
Influence of catalytic systems in Stille polymerization on the electrochromic performance of diketopyrrolopyrrole-based conjugated polymers†
Wei Teng
Neo
ab,
Qun
Ye
a,
Zugui
Shi
a,
Soo-Jin
Chua
ac and
Jianwei
Xu
 *ad
*ad
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, 138634, Singapore. E-mail: jw-xu@imre.a-star.edu.sg
bNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, 28 Medical Drive, 117456, Singapore
cDepartment of Electrical and Computer Engineering, National University of Singapore, 4 Engineering Drive 3, 117583, Singapore
dDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore
First published on 13th December 2017
Abstract
Two diketopyrrolopyrrole- (DPP) and thienothiophene-based donor–acceptor (D–A) type conjugated polymers are separately synthesized using Stille coupling reactions with Pd(PPh3)4 and PdCl2(PPh3)2 catalytic systems, respectively. The influence of the catalyst employed on the structural regularity of the resulting polymers, and their subsequent electrochromic (EC) performances are investigated. Marked differences in the optical contrasts, switching times, coloration efficiencies and redox stabilities of the polymers are uncovered. For example, polymer P1, which is prepared using Pd(PPh3)4 as a catalyst, exhibited a larger optical contrast and coloration efficiency by 40% and 79%, respectively, and a faster coloration time by 55%, at 825 nm, than its counterpart polymer P2, which is prepared using PdCl2(PPh3)2. The intrinsic optical, electrochemical and morphological properties of the polymers are characterized, revealing that the physical conformation and packing of the polymer chains are the main factors causing the discrepancy in EC performances.
Introduction
Electrochromic (EC) polymers belong to a class of smart materials that respond to an electrical stimulus by displaying colour or optical change reversibly. Such materials are highly suitable for numerous consumer products such as windows,1 electronic displays,2 and wearables like ophthalmic glasses3 and chameleonic fabrics.4Through the collective efforts of various research groups, EC polymers that display a wide range of hues and can operate with high optical contrasts, rapid switching speeds, good stabilities and low power consumptions are available today.5–8 This is achieved through ingenious chemical structural modifications on the polymers, which have been made possible following the continuous study and understanding of the structure–property relationship.9–17 Based on current knowledge, the colour-switching capability of EC polymers is highly dependent on both their chemical and physical properties. Besides the intrinsic chemical composition, bandgap and frontier molecular orbital energies, which affect the polymer's susceptibility towards redox reaction and degradation,18 as well as its ability to stabilize charged, doped states,19 the polymer chain packing and thin film morphology20,21 are also critical as they are expected to affect the manner in which the active polymeric layer interacts with constituting components such as the electrolyte during operation. To this end, the structural regularity and molecular weight of the polymers may thus be the key underlying factors which have not yet been well established.
In other areas of organic electronics research, the impact of structural regularity and molecular weights of conjugated polymers on device performances has been tremendous.22–24 For instance, the presence of intrachain homocoupling defects in polymer chains has, in general, led to detrimental effects on the performance of photovoltaic cells.23,25–27 Similarly, while an increase in molecular weight has largely been observed to yield higher solar cell power conversion efficiency,28–30 its effect on the charge carrier mobilities of organic thin-film transistors is more conflicting as reports documenting either its positive31 or negative influence32 have surfaced. Curiously, there has been, however, no similar study in the field of electrochromics. Hence, it will be of interest to explore and exact such influences.
Stille coupling reactions widely employed in the synthesis of conjugated polymers are conventionally assumed to occur in an ideal, perfect fashion where new carbon–carbon bonds are formed only between stannanes and halides. Nonetheless, studies have shown that homocoupling occurs rather readily during the polymerization process, with the extent of induced structural defects dependent on the catalytic system employed.33 In this work, we focused our attention on a diketopyrrolopyrrole (DPP)-based D–A copolymer (DPPT–TT)31,34 as the electrochromic material. To study the influence of the structural regularity of the polymers on their electrochromic properties, copolymers P1 and P2 were synthesized by employing different catalysts – Pd(PPh3)4 and PdCl2(PPh3)2, respectively – for their polymerization reactions.
Results and discussion
Synthesis and polymer characterization
The synthetic routes for polymers P1 and P2 are shown in Scheme 1. Tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] and bis(triphenylphosphine)palladium(II) dichloride [PdCl2(PPh3)2], commonly used in the synthesis of conjugated polymers via Stille coupling, were selected as catalysts for the polymerization reaction of monomers 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-octyldodecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (Br-T-DPP-T-Br) (1) and 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene (Me3Sn-TT-SnMe3) (2) to yield P1 and P2, respectively. It has been well recognized that the amount and type of catalyst play a critical role in the control of polymer structural regularity and molecular weight.31,33,35,36 In particular, structural defects have been reported in many DPP-based polymers in the literature. These structural defects are formed by the side reaction of the Stille coupling polymerization involving homocoupling in the Pd-catalyzed carbon–carbon formation reaction.37–39 For instance, the presence of an excess amount of Pd(II) catalyst employed for P2 will consume part of the distannyl monomer (monomer 2: Me3Sn-TT-SnMe3) during the initiation step of the Stille coupling reaction, leading to a stoichiometric imbalance of the participating monomers (a greater amount of monomer 1 (Br-T-DPP-T-Br) compared to that of monomer 2) and hence a higher degree of structural defects consisting of mainly TT-TT segments and therefore lower molecular weights in the resulting polymers. The effect of the type of catalyst on the structural defects in T-DPP-T based copolymers has also been established, evidently revealing that the T-DPP-T based polymer prepared using the Pd(PPh3)4 catalyst shows better structural regularity (fewer defects) than those prepared using Pd(PPh3)2Cl2 and Pd2(dba)3/P(o-tol)3 catalysts.33 The temperature-dependent 1H NMR spectra of two polymers P1 and P2 in deuterated chlorobenzene were examined, but no clear evidence on their structural regularity was confirmed due to the difficulty in identifying and then extracting defect segment aromatic protons from overlapped signals of thiophene and thieno[3,2-b]thiophene moieties as well as solvent. Although we do not have direct evidence on the structural regularity of polymers P1 and P2, indirect proof in terms of a reduction of the bandgap, a higher highest occupied molecular orbital (HOMO) level, and a lower lowest unoccupied molecular orbital (LUMO) level (vide infra)23,40 strongly supports that polymer P1 has fewer defects than P2 in their respective polymer backbones. In this work, gel permeation chromatography reveals different molecular weights for the polymers with P1 having Mn and Mw values that are approximately 30% and 50% larger (Table 1). While both polymers reveal comparable solubility in halogenated solvents such as chloroform and chlorobenzene, the difference in solubility is markedly observed when employing solvents such as tetrahydrofuran. While P2 is readily soluble at a concentration of 5 mg mL−1 under stirring at room temperature, P1 is only sparingly soluble even at elevated temperatures.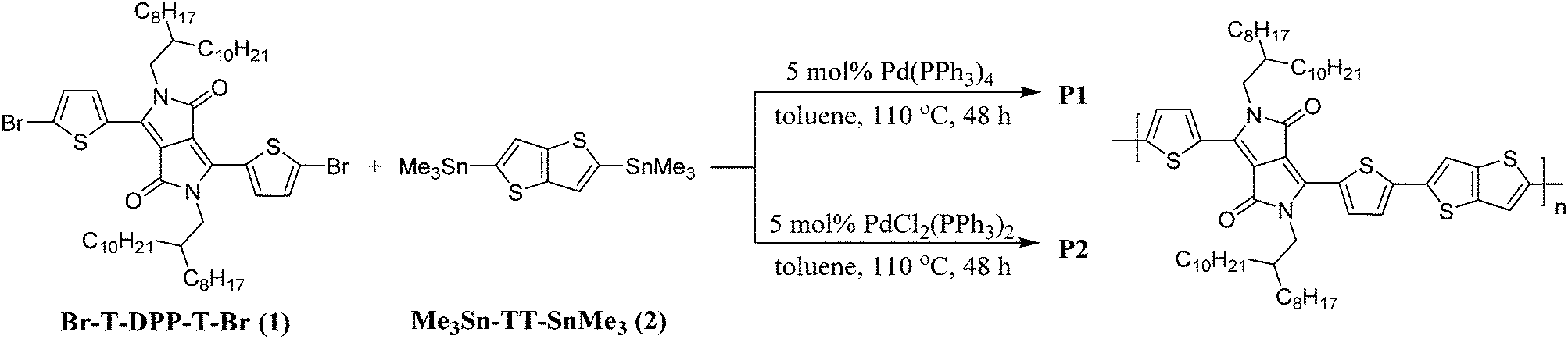 | ||
| Scheme 1 Synthetic routes leading to polymers P1 and P2. | ||
Optical and electrochemical properties
The optical behaviours of polymers P1 and P2 were probed using UV-vis spectroscopy. The molar absorptivities, ε, of the polymers were approximated using the mass of the repeating unit of the polymers for the computation of their concentrations. As illustrated in Fig. 1a, the molar absorptivities of the polymers in dilute chlorobenzene are noticeably different. Compared to P2, the molar absorptivity of P1 is about 60% larger (83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) than that of P2 (53000 M−1 cm−1) at 824 nm (near their absorption maxima). The difference in the molar absorptivities of the polymers can likely be attributed to the variations in the distributions of their physical conformations.41 While the higher molecular weight polymer P1 is expected to exist as more linear chains due to better chain-to-chain interactions, P2 may largely adopt a coiled structure as a result of greater chain–solvent interactions. While the oscillator strength is typically the highest in the lowest electronic transition (corresponding to the peak at 825 nm in this case) of linear polymer chains, it is transferred to higher electronic transitions (lower wavelengths) in coiled polymer chains.42
000 M−1 cm−1) than that of P2 (53000 M−1 cm−1) at 824 nm (near their absorption maxima). The difference in the molar absorptivities of the polymers can likely be attributed to the variations in the distributions of their physical conformations.41 While the higher molecular weight polymer P1 is expected to exist as more linear chains due to better chain-to-chain interactions, P2 may largely adopt a coiled structure as a result of greater chain–solvent interactions. While the oscillator strength is typically the highest in the lowest electronic transition (corresponding to the peak at 825 nm in this case) of linear polymer chains, it is transferred to higher electronic transitions (lower wavelengths) in coiled polymer chains.42
 | ||
| Fig. 1 (a) Molar absorptivities of P1 and P2 in dilute chlorobenzene. (b) Normalized UV-vis absorption spectra of P1 and P2 in chlorobenzene (solid lines) and as thin films (dashed lines). | ||
The variations in the absorption behaviours of the polymers in their solution states can be distinctly seen from the normalized absorption profiles (Fig. 1b), suggesting differences in their conjugated backbone structures as well. While P2 exhibits an expected blue shift in the absorption maximum compared to P1 (810 vs. 825 nm) probably due to its relatively lower molecular weight, more curious broadening and longer tailing of its absorption into the near-infrared (NIR) region were also observed. This is accompanied by the presence of a low-energy shoulder which is a signature of undesirable homocoupled DPPT–TDPP defects (coupling of monomer 1) along the polymer chain.23 Moreover, the pronounced and dramatic broadening and bathochromic-shift of the absorption of P2 in its thin film state compared to the solution state seem to confirm the presence of homocoupling defects. While shifts in the absorption maximum and onset of 75 and 84 nm were observed, respectively, the major absorption band of P2 broadens from the 680–890 nm to 600–960 nm range. This causes P2 to possess a smaller band gap.
The redox behaviours of P1 and P2 thin films were also probed, and both polymers were found to reveal two quasi-reversible redox couples under oxidative potentials (Fig. S5, ESI†). In general, the redox profiles are comparable although sharper and more distinct peaks are observed in P1. This indicates that the oxidation and reduction processes can be completed within a smaller potential window. For both polymers, the onset potentials for oxidation are found to be similar. Based on the onset potentials, the HOMO level of P1 lies at −5.19 eV, while that of P2 is slightly deeper at −5.20 eV. Moreover, from the HOMO level and the measured optical bandgap, P2 exhibits a LUMO level that is deeper than P1 (−3.99 eV vs. −3.84 eV), providing additional evidence that P2 has a larger extent of structural irregularity compared to P1. In addition, the presence of electron-withdrawing Br groups at the terminal ends of the polymer chains due to the excess amount of the bromine-containing monomer in the Stille polymerization reaction as discussed earlier may partially contribute to the lower LUMO level of P2. A summary of the optical and electrochemical properties of the polymers is shown in Table 2.
| Polymer | Molar absorptivity, εa (M−1 cm−1) | λ max (nm) | λ onset (nm) |
E
optgb (eV) |
E ox,onset (V) | HOMOd (eV) | LUMOe (eV) | ||
|---|---|---|---|---|---|---|---|---|---|
| Solution | Film | Solution | Film | ||||||
| a Values are calculated at 824 nm in dilute chlorobenzene. b E optg = 1240/λonset,film. c Values are calculated vs. ferrocene. d E HOMO = −(Eox,onset vs. ferrocene) – 4.8 eV. e E LUMO = EHOMO + Eoptg. | |||||||||
| P1 | 83000 |
424, 749 (sh), 825 | 426, 744, 821 | 899 | 916 | 1.35 | 0.39 | −5.19 | −3.84 |
| P2 | 53000 |
431, 810 | 435, 885 | 938 | 1022 | 1.21 | 0.40 | −5.20 | −3.99 |
Thin film morphology
The surface morphologies of spin-coated P1 and P2 films were imaged using atomic force microscopy (AFM) and Fig. 2a and b show the respective height images. The morphology of P1 generally adopts a loosely-packed structure with amorphous regions surrounding highly aligned crystallites which exist as well-defined fibrillar-like structures. In contrast, such features are absent in the P2 film, which is largely composed of aggregated, micron-sized globules with no specific directionality. The loss of the fibrillar motifs in P2 may presumably be due to the “impurities” arising from the defect conjugated segments which disrupt the effective packing and interchain interactions among neighbouring polymer chains. Further evidence highlighting the difference in morphologies between the polymer films can be seen from the height plots obtained from surface profilometry studies (Fig. 2c and d). While the film thicknesses of both polymers are around the same (P1: 157 nm and P2: 155 nm), the surface topographies are drastically different. The P1 film is significantly smoother with vertical displacements in the range of ±10 nm from the mean thickness, whereas for P2 variations as large as ±50 nm are observed.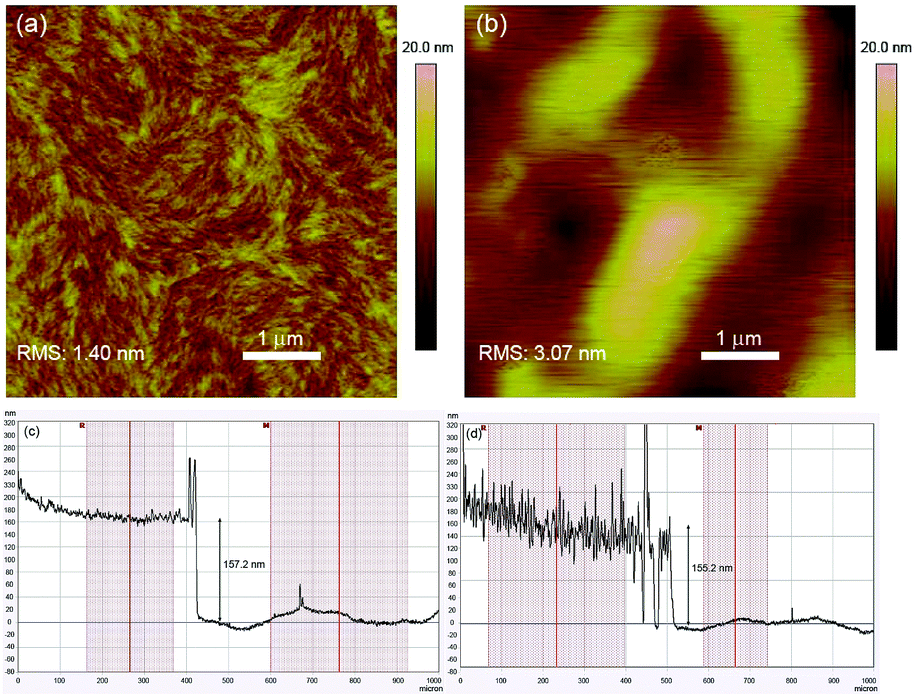 | ||
| Fig. 2 2D AFM height images of (a) P1 and (b) P2 thin films. Surface profiles (height plots) of (c) P1 and (d) P2 thin films. | ||
X-ray diffraction (XRD) was subsequently carried out on drop-cast polymer films and the recorded spectra are illustrated in Fig. S6 (ESI†). For both polymers, distinct (100) reflections were observed and the 2θ angles corresponding to the peak maxima are highly similar (P1: 4.3° and P2: 4.4°). The in-plane d-spacings (d(100)) for P1 and P2 were thus calculated to be 20.5 nm and 20.1 nm respectively. While the peak corresponding to the secondary (200) reflection is less obvious in P2, it is highly visible for P1 at 8.6°. This indicates a higher degree of molecular ordering and crystallinity in P1, which could have been brought on by the stronger interactions and alignment among the linear polymer chains43 as a consequence of the larger extent of structural regularity.
Electrochromic performance
The electrochromic properties of P1 and P2 were investigated in absorption/transmission type electrochromic devices (ECDs). For a fair analysis of the electrochromic behaviours of the polymers, the mass of the active polymeric layer was kept constant for both polymers at around 0.10 mg (see the Experimental section).The spectroelectrochemical graphs of the polymers are illustrated in Fig. 3a and b. In their neutral states, P1 and P2 possess the characteristic dual absorption bands and display either green or bluish-green hues. The prominent difference between the polymers is the optical densities of their films as a result of the difference in their molar absorptivities as discussed earlier. For similar film thicknesses, the absorbance is as high as 0.9 a.u. for P1, while P2 manages only a value of around 0.6 a.u. Under progressive oxidation, the visible-wavelength absorptions begin to deplete steadily, while a new NIR absorption band peaking at around 1285 nm is formed concurrently. In their fully oxidized states, both polymers reveal a transmissive grey shade. An illustration of the device in its neutral and oxidized states is provided using P1, and the photographs are shown in Fig. 3c. The measured colorimetric values of both P1 and P2 are also presented (Fig. 3d).
 | ||
| Fig. 3 Spectroelectrochemical graphs of (a) P1 and (b) P2 ECDs at various applied potentials. (c) Photographs of the fabricated P1 ECD and (d) comparison of colours and L*, a*, b* colour coordinates of P1 and P2 devices in their neutral and oxidized states. | ||
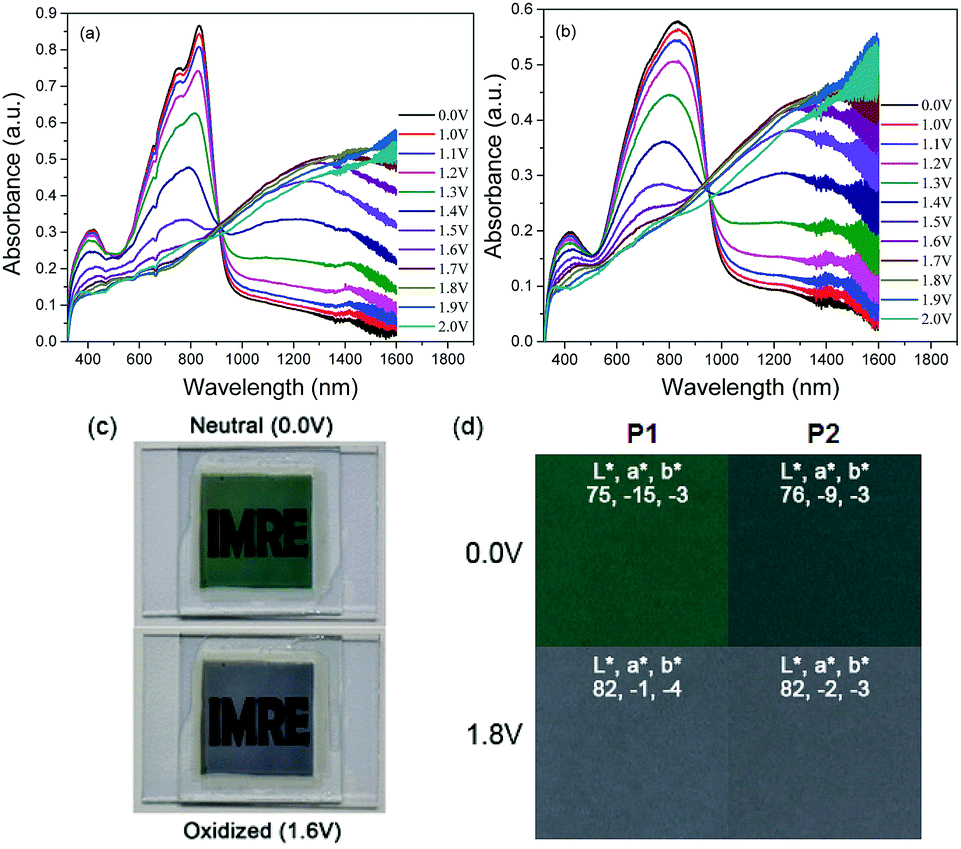
The switching properties of P1 and P2 ECDs were then probed using in situ square-wave potential step absorptiometry. The colour-changing behaviours were monitored at both 825 and 1285 nm, and potential pulses of +1.6 and −1.6 V were employed. Fig. 4 shows the switching cycles of the polymers. Evidently, P1 exhibited superior optical contrasts. Transmittance changes as large as 36 and 49% were at 825 and 1285 nm, respectively, which are approximately 1.3–1.4 times those of P2 (25 and 38%). By measuring the colour contrast using the absolute changes in absorbances at 825 nm (P1: Δabs = 0.64 a.u. and P2: Δabs = 0.35 a.u.) and taking into account the difference in molar absorptivities, P1 still reveals a higher degree of optical change. This seems to suggest that the ability of the polymer to convert from neutral to charged species may be related to its physical conformation, particularly its linearity and structural regularity. In this case, the more linear and crystalline P1 may have enabled a higher degree of polaron or bipolaron delocalization and stabilization under oxidation, allowing positively-charged species to reside along the polymer chain. On the other hand, the structural defects on P2 may have acted as charge traps,23 which reduce the charge movement and delocalization.
 | ||
| Fig. 4 Switching cycles of (a) P1 and (b) P2 ECDs cycled between +1.6 and −1.6 V at 825 and 1285 nm. | ||
P1 was also found to exhibit faster switching speeds and higher coloration efficiencies compared to P2. By and large, the switching times were reduced by around 1–4 s, while the coloration efficiencies were found to be larger by 99–293 cm2 C−1. The favourable electrochromic properties of P1 over P2 can most likely be attributed to its more open and porous morphology which provides not only more redox-active polymer sites but also most facilitated ion movement. To ensure the reproducibility and reliability of these findings, three separate trials were carried out for each polymer at the respective wavelengths. A summary of the electrochromic performance of P1 and P2 ECDs is presented in Table 3.
| Polymer | λ 1 (825 nm) | λ 1 (1285 nm) | ||||||
|---|---|---|---|---|---|---|---|---|
| OCb (%) | τ b (s) | τ c (s) | CEe (cm2 C−1) | OCb (%) | τ b (s) | τ c (s) | CEe (cm2 C−1) | |
| a Statistics reported are averages and standard deviations are based on 3 trials. b Optical contrast. c Bleaching time, where bleaching refers to the process in which the percent transmittance changes from a lower to higher value. d Coloration time, where coloration refers to the process in which the percent transmittance changes from a higher to lower value. e Coloration efficiency. | ||||||||
| P1 | 35 ± 1 | 40.6 ± 1.3 | 2.2 ± 0.4 | 665 ± 48 | 47 ± 2 | 4.1 ± 0.3 | 27.1 ± 1.1 | 503 ± 38 |
| P2 | 25 ± 0 | 41.8 ± 0.9 | 3.4 ± 0.4 | 372 ± 15 | 38 ± 0 | 6.1 ± 0.5 | 31.8 ± 0.7 | 404 ± 2 |
The redox stabilities of P1 and P2 were subsequently analyzed by measuring the changes in optical contrasts at 1285 nm as the devices were subjected to repeated cycling between +1.6 and −1.6 V with a cycling time of 20 s. Both polymers reveal similar natures of degradation in the initial stages (Fig. S7, ESI†), with the optical contrast decreasing steadily during the first 50 redox cycles. Beyond the 50th cycle, the optical contrast of P1 remains relatively constant and about 60% of the initial contrast is sustained after a total of 600 cycles. On the other hand, P2 is observed to degrade continuously up to the 150th cycle before levelling off. At the end of 600 potential cycles, approximately 50% of the initial contrast is sustained.
A closer look at the degradation behaviours of P1 and P2 (monitored in the NIR region at 1285 nm) reveals that the gradual loss in optical contrasts of the polymers arises from their inability to be oxidized to the level of the preceding cycles. As seen from Fig. 5a with P1 as the illustration, while the transmittance values in the reduced state remains relatively constant throughout the entire cycling period, the transmittance values in the oxidized state is found to increase over repeated cycles. Evidently, one of the major factors causing the loss in optical contrasts is the slowing down of the switching speeds such that the 20 s cycling time is not sufficient for a complete switch (Fig. 5b). This is confirmed by a set of experiments (Fig. 5c) using P1 as an example. When the duration of electrical bias is gradually extended from 20 to 80 s after the device has seemingly degraded after undergoing 50 redox cycles, the optical contrast attainable becomes closer to the initial maximum value. The above observation seems to suggest that the loss in the electrochromic performances of the polymers in the initial stages may stem from an impediment in the physical processes involved and less of an irreversible chemical structural change in the polymer (such as the breaking of conjugation in the polymer backbone44) which has commonly been accepted as one of the key degradation mechanisms. To account for the degradation behaviour of the electrochromic polymers, a reasoning which has also recently been proposed by Xu's group45 is provided. During the polymer doping/de-doping processes, charge balancing of the system is achieved by the ingress and egress of perchlorate ClO4− ions. One of the plausible pathways in which the susceptibility towards the oxidative (doping) process is hindered is the physisorption of the counterions onto the electrically-active sites, which reduces the number of addressable sites available for the next redox cycle. Moreover, the adhesion and trapping of the bulky counterions may block the movement of other ions present within the electrolyte. The probability of such occurrences may be similar for both polymers in the initial stages, which resulted in similar rates of loss in optical contrasts. Over time, the more compact morphological structure of P2 may have experienced greater charge trapping compared to that of P1, giving rise to an overall higher amount of inactive polymer sites and greater degree of optical contrast loss (50% vs. 40%). As observed in Fig. 5c, increasing the cycling time may have allowed more time for the trapped ions to diffuse away or mobile ions to diffuse to electrically-addressable polymer sites, enabling more redox reaction and a greater extent of electrochromic switching.
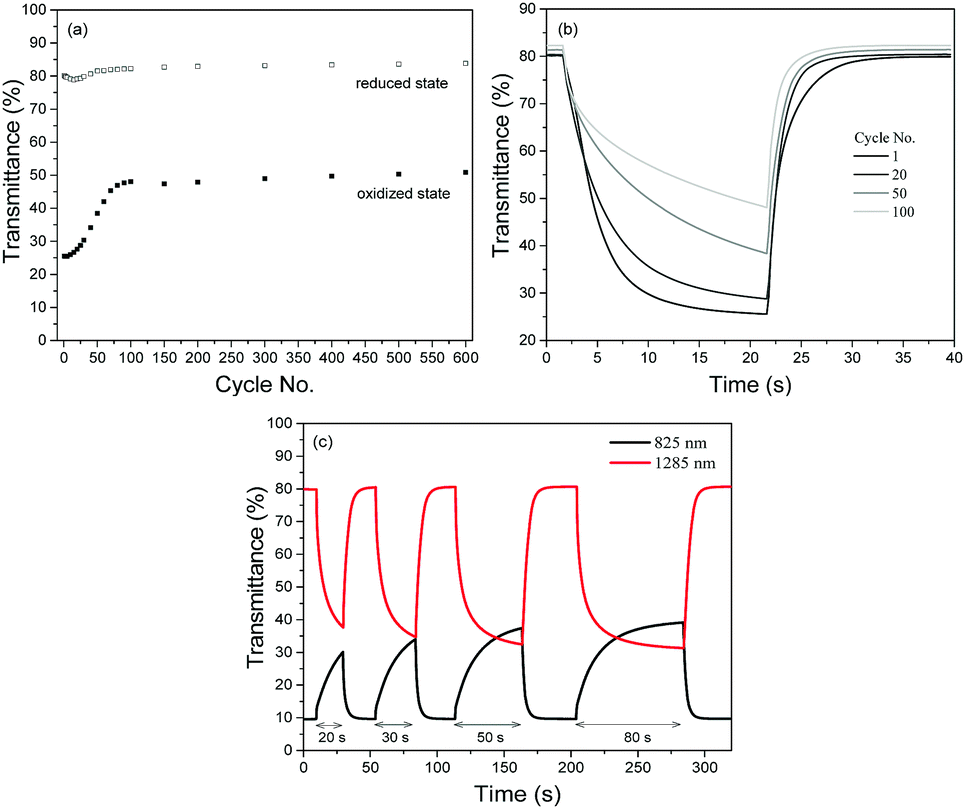 | ||
| Fig. 5 (a) Evolution of the percent transmittances of P1 ECDs in their reduced and oxidized states monitored at 1285 nm when subjected to repeated redox cycles between +1.6 V and −1.6 V. (b) Switching behaviour of P1 ECDs with cycle numbers 1, 20, 50 and 100 for a residence time of 20 s. (c) Evolution of the optical contrasts of P1 ECDs at 825 and 1285 nm when subjected to repeated redox cycles with increasing residence times between +1.6 V and −1.6 V. The data were recorded after the device underwent 50 repeated redox cycles between +1.6 V and −1.6 V. | ||
Conclusions
DPP-based polymers were obtained from Stille polymerization processes with two types of catalysts, leading to polymers with varying structural regularities. This in turn resulted in polymers displaying drastically different optical and morphological properties, and hence dissimilar electrochromic performances. In general, the experimental findings point to the conclusion that the polymer with a higher molecular weight (P1) and a lesser extent of coupling defects fares better in terms of optical contrasts, switching speeds, coloration efficiencies and redox stability. The major factor accounting for the differences in electrochromic properties is believed to be the type of physical conformation and packing nature adopted by the polymers in their solid states. For P1 which exists largely as linear chains with a higher degree of crystallinity in its thin film form, more favourable intrinsic properties such as stronger molar absorptivity and greater susceptibility towards redox reaction are revealed. More critically, the well-ordering and alignment of the polymer chains enable the film morphology to adopt a homogeneously structured yet loosely-packed configuration with amorphous regions surrounding the defined fibrillar-like crystallites, providing more redox-active polymer sites and unobstructed pathways for ion movement. In contrast, the polymer chains of P2 are more coiled up and aggregate strongly into micron-sized globules in its solid state. The compact film structure not only reduces the number of active polymeric sites per unit volume but also causes more significant charge trapping during the electrochromic operation, leading to comparatively poorer redox cyclability. These findings on the relationship between polymer regularity, polymer chain packing and electrochromic performances would be very useful as guidelines for the choice of synthetic methods to prepare electrochromic conjugated polymers with desired electrochromic properties.Experimental
Materials
3,6-Bis(5-bromothiophen-2-yl)-2,5-bis(2-octyldodecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1) was prepared according to the literature.46 Other chemicals were purchased from Sigma-Aldrich and were used without further treatments. ITO-coated glass substrates (15 Ω sq−1, 35 × 30 × 1.1 mm) were purchased from Xinyan Technology Ltd.Synthesis of polymers
Tetrakis(triphenylphosphine)palladium (0) (0.005 mmol, 5.5 mg), 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-octyldodecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1) (0.1 mmol, 102 mg) and 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene (2) (0.1 mmol, 47 mg) were dissolved in degassed dry toluene (5 mL) under nitrogen protection. The reaction was then sealed, stirred and heated at 110 °C for 48 h. The hot polymer solution was precipitated into methanol and stirred overnight. A black solid polymer was collected by filtration, followed by successive Soxhlet extraction with methanol, ethyl acetate, hexane and chloroform. The chloroform portion was then concentrated to a few millilitres and precipitated into methanol. After filtration and drying in a vacuum oven, pure polymer P1 was obtained. Polymer P2 was obtained using a similar procedure with bis(triphenylphosphine) palladium(II) dichloride (0.005 mmol, 3.5 mg) as the catalyst. 1H NMR (CDCl3, 400 MHz) δ = 9.35–8.60 (br), 7.15–6.42 (br), 4.14–3.16 (br), 2.10–0.94 (m), 0.90–0.65 (m).Electrochromic device fabrication
ITO/glass substrates were cleaned by successive ultrasonication in acetone, isopropyl alcohol and distilled water, and blown dry with nitrogen prior to use. Stock polymer solutions of P1 and P2 were prepared at concentrations of 10 and 15 mg mL−1, respectively, in a mixed chloroform:chlorobenzene (4:6 v/v) solvent system. Thin polymer films on the ITO/glass substrates were obtained by spin-coating at 800 rpm for 60 s. To yield an active area of 2 × 2 cm2, extra polymer edges were removed by swabbing with chloroform. The final mass of the polymer film was kept constant at around 0.10 mg. On another piece of the ITO/glass substrate, Parafilm which acts as both a spacer and a barrier layer was lined along a 2 × 2 cm2 area. Within the blocked-out area, 250 μL of the gel electrolyte (0.512 g of lithium perchlorate and 2.8 g of poly(methyl methacrylate) (MW = 120 000 g mol−1) in 6.65 mL of propylene carbonate and 28 mL of dry acetonitrile (ACN)) was dispensed and left to dry for 5 minutes. The device was assembled on the bench-top by sandwiching both ITO/glass substrates together with the polymer film and gel electrolyte in contact.
Instrumentation
Gel permeation chromatography was carried out at an elevated temperature of 160 °C using an Alliance PL 200 system, with 1,2,4-trichlorobenzene as the eluent and PMMA as the standard. Measurements of the mass of the active polymer layer were performed on a Sartorius MC5 microbalance. All UV-vis-NIR absorption spectra were recorded on a Shimadzu UV-3600 UV-vis-NIR spectrophotometer. An Autolab PGSTAT128N potentiostat was employed for cyclic voltammetry experiments. In the set-up, a three-electrode cell configuration with a polymer-coated glassy carbon electrode, Pt wire and Ag wire as the working, counter and pseudo-reference electrodes, respectively, was utilized. A 0.1 M LiClO4/ACN electrolyte/solvent couple was employed and all measurements were recorded at a scan rate of 50 mV s−1. Calibration of the pseudo-reference electrode was done against the ferrocene/ferrocenium redox couple. All electrochromic characterization studies were performed in situ, using both the spectrophotometer and potentiostat. Atomic force microscopy (AFM) images were obtained in the tapping mode with a Veeco Dimension 3100 atomic force microscope. Polymer films for AFM analysis were prepared by spin-coating the polymer solutions onto ITO/glass substrates as described in the ‘Electrochromic device fabrication’ section. A KLA Tencor Alpha-Step D-120 surface profiler was employed for surface profilometry studies. X-ray diffraction (XRD) was performed in reflection mode with a Cu-Kα radiation source (λ = 0.15406 nm) on a Bruker D8 General Area Detector Diffraction System. The CIE L*, a*, b* values of the devices were measured on a Hunterlab ColorQuest XE and were reported under outdoor daylight illumination (65/10°).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Agency for Science, Technology and Research (A*STAR) and the Ministry of National Development (MND) Green Building Joint Grant (No. 1321760011).References
- H. Shin, S. Seo, C. Park, J. Na, M. Han and E. Kim, Energy Environ. Sci., 2016, 9, 117–122 Search PubMed.
- T. Xu, E. C. Walter, A. Agrawal, C. Bohn, J. Velmurugan, W. Zhu, H. J. Lezec and A. A. Talin, Nat. Commun., 2016, 7, 10479 CrossRef CAS PubMed.
- M. Sassi, M. M. Salamone, R. Ruffo, G. E. Patriarca, C. M. Mari, G. A. Pagani, U. Posset and L. Beverina, Adv. Funct. Mater., 2016, 26, 5240–5246 CrossRef CAS.
- H. Yu, S. Shao, L. Yan, H. Meng, Y. He, C. Yao, P. Xu, X. Zhang, W. Hu and W. Huang, J. Mater. Chem. C, 2016, 4, 2269–2273 RSC.
- G. Atakan and G. Gunbas, RSC Adv., 2016, 6, 25620–25623 RSC.
- L. Ji, Y. Dai, S. Yan, X. Lv, C. Su, L. Xu, Y. Lv, M. Ouyang, Z. Chen and C. Zhang, Sci. Rep., 2016, 6, 30068 CrossRef PubMed.
- P. Xu, I. Murtaza, J. Shi, M. Zhu, Y. He, H. Yu, O. Goto and H. Meng, Polym. Chem., 2016, 7, 5351–5356 RSC.
- S.-H. Hsiao, Y.-H. Hsiao and Y.-R. Kung, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1289–1298 CrossRef CAS.
- W. T. Neo, Q. Ye, S.-J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 7364–7376 RSC.
- W. T. Neo, C. M. Cho, Z. Shi, S.-J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 28–32 RSC.
- C. M. Cho, Q. Ye, W. T. Neo, T. Lin, X. Lu and J. Xu, Polym. Chem., 2015, 6, 7570–7579 RSC.
- Q. Ye, W. T. Neo, T. Lin, J. Song, H. Yan, H. Zhou, K. W. Shah, S. J. Chua and J. Xu, Polym. Chem., 2015, 6, 1487–1494 RSC.
- Q. Ye, W. T. Neo, C. M. Cho, S. W. Yang, T. Lin, H. Zhou, H. Yan, X. Lu, C. Chi and J. Xu, Org. Lett., 2014, 16, 6386–6389 CrossRef CAS PubMed.
- E. Karabiyik, E. Sefer, F. Baycan Koyuncu, M. Tonga, E. Özdemir and S. Koyuncu, Macromolecules, 2014, 47, 8578–8584 CrossRef CAS.
- E. K. Unver, S. Tarkuc, D. Baran, C. Tanyeli and L. Toppare, Tetrahedron Lett., 2011, 52, 2725–2729 CrossRef CAS.
- S. Ozdemir, M. Sendur, G. Oktem, O. Dogan and L. Toppare, J. Mater. Chem., 2012, 22, 4687–4694 RSC.
- H. Zhang, H. Qu, H. Lv, S. Hou, K. Zhang, J. Zhao, X. Li, E. Frank and Y. Li, Chem. – Asian J., 2016, 11, 2882–2888 CrossRef CAS PubMed.
- W. T. Neo, K. H. Ong, T. T. Lin, S.-J. Chua and J. Xu, J. Mater. Chem. C, 2015, 3, 5589–5597 RSC.
- C. Lin, T. Endo, M. Takase, M. Iyoda and T. Nishinaga, J. Am. Chem. Soc., 2011, 133, 11339–11350 CrossRef CAS PubMed.
- R. J. Mortimer, K. R. Graham, C. R. G. Grenier and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2009, 1, 2269–2276 Search PubMed.
- J. Padilla, A. M. Österholm, A. L. Dyer and J. R. Reynolds, Sol. Energy Mater. Sol. Cells, 2015, 140, 54–60 CrossRef CAS.
- H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS PubMed.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed.
- T. Vangerven, P. Verstappen, N. Patil, J. D’Haen, I. Cardinaletti, J. Benduhn, N. Van den Brande, M. Defour, V. Lemaur, D. Beljonne, R. Lazzaroni, B. Champagne, K. Vandewal, J. W. Andreasen, P. Adriaensens, D. W. Breiby, B. Van Mele, D. Vanderzande, W. Maes and J. Manca, Chem. Mater., 2016, 28, 9088–9098 CrossRef CAS.
- F. Lombeck, H. Komber, D. Fazzi, D. Nava, J. Kuhlmann, D. Stegerer, K. Strassel, J. Brandt, A. D. de Zerio Mendaza, C. Müller, W. Thiel, M. Caironi, R. Friend and M. Sommer, Adv. Energy Mater., 2016, 6, 1601232 CrossRef.
- S. Chen, K. C. Lee, Z.-G. Zhang, D. S. Kim, Y. Li and C. Yang, Macromolecules, 2016, 49, 527–536 CrossRef CAS.
- M. Wakioka, S. Ishiki and F. Ozawa, Macromolecules, 2015, 48, 8382–8388 CrossRef CAS.
- Z. Ding, J. Kettle, M. Horie, S. W. Chang, G. C. Smith, A. I. Shames and E. A. Katz, J. Mater. Chem. A, 2016, 4, 7274–7280 RSC.
- C. Liu, K. Wang, X. Hu, Y. Yang, C.-H. Hsu, W. Zhang, S. Xiao, X. Gong and Y. Cao, ACS Appl. Mater. Interfaces, 2013, 5, 12163–12167 Search PubMed.
- Z. Xiao, K. Sun, J. Subbiah, T. Qin, S. Lu, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2015, 6, 2312–2318 RSC.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 CrossRef PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- W. Hong, S. Chen, B. Sun, M. A. Arnould, Y. Meng and Y. Li, Chem. Sci., 2015, 6, 3225–3235 RSC.
- Y. Li, S. P. Singh and P. Sonar, Adv. Mater., 2010, 22, 4862–4866 CrossRef CAS PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- A. L. Casado, J. A. Casares and P. Espinet, Organometallics, 1997, 16, 5730–5736 CrossRef CAS.
- R. van Asselt and C. J. Elsevier, Organometallics, 1994, 13, 1972–1980 CrossRef CAS.
- F. Ozawa, T. Hidaka, T. Yamamoto and A. Yamamoto, J. Organomet. Chem., 1987, 330, 253–263 CrossRef CAS.
- L. Lu, T. Zheng, T. Xu, D. Zhao and L. Yu, Chem. Mater., 2015, 27, 537–543 CrossRef CAS.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dorling, R. S. Ashraf, A. R. Goni, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746–753 CrossRef CAS PubMed.
- W. Barford and M. Marcus, J. Chem. Phys., 2014, 141, 164101 CrossRef PubMed.
- A. Gasperini, X. A. Jeanbourquin and K. Sivula, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2245–2253 CrossRef CAS.
- M. Schmittel and A. Burghart, Angew. Chem., Int. Ed., 1997, 36, 2550–2589 CrossRef.
- S. Guan, A. S. Elmezayyen, F. Zhang, J. Zheng and C. Xu, J. Mater. Chem. C, 2016, 4, 4584–4591 RSC.
- W. T. Neo, Z. Shi, C. M. Cho, S.-J. Chua and J. Xu, ChemPlusChem, 2015, 80, 1298–1305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and GPC of polymers, cyclic voltammograms and XRD spectra of polymer thin films, and redox stabilities of electrochromic devices. See DOI: 10.1039/c7qm00377c |
| This journal is © the Partner Organisations 2018 |