
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric addition of α-branched cyclic ketones to allenamides catalyzed by a chiral phosphoric acid†
Xiaoyu
Yang
and
F. Dean
Toste
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@Berkeley.edu
First published on 19th January 2016
Abstract
We describe the asymmetric addition of unactivated α-branched cyclic ketones to allenamides catalyzed by a chiral phosphoric acid, generating an all-carbon quaternary stereocenter with broad substrate scope and high enantioselectivity. The products are easily transformed into their corresponding 1,5- and 1,4-ketoaldehyde derivatives, which are both important building blocks in organic synthesis.
Cyclic ketones bearing an all-carbon quaternary stereocenter at the α-position are versatile building blocks in organic synthesis and exist in a variety of natural products.1 While allylic alkylation and direct alkylation have been applied to the enantioselective synthesis these compounds,2–4 Michael addition to conjugated olefins represents one of the most attractive methods for the formation of C–C bonds adjacent to ketones.5 Among the several types of Michael acceptors, α,β-unsaturated carbonyl compounds have attracted significant research interest because of their wide availability in organic synthesis. Therefore, many asymmetric catalytic systems using ketones as nucleophiles have been developed, including Lewis acid,6 Brønsted base,7 phase-transfer,8 phosphine9 and phosphoric acid or phosphate catalysis.10 Despite these advances, the majority of ketone substrates have been limited to activated functionality, such as β-keto esters, 2-oxindoles and others. For unactivated α-branched cyclic ketones, use of a preformed imine as a Michael donor is a reliable solution;11 however, the requirement for stoichiometric amounts of chiral amine and the additional steps of imine preparation and hydrolysis limit the utility of this protocol. A catalytic version of this reaction was recently achieved by Carter and co-workers, who reported the asymmetric addition of 2-alkyl cyclic ketones to alkyl acrylates catalyzed by a bifunctional catalyst system.12 However, the cyclic ketones were limited to 2-methyl and 2-ethyl substitution, which may be attributed to the steric difficulties for formation of the enamine intermediate.
Very recently, we reported on the direct asymmetric amination of α-branched cyclic ketones catalyzed by a chiral phosphoric acid to generate N-containing quaternary stereocenters from unactivated cyclic ketones.13 With our continuing interest in asymmetric functionalization of α-branched cyclic ketones,13,14 we turned our attention to potential Michael addition of these substrates to conjugated olefins. Unfortunately, addition of 2-phenylcyclohexanone to acrolein or methyl acrylate using (S)-TRIP as catalyst did not afford the desired addition product (Fig. 1a). Changing the acceptor to methyl vinyl ketone provided the Michael addition product in 44% yield with 19% ee. During the course of this work, List and co-workers reported on the asymmetric addition of α-branched cyclic ketones to alkyl vinyl ketones using (S)-TRIP as catalyst (Fig. 1b).15 However, bulky tert-butyl or isopropyl-substituted enones were required in order to achieve high enantioselectivities.
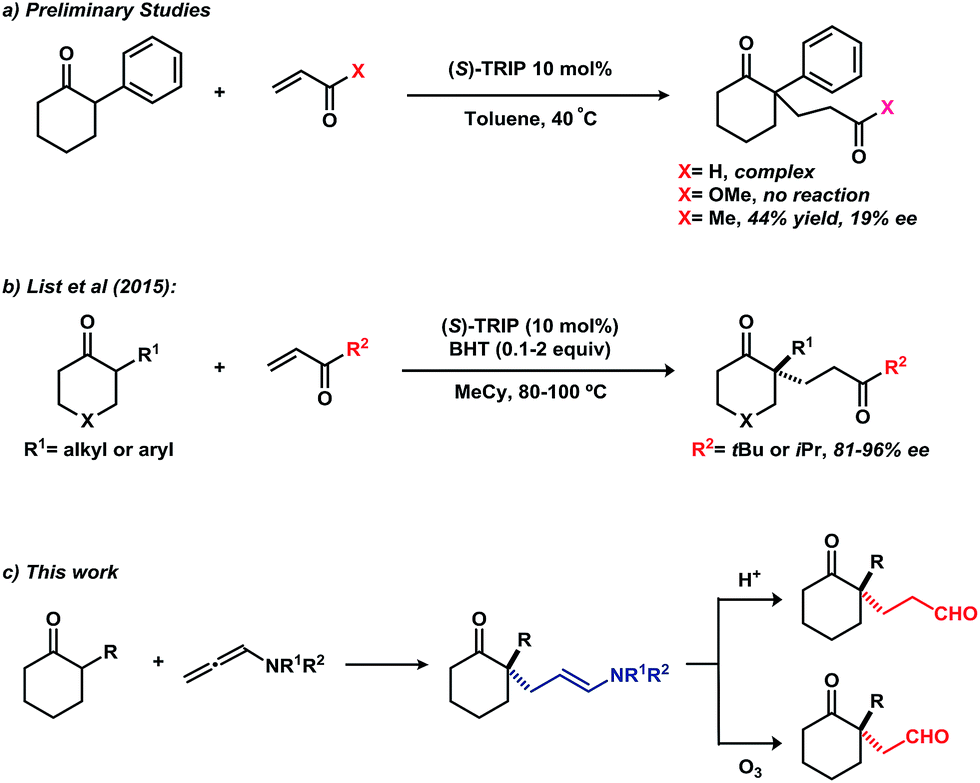 | ||
| Fig. 1 (a) Initial results on the addition of branched cyclic ketone to acrolein, methyl acrylate, and methyl vinyl ketone. (b) List's reports of asymmetric addition of branched cyclic ketones to tert-butyl vinyl ketone and isopropyl vinyl ketone. (c) Our proposal for the asymmetric addition of α-branched cyclic ketones to an allenamide and further transformation of the products. | ||
We hypothesized that using a Michael acceptor surrogate might overcome these limitations. Inspired by the work of Bandini,16 we envisioned an allenamide might serve this role in a chiral phosphoric acid catalysed addition of α-branched cyclic ketones (Fig. 1c). Hydrolysis of the enamide moiety in the product would give the 1,5-keto aldehyde; the product of a formal Michael addition to an acrolein acceptor (vide supra). Furthermore, 1,4-keto aldehydes could be obtained by the oxidative cleavage of the enamide moiety. Together, these products represent important building blocks in synthesis of complex molecular structures.
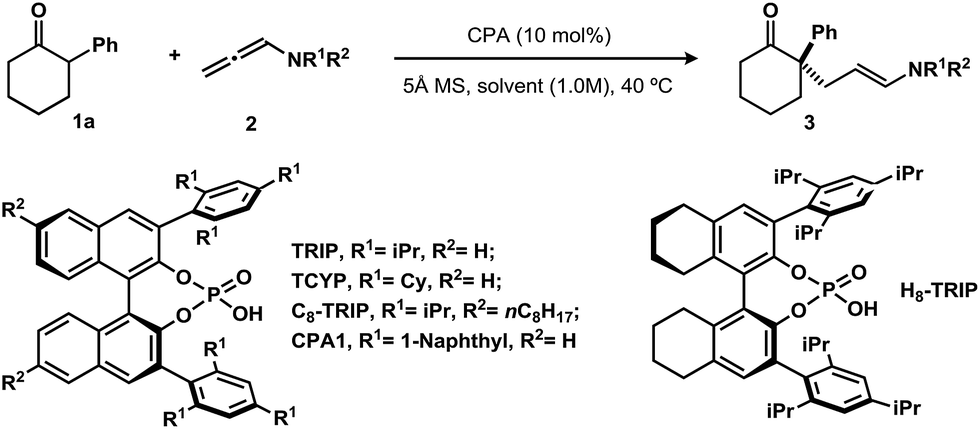
We selected 2-phenylcyclohexanone (1a) as a model substrate. Various N-phenyl-allenamides (2, 2.0 equiv.) were first screened using (S)-TRIP (10 mol%) as a catalyst in toluene (1.0 M) at 40 °C, in the presence of 5 Å molecular sieves to inhibit the hydrolysis of the allenamides under these conditions (Table 1). The N-Ts- or N-Boc-N-phenyl-allenamides gave the products in very low yields under these conditions (entries 1 and 2). The N-Bz-N-phenyl-substituted allenamide provided the product in 52% yield with 70% ee (entry 3). Replacement of N-phenyl by N-benzyl-allenamide failed to afford desired product (entry 4). In contrast, the desired adduct was isolated in 56% yield with 82% ee when the amide group was changed from a benzamide to an acetamide (entry 5). The enantioselectivity was further improved by increasing the steric bulk on the amide moiety. When N-pivaloyl-N-phenyl-allenamide was used, the product was obtained in 68% yield with 94% ee (entry 7). An examination of various solvents revealed that nonpolar solvents, such as benzene and xylenes, furnished the product with similar levels of selectivity to toluene, albeit in a slightly diminished yields (entries 8 and 9). Using dichloromethane as solvent or conducting the reaction neat, led to much lower yields of the products (entries 10 and 11). Chiral phosphoric acid catalysts were also evaluated; H8-TRIP or C8-TRIP gave the product with the identical enantioselectivity to TRIP, albeit in lower yield (entries 12 and 13). Changing the 3,3′-substitutions on phosphoric acids (CPA1 or TCYP) decreased the yield of the product (entries 14 and 15). Finally, in order to avoid diminished yields due to slow hydrolysis of the allenamide, we found adding the allenamide in two portions improved the yield to 72% (entry 16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Chiral phosphoric acids | Solvents | Yieldb [%] | eec [%] |
| a Reaction conditions, unless otherwise specified: 1a (0.1 mmol). 2 (0.2 mmol), catalyst (10 mol%), 30 mg 5 Å molecular sieves, solvent (0.1 mL), 40 °C, 24–48 h. b Isolated yield. c The ee value was determined by HPLC analysis on a chiral stationary phase. d The allenamide was added in two portions (1.0 equiv. at the beginning of the reaction, another 1.0 equiv. after 8 h). | ||||||
| 1 | Ph | Ts | (S)-TRIP | Toluene | 7% | ND |
| 2 | Ph | Boc | (S)-TRIP | Toluene | 17% | ND |
| 3 | Ph | Bz | (S)-TRIP | Toluene | 52% | 70% |
| 4 | Bn | Bz | (S)-TRIP | Toluene | NR | ND |
| 5 | Ph | Ac | (S)-TRIP | Toluene | 56% | 82% |
| 6 | Ph | iPrCO- | (S)-TRIP | Toluene | 68% | 90% |
| 7 | Ph | Piv | (S)-TRIP | Toluene | 68% | 94% |
| 8 | Ph | Piv | (S)-TRIP | Benzene | 57% | 94% |
| 9 | Ph | Piv | (S)-TRIP | Xylenes | 62% | 94% |
| 10 | Ph | Piv | (S)-TRIP | DCM | 15% | 86% |
| 11 | Ph | Piv | (S)-TRIP | -none- | 15% | 86% |
| 12 | Ph | Piv | (R)-H8-TRIP | Toluene | 62% | −94% |
| 13 | Ph | Piv | (R)-C8-TRIP | Toluene | 43% | −94% |
| 14 | Ph | Piv | (S)-CPA1 | Toluene | 19% | 73% |
| 15 | Ph | Piv | (R)-TCYP | Toluene | <10% | ND |
| 16 | Ph | Piv | (S)-TRIP | Toluene | 72% | 94% |
With the optimized conditions in hand, the substrate scope was explored (Scheme 1). Various substituted arenes were well tolerated at the 2-position of cyclohexanone (including electron–neutral, electron-donating and electron-withdrawing arenes, 3a–3h). The absolute configuration of the products was assigned as S by analogy to 3g, whose configuration was established by comparison of the optical rotation of the aldehyde resulting from oxidative cleavage to that previously reported.17 The more challenging o-tolyl substituted cyclohexanone also gave the desired product (3i) with 87% ee, although 20 mol% of (S)-TRIP was needed in order to obtain high conversion. A heteroaryl-substituted analogue worked well, yielding the product (3j) in excellent yield and enantioselectivity. In contrast to our previous work in the amination of the thiophenylketone, no kinetic resolution of the starting ketone was observed under these conditions.13 The cyclopentanone (3k) and 4-pyranone (3l) derived substrates were also well tolerated in this transformation.
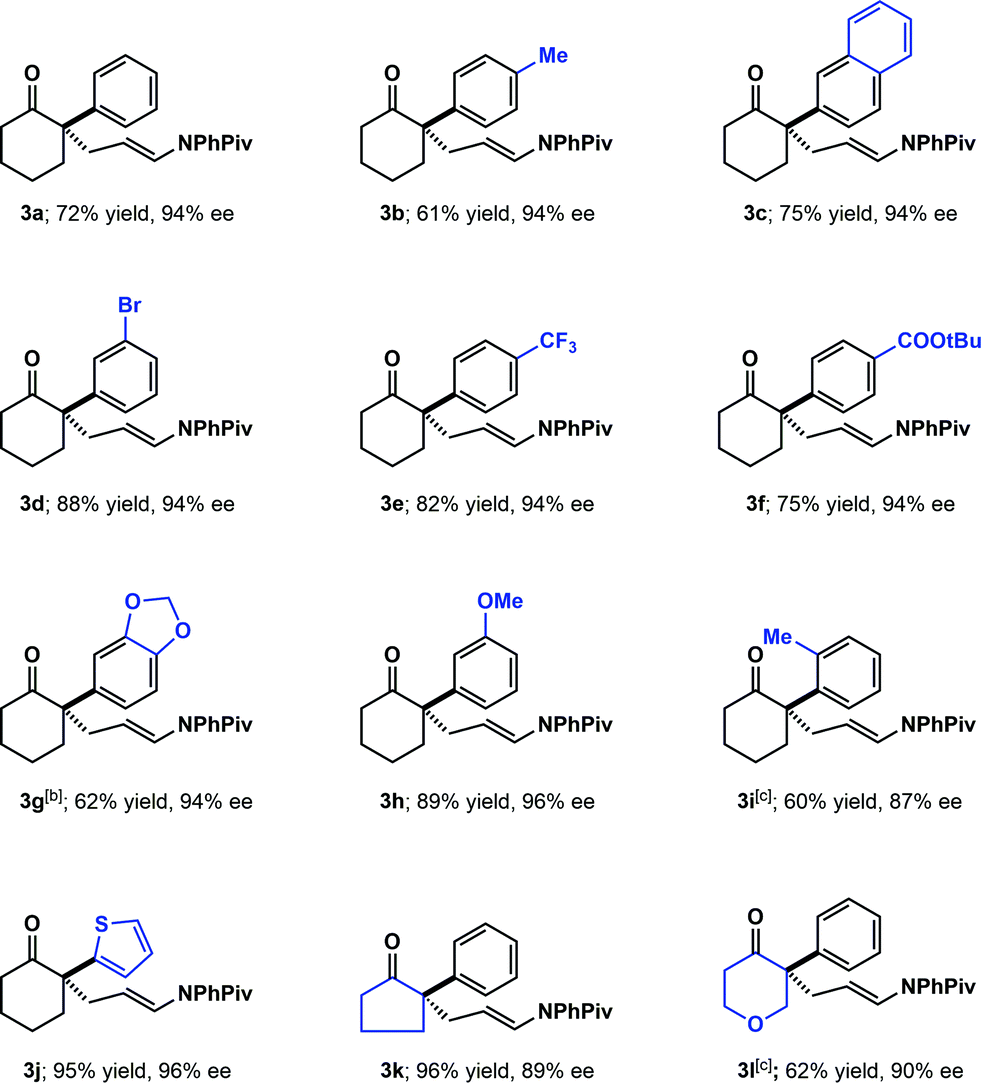 | ||
| Scheme 1 Substrate scope with α-aryl substitution. Reaction conditions, unless otherwise specified: 1 (0.3 mmol). 2 (0.6 mmol, added in two portions, 1.0 equiv. at the beginning of the reaction, another 1.0 equiv. after 8 h), (S)-TRIP (10 mol%), 90 mg 5 Å molecular sieves, toluene (0.3 mL), 40 °C, 24–48 h. Yields are isolated yields after chromatography. [b] The absolute configuration was determined by analogy to the derivative of 3g, see SI.† [c] 20 mol% (S)-TRIP was used. | ||
Having explored the compatibility of aryl substitution and modification of the cyclic ketone ring, we turned our attention to other substitution at the 2-position of cyclohexanone (Scheme 2). Various alkenyl substitutions were first investigated. The trans-styrenyl substituted ketone worked well, giving the desired product (3m) in 91% yield with 94% ee. The simple vinyl and 2-methyl propenyl-substituted substrates were compatible with these conditions (3o and 3p). Products (3n and 3q) derived from the sterically hindered cis-styrenyl and cyclohexenyl substituted ketones were also formed with excellent enantioselectivities, albeit in decreased yields. The 2-alkynyl substituted cyclohexanones were also evaluated under the standard conditions and found to provide the desired adducts (3r and 3s) in good yields with high enantioselectivities. Lastly, we examined 2-alkyl cyclohexanones, which present challenges due to issues with regioselectivity and reactivity. Under the standard conditions, 2-methyl- and 2-butylcyclohexanone gave the desired products (3t and 3u) in 70% and 78% ee, respectively; notably, we could not detect any of the undesired 6-substituted regioisomer.
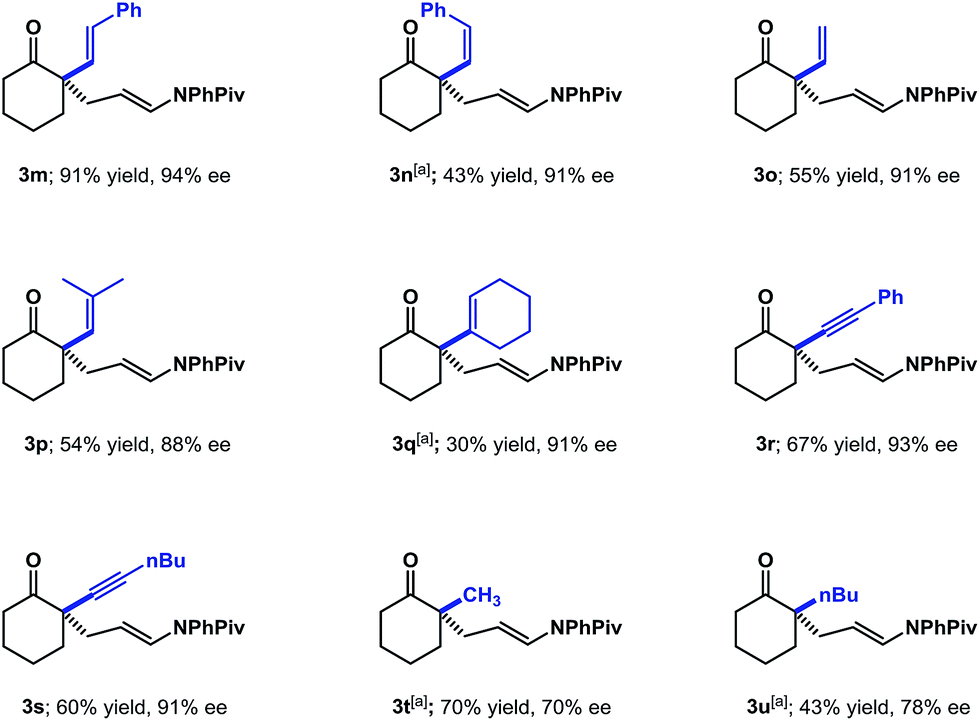 | ||
| Scheme 2 Substrate scope with α-alkenyl, alkynyl and alkyl substitution. Reaction conditions as indicated in Scheme 1. [a] 20 mol% (S)-TRIP was used. | ||
To explore viability of this approach as an alternative to the direct enantioselective Michael reaction, the application of the adducts to the asymmetric synthesis of structurally diverse compounds was examined (Scheme 3). As expected, treatment of 3a with 1 N HCl in Et2O for 3 min. afforded 1,5-keto aldehyde 4a in 92% yield without any loss of enantioselectivity. Further intramolecular aldol reaction of 4a under acid catalysis, followed by Dess–Martin periodinane-mediated oxidation furnished bicyclo[3.3.1]nonane185a in 73% yield. Compound 4a was transformed into bicyclic δ-lactone 6a in 84% yield with 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity using catalytic amount of SmI2 and iPrSH.19 The relative configuration of major diastereoisomer was assigned as cis by NOESY analysis. Oxidative cleavage of the enamide C
:1 diastereoselectivity using catalytic amount of SmI2 and iPrSH.19 The relative configuration of major diastereoisomer was assigned as cis by NOESY analysis. Oxidative cleavage of the enamide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with O3/Me2S provided 1,4-keto aldehyde 7a in 82% yield. Under reductive amination conditions, 7a was smoothly transformed into the cis-aryl hydroindole (8a) core structure of a variety of Amaryllidaceae-type alkaloids.4,20
C bond with O3/Me2S provided 1,4-keto aldehyde 7a in 82% yield. Under reductive amination conditions, 7a was smoothly transformed into the cis-aryl hydroindole (8a) core structure of a variety of Amaryllidaceae-type alkaloids.4,20
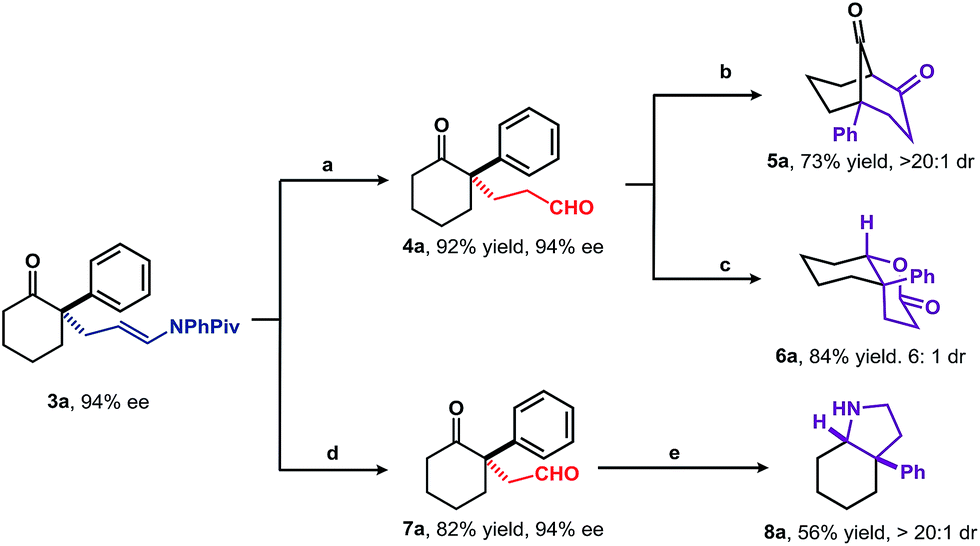 | ||
| Scheme 3 Synthetic transformations of the product 3a. Reaction conditions: (a) 1 N HCl, Et2O, rt, 3 min; (b) (1) 2 N HCl, Et2O, rt, overnight; (2) DMP, DCM, rt; (c) SmI2, iPrSH, THF, rt; (d) O3, Me2S, DCM, −78 °C ∼ rt; (e) NH4OAc, NaBH3CN, HOAc, EtOH, rt. DMP = Dess–Martin periodinane. | ||
Conclusions
In summary, we have achieved the asymmetric addition of unactivated α-branched cyclic ketones to allenamides catalyzed by a chiral phosphoric acid catalyst. The reaction generates a chiral quaternary stereocenter with broad substrate scope, including various aryl, alkenyl, alkynyl, and alkyl substitution as well as modification of the cyclic ketone ring. The products are easily transformed into their corresponding 1,4- and 1,5-ketoaldehyde derivatives, which are both important building blocks in organic synthesis. Moreover, the two-step protocol to produce 1,5-ketoaldehyes represents a viable entry into the direct enantioselective Michael addition of unactivated ketones to acrolein.Acknowledgements
We are grateful for financial support from the NIHGMS (RO1 GM073932). X. Y. acknowledges SIOC, Zhejiang Medicine, and Pharmaron for a fellowship. We thank Dillon H. Miles and Andrew J. Neel for manuscript revision.Notes and references
- For recent reviews on asymmetric synthesis of all-carbon stereocenters see: (a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed; (b) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473–1482 CrossRef CAS; (c) B. M. Trost, Synthesis, 2006, 369–396 CrossRef CAS; (d) M. Bella and T. Gasperi, Synthesis, 2009, 1583–1614 CrossRef CAS; (e) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC; (f) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- (a) B. M. Trost and G. M. Schroeder, J. Am. Chem. Soc., 1999, 121, 6759–6760 CrossRef CAS; (b) B. M. Trost, G. M. Schroeder and J. Kristensen, Angew. Chem., Int. Ed., 2002, 41, 3492–3495 CrossRef CAS; (c) B. M. Trost and G. M. Schroeder, Chem.–Eur. J., 2005, 11, 174–184 CrossRef PubMed.
- (a) D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 15044–15045 CrossRef CAS PubMed; (b) J. T. Mohr, D. C. Behenna, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2005, 44, 6924–6927 CrossRef CAS PubMed; (c) D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marinescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novák, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist, D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem.–Eur. J., 2011, 17, 14199–14223 CrossRef CAS PubMed.
- T. Kano, Y. Hayashi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 7134–7137 CrossRef CAS PubMed.
- (a) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701–1716 CrossRef CAS; (b) S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123–3135 RSC; (c) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 1279–1300 CrossRef CAS; (d) D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299–365 CrossRef; (e) M. Thirumalaikumar, Org. Prep. Proced. Int., 2011, 43, 67–129 CrossRef CAS; (f) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42–53 RSC.
- (a) M. Nakajima, Y. Yamaguchi and S. Hashimoto, Chem. Commun., 2001, 1596–1597 RSC; (b) Y. Hamashima, D. Hotta and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 11240–11241 CrossRef CAS PubMed; (c) F. Santoro, M. Althaus, C. Bonaccorsi, S. Gischig and A. Mezzetti, Organometallics, 2008, 27, 3866–3878 CrossRef CAS.
- (a) F. Wu, R. Hong, J. Khan, X. Liu and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 4301–4305 CrossRef CAS PubMed; (b) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem.–Eur. J., 2006, 12, 6039–6052 CrossRef CAS PubMed; (c) X. Li, Z.-G. Xi, S. Luo and J.-P. Cheng, Org. Biomol. Chem., 2010, 8, 77–82 RSC; (d) E. Badiola, B. Fiser, E. Gómez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. García, J. M. Odriozola, J. Razkin, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2014, 136, 17869–17881 CrossRef CAS PubMed.
- (a) W. Nerinckx and M. Vandewalle, Tetrahedron: Asymmetry, 1990, 1, 265–276 CrossRef CAS; (b) T. Ooi, T. Miki, M. Taniguchi, M. Shiraishi, M. Takeuchi and K. Maruoka, Angew. Chem., Int. Ed., 2003, 42, 3796–3798 CrossRef CAS PubMed; (c) R. He, C. Ding and K. Maruoka, Angew. Chem., Int. Ed., 2009, 48, 4559–4561 CrossRef CAS PubMed.
- (a) F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Angew. Chem., Int. Ed., 2013, 52, 943–947 CrossRef CAS PubMed; (b) T. Wang, W. Yao, F. Zhong, G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964–2968 CrossRef CAS PubMed.
- (a) T. Akiyama, T. Katoh and K. Mori, Angew. Chem., Int. Ed., 2009, 48, 4226–4228 CrossRef CAS PubMed; (b) W. Zheng, Z. Zhang, M. J. Kaplan and J. C. Antilla, J. Am. Chem. Soc., 2011, 133, 3339–3341 CrossRef CAS PubMed.
- (a) M. Pfau, G. Revial, A. Guingant and J. d'Angelo, J. Am. Chem. Soc., 1985, 107, 273–274 CrossRef CAS; (b) T. Volpe, G. Revial, M. Pfau and J. d'Angelo, Tetrahedron Lett., 1987, 28, 2367–2370 CrossRef CAS; (c) J. d'Angelo, D. Desmaële, F. Dumas and A. Guingant, Tetrahedron: Asymmetry, 1992, 3, 459–505 CrossRef; (d) M. Pfau, I. Jabin and G. Revial, J. Chem. Soc., Perkin Trans. 1, 1993, 1935–1936 RSC; (e) I. Jabin, G. Revial, A. Tomas, P. Lemoine and M. Pfau, Tetrahedron: Asymmetry, 1995, 6, 1795–1812 CrossRef CAS.
- J. Y. Kang and R. G. Carter, Org. Lett., 2012, 14, 3178–3181 CrossRef CAS PubMed.
- (a) X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS PubMed; (b) G. A. Shevchenko, G. Pupo and B. List, Synlett, 2015, 26, 1413–1416 CrossRef CAS.
- (a) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225–5228 CrossRef CAS PubMed; (b) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed; (c) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376–8379 CrossRef CAS PubMed.
- (a) I. Felker, G. Pupo, P. Kraft and B. List, Angew. Chem., Int. Ed., 2015, 54, 1960–1964 CrossRef CAS PubMed; (b) A. R. Burns, A. G. E. Madec, D. W. Low, I. D. Roy and H. W. Lam, Chem. Sci., 2015, 6, 3550–3555 RSC.
- (a) C. Romano, M. Jia, M. Monari, E. Manoni and M. Bandini, Angew. Chem., Int. Ed., 2014, 53, 13854–13857 CrossRef CAS PubMed; (b) E. Manoni, A. Gualandi, L. Mengozzi, M. Bandini and P. G. Cozzi, RSC Adv., 2015, 5, 10546–10550 RSC. For reviews on allenamides in organic synthesis see: (c) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862–4904 CrossRef CAS PubMed; (d) L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773–782 CrossRef CAS PubMed.
- See ESI†.
- For examples of this core in natural products, see: (a) E. Butkus, Synlett, 2001, 1827–1835 CrossRef CAS; (b) M. Ruiz, P. Lopez-Alvarado, G. Giorgi and J. C. Menendez, Chem. Soc. Rev., 2011, 40, 3445–3454 RSC; (c) J.-A. Richard, R. H. Pouwer and D. Y. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 4536–4561 CrossRef CAS PubMed.
- J.-L. Hsu, C.-T. Chen and J.-M. Fang, Org. Lett., 1999, 1, 1989–1991 CrossRef CAS.
- (a) Z. Jin, Nat. Prod. Rep., 2009, 26, 363–381 RSC; (b) M.-X. Wei, C.-T. Wang, J.-Y. Du, H. Qu, P.-R. Yin, X. Bao, X.-Y. Ma, X.-H. Zhao, G.-B. Zhang and C.-A. Fan, Chem.–Asian J., 2013, 8, 1966–1971 CrossRef CAS PubMed; (c) M. K. Das, S. De, S. Bisai and A. Bisai, Org. Biomol. Chem., 2015, 13, 3585–3588 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04202j |
| This journal is © The Royal Society of Chemistry 2016 |