
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot palladium-catalyzed synthesis of sulfonyl fluorides from aryl bromides†
Alyn T.
Davies
a,
John M.
Curto
b,
Scott W.
Bagley
 *b and
Michael C.
Willis
*a
*b and
Michael C.
Willis
*a
aDepartment of Chemistry, University of Oxford, Chemical Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
bCVMET Medicinal Chemistry, Pfizer Inc., Eastern Point Road, Groton, Connecticut 06340, USA. E-mail: scott.w.bagley@pfizer.com
First published on 11th October 2016
Abstract
A mild, efficient synthesis of sulfonyl fluorides from aryl and heteroaryl bromides utilizing palladium catalysis is described. The process involves the initial palladium-catalyzed sulfonylation of aryl bromides using DABSO as an SO2 source, followed by in situ treatment of the resultant sulfinate with the electrophilic fluorine source NFSI. This sequence represents the first general method for the sulfonylation of aryl bromides, and offers a practical, one-pot alternative to previously described syntheses of sulfonyl fluorides, allowing rapid access to these biologically important molecules. Excellent functional group tolerance is demonstrated, with the transformation successfully achieved on a number of active pharmaceutical ingredients, and their precursors. The preparation of peptide-derived sulfonyl fluorides is also demonstrated.
Introduction
The sulfonyl fluoride functional group has become widely adopted throughout the field of chemical biology due to its unique balance between reactivity and stability under physiological conditions.1 This is exemplified by the widespread use of (2-aminoethyl)benzenesulfonyl fluoride (AEBSF), its HCl salt Pefabloc®, and phenylmethylsulfonyl fluoride (PMSF) as serine protease inhibitors which are widely used in the preparation of cell lysates (Fig. 1).2 Sulfonyl fluorides are also used as chemical biology probes for targeting nucleophilic amino acids;3 for example, 5′-fluorosulfonylbenzoyl 5′-adenosine (FSBA), an ATP-binding protein inhibitor, covalently binds to lysine residues.4 Sulfonyl fluorides have also shown promising results as fluorinating reagents.5 In particular, the recent work of Doyle et al. has shown the potential of 2-pyridinesulfonyl fluoride (PyFluor) to act as a selective, thermally stable and inexpensive deoxyfluorination reagent (Fig. 1).5c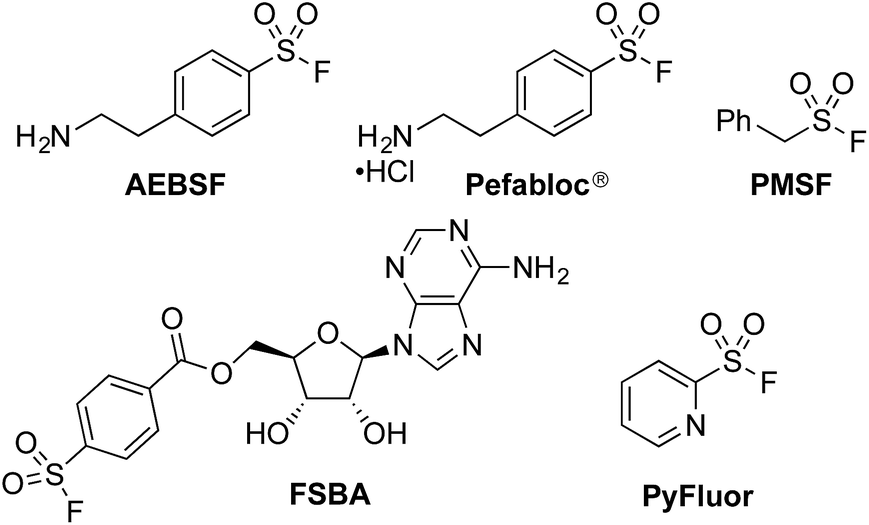 | ||
| Fig. 1 A selection of sulfonyl fluorides used as biological probes and chemical reagents. | ||
Despite their clear utility, direct synthetic approaches towards sulfonyl fluorides are limited. One commonly utilized method of sulfonyl fluoride synthesis is through 1,4-addition type reactions using the highly reactive Michael acceptor ethenesulfonyl fluoride (ESF).1,6 These reactions proceed in excellent yields, however, due to the nature of the reagent, the products are limited to ethyl-linked sulfonyl fluorides (Scheme 1a). Preformed sulfonates can also be converted to the corresponding sulfonyl fluorides in moderate yields by treatment with DAST.7 Alternatively, sulfonyl fluorides can be accessed through the corresponding sulfonyl chlorides; combination of the chloride with toxic potassium bifluoride is the most useful process,1 although alternative reagents, including KF/18-crown-6,8 are also known (Scheme 1b). Unfortunately, sulfonyl chlorides can often be challenging to prepare in their own right, and their inherent high reactivity means that they are poor candidates to survive intact for even a short synthetic sequence. In addition, these methods often prove incompatible with the highly functionalized bioactive molecules to which sulfonyl fluorides are commonly attached, and as such alternative methods for their synthesis are highly desirable. A mild, late stage functionalization approach would address these issues, and in particular would allow significantly greater tolerance towards sensitive functional groups. In this Edge Article we document the realization of such an approach, based on the use of palladium catalysis and readily available aryl- and heteroaryl bromide substrates (Scheme 1c).
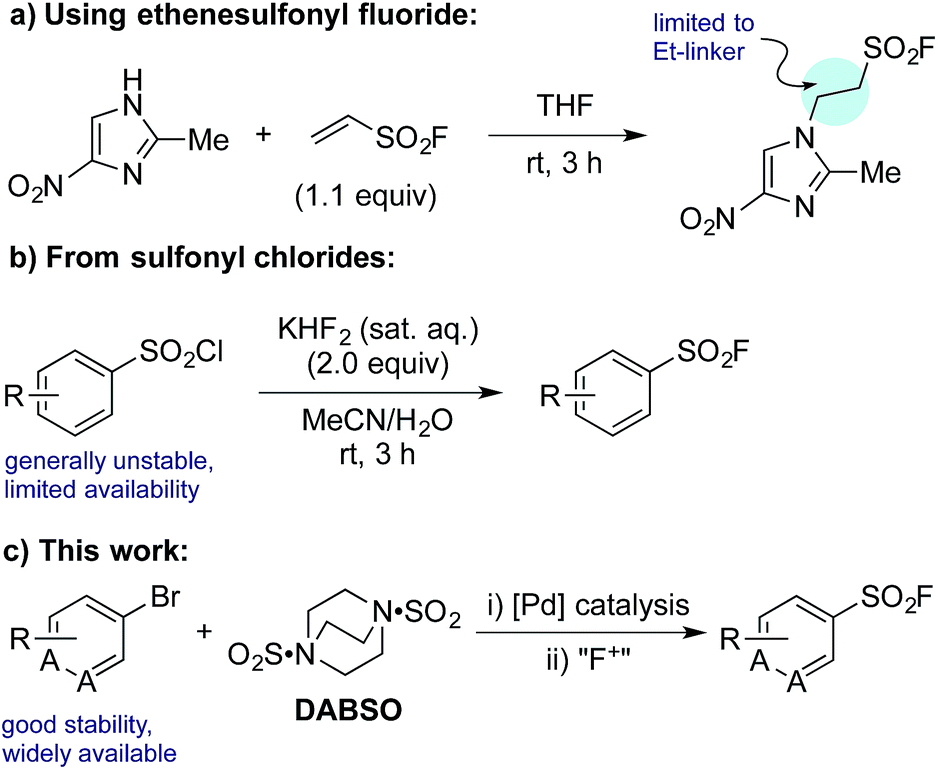 | ||
| Scheme 1 Representative examples of common synthetic routes to sulfonyl fluorides and the approach reported in this Edge Article. | ||
Results and discussion
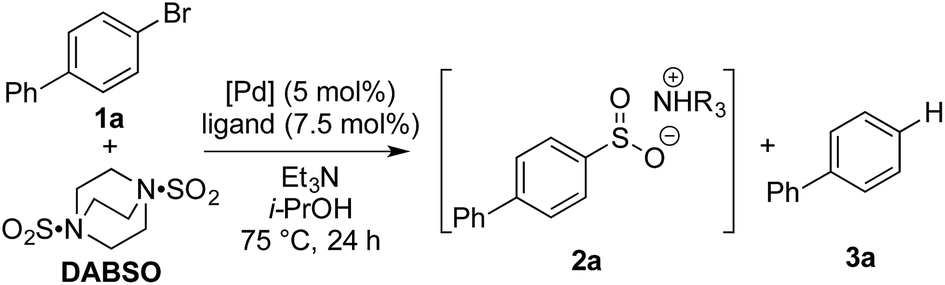
Our laboratories have shown a particular interest in the chemistry of sulfur-containing compounds, and we have previously described a variety of methods for the synthesis of sulfones, sulfonamides, sulfoxides and many other sulfur-based functional groups.9 The driving force behind this chemistry has been the recent emergence of reagents that function as sulfur dioxide surrogates, such as DABSO (1,4-diazabicyclo[2.2.2]octane-bis(sulfur dioxide)) and K2S2O5, which have proven to be a versatile reagents for the introduction of SO2.9o,10 We set out to exploit our experience with SO2 chemistry to develop a synthetic procedure which could alleviate many of the issues associated with sulfonyl fluoride synthesis, focusing on generating a method tolerant to the wide variety of functional groups often present in sulfonyl fluoride-based biological probes. In order to deliver a general method, aryl bromides were targeted as starting materials over the more reactive, but less readily available aryl iodides. Although there are limited reports of the successful use of aryl bromides as substrates in sulfonylation processes, the yields achieved are at best moderate.11Our investigation began by exploring the reactivity of 4-bromobiphenyl under previously reported conditions for sulfinate synthesis from aryl iodides, and pleasingly 33% consumption of 1a was observed (Table 1, entry 1). Consumption of the aryl bromide 1a was used as an indication of reactivity; the insolubility of sulfinate 2a prevented an accurate determination of conversion relative to an internal standard (benzophenone). In order to avoid undesired reactivity, the reaction was monitored for the reduction product, biphenyl 3a. An evaluation of electron-rich phosphine ligands was subsequently undertaken, as previous work had shown these were the most active in similar systems (entries 2–7).12 To our delight, the AmPhos ligand (L6) in combination with palladium(II) acetate showed excellent consumption of the aryl bromide 1a with negligible amounts of the reduction product being formed (entry 7). By comparison, the Shavnya conditions11 using K2S2O5 as the SO2 source proved highly active, however, significant quantities of 3a were observed (entry 8). For convenience in experimental set up, the reaction was attempted with commercial preformed Pd/AmPhos complex and consumption of 1 was increased to 91% (entry 9). With efficient conditions for converting aryl bromides to aryl sulfinates achieved, these conditions were then examined in potential fluorination processes. Fluorination of sulfinate intermediate 2a proved to be straightforward, and could be accomplished in a one-pot procedure with no change of solvent. Aryl bromide 1a was subjected to the previously optimized reaction conditions to generate the intermediate sulfinate 2a, subsequent treatment with 1.5 equivalents of NFSI produced the desired sulfonyl fluoride 4a in 84% isolated yield. As a result, these conditions were taken forward to evaluate the substrate scope for this transformation.
|
|
||||
|---|---|---|---|---|
| Entry | [Pd] | Ligand | Consumption of 1b | Reduction to 3b |
| a Reaction conditions: aryl bromide (0.4 mmol), [Pd] (5 mol%), ligand (7.5 mol%), DABSO (0.6 equiv.), Et3N (3.0 equiv.), i-PrOH [0.2 M], 75 °C, 24 h. b Consumption of 1a and reduction to 3a measured by HPLC relative to benzophenone as an internal standard. c K2S2O5 (2.0 equiv.), TBAB (1.1 equiv.), NaCO2H (2.2 equiv.) and MeCN (1.4 mL) at 70 °C. | ||||
| 1 | Pd(OAc)2 | PAd2n-Bu | 33% | 2% |
| 2 | Pd(OAc)2 | L1 | 37% | 3% |
| 3 | Pd(OAc)2 | L2 | 28% | 2% |
| 4 | Pd(OAc)2 | L3 | 33% | 2% |
| 5 | Pd(OAc)2 | L4 | 58% | 2% |
| 6 | Pd(OAc)2 | L5 | 77% | 10% |
| 7 | Pd(OAc)2 | L6 | 83% | 3% |
| 8c | Pd(OAc)2 | PPh3/1,10-phen | 98% | 29% |
| 9 | PdCl2(AmPhos)2 | n/a | 91% | 1% |
|
||||
Upon examination the reaction proved tolerant of a wide variety of electron-donating and electron-withdrawing functional groups (Table 2). The para-chloro substitution (4i) is of particular interest as a handle for further manipulation. A number of sensitive moieties could also be incorporated using this chemistry, such as silane (4k), Weinreb amide (4m), indole (4n), 4-quinazolinone (4o) and indazole (4p) functionalities. In addition to aryl bromides, the use of aryl iodide substrates is also facilitated, as exemplified by para-tolyl sulfonyl fluoride (4f).
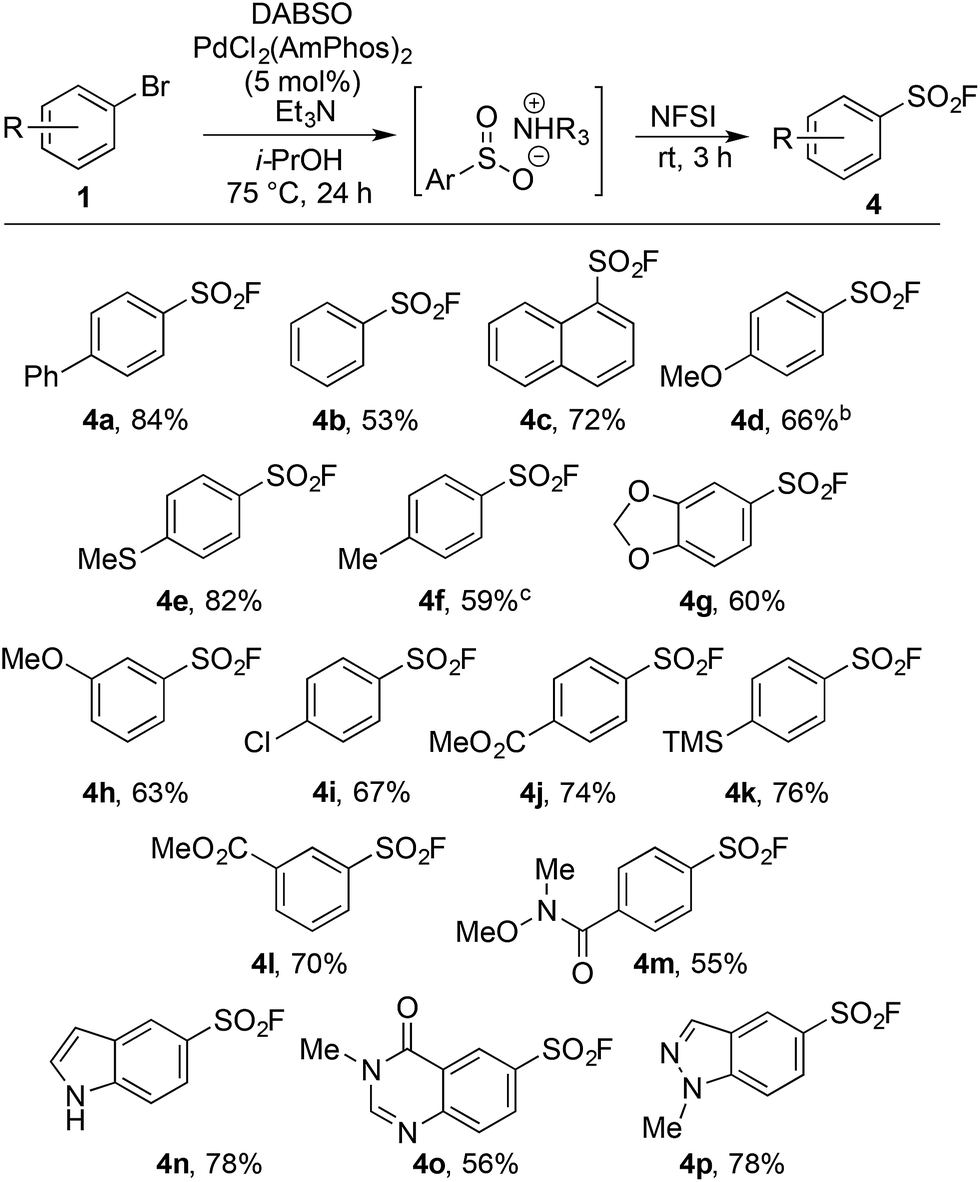
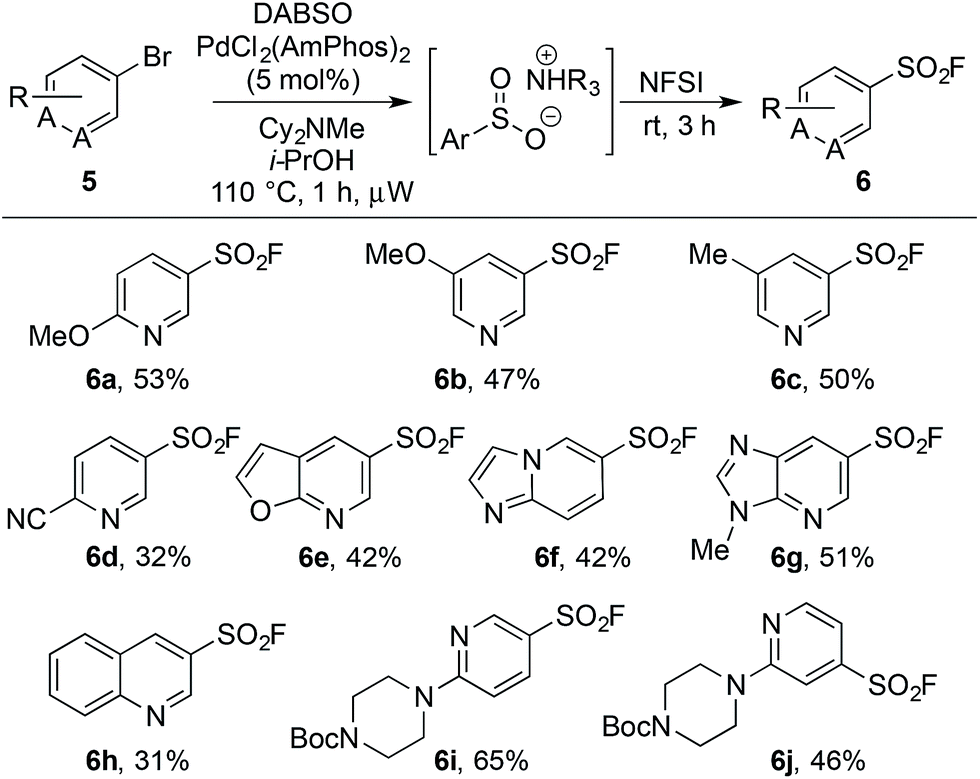
An on-going challenge in the synthesis of sulfur-containing compounds is the functionalization of heteroaromatics.9g,h,13 In the present chemistry hetero-aryl substrates were tolerated when the bromine was positioned on a benzenoid ring (Table 2), however, undertaking the reaction directly on a heteroaromatic ring proved difficult. Bromide 5a, when subjected to the conditions in Table 2, provided sulfonyl fluoride 6a in only 15%, with undesired biaryl dimer and palladium black being observed.12 Microwave conditions were investigated to achieve higher reaction temperatures in the presence of i-PrOH, producing 6a in 35% at 110 °C. Alternative bases were explored and methyl(dicyclohexyl)amine provided 6a in 53% (Table 3). The increased steric bulk of methyl(dicyclohexyl)amine is believed to decrease the rate of homo cross-coupling and accelerate the formation of palladium(0).14 These conditions proved general for a variety of substituted bromo-pyridines, providing sulfonyl fluorides in adequate yields (Table 3). Non-pyridine heteroaromatics, which are common in drug-like molecules, also formed sulfonyl fluorides efficiently (Table 3, 6e–6h), yet bromo-imidazoles, -pyrazoles and -pyrimidines proved recalcitrant for sulfonyl fluoride formation.12
| a Reaction conditions: (i) aryl bromide (0.4 mmol), PdCl2(AmPhos)2 (5 mol%), DABSO (1.0 equiv.), Cy2NMe (3.0 equiv.), i-PrOH [0.2 M], 110 °C, 1 h, μW heating. (ii) NFSI (1.5 equiv.), rt, 3 h. Isolated yields. |
|---|
|
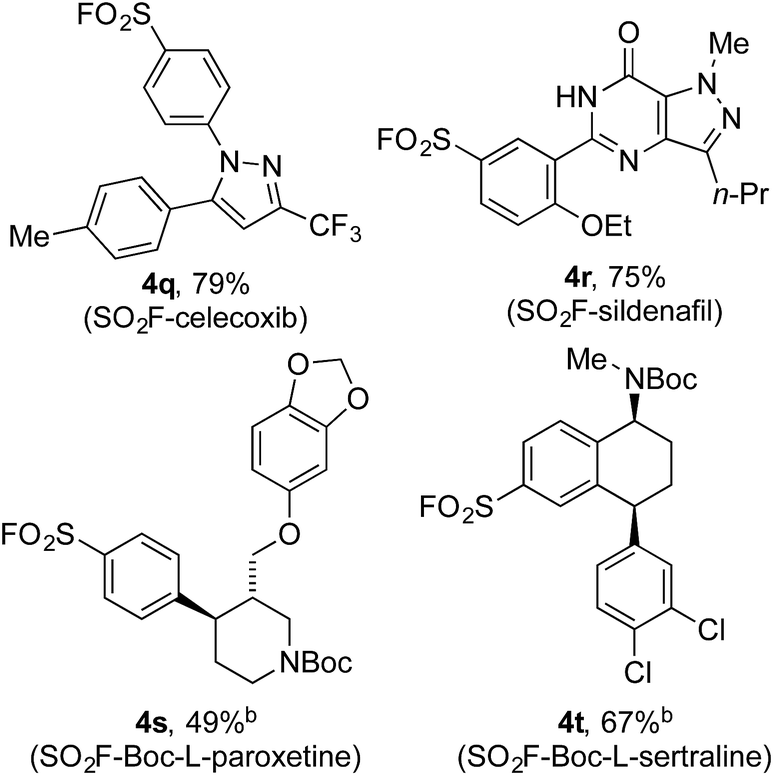
Encouraged by the scope of this method the sulfonyl fluoride moiety was incorporated into late-stage pharmaceutical intermediates (Table 4). The mild nature of the reaction allowed the straightforward preparation of sulfonyl fluoride derivatives of celecoxib (4q), sildenafil (4r), N-Boc paroxetine (4s) and N-Boc sertraline (4t) in good yields from the appropriately halogenated intermediates. The ability to functionalize highly advanced compounds with a sulfonyl fluoride handle showcases the utility of this methodology to quickly prepare chemical biology probes or templates for further elaboration (e.g. sulfonamides),15 and would be extremely challenging using existing methods.
To date, the synthesis of sulfonyl fluoride labelled amino acids and peptides is limited to incorporation of fragments previously modified with SO2–X functionality.16 Application of our Pd-catalyzed method to N-Boc-L-4-halophenylalanine methyl esters successfully delivered the derivatized analogue 4u in 61% and 73% yield from the 4-bromo and 4-iodo derivatives, respectively (Scheme 2a). Subsequently, compound 4u was reacted with N-Boc-L-lysine methyl ester to deliver the tail-to-tail linked sulfonamide (7) in 84% yield, thus validating the ability of the sulfonyl fluoride moiety to react with nucleophilic residues, central to their use as chemical biology probes. Halogenated tetramer (8),17 prepared via HATU-mediated peptide coupling, was gratifyingly converted to its sulfonyl fluoride derivative (4v) in 65% yield (Scheme 2b). The ability to quickly prepare peptide-based sulfonyl fluoride probes could lead to increased exploration of protein binding pockets containing nucleophilic residues such as cysteine, tyrosine, lysine, serine and threonine3a or for use as irreversible inhibitors.18
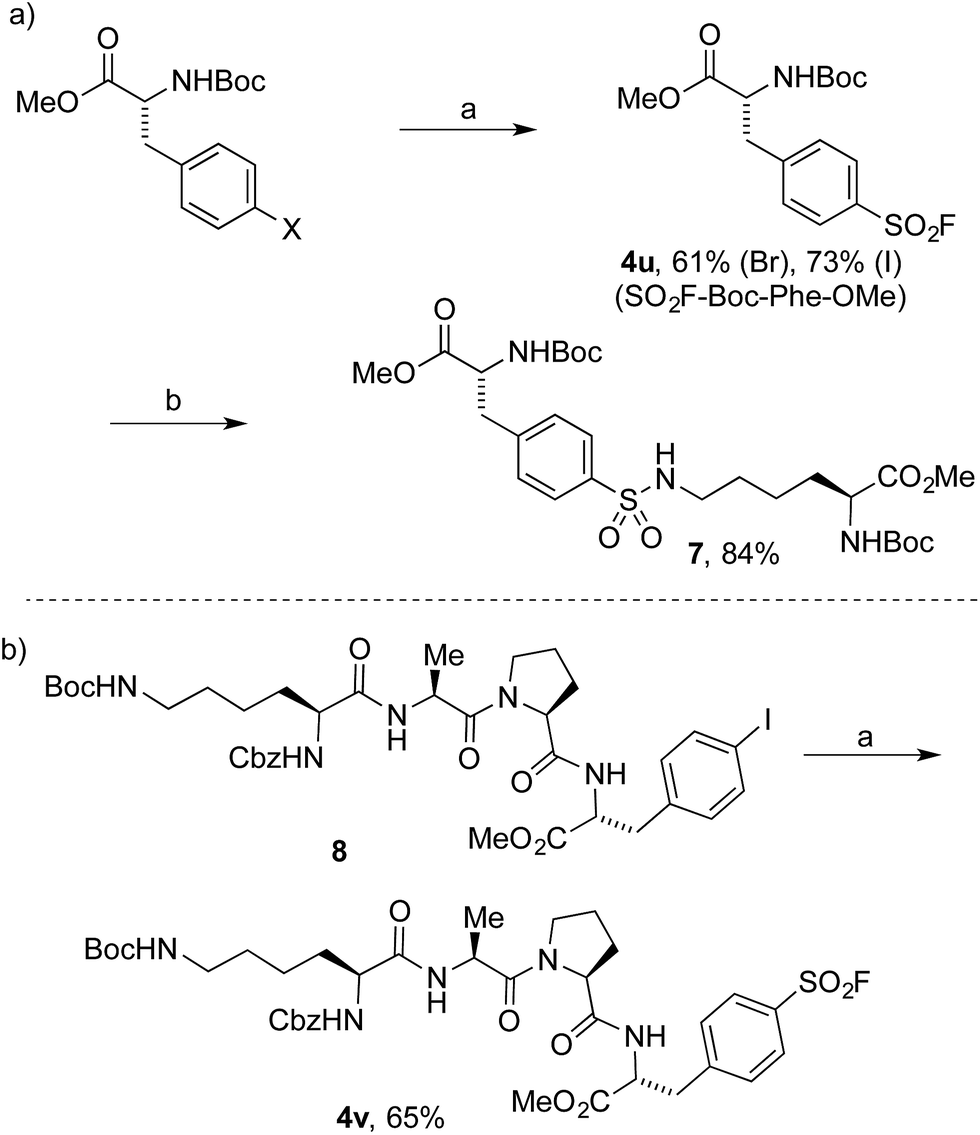 | ||
| Scheme 2 Synthesis of amino acid and peptidic sulfonyl fluorides. (a) Reaction conditions: (i) aryl halide (0.4 mmol), PdCl2(AmPhos)2 (5 mol%), DABSO (0.6 equiv.), Et3N (3.0 equiv.), i-PrOH [0.2 M], 75 °C, 16 h. (ii) NFSI (1.5 equiv.), rt, 3 h. (b) N-Boc-L-Lys-OMe, i-Pr2NEt, DMSO, 100 °C, 15 h. Isolated yields. | ||
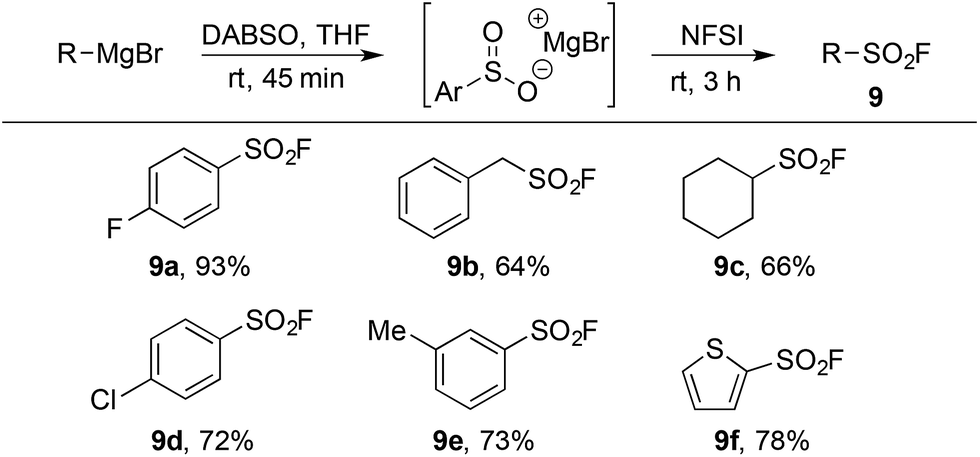
The limitations inherent to using ESF to prepare sulfonyl fluorides (Scheme 1) highlight the unmet need for methods to generate diverse alkyl sulfonyl fluorides. Previous work in our groups has shown that organometallic reagents and DABSO can combine to generate sulfinate intermediates.9j,9l This method, complementary to the conditions developed in Table 1, provides an opportunity to access a variety of alkyl and aryl sulfonyl fluorides. Utilizing the fluorination conditions in Table 2, the sulfinate intermediate derived from reaction of DABSO with aryl, benzylic and alkyl Grignard reagents provided sulfonyl fluorides in excellent yield (Table 5). On-going work in our laboratory is focusing on expanding this orthogonal method to additional organometallic substrates.
| a Reaction conditions (i) R–MgBr (2.0 mmol), DABSO (1.0 equiv.), THF [0.5 M], rt, 45 min. (ii) NFSI (1.5 equiv.), 0 °C – rt, 3 h. Isolated yields. |
|---|
|
Conclusions
A palladium-catalyzed approach to the synthesis of sulfonyl fluorides from aryl bromides has been developed. The initial step of this sequence represents the first general method for the sulfonylation of aryl bromides. The overall transformation proceeds smoothly on a wide variety of substrates and minor modifications of the reaction conditions allow the use of heteroaryl bromides and organometallic reagents. Of particular note is the tolerance of different functional groups, which allows the transformation to be undertaken on complex molecules, such as active pharmaceutical ingredients, their precursors and peptides. This catalytic, operationally simple method has the ability to expand the scope of sulfonyl fluorides available for investigation as biological probes and has the potential to become a preferred method for complex sulfonyl fluoride synthesis.Acknowledgements
ATD and MCW thank the EPSRC for support of this study, and Dr D. R. Owen (Pfizer) for stimulating discussions. SWB and JMC would like to thank Pfizer colleagues Dr V. Mascitti for support, Dr R. Singer for helpful discussions, A. Shavnya for key intermediates and Dr J. Stroh for HRMS data.Notes and references
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- (a) J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed; (b) D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS; (c) M. O. Lively and J. C. Powers, Biochim. Biophys. Acta, Enzymol., 1978, 525, 171–179 CrossRef CAS.
- (a) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC; (b) O. Fadeyi, M. D. Parikh, M. Z. Chen, R. E. Kyne Jr, A. P. Taylor, I. O'Doherty, S. E. Kaiser, S. Barbas, S. Niessen, M. Shi, S. L. Weinrich, J. C. Kath, L. H. Jones and R. P. Robinson, ChemBioChem, 2016 DOI:10.1002/cbic.201600427.
- (a) P. K. Pal, W. J. Wechter and R. F. Colman, J. Biol. Chem., 1975, 250, 8140–8147 CAS; (b) R. F. Colman, Annu. Rev. Biochem., 1983, 52, 67–91 CrossRef CAS PubMed; (c) M. P. Kamps, S. S. Taylor and B. M. Sefton, Nature, 1984, 310, 589–592 CrossRef CAS PubMed; (d) M. W. Russo, T. J. Lukas, S. Cohen and J. V. Staros, J. Biol. Chem., 1985, 260, 5205–5208 CAS.
- (a) B. Bennua-Skalmowski and H. Vorbrüggen, Tetrahedron Lett., 1995, 36, 2611–2614 CrossRef CAS; (b) J. Yin, D. S. Zarkowsky, D. W. Thomas, M. M. Zhao and M. A. Huffman, Org. Lett., 2004, 6, 1465–1468 CrossRef CAS PubMed; (c) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed.
- J. J. Krutak, R. D. Burpitt, W. H. Moore and J. A. Hyatt, J. Org. Chem., 1979, 44, 3847–3858 CrossRef CAS.
- A. J. Brouwer, T. Ceylan, A. M. Jonker, T. van der Linden and R. M. J. Liskamp, Bioorg. Med. Chem., 2011, 19, 2397–2406 CrossRef CAS PubMed.
- T. A. Bianchi and L. A. Cate, J. Org. Chem., 1977, 42, 2031–2032 CrossRef CAS.
- (a) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372–16373 CrossRef CAS PubMed; (b) H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876–4878 CrossRef CAS PubMed; (c) E. J. Emmett, C. S. Richards-Taylor, B. Nguyen, A. Garcia-Rubia, B. R. Hayter and M. C. Willis, Org. Biomol. Chem., 2012, 10, 4007–4014 RSC; (d) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 CrossRef CAS PubMed; (e) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 46, 2701–2710 CrossRef; (f) A. S. Deeming, C. J. Russell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150–153 CrossRef CAS PubMed; (g) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222–228 RSC; (h) E. J. Emmett and M. C. Willis, Org. Synth., 2014, 91, 125–136 CrossRef CAS; (i) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS; (j) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2015, 54, 1168–1171 CrossRef CAS PubMed; (k) E. Ferrer Flegeau, J. M. Harrison and M. C. Willis, Synlett, 2016, 27, 101–105 Search PubMed; (l) D. C. Lenstra, V. Vedovato, E. Ferrer Flegeau, J. Maydom and M. C. Willis, Org. Lett., 2016, 18, 2086–2089 CrossRef CAS PubMed; (m) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747–750 CrossRef CAS PubMed; (n) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404–4407 CrossRef CAS PubMed; (o) A. S. Tsai, J. M. Curto, B. N. Rocke, A.-M. R. Dechert-Schmitt, G. K. Ingle and V. Mascitti, Org. Lett., 2016, 18, 508–511 CrossRef CAS PubMed.
- (a) X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056–4058 CrossRef CAS PubMed; (b) S. Ye, H. Wang, Q. Xiao, Q. Ding and J. Wu, Adv. Synth. Catal., 2014, 356, 3225–3230 CrossRef CAS; (c) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451–2454 CrossRef CAS PubMed; (d) B. Skillinghaug, J. Rydfjord and L. R. Odell, Tetrahedron Lett., 2016, 57, 533–536 CrossRef CAS; (e) Y. An, H. Xia and J. Wu, Org. Biomol. Chem., 2016, 14, 1665–1669 RSC.
- A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226–6229 CrossRef CAS PubMed.
- See ESI for further details.†.
- E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed.
- C. Gürtler and S. L. Buchwald, Chem.–Eur. J., 1999, 5, 3107–3112 CrossRef.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne Jr, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- A. J. Brouwer, A. Jonker, P. Werkhoven, E. Kuo, N. Li, N. Gallastegui, J. Kemmink, B. I. Florea, M. Groll, H. S. Overkleeft and R. M. J. Liskamp, J. Med. Chem., 2012, 55, 10995–11003 CrossRef CAS PubMed.
- The 4-iodo-Phe derivative was used for preparation of the peptide example due to wider availability versus the 4-Br Phe analog.
- (a) L. H. Jones, Mol. BioSyst., 2016, 12, 1728–1730 RSC; (b) C. Dubiella, H. Cui, M. Gersch, A. J. Brouwer, S. A. Sieber, A. Krüger, R. M. J. Liskamp and M. Groll, Angew. Chem., Int. Ed., 2014, 53, 11969–11973 CrossRef CAS PubMed; (c) M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072–10079 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting characterisation data. See DOI: 10.1039/c6sc03924c |
| This journal is © The Royal Society of Chemistry 2017 |