
A single palladium site catalyst as a bridge for converting homogeneous to heterogeneous in dimerization of terminal aryl acetylenes†

Abstract
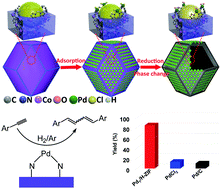
Herein, we utilize the surface dangling bond of MOFs as anchoring sites to access single Pd sites embedded on a hollow MOF nanobox. The stabilization of isolated Pd1 species is based on the strong coordination of the surface dangling bond of MOFs, followed by sequential reduction and phase transfer processes. The supported isolated single Pd sites can effect the highly active and selective process to produce conjugated dienes towards the dimerization of terminal aryl acetylenes, which has been previously only catalyzed by homogeneous catalysts. Unlike the commercial Pd/C and nanoparticles (NPs), the heterolytic cleavage of H2 and C–H bond efficient cleavage of terminal alkynes on atomically dispersed Pd1 sites ensure the high selectivity process and prevent the generation of styrene.
- This article is part of the themed collection: 2017 Emerging Investigators by MCF
 Please wait while we load your content...
Please wait while we load your content...

