 core was gradually dissolved after the phase change process, leaving a stable hollow residue (Pd1/H-ZIF). The obtained Pd1/H-ZIF catalyst has a uniform hollow rhombic dodecahedron shell structure, without the formation of Pd NPs. Energy dispersive X-ray spectroscopy (EDS) analysis in a scanning transmission electron microscope (STEM) identified that the Pd atoms were successfully anchored within the remaining shell (Fig. 1d). This result indicated that the Pd–N coordination is more stable than Co–N coordination in the presence of water (Fig. 2a). To further elucidate the existence form of the as-grown Pd atoms, aberration corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1e and Fig. S3, ESI†) was conducted. As a result, the isolated Pd atoms with brighter contrast could be discerned (Fig. 1f), underlying the atomic dispersion and reinforcing the stability of Pd–N coordination. The loading of Pd was 1.81%, which was measured by inductively coupled plasma atomic emission spectrometry (ICP-AES). The intensity profiles obtained from the yellow area in Fig. 1f clearly illustrate that the intensity of the two bright dots is obviously different from the neighbouring atoms due to their higher Z-contrast, further proving the isolated Pd atoms in Pd1/H-ZIF (Fig. 1g).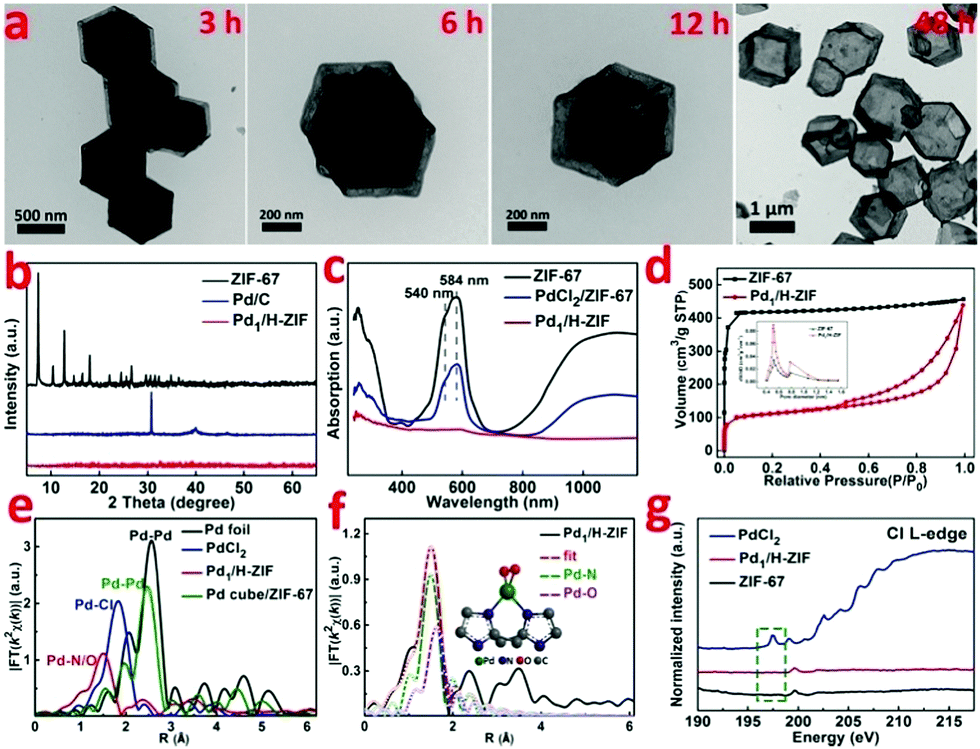 | ||
| Fig. 2 (a) The phase change process of Pd1/H-ZIF at room temperature, (b) XRD, (c) UV/Vis diffuse reflection spectrum, (d) nitrogen adsorption isotherm and desorption isotherm, (e) the k3-weighted χ(k)-function of the EXAFS spectra, (f) the corresponding Pd K-edge EXAFS fitting curves of Pd1/H-ZIF, and (g) Cl L-edge spectra. | ||
The representative steps for the synthesis of Pd1/H-ZIF and the corresponding crystalline phase evolution were firstly investigated by X-ray diffraction (XRD). As shown in Fig. 2b, the adsorption of Pd2+ would not induce structural damage of the ZIF-67, because most of the Pd2+ only coordinated with the surface N atoms from the ligands. However, after the hydrothermal treatment, the characteristic peaks of ZIF-67 completely disappeared when compared to the as-prepared Pd1/H-ZIF, evidencing the dissolution of the ZIF-67 core. Moreover, there were no observable characteristic crystal peaks of Pd NPs in the XRD patterns of Pd1/H-ZIF as compared to Pd/C, further excluding the formation of large particles (Fig. 2b and Fig. S7, ESI†). The phase change coincided well with the results obtained by UV/Vis diffuse reflection spectroscopy (Fig. 2c). That is, the peaks at 584 nm and 540 nm are characteristic of Co2+ in tetrahedral coordination with N in ZIF-67. The evanishment of these peaks suggested the degradation of the Co–N coordination mode along with the phase transformation. It was proposed that water molecules may be involved in the unit cell by forming H-bonds. This interaction may affect the coordination mode between Co2+ and 2-methylimidazole, which drives the structural evolution from ZIF-67 to a hollow rhombic dodecahedron shell structure. Surprisingly, the surface Pd–N coordination is much stronger than Co–N coordination, guaranteeing the robustness of the hollow rhombic dodecahedron shell structure under hydrothermal conditions. ICP indicated that the residual content of Co was 5.63%, which was used to maintain the hollow shell structure. A color variation from purple to gray was also observed for samples collected during the evolution process from ZIF-67 to Pd1/H-ZIF (Fig. S8, ESI†). Thermogravimetric analysis (TGA) demonstrated that the stability of the Pd1/H-ZIF improves obviously after the phase transformation (Fig. S9, ESI†). Brunauer–Emmett–Teller (BET) adsorption–desorption isotherms indicated that the surface area of the hollow Pd1/H-ZIF was 355.4 m2 g−1 (Fig. 2d).
The X-ray absorption near edge structures (XANES) of the Pd K-edge for the Pd1/H-ZIF catalyst and reference materials of Pd foil, a Pd cube and PdO are shown in Fig. S10 and S11 (ESI†). In comparison with the Pd foil and Pd cube, the white line of Pd1/H-ZIF shows a small shift to higher energy for the absorption edge (E0), and the near-edge structure was similar to PdO, suggesting that the valence state of Pdδ+ in Pd1/H-ZIF is more positive than Pd0 and negative than Pd2+. This conclusion was corroborated by the X-ray photoelectron spectroscopy (XPS) measurements, which also revealed that the valence states of Pd were positively charged between 0 and +2 (Fig. S12, ESI†). To further analyze the atomic structure of Pd1/H-ZIF, we performed extend X-ray absorption fine structure (EXAFS) spectroscopy at the Pd K-edge to probe the detailed electronic structure and coordination environment (Fig. 2e). Different from the Pd foil and Pd cube, both of which have obvious peaks in the region of 2 to 3 Å assigned to the Pd–Pd contribution, there was only one notable peak in the region of 1 to 2 Å for Pd1/H-ZIF. In contrast to the Pd–Cl bond contribution in PdCl2, the peak of Pd1/H-ZIF with a smaller R value should result from the mixture of the Pd–N and Pd–O contributions (Fig. 2f). Meanwhile, according to the fitting results presented in Table S1 and Fig. S13 (ESI†), the coordination numbers of Pd–N and Pd–O were 2 and 2, respectively. The surface N atoms of the support bridge the Pd atoms with a Pd–N bond and the oxygen coordinates with a Pd–O bond, as shown in the calculated structure model in Fig. 2e. In addition, the Cl L-edge X-ray absorption spectroscopy (XAS) spectra confirmed that no Pd–Cl bond signal was found for Pd1/H-ZIF (Fig. 2g), in line with the EXAFS spectra.
Catalytic measurements of Pd1/H-ZIF
Table S2 (ESI†) shows the catalytic performances of a wide range of Pd-based homogeneous and heterogeneous catalysts including salts, complexes, and different Pd supported species for the dimerization of phenylacetylene to generate conjugated dienes. The result is that only Pd1/H-ZIF exhibited high selectivity to access conjugated dienes in high yield (entry 1). As indicated by GC-MS and 1H NMR spectroscopy, 1,4-diphenyl-1,3-butadiene ((E,E) isomer) was observed as a major product, along with by products such as styrene and cyclotrimerization products. Meanwhile, GC-MS revealed that two additional species, with the same mass and similar GC retention time to the (E,E) isomer (presumably the (E,Z) and (Z,Z) isomers), were present in all reaction samples in a low concentration (13% of the (E,E) isomer concentration).For tests on different Pd salts and complexes (entries 9–14), we notice that such a reaction process was catalyzed by Pd2+ rather than Pd0, which is in good agreement with the existence of Pdδ+ for Pd1/H-ZIF. By contrast, the ZIF-67 did not show any observable catalytic activity, implying the crucial role of Pdδ+ centers instead of the MOF substrate (entry 8). It is reasonable that the hydrogenation reaction catalyzed by the heterogeneous Pd NPs mainly delivers the full hydrogenation products of styrene (entries 2–4). This poor selectivity to conjugated dienes was attributed to the complex reactive sites on Pd NPs and the easy cleavage of a H–H bond on a Pd surface. The Pd cube@ZIF-67 exhibited negligible catalytic activity as compared with Pd cube/ZIF-67, which demonstrated that the catalysis process occurs on the surface of the catalyst (entry 5).
The variation of conversion and selectivity as a function of reaction time were further determined to track the reaction kinetics (Fig. 3a and b). In contrast to Pd1/H-ZIF, the PdCl2 and commercial Pd/C both exhibited poor selectivity toward conjugated dienes, leading to the formation of styrene and ethylbenzene as the main products. When phenylacetylene was fully converted, the yields of conjugated diene were only 13.1% and 9.8%, respectively (Fig. 3b). To strengthen the general applicability, we further adopted a series of terminal aryl acetylenes with different substitutions (Fig. 3c), including 1-fluoro-4-phenylacetylene, 1-methyl-4-phenylacetylene, 1-methoxy-4-phenylacetylene, 1-hydroxy-4-phenylacetylene, 1-amino-4-phenylacetylene, and 1-trifluoromethyl-4-phenylacetylene, as the substrates under the catalysis of Pd1/H-ZIF. As a new promising heterogeneous catalyst, the Pd1/H-ZIF also exhibited excellent stability and recyclability, as verified by invariant activity and selectivity for more than six cycles of catalysis (Fig. 3d).
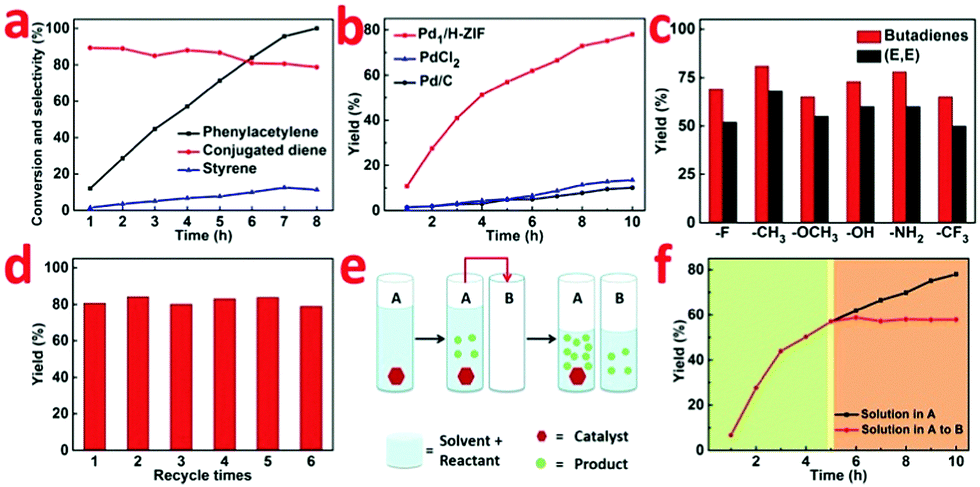 | ||
| Fig. 3 (a) Time-dependent catalysis by the Pd1/H-ZIF. (b) Reaction performances for Pd1/H-ZIF, Pd/C and PdCl2 in dimerization of phenylacetylene to generate a conjugated diene. (c) Catalytic performances for dimerization of substituted phenylacetylene over the Pd1/H-ZIF. (d) The recyclability of Pd1/H-ZIF. (e and f) Hot filtration test. | ||
To further confirm that the catalysis over Pd1/H-ZIF underwent a heterogeneous process and the active Pd1 species were not leaching to form a homogeneous active catalyst, a hot Filtration test was performed. A mixture of the substrate, Pd1/H-ZIF, and solvent were added to a vessel (A) to generate products (Fig. 3e and f). When the reaction in vessel (A) reached 57% yield, half of the solution after the removal of the catalysts was placed into a new vessel (B) without catalyst. The catalyst of Pd1/H-ZIF was preserved in (A). Both vessels A and B were then stirred and monitored under the same reaction conditions. The yield in B remained constant (red section), whereas the reaction in (A) continued to generate the product (black section). This further indicated that the active species was a heterogeneous catalyst rather than leached homogeneous Pd species. Elemental analysis by ICP of B also showed that there was no observable soluble Pd species detected above the instrument's detection limits. Together, this collection of experiments strongly supports that the catalytically active species is the heterogeneous Pd1/H-ZIF, rather than homogeneous species that were leached from the Pd1/H-ZIF during the reaction.
In the catalytic reaction process, slight 1,3-enyne intermediate was caught by GC-MS, which suggests that the activation of a terminal C–H bond in phenylacetylene might be a crucial step for the C–C coupling (Fig. 4a). To verify this hypothesis and further explore the reaction mechanism, we used 1,3-enyne as the reactant under the same hydrogenation conditions. As shown in Fig. 4b, 1,3-enyne was rapidly catalyzed to generate a conjugated diene with a higher TOF value of 476.4 h−1 (Route 2), whereas the starting material of phenylacetylene only delivered a TOF of 70.6 h−1 (Route 1). For these two different reaction routes, the corresponding activation energy (Ea) calculated based on the Arrhenius plot were 80.9 ± 12.1 KJ mol−1 (Route 1) and 46.8 ± 3.7 KJ mol−1 (Route 2), respectively (Fig. 4c). This indicated that the generation of 1,3-enyne is much slower than the following hydrogenation process to conjugated diene. Based on the above findings, we find the sequential C–C coupling of phenylacetylene and hydrogenation of 1,3-enyne. The selectivity of the hydrogenation of phenylacetylene catalyzed by Pd1/H-ZIF (Route 1) was highly sensitive to the concentration of hydrogen (Fig. 4d). That is, the dimerization of phenylacetylene was mainly accessed under the 10% H2/Ar conditions with up to 80.7% yield and the yield to styrene was restrained to 8.0%. If the hydrogenation atmosphere was changed to 20% H2/Ar or pure H2, the yield to styrene would significantly increase to 12.5% and 90.3%, respectively (Table S3, ESI†). Under a H2-free environment, the cyclotrimerization of phenylacetylene would dominantly occur with a quite sluggish rate (19.87% yield in 10 h). In contrast, the selectivity of hydrogenation of 1,3-enyne (Route 2) almost remained unchanged under different H2 concentrations (Fig. 4d). These results implied that the competition between the coupling of two phenylacetylenes and the hydrogenation of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C was crucial for the selectivities towards conjugated diene and styrene, which was strictly regulated by the isolated Pd1 sites and hydrogen concentration.
C was crucial for the selectivities towards conjugated diene and styrene, which was strictly regulated by the isolated Pd1 sites and hydrogen concentration.
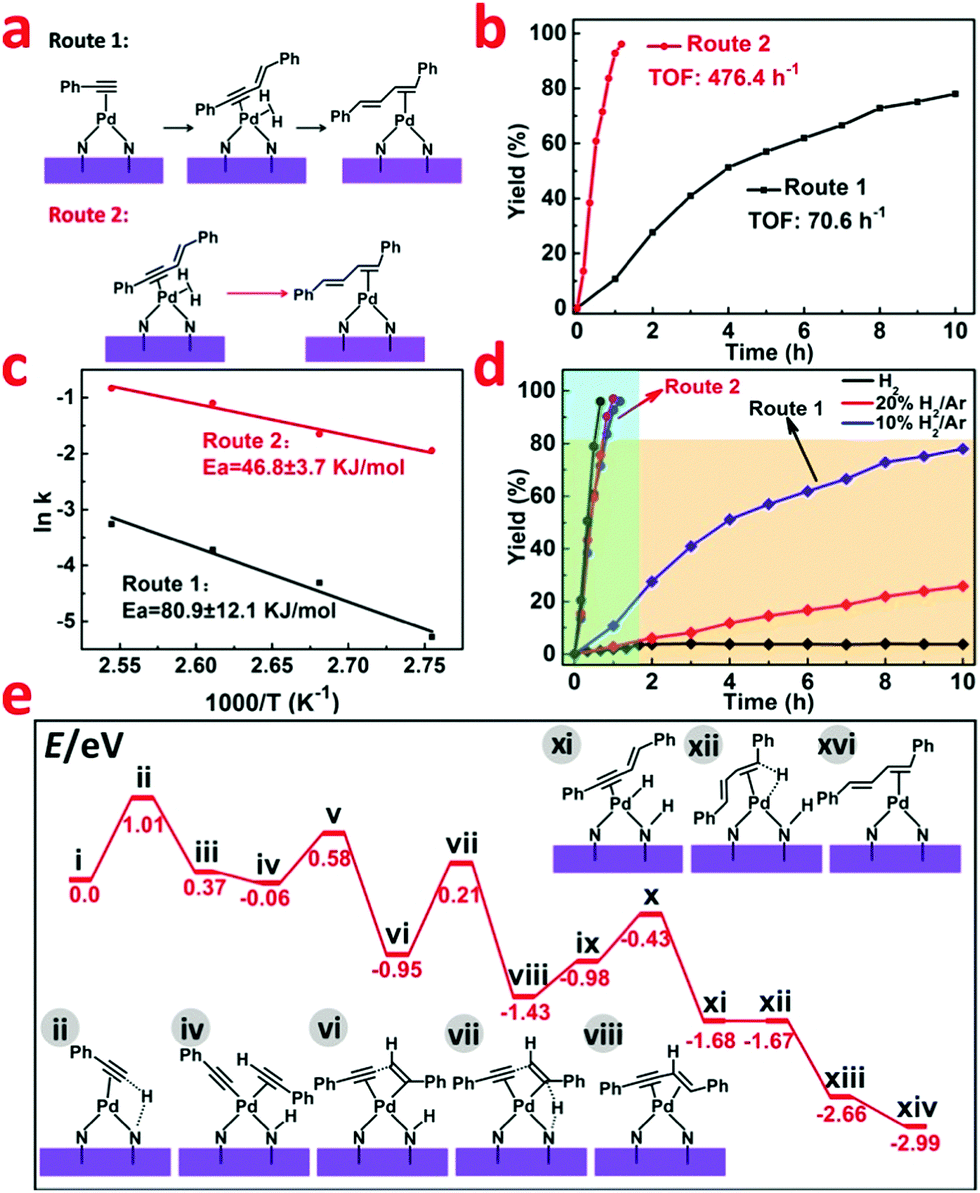 | ||
| Fig. 4 (a and b) Different routes in the reaction process. (c) Arrhenius plot of the different reaction routes for Pd1/H-ZIF. (d) The influence of different atmospheres in different routes. (e) Energies of intermediates and transition states in the mechanism of phenylacetylene dimerization at the Pd1/H-ZIF from DFT calculations. | ||
Discussion
According to our DFT calculations, the N group on the support could act as a flexible H-transfer agent throughout the reaction process. More importantly, the activation of H2 in a heterolytic pathway over the isolated atomic Pd1 sites on Pd1/H-ZIF ensured the high selectivity to conjugated diene and styrene generation. As shown in Fig. 4e and Fig. S16 (ESI†), at first, the C–H bond efficient activation of terminal alkynes occurred on Pd1 sites. And the coupling is initiated by the H-transfer from the alkyne to the N group via the transition state ii. The energy barrier for this step is calculated to be 1.01 eV. After that, the formed intermediate iii easily undergoes the coordination of another alkyne substrate to form the intermediate iv. The C–C bond formation and the H-transfer (from the N group to the alkene group) occur on single Pd1 sites successively to generate the 1,3-enyne coordinated intermediate viii. Throughout the transformation from i to viii, the second H-transfer is the rate-determining step, and the overall activation energy barrier is determined to be 1.16 eV (vi → vii → viii). From viii, the following hydrogenation of the alkyne group was also facilitated by cleavage of H2 over a Pd–N site. As shown in Fig. S16 (ESI†), after the coordination of one H2 molecule, the H-transfer to the N group occurs prior to the hydrogenation step (ix → x → xi → xii → xiii). Finally, a subsequent H-transfer (from the N group) to the internal C atom of the enyne substrate (xiii → xiv) occurs to generate the formation of the coupling product. From Fig. 4e, the overall activation energy barrier for the transformation from xi to xiv is 1.0 eV. The relatively low energy barrier in the hydrogenation step of 1,3-enyne (compared to that in the 1,3-enyne formation step, 1.16 eV) indicates that the step of C–C bond formation dominates the overall reaction rates. This conclusion correlates well with the experimental observations.Conclusions
In summary, we successfully synthesized Pd1/H-ZIF by utilizing the surface unsaturated dangling bond of MOFs. Such a single site catalyst achieves a heterogeneous catalytic process with high selectivity in dimerization of terminal aryl acetylenes, which has been previously only catalyzed by homogeneous catalysts. This work can provide a framework for rationally designing highly efficient heterogeneous catalysts, and further insight into the relationship of the catalytic mechanism and structure of catalysts at the atomic scale.Author contributions
Y. E. W. and Y. D. L. conceived the idea and co-wrote the paper. C. Z. carried out the sample synthesis, characterization and measurement. J. W, Z. J. L, M. C and J. Y helped with the characterization and measurement. T. Y., W. C., W. S. Y. and S. Q. W. helped with the XAFS measurements and discussion. H. Z. Y. performed DFT calculations. All the authors contributed to the overall scientific interpretation and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China 2017YFA (0208300), and the National Natural Science Foundation of China (21522107, 21671180, 21521091, 21390393, and U1463202). This work made use of the resources and finance support of the National Synchrotron Radiation Laboratory in Beijing and Shanghai. We thank the Photoemission End stations (BL10B) in the National Synchrotron Radiation Laboratory (NSRL) for help in characterization.Notes and references
- H. Cong and J. A. Porco Jr, ACS Catal., 2012, 2, 65 CrossRef CAS PubMed.
- M. Boronat and A. Corma, Acc. Chem. Res., 2014, 47, 834 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Mao, H. Fan, X. Pan and X. Bao, Science, 2014, 344, 616 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189 CrossRef CAS PubMed.
- J. H. Kwak, J. Mei, D. Hu, C. W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, M. Mavrikakis and M. Flytzani-Stephanopoulos, Nat. Chem., 2010, 2, 36 CrossRef PubMed.
- J. Zhang, B. An, Y. Hong, Y. Meng, X. Hu, C. Wang, J. Lin, W. Lin and Y. Wang, Mater. Chem. Front., 2017, 1, 2405 RSC.
- Y. Lei, Y. Wan, G. Li, X. Zhou, Y. Gu, J. Feng and R. Wang, Mater. Chem. Front., 2017, 1, 1541 RSC.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and G. Gascon, Mater. Chem. Front., 2017, 1, 1709 RSC.
- A. Balram, H. Zhang and S. Santhanagopalan, Mater. Chem. Front., 2017, 1, 2376 RSC.
- M. Asnavandi and C. Zhao, Mater. Chem. Front., 2017, 1, 2541 RSC.
- C. A. Witham, W. Huang, C. K. Tsung, J. N. Kuhn, G. A. Somorjai and F. D. Toste, Science, 2009, 323, 760 CrossRef PubMed.
- W. Huang, J. H. Liu, P. Alayoglu, Y. Li, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 16771 CrossRef CAS PubMed.
- R. Ye, A. V. Zhukhovitskiy, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Acc. Chem. Res., 2017, 50, 1894 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- Z. Wang, L. Gu, L. Song, H. Wang and R. Yu, Mater. Chem. Front., 2018 10.1039/c8qm00081f.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, G. Fu and N. F. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800 CrossRef CAS PubMed.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419 CrossRef CAS PubMed.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang and J. L. Lu, J. Am. Chem. Soc., 2015, 137, 10484 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740 CrossRef CAS PubMed.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. Cui and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054 CrossRef CAS PubMed.
- A. Schmid, Nature, 2001, 409, 258 CrossRef CAS PubMed.
- D. H. Wang, K. M. Engle, B.-F. Shi and J. Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed.
- G. F. Swiegers, Mechanical Catalysis: Methods of Enzymatic, Homogeneous, and Heterogeneous Catalysis, JohnWiley, 2008 Search PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078 CrossRef CAS PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058 CrossRef CAS PubMed.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo and J. T. Miller, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, G. Fu and N. F. Zheng, Nat. Mater., 2016, 15, 564 CrossRef CAS PubMed.
- S. A. Rao and M. Periasamy, J. Chem. Soc., Chem. Commun., 1987, 495 RSC.
- Y. Yamamoto, H. Yatagai, K. Maruyama, A. Sonoda and S. I. Murahashi, J. Am. Chem. Soc., 1977, 99, 5652 CrossRef CAS.
- M. Wilklow-Marnell, B. Li, T. Zhou, K. Krogh-Jespersen, W. W. Brennessel and T. J. Emge, J. Am. Chem. Soc., 2017, 139, 8977 CrossRef CAS PubMed.
- R. C. Larock, J. Org. Chem., 1976, 41, 2241 CrossRef CAS.
- N. Satyanarayana and M. Periasamy, Tetrahedron Lett., 1986, 27, 6253 CrossRef CAS.
- S. Ren, T. Seki, D. Necas, H. Shimizu, K. Nakajima and K.-i. Kanno, Chem. Lett., 2011, 40, 1443 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00095f |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2018 |