
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic framework-based CoP/reduced graphene oxide: high-performance bifunctional electrocatalyst for overall water splitting†
Long
Jiao
,
Yu-Xiao
Zhou
and
Hai-Long
Jiang
 *
*
Hefei National Laboratory for Physical Sciences at the Microscale, Key Laboratory of Soft Matter Chemistry, Chinese Academy of Sciences, Collaborative Innovation Center of Suzhou Nano Science and Technology, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: jianglab@ustc.edu.cn
First published on 12th January 2016
Abstract
Efficient and cost-effective electrocatalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), especially bifunctional catalysts for overall water splitting, are highly desired. In this work, with rationally designed sandwich-type metal–organic framework/graphene oxide as a template and precursor, a layered CoP/reduced graphene oxide (rGO) composite has been successfully prepared via pyrolysis and a subsequent phosphating process. The resultant CoP/rGO-400 exhibits excellent HER activity in acid solution. More importantly, the catalyst manifests excellent catalytic performances for both the HER and OER in basic solution. Therefore, it can be utilized as a bifunctional catalyst on both the anode and cathode for overall water splitting in basic media, even displaying superior activity to that of the integrated Pt/C and IrO2 catalyst couple.
Introduction
With increasing environmental concerns and consumption of fossil fuels, there is an urgent demand to develop alternative clean energies, such as, hydrogen. Electrocatalytic water splitting, a combination of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) to generate hydrogen and oxygen, respectively, has been recognized to be one of the most promising ways for energy conversion.1 To reach this goal, the development of efficient catalysts to accelerate the kinetics of both half-cell reactions is a prerequisite.1 Precious metals (Pt) and noble metal oxides (RuO2 and IrO2) possess the best activity for the HER and OER, respectively, while their large-scale industrial application is impeded by their high cost and scarcity.1b,f It is highly desirable to develop cost-effective catalysts with high activity and durability. Currently, much effort has been devoted to the exploration of the half-cell reactions and great progress has been achieved, for example, transition-metal sulfides, selenides, carbides and phosphides exhibit stable catalytic activity for the HER in acid solution,2 and transition metal oxides/hydroxides are found to be active for the more challenging OER in basic solution.3 To meet the practical application, the HER and OER should be conducted in the same electrolyte based on a single catalyst to achieve the overall water splitting. However, the current prevailing water splitting requires the integration of two types of catalysts that are specially suitable for the HER and OER, respectively. Such a combination is usually incompatible and thus results in a poor overall performance. Therefore, the development of bifunctional catalysts, highly active for both the HER and OER, is of prime importance for overall water splitting, which remains a significant challenge and has been rarely reported thus far.1f,g,4To achieve high catalytic activity, nanostructuring the catalysts with porous character would be an essential way to expose active sites as far as possible to the electrolyte and substrate. In this context, metal–organic frameworks (MOFs), a family of crystalline porous materials with well-organized structures, should be ideal precursors.5 Recently, MOFs as templates/precursors have been demonstrated to afford various porous nanostructured carbon/metal oxides with good catalytic performances.6,7 Despite this, the poor electrical conductivity of MOF-derived nanocomposites is unfavorable for the electrocatalysis. Given that graphene oxide (GO) has been well documented to be an excellent conductor, the formation of hybrid structures between the MOF-derived nanocomposites and GO might effectively exert their synergistic effect by taking advantage of their respective advantages.7
Among the different electrocatalysts for the HER or OER, transition metal phosphides (TMP) are very promising not only because of their high abundance and low cost but also owing to their great acid–base stability in the pH range of 0–14. In this work, we have fabricated a MOF/GO sandwich-type composite with a sheet-like structure based on the template role of GO via a facile one-step room-temperature reaction, without any bridging agent. Upon pyrolysis, the Co-based MOF/GO was converted to porous Co3O4/rGO, which was further phosphated to afford a porous CoP/rGO nanocomposite with retained sheet-like morphology (Scheme 1). The MOF-derived porous crystalline CoP nanostructure guarantees highly exposed active sites, and the close contact between CoP and rGO contributes to a consecutive conductive network, which is crucial for electron transfer. As a result, the CoP/rGO nanocomposite exhibits excellent HER activity in a wide pH range of 0–14 with overpotentials of 105 mV in acidic solution and 150 mV in basic solution at 10 mA cm−2. In addition, it also displays outstanding OER activity in basic solution with an overpotential of 340 mV at 10 mA cm−2. When CoP/rGO was employed as a bifunctional catalyst in both the anode and cathode, overall water splitting in basic solution was achieved with high efficiency, which is even superior to the integrated Pt/C and IrO2 catalyst couple.
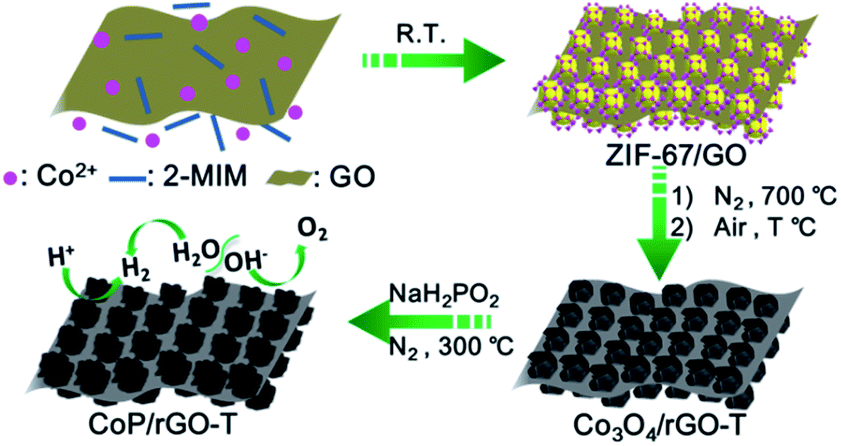 | ||
| Scheme 1 Illustration of the fabrication procedure for the CoP/rGO-T nanocomposite. | ||
Experimental
Materials and instrumentation
All chemicals were from commercial sources and used without further purification. Deionized water with a specific resistance of 18.2 MΩ cm was obtained using reversed osmosis followed by ion-exchange and filtration.Powder X-ray diffraction (XRD) studies were carried out on a Japan Rigaku SmartLab™ rotation anode X-ray diffractometer or a Holland X'Pert PRO fixed anode X-ray diffractometer equipped with graphite monochromatized Cu Kα radiation (λ = 1.54 Å). Field-emission scanning electron microscopy (FE-SEM) was carried out with a field emission scanning electron microanalyzer (Zeiss Supra 40 scanning electron microscope at an acceleration voltage of 5 kV). The transmission electron microscopy (TEM) images and high-resolution TEM images were acquired on a JEOL-2100F with an electron acceleration energy of 200 kV. The content of nitrogen was measured using a VarioEL III Elemental analyzer. The X-ray photoelectron spectroscopy (XPS) measurements were performed using an ESCALAB 250Xi high-performance electron spectrometer using monochromatized Al Kα (hν = 1486.7 eV) as the excitation source. The nitrogen sorption isotherms were measured using automatic volumetric adsorption equipment (Micrometritics ASAP 2020). Prior to the nitrogen adsorption/desorption measurement, the samples were dried overnight at 150 °C under vacuum.
Synthesis
Electrochemical measurements
Electrochemical measurements were performed with a CHI 760E electrochemical analyzer (CH Instruments, Inc., Shanghai) and a rotating disk electrode (RDE) (Pine Instruments, Grove City, PA) with a speed of 1600 rpm. All electrochemical measurements were conducted in a typical three-electrode setup with a Pt counter electrode and Ag/AgCl reference electrodes. The 0.5 M H2SO4 solution and 1 M KOH solution were used for the electrochemical measurements and the solutions were purged with N2 for 30 min prior to the HER or overall water splitting test or with O2 prior to the OER test. Before recording the electrochemical activity of the catalyst, the catalyst was activated using 20 cyclic voltammetry scans at a scan rate of 100 mV s−1. LSV measurements were conducted with a scan rate of 5 mV s−1. All potentials reported in this paper were converted from vs. Ag/AgCl to vs. RHE by adding a value of 0.197 + 0.059 × pH. All data were presented without iR compensation.The catalyst ink was prepared by dispersing 2 mg of catalyst into 1 mL of ethanol solvent containing 10 μL of 5 wt% Nafion and sonicating for 30 min. Then 28 μL of the catalyst ink was loaded onto a GCE of 5 mm diameter (loading amount: ∼0.28 mg cm−2).
To evaluate the bifunctionality of CoP/rGO in alkaline solutions, the catalyst was also loaded on two 1 × 1 cm carbon fiber papers (loading amount: ∼0.28 mg cm−2).
Results and discussion
The zeolite-type MOF, Co(2-MIM)2 (called ZIF-67, 2-MIM = 2-methylimidazole),8 featuring a three-dimensional (3D) network with 1.1 nm cages and a large surface area (BET, >1500 m2 g−1), is very suitable for acting as a template/precursor and can be converted to Co or a CoOx-based composite upon pyrolysis.9 The introduction of a GO template into the growth solution for ZIF-67 in mixed solvents leads to the successful fabrication of ZIF-67/GO with the inherited sheet-like morphology of GO. It is clearly visible that ZIF-67 particles in 100–200 nm size are grown on both surfaces of GO to give a sandwich-type structure, clearly demonstrating the template role of GO. The thickness of the ZIF-67/GO layer with rough surfaces is 200–400 nm and the successful synthesis of ZIF-67 is supported by the powder X-ray diffraction (XRD) pattern (Fig. 1a and S1†). It is noteworthy that, although a few MOF–GO composites have been synthesized by the direct growth of MOFs in the presence of GO, no sheet-like morphology was reported for their resultant products;7a,10 a ZIF-8/GO layered structure can be obtained only in the presence of PVP as an additional binder.7c This is a very rare synthesis of MOF/GO sheets based on a GO template without any bridging agent. | ||
| Fig. 1 Scanning electron microscope (SEM) images of (a) ZIF-67/GO and (b) CoP/rGO-400. (c) Transmission electron microscopy (TEM) and (d) high-resolution TEM images of CoP/rGO-400 (inset in (d): SAED pattern for CoP nanocrystal). | ||
The ZIF-67/GO underwent pyrolysis at 700 °C in a N2 atmosphere and subsequent oxidation at different temperatures to afford Co3O4/rGO-T (T represents oxidation temperature) with a retained layered structure (Fig. S2†), the BET surface area of which reached 40 m2 g−1, thanks to the high porosity of ZIF-67 (Fig. S3 and S4†). Upon the further phosphating process for the cobalt oxides, CoP/rGO-T (T = 350, 400, 450) were obtained and the presence of the CoP species was proven by the powder XRD profiles (Fig. S5†). As a representative, CoP/rGO-400 has been found to mostly retain the sheet-like morphology (Fig. 1b and S6†), although the thickness shrinks to ∼200 nm due to the removal of organic species during the heat treatment. As expected, CoP/rGO-400 exhibits a porous character and possesses hierarchical pores, especially macropores (Fig. S7†), which is in good agreement with the TEM result (Fig. 1c). The high-resolution TEM (HRTEM) image reveals clear lattice fringes with an interplanar spacing of 0.19 nm that corresponds to the (211) planes of CoP, further evidencing the formation of a crystalline CoP species. In addition, the selected area electron diffraction (SAED) pattern exhibits the individual spots associated with concentric rings indexed to the (011), (111), (211) and (301) planes of orthorhombic CoP (Fig. 1d).2f,11 The GO was reduced during the thermal treatment, as indicated by the weakened intensity of the oxygen-containing bonds in the X-ray photoelectron spectroscopy (XPS) survey (Fig. S8†).
To evaluate the electrocatalytic HER activity, the catalysts were deposited on the RDE with a fixed mass loading (∼0.28 mg cm−2). The HER performances were measured in acid (0.5 M H2SO4) and alkaline (1 M KOH) solutions (Fig. 2). Bare GCE, rGO, CoP and commercial Pt/C (20 wt%) were also examined for comparison. As expected, Pt/C shows the most excellent activity while bare GCE and rGO have very poor HER performances. Surprisingly, ZIF-67-derived CoP exhibits very high activity and CoP/rGO-400 is more active, possibly due to the synergistic effect between the porous CoP and conductive rGO, the latter of which significantly lowers the impedance of CoP/rGO-400 (Fig. S9†). As determined from the linear sweep voltammetry (LSV) in acid solution (Fig. 2a), the HER onset potential of CoP/rGO-400 is ∼−13 mV and the kinetic current density reaches 10 mA cm−2 at an overpotential of 105 mV. The corresponding Tafel slope is 50 mV dec−1 (Fig. 2c). The very low overpotential and Tafel slope indicate a higher HER activity than almost all state-of-the-art HER catalysts in acid solution, upon considering the same catalyst loading (Table S1†).12 Moreover, its polarization curve after 3000 cycles of continuous CV scanning shows negligible difference with the initial one in 0.5 M H2SO4 (Fig. 2e). The chronoamperometry test for CoP/rGO-400 at an overpotential of 105 mV suggests that the activity remains very good even after 22 hours (Fig. 2e, inset). In addition, similar results have been obtained when CoP/rGO-400 was applied under alkaline conditions. It exhibits a low overpotential of 150 mV at 10 mA cm−2, superior to those of CoP, rGO and bare GCE (Fig. 2b), and a Tafel slope of 38 mV dec−1 which is comparable to that of Pt/C (Fig. 2d). These values are much lower than those for other base metal HER catalysts in basic media (Table S2†). There is only a slight activity loss after 3000 cycles, while the chronoamperometry test for 22 hours presents its reliable stability at an overpotential of 150 mV (Fig. 2f). It is noteworthy that although Pt/C shows a very small onset potential, the catalytic current density of CoP/rGO-400 surpasses that of Pt/C when the overpotential exceeds 240 mV in alkaline solution. Taken together, the CoP/rGO-400 shows excellent HER activity and durability in both acidic and alkaline solutions.
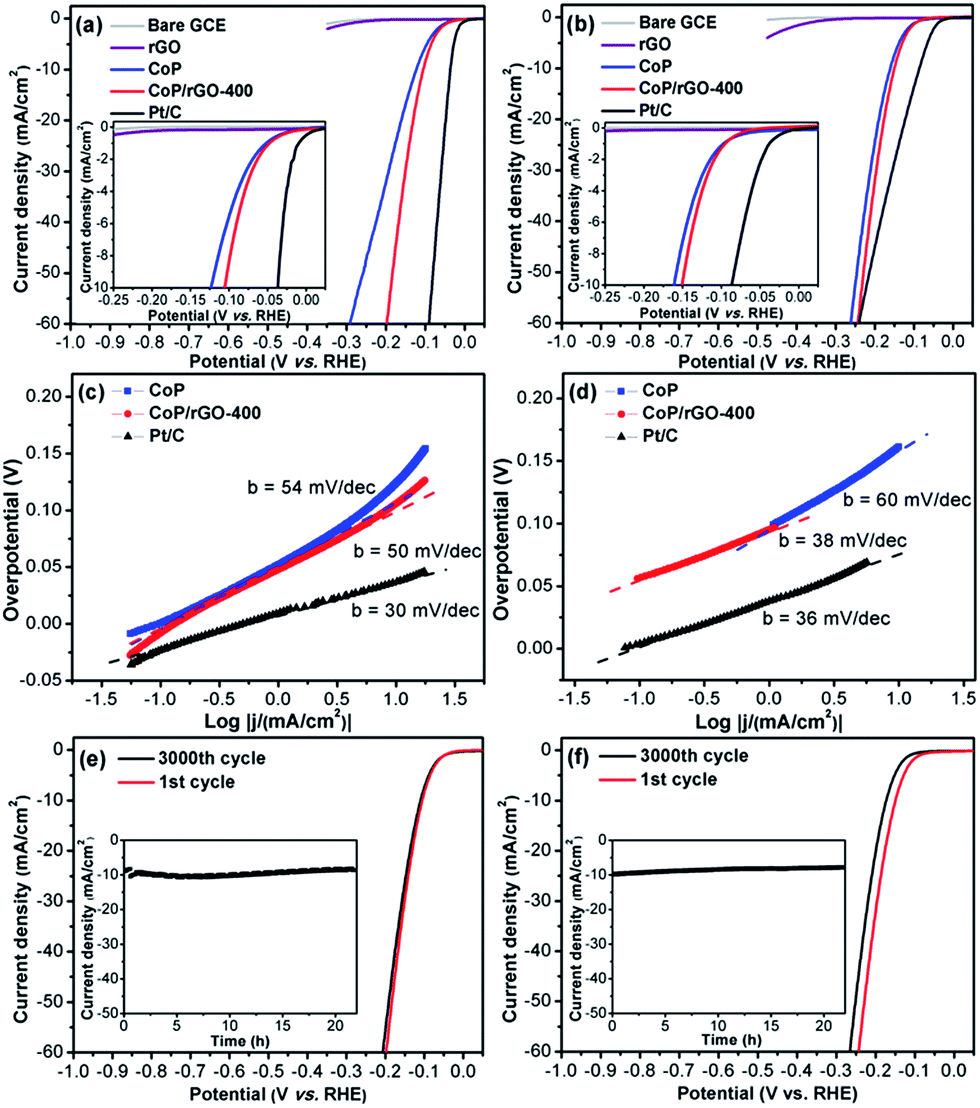 | ||
| Fig. 2 Electrochemical HER activity of CoP/rGO-400. (a) LSV curves, (c) Tafel slopes and (e) durability test in 0.5 M H2SO4 solution; (b) LSV curves, (d) Tafel slopes and (f) durability test in 1 M KOH solution. Insets in (e and f): time-dependent current density curves under static overpotentials of 105 mV in 0.5 M H2SO4 and 150 mV in 1 M KOH solution, respectively. | ||
It is even more challenging to obtain an efficient catalyst for the OER as it is a very complex and energy-intensive process, in which the O–H bond breaking and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond formation as well as the four-electron transfer process have sluggish kinetics.1c,e It has been reported very recently that CoP can act as an OER catalyst via in situ transformation.4b,13 Therefore, the OER performance of CoP/rGO-400 was investigated in 1 M KOH solution. Delightfully, CoP/rGO-400 can reach a current density of 10 mA cm−2 at an overpotential of 340 mV with a Tafel slope of 66 mV dec−1, which is superior to CoP, rGO and even IrO2, the state-of-the-art OER catalyst (Fig. 3a and b). The durability test indicates that although there is a little regression after 3000 cycles, CoP/rGO-400 is pretty stable at an overpotential of 340 mV for 22 hours and remains at a very high activity (Fig. 3c). Control experiments for the CoP/rGO-T catalysts prepared at different oxidation temperatures have suggested the best electrochemical performances of CoP/rGO-400 for both the HER and OER (Fig. S10–12†). The XPS spectra for CoP/rGO-400 after the OER test clearly demonstrate the negligible incorporation of Fe species into the catalyst during the OER reaction in KOH solution, different from a recent report (Fig. S13†).14 In addition, given that Co3O4/rGO-400 is electrochemically active in alkaline solution, the comparison between CoP/rGO-400 and Co3O4/rGO-400 has been made and the result shows that CoP/rGO-400 is much more active than Co3O4/rGO-400 for the HER and OER in 1 M KOH (Fig. S14†).
O bond formation as well as the four-electron transfer process have sluggish kinetics.1c,e It has been reported very recently that CoP can act as an OER catalyst via in situ transformation.4b,13 Therefore, the OER performance of CoP/rGO-400 was investigated in 1 M KOH solution. Delightfully, CoP/rGO-400 can reach a current density of 10 mA cm−2 at an overpotential of 340 mV with a Tafel slope of 66 mV dec−1, which is superior to CoP, rGO and even IrO2, the state-of-the-art OER catalyst (Fig. 3a and b). The durability test indicates that although there is a little regression after 3000 cycles, CoP/rGO-400 is pretty stable at an overpotential of 340 mV for 22 hours and remains at a very high activity (Fig. 3c). Control experiments for the CoP/rGO-T catalysts prepared at different oxidation temperatures have suggested the best electrochemical performances of CoP/rGO-400 for both the HER and OER (Fig. S10–12†). The XPS spectra for CoP/rGO-400 after the OER test clearly demonstrate the negligible incorporation of Fe species into the catalyst during the OER reaction in KOH solution, different from a recent report (Fig. S13†).14 In addition, given that Co3O4/rGO-400 is electrochemically active in alkaline solution, the comparison between CoP/rGO-400 and Co3O4/rGO-400 has been made and the result shows that CoP/rGO-400 is much more active than Co3O4/rGO-400 for the HER and OER in 1 M KOH (Fig. S14†).
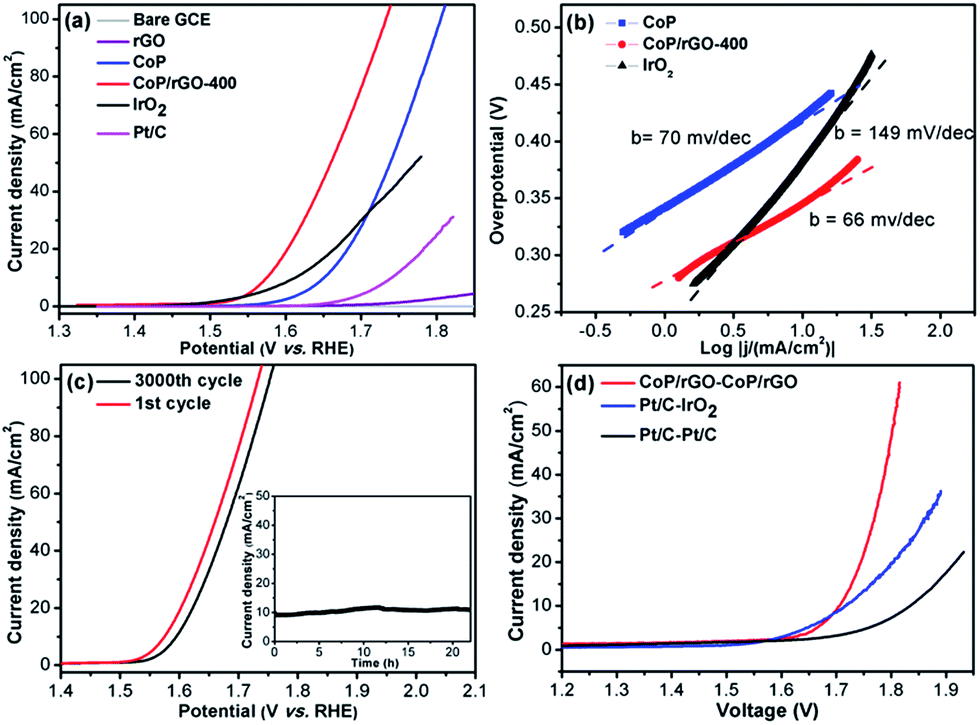 | ||
| Fig. 3 (a) LSV curves, (b) Tafel slopes, and (c) durability test for the electrochemical OER of CoP/rGO-400 (inset: time-dependent current density curve of CoP/rGO-400 under static overpotential of 340 mV) in 1 M KOH solution. (d) Bifunctional water electrolysis tested by LSV in 1 M KOH solution. | ||
To probe the composition change of CoP/rGO-400 after the HER and OER, high-resolution XPS spectra for the as-prepared, post-HER and post-OER catalysts were collected. For the as-prepared catalyst, the Co 2p spectra show two peaks at 793.4 and 778.4 eV, corresponding to Co 2p3/2 and Co 2p1/2 of metallic Co, respectively (Fig. S15a†);15 the P 2p spectra display two peaks at 129.4 eV and 130.2 eV related to the signals of phosphide, while the peak at 133.6 eV gradually decreasing along with Ar+ sputtering should be attributed to the superficial oxidation of CoP (Fig. S15b†).15,16 The peaks of Co 2p for the post-HER catalyst are similar to those of the as-prepared one, implying the retained metallic Co during the HER (Fig. S15c†). Interestingly, only the peak at 129.9 eV is observable while the phosphate peak at 133.6 eV is absent from the P 2p spectra of the post-HER sample, possibly due to the dissolution of cobalt phosphate under the cathodic conditions (Fig. S15d†).
In comparison, besides the metallic Co 2p peaks at 778.4 and 793.4 eV, two new peaks at 781.4 and 797.0 eV, as well as their satellite peaks at 786.8 and 803.6 eV, emerge in the Co 2p spectra for the post-OER catalyst and those can be assigned to Co3O4 (Fig. S15e†).4b The P 2p spectra also show a phosphate peak at 133.4 eV, together with the phosphide feature at 129.5 eV (Fig. S15f†). Different from those in the as-prepared CoP/rGO-400, the peaks for Co3O4 and phosphate in the post-OER catalyst remain strong even after long-time Ar+ sputtering, indicating that the CoP in the catalyst is partially oxidized to Co3O4 and cobalt phosphate during the OER. Such an in situ transformation explains the excellent OER activity of CoP/rGO-400 and that is in accordance with the reported results.4b,13
Encouraged by the above results, a two-electrode configuration was employed with CoP/rGO-400 as a bifunctional electrocatalyst to investigate its performance for overall water splitting. Since Pt/C is well-established for the HER and IrO2 for the OER, the integration of the Pt/C and IrO2 couple should provide a superb catalytic system, which, indeed, realizes water splitting with an overpotential of 480 mV at 10 mA cm−2 and a Tafel slope of 201 mV dec−1. When Pt/C was used as both electrodes, a much reduced catalytic performance was obtained (overpotential of 600 mV at 10 mA cm−2 and a Tafel slope of 251 mV dec−1). In stark contrast, CoP/GO-400, being applied as both electrodes, outperforms both systems and affords an overpotential of 470 mV at 10 mA cm−2 and a Tafel slope of 135 mV dec−1 (Fig. 3d and S16†). Particularly, the H2 and O2 yields during the water splitting over CoP/GO-400 were measured and the obtained molar ratio of H2/O2 fits well with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the total yield fits a 100% faradaic efficiency (Fig. S17†). The separated HER and OER as well as the overall water splitting performances of CoP/rGO-400, compared with Pt/C, IrO2 and their integrated couples, are shown in Table 1, clearly presenting the superb electrocatalytic activity of CoP/rGO-400.
:1 and the total yield fits a 100% faradaic efficiency (Fig. S17†). The separated HER and OER as well as the overall water splitting performances of CoP/rGO-400, compared with Pt/C, IrO2 and their integrated couples, are shown in Table 1, clearly presenting the superb electrocatalytic activity of CoP/rGO-400.
It should be noted that Sun et al. reported HER catalysts derived from ZIF-67 during the submission of our work.17 Apparent advantages of our work have been found after careful investigation into their work and the results reported herein. The detailed comparisons are listed as follows: (1) For catalysis, to clarify the real active site, a phase pure catalyst is highly desired unless one can demonstrate that a mixture possesses obvious advantages. The catalysts reported by Sun et al. are a mixture of CoP and Co2P, while no pure CoP or Co2P was obtained in their work. In contrast, pure CoP was obtained as the active site in our work (Fig. S5†). (2) The heterogeneous nucleation/growth of ZIF-67 on a substrate is very challenging as the nucleation of ZIF-67 is very fast and it is prone to self-nucleation.18 For the first time, we have successfully synthesized 2D ZIF-67/GO sheets with a sandwich-like morphology in the absence of a binding agent via a facile room-temperature route (Scheme 1). Our success could offer an important reference for the synthesis of other MOFs/GO composites. (3) To fabricate a highly active electrocatalyst, we have rationally combined ZIF-67 (as a porous CoP precursor) and GO (as a 2D hard template) to give 2D ZIF/GO sheets and the derived CoP/rGO-400 composite (Fig. 1a). As a result, with the help of GO, the conductivity and electrochemical activity of CoP/rGO-400 is much better than pure CoP, which fits well with our original expectation. Compared to our rational design and synthesis, Sun et al. just worked on the thermal conversion of single ZIF-67 to a Co–P mixture species. (4) Thanks to the rational design, the resultant CoP/rGO-400 catalyst exhibits superb HER and OER activity in 1 M KOH with an overpotential of 150 mV and 340 mV, respectively, at a mass loading of 0.28 mg cm−2 (Fig. 2b and 3a). In contrast, Sun's work is about 180 mV and 350 mV at 0.283 mg cm−2. Note that both the HER and OER activities of CoP/rGO-400 increase along with an increased mass loading (Fig. S18†). When the mass loading reached 0.42 mg cm−2, the overpotential of our CoP/rGO-400 for the HER and OER in 1 M KOH is 104 mV and 320 mV, respectively, which are even better than the result at 1 mg cm−2 in Sun's work (154 mV for HER and 319 mV for OER). (5) In our work, the H2 and O2 yields over time during the overall water splitting have demonstrated that the molar ratio of H2/O2 fits well with 2:1 and the total yield fits 100% faradaic efficiency (Fig. S17†), while similar tests are lacking in Sun's report. On the whole, our work is significantly different from Sun's work, not only the idea of the catalyst design, but also the catalyst fabrication, main active sites/composition (CoP and Co2P mixture for Sun et al., while CoP only for us) as well as the final catalytic performances.
Conclusions
In summary, we have developed CoP/rGO layered composites as bifunctional catalysts for overall water splitting, via a GO-templated MOF growth and subsequent pyrolysis and phosphating process. The resultant CoP/rGO-400 nanocomposite exhibits superior HER catalytic performance in acid solution. Moreover, it is able to behave as an electrocatalyst for both the HER and OER, in alkaline solution with great efficiency and durability. The excellent electrocatalytic performance might be attributed to the synergistic effect between the MOF-derived CoP and rGO in terms of the porous nanostructures, high electrical conductivity and stability against corrosion during the HER and OER. Significantly, CoP/rGO-400 can be directly employed as a catalyst for both electrodes to afford efficient H2 and O2 generation in a single electrolyzer, making it a promising overall water splitting catalyst. This study opens up an exciting avenue to the design of efficient electrocatalysts based on MOF–GO composites by integrating their respective merits. Given the huge diversity and tailorability of MOFs, the strategy presented herein holds great promise for electrocatalysis and studies along this line are ongoing in our laboratory.Acknowledgements
We are grateful to the reviewers for their insightful comments and valuable suggestions. This work is supported by the NSFC (21371162, 51301159 and 21521001), the 973 program (2014CB931803), the Recruitment Program of Global Youth Experts and the Fundamental Research Funds for the Central Universities (WK2060190026).Notes and references
- (a) J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed; (b) H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed; (c) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed; (d) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed; (e) R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550 CrossRef CAS PubMed; (f) Y. Yang, H. Fei, G. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175 CrossRef CAS PubMed; (g) H. Wang, H.-W. Lee, Y. Deng, Z. Lu, P.-C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS PubMed.
- (a) T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed; (b) Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed; (c) C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671 CrossRef CAS PubMed; (d) M.-R. Gao, J.-X. Liang, Y.-A. Zheng, Y.-F. Xu, J. Jiang, Q. Gao, J. Li and S.-H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed; (e) L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387 RSC; (f) Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 6710 CrossRef CAS PubMed; (g) E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427 CrossRef CAS PubMed; (h) D. Li, U. N. Maiti, J. Lim, D. S. Choi, W. J. Lee, Y. Oh, G. Y. Lee and S. O. Kim, Nano Lett., 2014, 14, 1228 CrossRef CAS PubMed.
- (a) T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925 CrossRef CAS PubMed; (b) Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950 CrossRef CAS PubMed; (c) H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef CAS PubMed; (d) L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071 CrossRef CAS PubMed.
- (a) C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351 CrossRef CAS PubMed; (b) N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251 CrossRef CAS PubMed; (c) Y.-P. Zhu, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337 CrossRef CAS.
- (a) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC; (b) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (c) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- (a) S. Ma, G. A. Goenaga, A. V. Call and D.-J. Liu, Chem.–Eur. J., 2011, 17, 2063 CrossRef CAS PubMed; (b) W. Zhang, Z.-Y. Wu, H.-L. Jiang and S.-H. Yu, J. Am. Chem. Soc., 2014, 136, 14385 CrossRef CAS PubMed; (c) Q. Lin, X. Bu, A. Kong, C. Mao, X. Zhao, F. Bu and P. Feng, J. Am. Chem. Soc., 2015, 137, 2235 CrossRef CAS PubMed; (d) Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010 CrossRef CAS PubMed; (e) W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837 RSC; (f) P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, Energy Environ. Sci., 2014, 7, 442 RSC; (g) D. Zhao, J.-L. Shui, L. R. Grabstanowicz, C. Chen, S. M. Commet, T. Xu, J. Lu and D.-J. Liu, Adv. Mater., 2014, 26, 1093 CrossRef CAS PubMed; (h) S. Zhao, H. Yin, L. Du, L. He, K. Zhao, L. Chang, G. Yin, H. Zhao, S. Liu and Z. Tang, ACS Nano, 2014, 8, 12660 CrossRef CAS PubMed; (i) G. Huang, Y.-Z. Chen and H.-L. Jiang, Acta Chim. Sinica, 2016 DOI:10.6023/A15080547 Search PubMed.
- (a) M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363 CrossRef CAS; (b) Y. Hou, T. Huang, Z. Wen, S. Mao, S. M. Cui and J. H. Chen, Adv. Energy Mater., 2014, 4, 1400337 Search PubMed; (c) H.-X. Zhong, J. Wang, Y.-W. Zhang, W.-L. Xu, W. Xing, D. Xu, Y.-F. Zhang and X.-B. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235 CrossRef CAS PubMed.
- (a) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS PubMed; (b) J. Qian, F. Sun and L. Qin, Mater. Lett., 2012, 82, 220 CrossRef CAS.
- (a) R. Wu, X. Qian, X. Rui, H. Liu, B. Yadian, K. Zhou, J. Wei, Q. Yan, X.-Q. Feng, Y. Long, L. Wang and Y. Huang, Small, 2014, 10, 1932 CrossRef CAS PubMed; (b) N. L. Torad, M. Hu, S. Ishihara, H. Sukegawa, A. A. Belik, M. Imura, K. Ariga, Y. Sakka and Y. Yamauchi, Small, 2014, 10, 2096 CrossRef CAS PubMed; (c) Y.-X. Zhou, Y.-Z. Chen, L. Cao, J. Lu and H.-L. Jiang, Chem. Commun., 2015, 51, 8292 RSC; (d) W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850 CrossRef CAS; (e) H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590 CrossRef CAS PubMed.
- (a) C. Petit and T. J. Bandosz, Adv. Mater., 2009, 21, 4753 CrossRef CAS; (b) M. Jahan, Q. Bao, J.-X. Yang and K. P. Loh, J. Am. Chem. Soc., 2010, 132, 14487 CrossRef CAS PubMed; (c) C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2011, 21, 2108 CrossRef CAS; (d) J. H. Lee, S. Kang, J. Jaworski, K. Y. Kwon, M. L. Seo, J. Y. Lee and J. H. Jung, Chem.–Eur. J., 2012, 18, 765 CrossRef CAS PubMed.
- Y. Li, M. A. Malik and P. O'Brien, J. Am. Chem. Soc., 2005, 127, 16020 CrossRef CAS PubMed.
- (a) Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693 CrossRef CAS; (b) C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC.
- (a) J. Ryu, N. Jung, J. H. Jang, H. J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066 CrossRef CAS; (b) J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638 CrossRef CAS PubMed.
- A. P. Grosvenor, S. D. Wik, R. G. Cavell and A. Mar, Inorg. Chem., 2005, 44, 8988 CrossRef CAS PubMed.
- F. H. Saadi, A. I. Carim, E. Verlage, J. C. Hemminger, N. S. Lewis and M. P. Soriaga, J. Phys. Chem. C, 2014, 118, 29294 CAS.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636 CrossRef.
- H. K. Kwon, H.-K. Jeong, A. S. Lee, H. S. An and J. S. Lee, J. Am. Chem. Soc., 2015, 137, 12304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of graphene oxide (GO), figures referred in the text and comparison of the literature catalytic parameters of various non-noble electrocatalysts. See DOI: 10.1039/c5sc04425a |
| This journal is © The Royal Society of Chemistry 2016 |