
Sterically crowded hydrogen-bonded hexagonal network frameworks†
Ichiro
Hisaki
 *a,
Nobuaki
Ikenaka
a,
Seiji
Tsuzuki
b and
Norimitsu
Tohnai
a
*a,
Nobuaki
Ikenaka
a,
Seiji
Tsuzuki
b and
Norimitsu
Tohnai
a
aGraduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: hisaki@mls.eng.osaka-u.ac.jp
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki 305-8568, Japan
First published on 12th December 2017
Abstract
A hydrogen-bonded hexagonal network (H-HexNet) composed of C3-symmetric π-conjugated molecules with six carboxyphenyl groups is one of the candidate platforms for porous organic materials. For formation of H-HexNets, a triangular cyclic structure, a so-called phenylene triangle (PhT) motif, composed of hydrogen bonded carboxyphenyl groups is a key structure. In this paper, we synthesized two derivatives of hexakis(carboxyphenyl)triphenylene TpMe and TpF, in which substituents (Me or F) were introduced in the ortho-positions of the carboxy groups, to investigate whether the PhT motif forms and how the structure and property of H-HexNets are affected by steric bulkiness around the hydrogen bonding moieties. Crystal structures, thermal stability, evaluation of permanent porosity by gas sorption experiments, and photochemical properties are revealed, which can contribute to establishment and fine-tuning of porous organic materials based on H-HexNets.
Introduction
A hydrogen-bonded (H-bonded) dimer of carboxy groups is one of the simplest and the most well-known supramolecular synthons,1 that is, structural units within supermolecules which can be formed and/or assembled by known or conceivable synthetic operations involving intermolecular interactions. The dimer, however, has been still an attractive H-bonding unit to achieve predesigned supramolecular architectures, due to its highly directional bonding nature and facile accessibility to a huge number of its derivatives.2Two dimensionally networked porous frameworks based on organic molecules have been intensively investigated from the viewpoints of not only gas storage and separation,3 catalysts4 and sensors,5 but also photoelectronic and (semi)conducting materials.6 They include metal–organic frameworks (MOFs),7 covalent organic frameworks (COFs),8 and H-bonded organic frameworks (HOFs).9 Introducing the carboxy groups into building block molecules with a well-defined geometry can yield such HOFs.10 The pioneering system is the 2D honeycomb framework of trimesic acid, reported by March10a and Herbstein.10b Kobayashi also reported the layered assembly of a H-bonded hexagonal network (LA-H-HexNet) composed of hexakis(4-carboxyphenyl)benzene.10c Recently, some HOFs with permanent porosity have been constructed with carboxylic acid.11 However, it is still challenging to establish a general way to access functional HOFs with tunable pore size, shape, and surface properties.
In connection with this, we have also demonstrated that a series of C3-symmetric planar π-conjugated hydrocarbons, such as triphenylene (Tp), possessing three 4,4′-dicarboxy-o-terphenyl moieties in the periphery form a dual-pored HexNet through a cyclic H-bonded motif called a phenylene triangle (PhT) (Fig. 1c), and that the H-HexNet stacks without interpenetration to give layered HOFs (LA-H-HexNets) with porosity (Fig. 1a).12
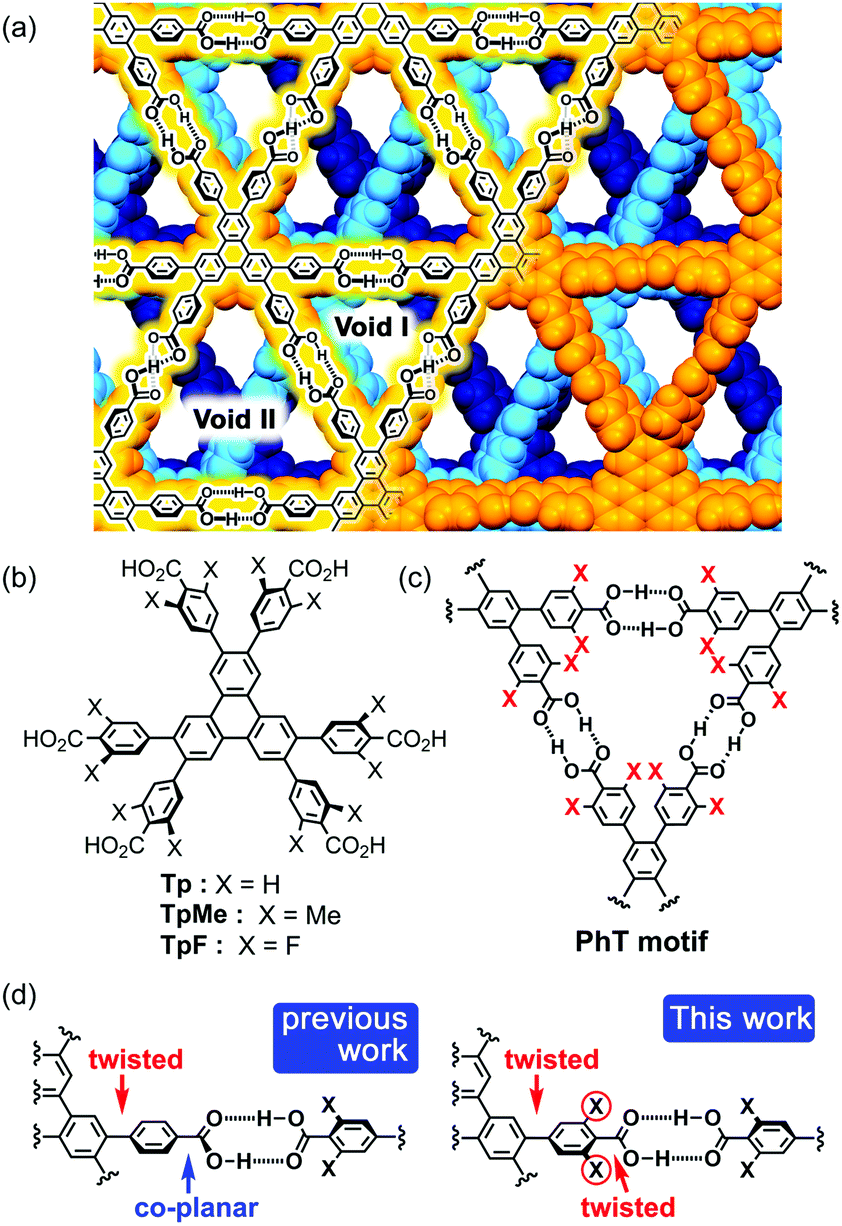 | ||
| Fig. 1 (a) Layered assembly of hydrogen-bonded hexagonal network (LA-H-HexNet) structure composed of Tp. (b) Triphenylene derivatives Tp, TpMe, and TpF. (c) Hydrogen-bonded motif, so-called phenylene triangle (PhT) motif. (d) Twisted or co-planar conformation at the peripheral carboxyphenyl groups. | ||
The important structural feature of the building block is that the peripheral carboxy phenyl groups are not coplanar but twisted against the plane of the central core (Fig. 1d, left). This twisted conformation enhances the solubility of the molecules in common organic solvents, provides shape-persistent pores surrounded by rigid phenylene walls,13 and plays a role in the stacking ways of H-HexNets to give layered structures.
In this study, we newly introduced substituents (Me or F) in the ortho-positions of the carboxy groups to force the carboxy and phenylene groups twisted (Fig. 1d, right), and investigated the effects of the substituents on molecular assemblies and properties. Regarding the steric effects of substituents on H-bonding, Ośmilałowsky demonstrated that bulkiness of substituents near the H-bonding moiety can both increase and decrease attractive interactions, depending on the situation.14 Thus, understanding of sterical effects on H-bonding is essential for constructing molecular architectures. Furthermore, in the case of a fluorine substituted derivative, electrostatic repulsion between oxygen and fluorine atoms might also have an effect on molecular conformation and packing.15
Herein, we describe the synthesis of hexakis(4-carboxy-3,5-dimethylphenyl)triphenylene TpMe and hexakis(4-carboxy-3,5-difluorophenyl)triphenylene TpF (Fig. 1b), crystal structures of their LA-H-HexNets, their thermal stability, evaluation of their permanent porosity by gas sorption experiments, and their photochemical properties. We revealed, for the first time, how the structure and property of H-HexNets are affected by sterically twisted conformation and bulkiness of the peripheral hydrogen bonding moieties.
Experimental
General
1H and 13C NMR spectra were measured using a Bruker (600 MHz for 1H, 150 MHz for 13C) or JEOL (400 MHz for 1H, 376 MHz for 19F) spectrometer. Residual 1H and 13C of the deuterated solvents were used as an internal standard [CDCl3: 7.27 ppm and DMSO-d6: 2.50 ppm for 1H, CDCl3: 77.00 ppm and DMSO-d6: 39.51 ppm for 13C]. For 19F NMR, 0.05%-C6H5CF3 in C6D6 was used as an external standard (−62.0 ppm). Mass spectrum data were obtained from a JEOL JMS-700 instrument or Autoflex III Bruker. FT-IR spectra of the synthesized compounds were recorded using a JASCO FT/IR-4200 spectrometer. Thermogravimetric (TG) and differential thermal (DT) analyses were performed using a Thermo Plus 8120 (Rigaku) instrument under N2 purge at a heating rate of 5 °C min−1. Emission and excitation spectra in solid states were measured using a JASCO FP-6500 spectrofluorometer. Powder X-ray diffraction (PXRD) data were collected on a Rigaku Ultima-IV using graphite-monochromatized Cu-Kα radiation (λ = 1.54187 Å) at room temperature. Gas sorption measurements were performed on BELSORP-max.Single crystal X-ray analysis
For crystals TpMe-1, TpMe-3, and TpF-1, diffraction data were collected on a two-dimensional X-ray detector (PILATUS 200K/R) equipped in a Rigaku XtaLAB P200 diffractometer using multi-layer mirror monochromated Cu-Kα radiation (λ = 1.54187 Å). The cell refinements were performed with the software CrysAlisPro 1.171.38.41o.16 For crystals TpMe-2, diffraction data were collected on a CCD (MX225HE, Rayonix) with synchrotron radiation (λ = 0.8000 Å) monochromated by the fixed exit Si (111) double crystal. The cell refinements were performed with HKL2000 software.17 Direct methods (SHELXT)18 were used for the structure solution of the crystals. All calculations were performed with the observed reflections [I > 2σ(I)] with the program CrystalStructure crystallographic software packages,19 except for refinement which was performed by SHELXL.20 All non-hydrogen atoms were refined with anisotropic displacement parameters, and hydrogen atoms were placed in idealized positions and refined as rigid atoms with the relative isotropic displacement parameters. The SQUEEZE function equipped in the PLATON program was used to remove disordered solvent molecules in voids.21 Crystal data are listed in Table 1.| TpMe-1 | TpMe-2 | TpMe-3 | TpF-1 | |
|---|---|---|---|---|
| Formula | C72H60O12⋯C8H8O2 | C72H60O12 | C72H60O12 | C60H24F12O12 |
| FW | 1253.41 | 1117.26 | 1117.26 | 1164.82 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | I2/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
I2/a |
| a [Å] | 27.8455(3) | 13.2528(3) | 14.5930(5) | 12.2591(3) |
| b [Å] | 22.4501(2) | 16.2730(4) | 15.3214(5) | 38.9347(8) |
| c [Å] | 29.8331(3) | 18.7976(4) | 20.3750(6) | 18.0305(4) |
| α [°] | 90 | 100.6586(8) | 102.970(2) | 90 |
| β [°] | 95.3352(9) | 93.1880(8) | 102.468(3) | 93.869(2) |
| γ [°] | 90 | 98.3918(14) | 97.238(3) | 90 |
| V [Å3] | 18568.9(3) | 3927.09(16) | 4261.2(3) | 8586.4(3) |
| Z/Z′ | 4/1 | 2/1 | 2/1 | 2/1 |
| R 1 [I < 2σ(I)] | 0.0883 | 0.1263 | 0.1052 | 0.1062 |
| R w (all) | 0.2926 | 0.4165 | 0.3608 | 0.3799 |
| Obs./unique reflns. | 54132/18891 | 20624/10145 | 40581/17192 | 41187/8961 |
| T/K | 213 | 213 | 213 | 213 |
| CCDC | 1578416 | 1578414 | 1578415 | 1578417 |
Variable temperature (VT) PXRD measurements
The crystalline bulk placed on an aluminum substrate was subjected to VT-PXRD measurements under the air atmosphere. PXRD data were collected on a Rigaku Ultima-IV using graphite-monochromatized Cu-Kα radiation (λ = 1.54187 Å). The temperature of the sample was increased from room temperature to 360 °C at a rate of 1 °C min−1. During the temperature increase, XRD patterns ranging from 3° to 18° were repeatedly recorded with a scan rate of 3° min−1. Therefore, one PXRD scan has a temperature width of 5 °C.Ab initio calculations
The geometries of 1-H, 1-Me, and 1-F (Chart 1) and their hydrogen bonded dimers were optimized at the MP2/6-311G** level using the Gaussian 09 program.22 Vibration analysis showed that the optimized structures are energy minimum structures. Conformational energies were calculated at the MP2/6-311G** level. The intermolecular interaction energies (Eint) were calculated at the MP2/cc-pVTZ level by the supermolecule method. The basis set superposition error (BSSE)23 was corrected for all the interaction energy calculations using the counterpoise method.24 The stabilization energy for forming a dimer from the isolated monomers (Eform) was calculated as the sum of Eint and the deformation energy (Edef), which is the sum of the increase in energy due to the deformation of the monomers during formation of the dimer. Here, Edef was calculated at the MP2/cc-pVTZ level.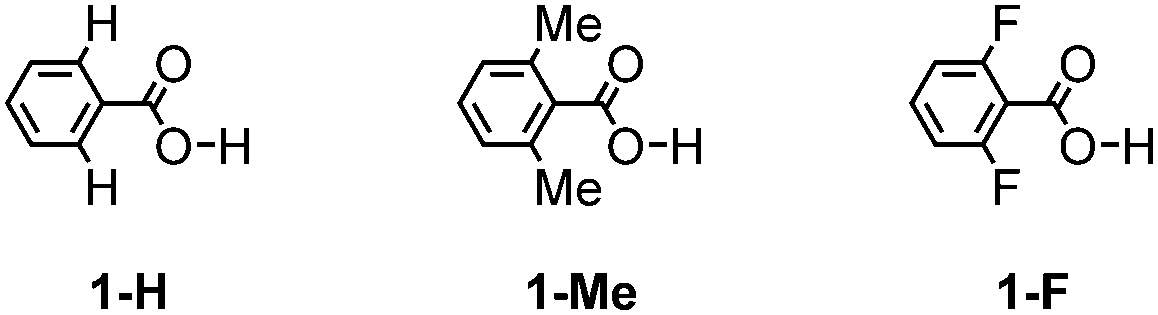 | ||
| Chart 1 Model compounds for calculation. | ||
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 5:1) to give 4 (1.08 g, 0.915 mmol, 64%) as a pale yellow solid. M.p.: 338 °C (decomp.). 1H NMR (600 MHz, CDCl3): δ 8.62 (s, 6H), 7.02 (s, 12H), 3.94 (s, 18H), 2.30 (s, 36H) ppm. 13C NMR (150 MHz, CDCl3): δ 19.87, 51.90, 125.50, 128.94, 129.23, 132.41, 135.08, 139.32, 142.24, 170.37 ppm. FT-IR (KBr): ν = 2947, 1717, 1606, 1570, 1502, 1437, 1378, 1266, 1190, 1115, 1093, 1078, 870, 822, 802, 784, 754, 732, 681 cm−1. HR-MS (FAB): m/z calc. for [M]+ C78H72O12 1200.502; measured 1200.502.
:AcOEt = 50:1) to give 5 (34.6 mg, 27.8 μmol, 34%) as a pale yellow solid. M.p.: 264 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 6H), 6.96 (d, 12H, J = 8.4 Hz), 3.99 (s, 18H) ppm. 13C NMR (150 MHz, CDCl3): δ 161.50, 159.82, 145.08, 137.59, 129.55, 125.93, 113.58, 110.22, 52.90 ppm. 19F NMR (367 MHz, CDCl3): δ −107.73, −107.75 ppm. FT-IR (KBr): ν = 2360, 1735, 1636, 1560, 1434, 1402, 1329, 1274, 1192, 1107, 1069, 1042, 884, 863, 827, 788, 592, 530 cm−1. HR-MS (FAB): m/z calc. for [M]+ C66H48F12O12 1248.202; measured 1248.202.
:hexane = 5:1) to give 4 (1.08 g, 0.915 mmol, 64%) as a pale yellow solid. M.p.: 338 °C (decomp.). 1H NMR (600 MHz, CDCl3): δ 8.62 (s, 6H), 7.02 (s, 12H), 3.94 (s, 18H), 2.30 (s, 36H) ppm. 13C NMR (150 MHz, CDCl3): δ 19.87, 51.90, 125.50, 128.94, 129.23, 132.41, 135.08, 139.32, 142.24, 170.37 ppm. FT-IR (KBr): ν = 2947, 1717, 1606, 1570, 1502, 1437, 1378, 1266, 1190, 1115, 1093, 1078, 870, 822, 802, 784, 754, 732, 681 cm−1. HR-MS (FAB): m/z calc. for [M]+ C78H72O12 1200.502; measured 1200.502.
:AcOEt = 50:1) to give 5 (34.6 mg, 27.8 μmol, 34%) as a pale yellow solid. M.p.: 264 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 6H), 6.96 (d, 12H, J = 8.4 Hz), 3.99 (s, 18H) ppm. 13C NMR (150 MHz, CDCl3): δ 161.50, 159.82, 145.08, 137.59, 129.55, 125.93, 113.58, 110.22, 52.90 ppm. 19F NMR (367 MHz, CDCl3): δ −107.73, −107.75 ppm. FT-IR (KBr): ν = 2360, 1735, 1636, 1560, 1434, 1402, 1329, 1274, 1192, 1107, 1069, 1042, 884, 863, 827, 788, 592, 530 cm−1. HR-MS (FAB): m/z calc. for [M]+ C66H48F12O12 1248.202; measured 1248.202.
Results and discussion
Theoretical calculation of hydrogen bonding
To investigate the ortho-substitution effects on the structures and magnitude of the interactions of the hydrogen bonded dimer, three model systems 1-H, 1-Me, and 1-F were subjected to theoretical calculation (Chart 1). In contrast to 1-H, which has a coplanar conformation (ω = 0°, where ω denotes dihedral angles between the benzene ring and the carboxy group), 1-Me and 1-F have a twisted conformation (ω = 53.8° and 54.5°, respectively) due to steric hindrance of the substituents. The co-planar conformers of 1-Me and 1-F are less stable than the corresponding twisted energy minimum conformers by as much as 2.37 kcal mol−1 and 1.86 kcal mol−1, respectively. The 1-Me and 1-F dimers have two nearly isoenergetic isomers shown in Fig. S1 (ESI†). The energy differences between the isomers are less than 0.1 kcal mol−1.Subsequently, the Eform for the dimers of 1-H, 1-Me, and 1-F was calculated (Table 2). Stabilization energies of the dimers of 1-H, 1-Me, and 1-F range from −14.8 to −15.4 kcal mol−1, indicating that substituent effects on hydrogen bonding energy are negligible. This result is consistent with the atomic charges on carbonyl oxygen atoms and hydrogen atoms of the carboxy group in 1-H, 1-Me and 1-F obtained by ab initio calculations (Table S2, ESI†). The charges on these atoms are nearly identical in the three molecules, in spite of the different o-substituents. To further elucidate the substituent effects on the hydrogen bonds of the carboxy group, we evaluated the interactions of carboxylic acids (1-H, 1-Me, 1-F) with Na+ and Cl− as shown in Fig. S2 and S3 (ESI†). No significant differences were observed among the interactions of these three carboxylic acids with Na+ and Cl−.
| Dimer | E int | E def | E form |
|---|---|---|---|
| a E int is the intermolecular interaction energy calculated for the hydrogen bonded dimer at the MP2/cc-pVTZ level. b E def is the deformation energy. c E form is the stabilization energy for the formation of a dimer, which is the sum of Eint and Edef. See text for details. | |||
| 1-H | −18.0 | 2.5 | −15.4 |
| 1-F | −17.1 | 2.3 | −14.8 |
| 1-Me | −17.7 | 2.5 | −15.2 |
Synthesis and crystallization of TpMe and TpF
Hexakis(methoxycarbonylphenyl)triphenylene derivatives 4 and 5 were synthesized in 64% and 37%, respectively, by Suzuki–Miyaura cross coupling of 2,3,6,7,10,11-hexabromotriphenylene and the corresponding boronic acid 2 or pinacol borate 3 in the presence of Pd(dppf)Cl2 and bases. Subsequently, hydrolysis of 4 and 5 was performed with CF3CO2H and KOH, respectively, to yield TpMe and TpF in 64% and 85% yields, respectively (Scheme 1a).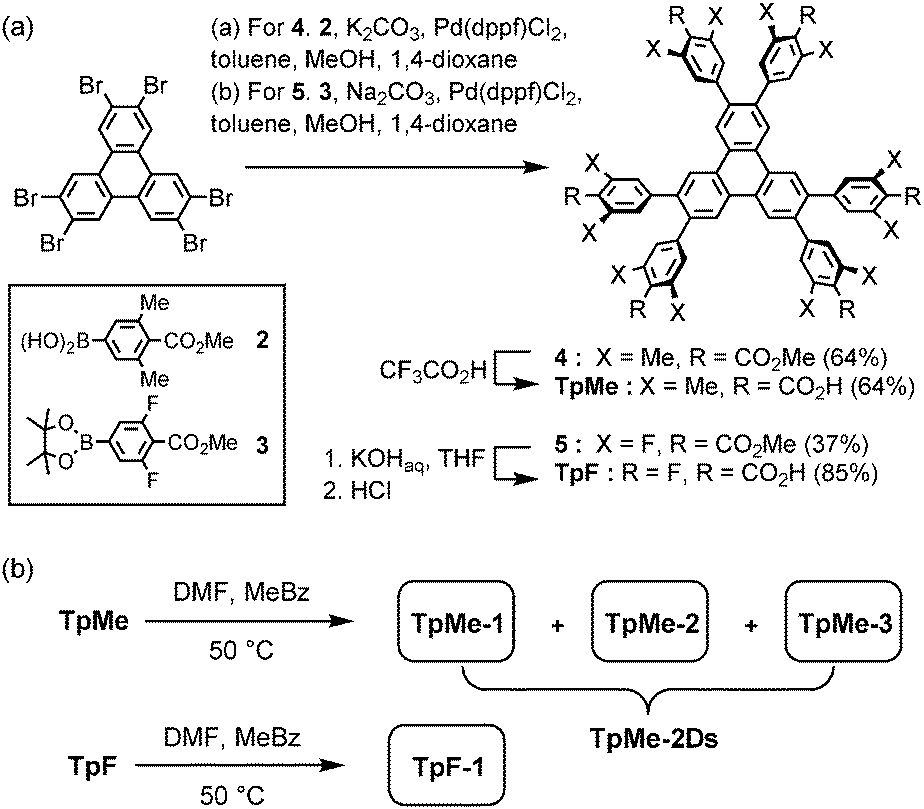 | ||
| Scheme 1 (a) Synthesis and (b) crystallization of hexasubstituted triphenylene derivatives TpMe and TpF. Abbreviations, DMF: N,N-dimethylformamide, MeBz: methyl benzoate. | ||
TpMe and TpF were recrystallized by slow evaporation of a mixed solution of DMF and methyl benzoate at 50 °C to yield single crystals suitable for X-ray crystallographic analysis (Scheme 1b). TpMe yielded totally three polymorphic crystals TpMe-1, TpMe-2, and TpMe-3 under the same conditions. However, it needs to be mentioned that it is not clear whether three polymorphs are always contained in the crystalline bulk formed in the same batch, because a general diffractometer in the laboratory provided only weak and ambiguous XRD patterns for the obtained crystalline materials due to the low electron density contrast generated by disordered methyl benzoate molecules within the pores, making it difficult to characterize the polymorphic forms. Therefore, such bulk crystals obtained by crystallization at 50 °C are called TpMe-2Ds. On the other hand, TpF yielded one crystalline form TpF-1.
Crystal structures of HexNet frameworks
In the crystal structures of TpMe-1, TpMe-2, and TpMe-3 (Fig. 2), TpMe forms a H-HexNet, which stacks in a non-interpenetrated manner to give layered assemblies (LA-H-HexNets). TpMe-1 shows conformational disorder at five carboxy groups due to steric hindrance of the o-substituents as shown by yellow atoms in Fig. 2a. This kind of rotational disorder was expected, as predicted by the theoretical calculation. However, to our surprise, the other polymorphs show no rotational disorder at the carboxy groups, and furthermore, all three polymorphs have no disorder at the phenylene groups, although their anisotropic displacement ellipsoids are more extended compared with those of the triphenylene core. The void ratios in TpMe-1, TpMe-2, and TpMe-3 are 45.2%, 35.9%, and 41.5%, respectively, which are relatively small compared with LA-H-HexNets of Tp (50.0–54.4%) due to the bulky substituents.12b In the void spaces, methyl benzoate molecules are accommodated, although they are severely disordered. | ||
| Fig. 2 Crystal structures of (a–c) TpMe-1, (d–f) TpMe-2, and (g–i) TpMe-3. (a, d and g) Molecular conformation drawn in anisotropic displacement ellipsoids with 50% probability. (d, e and h) Motif of HexNet frameworks. (c, f and i) Layered packing diagram composed of three HexNet sheets. Void spaces are filled by methyl benzoate used as a solvent, although most of them cannot be solved crystallographically due to severe disorder. | ||
The present polymorphs were caused by versatile conformations of the phenylene groups and slightly different stacking manners of the HexNet sheets. To evaluate the conformational versatility of the peripheral carboxy phenyl moieties, the following two parameters ϕ and ω were introduced (Fig. 3), where ϕ denotes the dihedral angle between the triphenylene core and the phenylene ring and ω denotes the dihedral angle between the phenylene ring and the carboxy group. The values are listed in Table 3. As we expected, dihedral angle ϕ in the TpMe polymorphs ranges from −75.8° to +55.7°, which is similar to that of Tp-1. On the other hand, dihedral angle ω in the TpMe polymorphs ranges from −87.0° to +84.9°, which is significantly larger compared to that of Tp-1 (i.e. −4.3° to +11.5°).12b
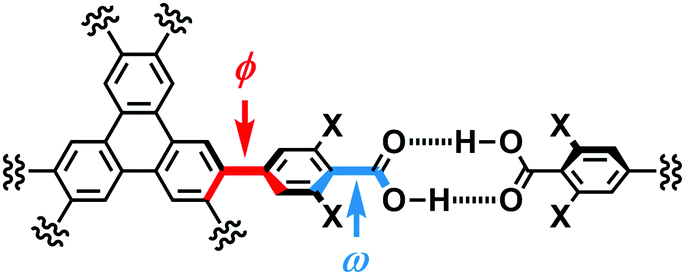 | ||
| Fig. 3 Definition of two parameters related to conformations of the phenylene and carboxy groups: dihedral angles ϕ and ω, respectively. | ||
| ϕ | TpMe-1 | TpMe-2 | TpMe-3 | TpF-1 | Tp-1 |
|---|---|---|---|---|---|
| a Two kinds of values are described due to the disorder of the phenylene groups. b Ref. 12b. c An asterisk (*) denotes that the conformational frustration is located in the corresponding peripheral group. d Classification of the conformers; for details, see ref. 12b. e For TpMe-1, only major conformation is applied to show the ω value. f Approximated range of torsion angles are listed due to highly elongated anisotropic displacement parameters. | |||||
| A | +55.7 | −75.8 | +49.8 | +59, −59 | +52.7 |
| B | +44.0 | −50.0 | +40.1 | +59, −59 | +52.4*c |
| C | −62.4 | −32.5 | +48.8 | −58, +62 | −49.6 |
| D | −47.4 | −65.6 | +47.7 | −59, +52 | −46.6 |
| E | −49.0 | +54.5 | −40.0 | −59, +52 | +67.4*c |
| F | −46.7 | +32.0 | −54.7 | −58, +62 | +52.8 |
| Conf.d | PMM | MMP | PPM | — | PMP |
In contrast to TpMe, TpF has yielded one crystalline form TpF-1 under the same crystallization conditions as with TpMe (Fig. 4). In the crystal structure, the TpF molecule with a two-fold axis forms a LA-H-HexNet with a stacking manner similar to that of TpMe-1. The void ratio of TpF-1 is 50.4% and severely disordered methyl benzoate molecules are accommodated in the void. Interestingly, all the phenylene groups and four of six carboxy groups are rotationally disordered into two positions and their anisotropic displacement ellipsoids are also elongated. The phenylene rings of the molecule in TpF-1 are twisted as observed in other TpMe and Tp-1 systems (see ϕ values in Table 3). The carboxy groups are also twisted against the plane of the phenylene rings: ω values range from 20° to 90°, although the exact values are not presented due to the relatively severe disorder of the carboxy groups.
 | ||
| Fig. 4 Crystal structure of TpF-1. (a) Molecular structure with anisotropic displacement ellipsoid with 50% probability, where the peripheral carboxy and difluorophenylene groups are severely disordered. (b) Rhombic motif. (c) Layered framework composed of three HexNet sheets, in which one of the disordered structures was applied for clarity. | ||
Versatile conformations of TpMe and TpF observed in crystalline states can be explained by the following interpretation. Namely, introduction of substituent groups into the ortho-positions of the carboxy group causes an increase of the rotationally variable range of the carbonyl groups, in addition to the phenylene groups. This wide range of conformations results in the generation of three polymorphs in the case of TpMe. However, the conformation of the TpMe molecule is fixed in one way in each of the polymorphs due to the bulkiness of the methyl group. In the case of TpF, on the other hand, the varied conformation of the peripheral groups causes no polymorphs but one crystalline form with significant disordered peripheral hydrogen-bonding groups (Fig. 4b). Probably, fluorine atoms at the ortho-positions are not bulky enough to affect the molecular packing and generation of polymorphs, and the varied rotational ability of the peripheral groups is neutralized by local rotational disorder.
Thermal behaviour of the frameworks
Freshly prepared crystalline bulks of TpMe-2Ds (which contain either one, two, or all form(s) of TpMe-1, TpMe-2, and TpMe-3) and TpF-1 were subjected to thermal analysis (Fig. 5). The thermogravimetric (TG) curve of TpMe-2Ds revealed that the included solvent was completely released at around 200 °C. The weight loss of 38 wt% indicates that the framework involved solvent molecules (methyl benzoate) with a host–guest ratio of 1:4, which is also supported by 1H NMR spectroscopy (Fig. S4 and S5, ESI†). The second weight loss starting from ca. 300 °C is assigned to thermal decomposition of the compound. TpF-1, the host–guest ratio of which is 1:7 (47 wt%) based on 1H NMR spectroscopic analysis (Fig. S5, ESI†), showed ambiguous TG curves involving several steps of the weight loss. Removal of the guest molecules and thermal decomposition may occur simultaneously.
 | ||
| Fig. 5 TG curves of TpMe-2Ds and TpF-1. | ||
To obtain structural information during desolvation by heating, powder X-ray diffraction (PXRD) patterns of crystalline bulks of TpMe-2Ds and TpF-1 were recorded with gradual heating (Fig. 6). The initial patterns are not clear, particularly in the case of TpMe-2Ds, due to the severe disorder of solvent molecules accommodated in voids. However, peaks gradually appeared upon heating.
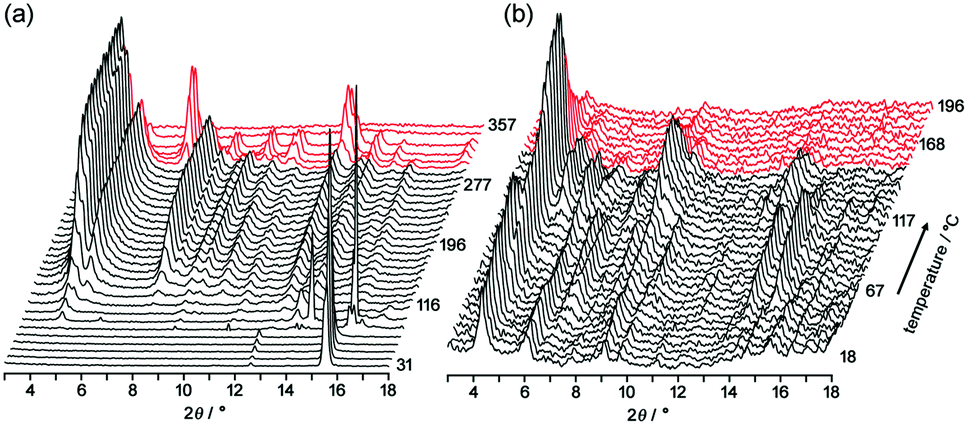 | ||
| Fig. 6 Changes of PXRD patterns of (a) TpMe-2Ds and (b) TpF-1 upon heating. | ||
Upon heating of TpMe-2Ds, obvious peaks started to grow at 4.54°, 5.18°, 7.96° and so on at 116 °C and plateaued at around 190 °C. The obtained pattern is not in agreement with the simulated pattern of either TpMe-1, TpMe-2, or TpMe-3, indicating that structural changes occurred. Complete removal of the solvent molecules was confirmed by 1H NMR spectroscopy. The pattern then started to change at 301 °C and completely disappeared at around 347 °C due to thermal decomposition of the compound.
In the case of TpF-1, the pattern change occurred in three steps, although the signal/noise ratio was relatively low. First, new peaks started to appear at around 6.9° and 7.2° at 55 °C. Then, other new peaks at 5.4° and 8.1° started to appear at 115 °C. Finally, the remaining peaks started to decay at around 155 °C and any peaks that remained disappeared at around 200 °C. This indicates that the TpF-1 crystal changes into an amorphous phase by heating or desolvation via two intermediate phases. These multi-step structural changes are also implied by the TG curve (Fig. 5b).
In this way, despite the fact that both compounds have similar molecular structures and form similar LA-H-HexNets, their structure-retention ability strongly depends on the substituents at the ortho-positions. Particularly, it was revealed that the HexNet framework of TpF-1 is not resistant to heat higher than ca. 150 °C.
Activation (desolvation with retaining pores) of TpMe-2Ds and TpF-1 crystalline bulks was preliminarily attempted by heating at 100 °C or 200 °C under vacuum conditions, resulting in a significant loss of crystallinity in the case of both TpMe-2Ds and TpF-1 at both temperatures. Consequently, activation was performed by a solvent exchange method. The PXRD pattern of as-formed crystalline bulks of TpMe again shows an ambiguous profile (Fig. 7a-ii), which is therefore difficult to be characterized when compared with the simulated pattern of TpMe-1, TpMe-2, and TpMe-3 (Fig. 7i-1, i-2 and i-3, respectively). The as-formed crystalline bulk of TpMe-2Ds was soaked in benzene solution to exchange the solvent molecules in the pores. After soaking for 48 h, we confirmed by 1H NMR spectroscopy that methyl benzoate was completely replaced with benzene (Fig. S6, ESI†). The resultant material showed an obvious PXRD pattern (Fig. 7a-iii). Subsequently, the materials were laid under vacuum conditions at 60 °C for 24 h, enabling complete desolvation to yield crystalline porous material TpMe-apo (Fig. 7a-iv and Fig. S7, ESI†). The PXRD pattern of TpMe-apo is in good agreement with that observed in VT-PXRD analysis (Fig. 6a).
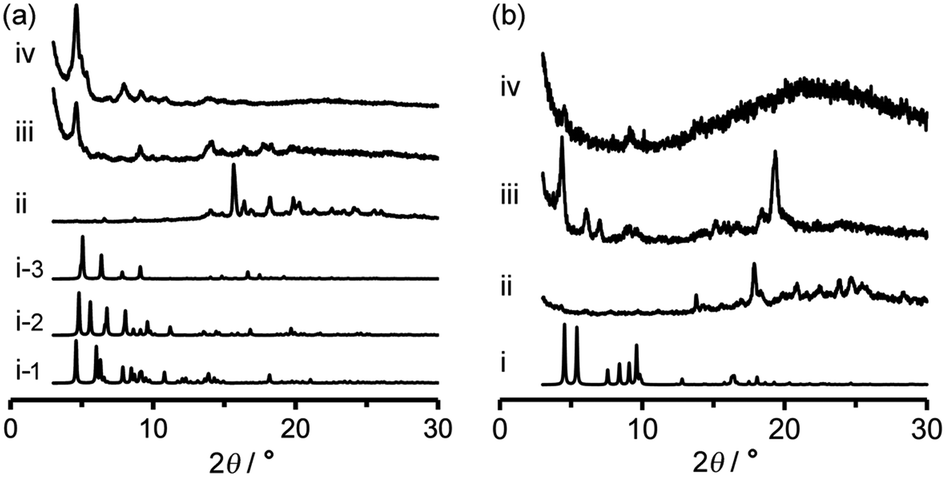 | ||
| Fig. 7 PXRD pattern changes of (a) TpMe-2Ds and (b) TpF-1 upon activation by a solvent exchange method. (i) Simulated PXRD patterns of TpMe-2Ds (i-1: TpMe-1, i-2: TpMe-2, i-3: TpMe-3) for (a) and TpF-1 for (b), respectively. (ii) As-formed crystalline bulks. (iii) Benzene-soaked materials. (iv) Desolvated materials TpMe-apo (a) and TpF-apo (b). | ||
Activation of TpF-1 was performed by the same procedure as in the case of TpMe-2Ds except for the complete removal of benzene, which needed additional heating at 100 °C for another 24 h under vacuum conditions, to yield a desolvated material TpF-apo (Fig. S8–S10, ESI†). However, the crystallinity of the resultant TpF-apo was very low (Fig. 7b-iv).
Gas sorption properties
To evaluate the permanent porosity of TpMe-apo and TpF-apo, N2, CO2, and H2 adsorption–desorption experiments were undertaken at 77 K, 195 K, and 77 K, respectively (Fig. 8). TpMe-apo shows a type-I sorption isotherm for all gasses, indicating that TpMe-apo has micropores within the framework. The amounts of uptake for N2, CO2, and H2 are 182 cm3 g−1, 157 cm3 g−1, and 81 cm3 g−1, respectively. The pore size distribution of TpMe-apo was calculated by the nonlocal density functional theory (NL-DFT) method (Fig. S11, ESI†), resulting in a diameter of 0.77 nm, which is in good agreement with the pore size provided by layered HexNet frameworks composed of triphenylene derivatives (Tp-apo). Indeed, the amounts of uptake and sorption selectively for gas species are quite similar between TpMe-apo and Tp-apo. TpF-apo shows quasi type-I sorption isotherms for CO2 with an uptake of 91 cm3 g−1, while it shows almost no adsorption for N2 and H2. This result, combined with PXRD changes upon heating, indicates that TpF-apo does not retain the LA-H-HexNet structure but is a less crystalline material with narrower void spaces. The surface area (SABET) calculated by the Brunauer–Emmett–Teller model for CO2 sorption isotherms is 561 m2 g−1 for TpMe-apo and 219 m2 g−1 for TpF-apo. It is noteworthy that TpF-apo exhibits selective sorption behavior for CO2, mainly due to the size effect of the narrow pores, and presumably due to the additional electrostatic effects of fluorine substituents, although the contribution of the latter effect is small.25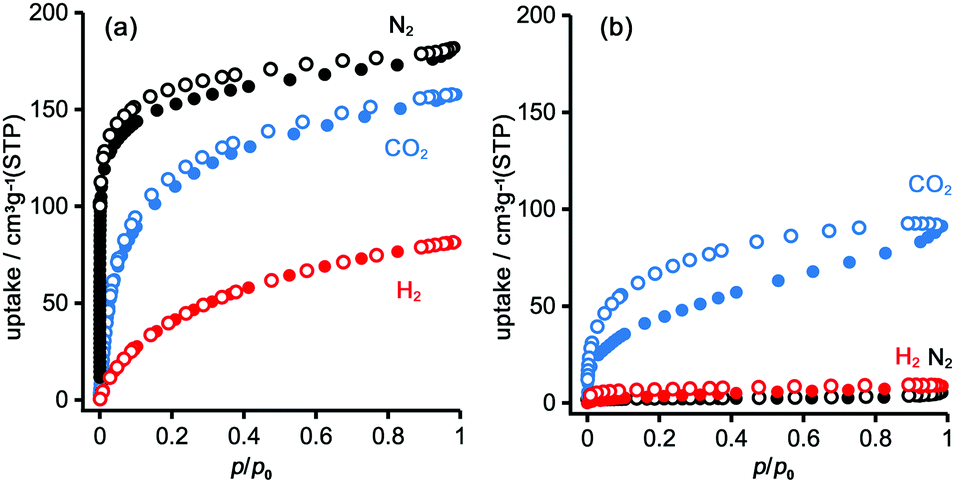 | ||
| Fig. 8 Gas adsorption isotherms of (a) TpMe-apo and (b) TpF-apo for CO2 (cyan) at 195 K, H2 (red) at 77 K and N2 (black) at 77 K. Solid and open symbols denote adsorption and desorption processes, respectively. | ||
Fluorescence properties in solid states
The photochemical properties of LA-H-HexNets of TpMe and TpF were investigated before and after desolvation. Fig. 9 shows the fluorescence and excitation spectra of the as-formed crystals (TpMe-2Ds and TpF-1) and the corresponding desolvated materials (TpMe-apo and TpF-apo). TpMe-2Ds exhibits a structureless emission band at 407 nm (ϕF = 0.17, where ϕF denotes the fluorescence quantum yield), which was slightly red-shifted by 4 nm after desolvation (λmaxem = 411 nm, ϕF = 0.09 for TpMe-apo). Excitation spectra are also red-shifted from 373 nm to 388 nm, indicating that intermolecular interactions between the adjacent triphenylene cores in TpMe-apo are stronger than those in TpMe-2Ds. TpF-apo also exhibits a structureless emission band at 423 nm (ϕF = 0.23), which was red-shifted by 12 nm compared with TpF-1 (λmaxem = 411 nm, ϕF = 0.16). The excitation maxima of TpF-1 and TpF-apo are 381 nm and 394 nm, respectively.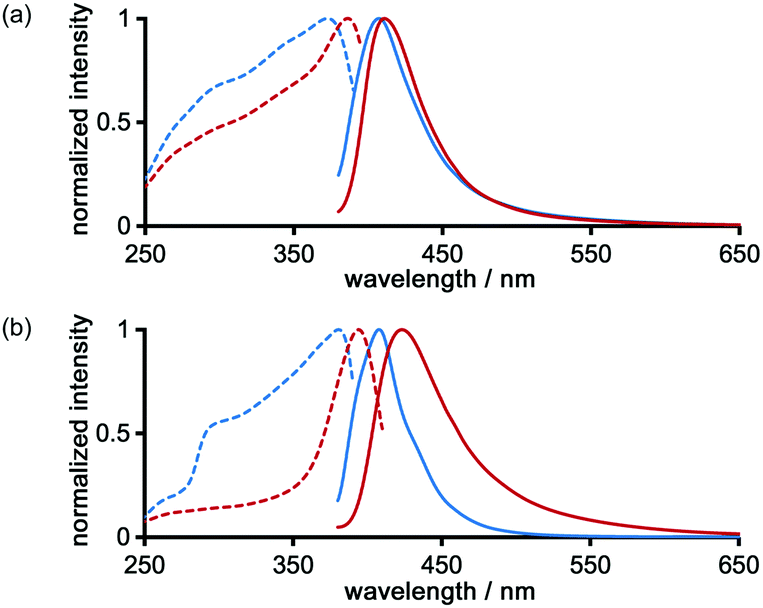 | ||
| Fig. 9 Solid state excitation (dashed line) and emission (solid line) spectra of (a) TpMe-2Ds and (b) TpF-1 before (blue) and after (red) desolvation [excitation wavelength (λex) = 365 nm; detected wavelength = λmaxem]. | ||
Conclusions
In this paper, we synthesized two derivatives of hexakis(carboxyphenyl)triphenylene TpMe and TpF, in which substituents (Me or F) were introduced in the ortho-positions of the carboxy groups, and investigated the substituent effects on the crystal structures of the LA-H-HexNets, thermal stability, and retention ability of permanent porosity. As a result, we revealed the following important features of the present system. (1) Substituent groups have no effects on the binding energy of hydrogen-bonded dimerization (∼15 kcal mol−1). (2) Methyl and fluorine substituents in the ortho-positions of the carboxy groups made both carboxy and phenylene groups twisted, resulting in an increase of variability of peripheral conformation. (3) Depending on the bulkiness of the substituents, TpMe yielded three polymorphs, while TpF yielded one crystalline form with highly disordered peripheral groups. (4) Thermal stability and permanent porosity were drastically changed by the substituents despite the fact that both the crystals have quite similar LA-H-HexNet structures. These results can contribute to the development of functional HOFs with tunable pores based on H-HexNet frameworks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by a Grant-in-Aid for Scientific Research (C) (JPT15K04591) and for Scientific Research on Innovative Areas: π-System Figuration (JP15H00998) from MEXT Japan. Synchrotron radiation experiments were undertaken at BL38B1 in SPring-8 with the approval of JASRI (proposal No. 2015B1397, 2016A1121, and 2016B1151).Notes and references
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- (a) G. R. Desiraju and T. Steiner, The weak hydrogen bond, Oxford University Press Inc., New York, 1999 Search PubMed; (b) O. Ivasenko and D. F. Perepichka, Chem. Soc. Rev., 2011, 40, 191 RSC.
- (a) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed; (b) C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS PubMed.
- (a) G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931 RSC; (b) Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613 RSC; (c) N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8704 CrossRef CAS PubMed; (d) Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698 CrossRef CAS PubMed; S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031 Search PubMed.
- (a) T. Kambe, R. Sakamoto, T. Kusamoto, T. Pal, N. Fukui, K. Hoshiko, T. Shimojima, Z. Wang, T. Hirahara, K. Ishizaka, S. Hasegawa, F. Liu and H. Nishihara, J. Am. Chem. Soc., 2014, 136, 14357 CrossRef CAS PubMed; (b) J. Guo., Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramono, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 Search PubMed; (c) M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920 CrossRef CAS PubMed; (d) M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802 CrossRef CAS PubMed.
- M. Zhao, Q. Lu, Q. Ma and H. Zhang, Small Methods, 2017, 1, 1600030 CrossRef.
- (a) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC; (b) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC; (c) N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- (a) J. Lu and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 9474 CrossRef PubMed; (b) A. R. A. Palmans, J. A. J. M. Vekemans, H. Kooijman, A. L. Spek and E. W. Meijer, Chem. Commun., 1997, 2247 RSC; (c) P. Sozani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816 CrossRef PubMed; (d) X.-Z. Luo, X.-J. Jia, J.-H. Deng, J.-L. Zhong, H.-J. Liu, K.-J. Wang and D.-C. Zhong, J. Am. Chem. Soc., 2013, 135, 11684 CrossRef CAS PubMed; (e) T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. Š. Miljanić, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed; (f) P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. O’Keeffe, Y. Han and B. Chen, Angew. Chem., Int. Ed., 2015, 54, 574 CAS.
- (a) D. J. Duchamp and R. Marsh, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 5 CrossRef CAS; (b) F. H. Herbstein, M. Kapon and G. M. Reisner, J. Inclusion Phenom., 1987, 5, 211 CrossRef CAS; (c) K. Kobayashi, T. Shirasaka, E. Horn and N. Furukawa, Tetrahedron Lett., 2000, 41, 89 CrossRef CAS; (d) I. Hisaki, N. Q. E. Affendy and N. Tohnai, CrystEngComm, 2017, 19, 4892 RSC.
- (a) C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. FitzGerald and J. L. C. Rowsell, Chem. Commun., 2015, 51, 11642 RSC; (b) S. Nandi, D. Chakraborty and R. Vaidhyanathan, Chem. Commun., 2016, 52, 7249 RSC; (c) F. Hu, C. Liu, M. Wu, J. Pang, F. Jiang, D. Yuan and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 2101 CrossRef CAS PubMed; (d) I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Chem. – Eur. J., 2017, 23, 11611 CrossRef CAS PubMed.
- (a) I. Hisaki, S. Nakagawa, N. Tohnai and M. Miyata, Angew. Chem., Int. Ed., 2015, 54, 3008 CrossRef CAS PubMed; (b) I. Hisaki, N. Ikenaka, N. Tohnai and M. Miyata, Chem. Commun., 2016, 52, 300 RSC; (c) I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, J. Am. Chem. Soc., 2016, 138, 6617 CrossRef CAS PubMed; (d) I. Hisaki, S. Nakagawa, H. Sato and N. Tohnai, Chem. Commun., 2016, 52, 9781 RSC; (e) I. Hisaki, H. Toda, H. Sato, N. Tohnai and H. Sakurai, Angew. Chem., Int. Ed., 2017, 56, 15294 CrossRef CAS PubMed.
- (a) M. Mastalerz and I. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252 CrossRef CAS PubMed; (b) W. Yan, X. Yu, T. Yan, D. Wu, E. Ning, Y. Qi, Y.-F. Han and Q. Li, Chem. Commun., 2017, 53, 366 Search PubMed.
- (a) B. Ośmilałowsky, E. Kolehmainen, R. Gawinecki, R. Dobosz and R. Kauppinen, J. Phys. Chem. A, 2010, 114, 12881–12887 CrossRef PubMed; (b) B. Ośmilałowsky, E. Kolehmainen and M. Kowalska, J. Org. Chem., 2012, 77, 1653 CrossRef PubMed.
- B. Ośmilałowsky, E. Kolehmainen, R. Gawinecki, R. Kauppinen, J. Koivukorpi and A. Valkonen, Struct. Chem., 2010, 21, 1061 CrossRef.
- Rigaku Oxford Diffraction (2015), Software CrysAlisPro 1.171.38.41o, Rigaku Corporation, Tokyo, Japan.
- HKL2000. Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- Rigaku (2015), CrystalStructure. Ver. 4.2, Rigaku Corporation, Tokyo, Japan.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- (a) P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef; (b) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- B. J. Ransil, J. Chem. Phys., 1961, 34, 2109 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2009, 130, 194703 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1578414–1578417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00544j |
| This journal is © the Partner Organisations 2018 |