
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerating chemoselective peptide bond formation using bis(2-selenylethyl)amido peptide selenoester surrogates†
Laurent
Raibaut‡
a,
Marine
Cargoët‡
a,
Nathalie
Ollivier
a,
Yun Min
Chang
a,
Hervé
Drobecq
a,
Emmanuelle
Boll
a,
Rémi
Desmet
a,
Jean-Christophe M.
Monbaliu
 *b and
Oleg
Melnyk
*a
*b and
Oleg
Melnyk
*a
aUMR CNRS 8161 CNRS, Université de Lille, Institut Pasteur de Lille, 1 rue du Pr Calmette, 59021 Lille Cedex, France. E-mail: oleg.melnyk@ibl.cnrs.fr
bCenter for Integrated Technology and Organic Synthesis, Department of Chemistry, University of Liège, Building B6a, Room 3/16a, Sart-Tilman, B-4000 Liège, Belgium
First published on 11th January 2016
Abstract
Given the potential of peptide selenoesters for protein total synthesis and the paucity of methods for the synthesis of these sensitive peptide derivatives, we sought to explore the usefulness of the bis(2-selenylethyl)amido (SeEA) group, i.e. the selenium analog of the bis(2-sulfanylethyl)amido (SEA) group, for accelerating peptide bond formation. A chemoselective exchange process operating in water was devised for converting SEA peptides into the SeEA ones. Kinetic studies show that SeEA ligation, which relies on an initial N,Se-acyl shift process, proceeds significantly faster than SEA ligation. This property enabled the design of a kinetically controlled three peptide segment assembly process based on the sequential use of SeEA and SEA ligation reactions. The method was validated by the total synthesis of hepatocyte growth factor K1 (85 AA) and biotinylated NK1 (180 AA) domains.
Introduction
The field of protein chemical synthesis has flourished substantially during the last decades to a point where the use of chemical methods is considered as a useful alternative to recombinant techniques for accessing small functional protein domains. Chemical synthesis is especially useful when proteins having unusual architectures1–6 or well-defined modifications at specific sites are desirable. An application of protein chemical synthesis which is of utmost importance in biological sciences is the study of some complex post-translational modifications such as glycosylation,7 ubiquitination8–10 or sumoylation.11,12Today, synthetic proteins are usually produced by assembling unprotected peptide segments in water. The native chemical ligation (NCL) is a popular chemoselective amide bond forming reaction in the field,13,14 although other ligations show great promise as well.15–19 The NCL reaction between a C-terminal peptide thioester and an N-terminal cysteinyl (Cys) peptide yields a native peptide bond to cysteine. Recently, C-terminal peptide selenoesters were also shown to be useful reactants for amide bond formation with the advantage of being significantly more reactive than peptide thioesters.20,21 Unfortunately, the application of peptide selenoesters for protein synthesis is difficult due to the limited access to these sensitive peptide derivatives, which were produced up to now using Boc solid phase peptide synthesis (SPPS) methods.20,22
The powerfulness of the NCL reaction has stimulated the development of several methods for accessing peptide thioesters using the Fmoc SPPS.23 In particular, several N,S-acyl shift systems relying on the N-(2-sulfanylethyl)amide structure were shown to rearrange into thioesters in acidic media.24–29 The fact that N-(2-sulfanylethyl)amides rearrange spontaneously into thioesters in strong aqueous acids has been noticed in the 1950s by Martin and coworkers30 using N-acetyl-β-mercaptoethylamine as a model system. This property is also shared by the peptide bond to cysteine.31 In contrast, the capacity of some N-(2-sulfanylethyl)amides32–35 such as the bis(2-sulfanylethyl)amido (SEA) group32,36 to rearrange into thioesters in water at neutral32 or mildly acidic37–39 pH is a recent observation (Scheme 1). The reaction of a SEA peptide with an N-terminal cysteinyl peptide yields a native peptide bond to cysteine and proceeds efficiently in the presence of 4-mercaptophenylacetic acid (MPAA40). The usefulness of the SEA ligation has been demonstrated by the total synthesis of several functional proteins.11,12,41–45
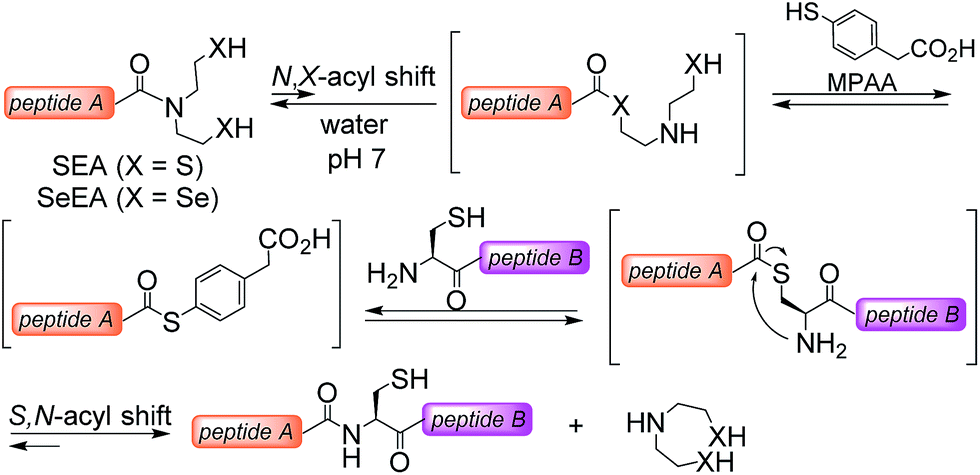 | ||
| Scheme 1 Principle of SEA (X = S) and SeEA (X = Se) chemoselective ligations. | ||
Given the promise of peptide selenoester chemistry for protein total synthesis, we sought to explore the potential of the bis(2-selenylethyl)amido (SeEA) amido group,46i.e. the selenium analog of the SEA group (Scheme 1), for accelerating the chemoselective formation of the peptide bond to Cys. We first developed a mild and chemoselective exchange process for converting SEA peptides into SeEA derivatives in water. Importantly, SeEA peptides proved to be more reactive than classical peptide alkylthioesters and significantly more reactive than SEA peptides in model ligation experiments conducted at neutral pH. This property enabled designing a one-pot kinetically controlled47 three peptide segment assembly process involving the sequential use of SeEA and SEA ligations.
Synthesis of SeEA peptides
SEA peptides are easily accessible by the Fmoc SPPS using SEA polystyrene32,48 or PEG-based resins.11,12 We therefore sought to convert SEA peptides 1 into SeEA derivatives 5 by using the exchange reaction shown in Scheme 2, which takes advantage of the capacity of SEA amide form 1 to equilibrate in water with the SEA transient thioester form 2.29,32,49 We reasoned that the latter might participate to a selenol–thioester exchange reaction with an excess of the bis(2-selenylethyl)amine 3 to produce, after an Se,N-acyl shift, the target SeEA peptide 5. Overall, the exchange process shown in Scheme 2 corresponds to a chemoselective transamidation reaction36 which is applied to unprotected SEA peptide segments.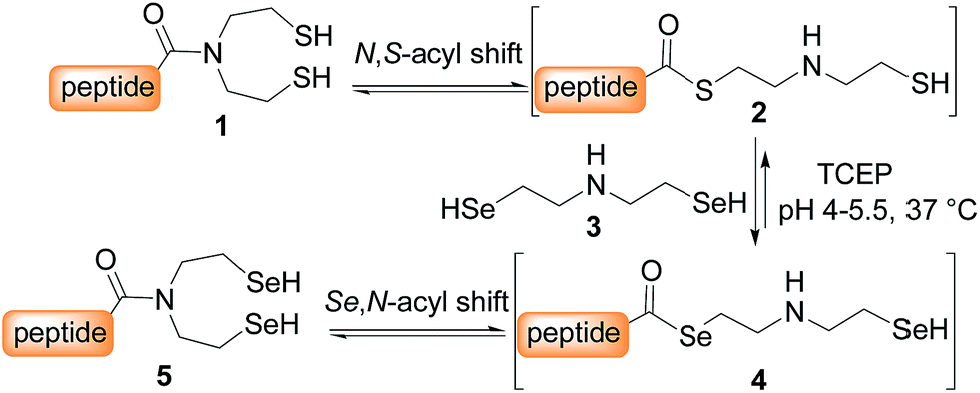 | ||
| Scheme 2 Conversion of SEA peptides into SeEA derivatives by transamidation. | ||
The trifluoroacetate salt of cyclic diselenide 7, i.e. 1,2,5-diselenazepane, was envisioned as a good precursor for diselenol 3. Its synthesis has been described elsewhere46 by reducing metallic selenium with sodium borohydride and reacting the resulting diselenide dianion Se2− with bis(2-chloroethyl)amine hydrochloride 6 (Scheme 3). The reaction yielded also the trifluoroacetate salt of triselenide 8, i.e. 1,2,3,6-triselenazocane, which was isolated and characterized in this work.
 | ||
| Scheme 3 Preparation of cyclic di or triselenides 7 and 8. | ||
The first attempts to transamidate model peptide 9a with diselenide 7 in the presence of TCEP yielded only trace amounts of the target SeEA peptide 10a (Fig. 1A and B). Instead, we observed the formation of a compound whose molecular mass corresponded to peptide 10a minus one selenium atom and to whom the structure of peptide 11a was assigned on the basis of the following arguments.
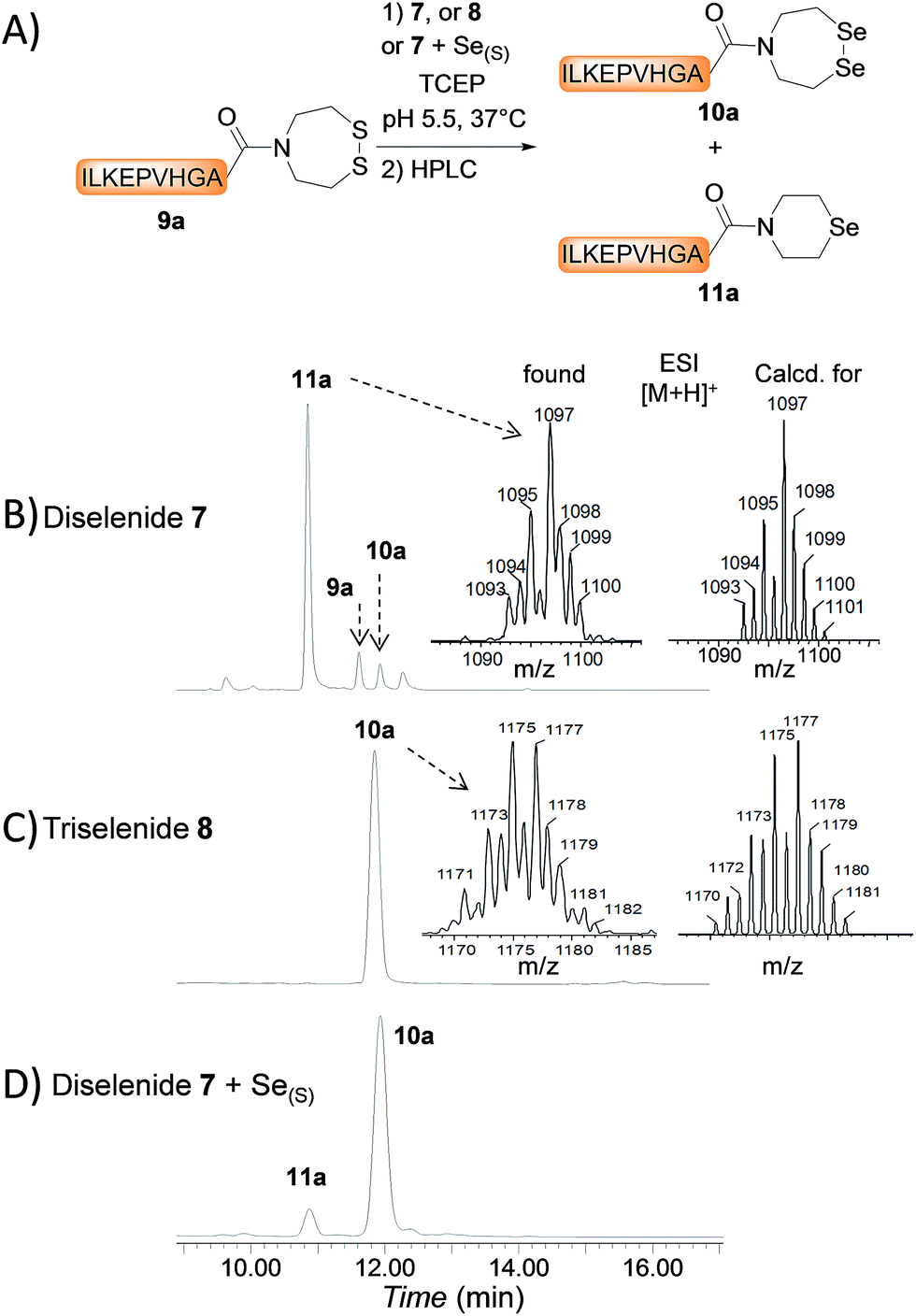 | ||
| Fig. 1 Conversion of SEA peptides into SeEA derivatives by transamidation (A). HPLC of the crude exchange reaction with diselenide 7 (10 equiv) (B), triselenide 8 (10 equiv) (C) or diselenide 7 (10 equiv) plus metallic selenium (2 equiv) (D). Peptide 9a 10 mM, TCEP 100 mM, pH 5.5, 37 °C, 24 h. | ||
The deselenization of selenocysteine into alanine by TCEP, which is reminiscent of the desulfurization of thiols by phosphites,50,51 is a well-known process which has been noticed as a serious side-reaction or utilized as a useful synthetic transformation.52–54 However, a similar deselenization process when applied to SeEA peptide 10a would yield a peptide featuring a C-terminal N-(2-selenylethyl)-N-ethyl amide moiety, whose molecular weight is 2 units greater to those measured for 11a. This mode of deselenization can thus be ruled out.
In contrast, the extrusion of selenium from diselenides to produce selenoethers has received much attention in the past and is a simple explanation to the formation of peptide 11a from diselenide 10a.55 Since this free-radical process is catalysed by tertiary phosphines,56 a potential solution to this problem was to replace TCEP by dithiothreitol (DTT) in the exchange reaction. However, while DTT is capable of reducing acyclic diselenides such as selenocystine bonds,57 it is unable to reduce cyclic diselenide 7.46 This fact imposed the use of TCEP in the exchange reaction and thus to find solution for avoiding the extrusion of selenium. Gratefully, the use of triselenide 8 as a precursor for diselenol 3 yielded exclusively the target SeEA peptide 10a (51%, Fig. 1C and Scheme 4). The exchange procedure using triselenide 8 and TCEP enabled also the successful preparation of several SeEA peptides in good yield such as SeEA peptide 10b which features a sterically demanding valine residue at the C-terminus or the 24 amino acids SeEA peptide 10c which includes an internal cysteine residue.
 | ||
| Scheme 4 Synthesis of SeEA peptides. *The Cys residue is temporarily protected by a tert-butylsulfenyl (tBuS) group in 9c. It is removed during the exchange process. | ||
The triselenide 8 is reduced in situ by TCEP into diselenol 3 with the concomitant production of the selenophosphine derived from TCEP, i.e. Se = TCEP, which is indeed easily detected by LC-MS (see ESI†). We have recently shown that Se = TCEP can inhibit the free-radical process leading to the deselenization of Sec by TCEP.58 Therefore, the presence of Se = TCEP in the exchange mixture might inhibit the formation of peptide 11a as well. In support of this hypothesis, the combined use of diselenide 7 and of metallic selenium as an alternative for the in situ formation of Se = TCEP led to a significant decrease in the proportion of peptide 11a in the exchange mixture and allowed the isolation of 10a in good yield (10a 56%, Fig. 1D and Scheme 4).§
Interestingly, SeEA peptides could also be prepared from C-terminal peptide thioesters by using triselenide 8 (12a → 10aScheme 4) or diselenide 7 and metallic selenium (29 → 30, Scheme 6). This exchange reaction is an interesting alternative for accessing SeEA peptides, since a large variety of synthetic methods are now available for producing peptide thioesters.23 Note that although SeEA peptides are probably produced in the exchange mixture in their reduced diselenol form due to the excess of TCEP, they eluted during analytical or preparative HPLC only as cyclic diselenides such as 10. This is probably due to the rapid air oxidation of the diselenols into the corresponding cyclic diselenides during workup.
The last synthetic access to SeEA peptides which is depicted in Scheme 4 is based on the coupling of diselenide 7 to a peptide acid using PyBOP/DIEA activation (13d → 10d, Scheme 4). This method which has been described elsewhere46 is not compatible with peptides featuring functional residues. It is thus of limited scope in comparison with the exchange processes discussed above.
To summarize at this stage, the chemoselective exchange of SEA peptides or peptide thioesters by bis(2-selenylethyl)amine 3 generated in situ by reduction of diselenide 7 or triselenide 8 constitutes a mild access to SeEA peptides. The high stability of SeEA peptides during the HPLC purification step and upon storage thanks to their tertiary amide structure is also worth mentioning.
Kinetic studies
We next determined the kinetic rates of the reaction of SEA peptides 9a,b, SeEA peptides 10a,b or MPA peptide thioesters 12a,b with Cys peptide 14 (Fig. 2A). The ligations with SeEA peptides 10a,b were performed in the presence of Se = TCEP to avoid the deselenization of the SeEA group by TCEP.58 The fraction ligated was quantified by HPLC (Fig. 2B). The data were nicely fitted to a first order kinetic law from which the t1/2 were determined (see the insert in Fig. 2B). Interestingly, the SeEA peptides proved to be significantly more reactive than SEA peptides. For example, the t1/2 for SEA peptide 9a with a C-terminal alanine residue was 3.3 h, while the t1/2 for the SeEA analog 10a was 0.24 h only, that is ∼14 fold lower. Peptide 15a produced by reacting SeEA peptide 10a with Cys peptide 14 was isolated by HPLC with a 56% yield. No hydrolysis of the SeEA peptide nor side-reactions with nucleophilic residues such as lysine could be detected by LC-MS. Moreover, analysis of peptide 15a by chiral GC-MS after total acid hydrolysis showed a D-Ala content <0.43%.59 Similarly, an ∼8 fold decrease of the t1/2 was observed for valine as C-terminal residue (SEA peptide 9bt1/2 23 h, SeEA peptide 10bt1/2 2.9 h). Interestingly, SeEA peptides 10a,b were also more reactive than the corresponding MPA peptide thioesters 12a,b (Fig. 2). Therefore, the observed order of reactivity at pH 7 was SeEA peptides > MPA peptide thioester ≫ SEA peptide. | ||
| Fig. 2 Kinetic rates for the reaction of SEA peptides 9a,b, SeEA peptides 10a,b or MPA peptide thioesters 12a,b with Cys peptide 14. The data were fitted to a first order kinetic law (continuous curves). The half-times (t1/2) of the reactions are indicated in the insert. Peptide concentration was 3.5 mM. | ||
We further performed the experiment depicted in Fig. 3 with the aim to identify the rate limiting step of the SEA ligation process, and propose an explanation for the significant impact of the S → Se substitution on the reactivity of the bis(2-chalcogenoethyl)amido group. The first experiment corresponds to a typical SEA ligation process (Fig. 3A, condition A). In these conditions, the SEA amide dithiol form is the only species observed for the reduced SEA peptide 9b during ligation. In the second experiment (Fig. 3A, condition B), the SEA peptide 9b was first reduced and incubated at pH 1 and 37 °C for 8 h to convert the SEA amide form into the SEA thioester form which represents ∼90% at equilibrium.29 Then, peptide 14 was added together with MPAA and the pH was quickly adjusted to 5.5. Fig. 3B shows the kinetic rate for these two ligation reactions as determined by HPLC. The data obtained using condition A obeys to a first-order kinetic law with a reaction half-life (t1/2) of 27.8 h. In contrast, the kinetic data obtained using condition B clearly show an initial burst of ligation product 15b formation followed by a slower phase. The first phase is due to the rapid consumption of the SEA thioester form produced during the pre-equilibration step. However, it is also competitively converted back to the SEA amide form, which is significantly less reactive and at the origin of the slower phase. The significant increase of the ligation rate which is observed when the SEA amide form is pre-equilibrated into the SEA thioester form supports the N,S-acyl shift of the SEA group to be the rate limiting step of the SEA ligation process.
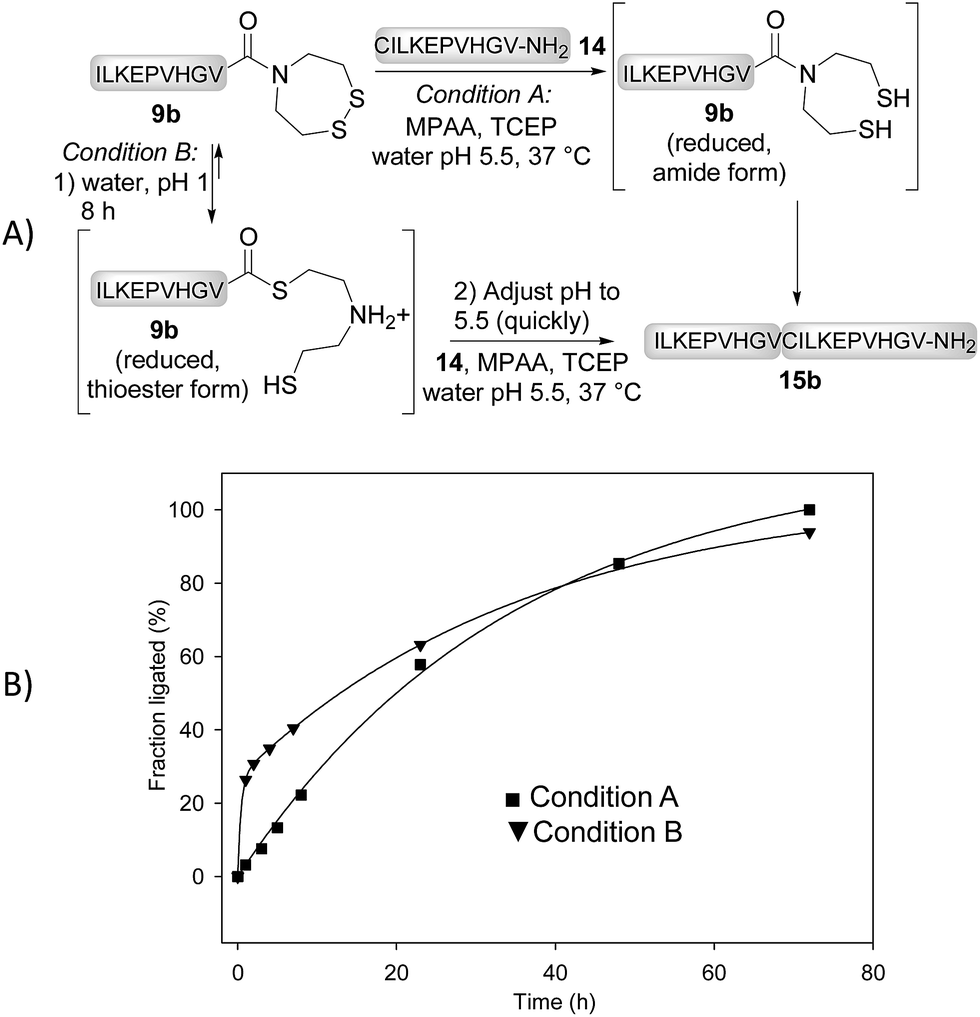 | ||
| Fig. 3 SEA ligation without (condition A) or with pre-equilibration of the SEA amide form into the SEA thioester form (condition B). | ||
Another interesting observation is that the rate of SEA ligation is accelerated while the pH is decreased from pH 7.2 to 5.5 (Fig. 4). The significant increase of the SEA ligation rate upon acidification, which has also been observed with peptidyl prolyl SEA peptides38 or with peptides featuring a SEA group on an aspartic or glutamic side-chain,37 shows that the transfer of a proton occurs in the transition state of the rate limiting step, that is the N,S-acyl shift.
 | ||
| Fig. 4 The effect of the pH on the rate of SEA ligation (peptide 9a or 9b 7 mM, peptide 14 10.5 mM, MPAA 200 mM, TCEP 200 mM, 37 °C). | ||
Preliminary DFT computations using N-formyl Gly-SEA 16a as a model for SEA peptides revealed that amongst the different plausible neutral or anionic transitions states which were identified up to now for the N,S-acyl shift, the anionic and concerted transition state TSSEA shown in Fig. 5A is by far the one displaying the lowest barrier of activation. In TSSEA, the neutral 2-sulfenylethyl limb is involved in an intramolecular S–H–N interaction. The intramolecular transfer of the thiol proton to the amide nitrogen in TSSEA is fully consistent with the experimentally observed pH-dependency of the SEA ligation reaction. The N-protonation of amides is known to weaken the nN → πC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O* conjugation, to disrupt the amide bond planarity and thus to increase the electrophilicity of the carbonyl toward nucleophiles.60 N-protonation is often used by biocatalysts for catalyzing biochemical processes such as amide bond isomerization,61 hydrolysis or cleavage.62 The N-protonation of amides (pKNa ∼ −7 (ref. 63)), although thermodynamically unfavourable over O-protonation (pKOa ∼ 0), can be facilitated within constrained systems which favour a hydrogen bond donation to the amide nitrogen.61
O* conjugation, to disrupt the amide bond planarity and thus to increase the electrophilicity of the carbonyl toward nucleophiles.60 N-protonation is often used by biocatalysts for catalyzing biochemical processes such as amide bond isomerization,61 hydrolysis or cleavage.62 The N-protonation of amides (pKNa ∼ −7 (ref. 63)), although thermodynamically unfavourable over O-protonation (pKOa ∼ 0), can be facilitated within constrained systems which favour a hydrogen bond donation to the amide nitrogen.61
 | ||
| Fig. 5 (A) Hypothetical anionic transition state structure for the intramolecular N-to-S acyl transfer of the SEA group. (B) pKas for the SEA thiol or SeEA selenol groups were estimated using ACS/Labs software. | ||
Importantly, the intramolecular N-protonation/nucleophilic attack mechanism permitted by the presence of two N-mercaptoethyl groups in close proximity to the amide group might explain the capacity of the SEA group to act as a good thioester surrogate at neutral pH,32 in comparison with related N,S-acyl shift systems which feature only one mercaptoethyl limb such as N-methyl cysteine25 or N-butyl-N-(2-sulfanylethyl)amido27 thioester surrogates. It also allows proposing an explanation for the greater reactivity of the SeEA group compared to the SEA one. Indeed, the pKa of selenols such as selenocysteamine or selenocysteine (pKa ∼ 5.5 (ref. 64)) are about 3 pKa units lower than the pKa of the sulfur analogs, cysteamine or cysteine (pKa ∼ 8.2 (ref. 65 and 66)). This difference in pKa, which is reminiscent of the higher acidity of hydrogen selenide (pKa 3.8) compared to hydrogen sulfide (pKa 7.0), is also found in S(Se)EA models 16a,b those pKa was estimated using ACD/Labs software (Fig. 5B).67 Therefore, the intramolecular protonation of the amide nitrogen and thus the activation of the amide carbonyl might be more pronounced for SeEA peptides. Moreover, the SEA group is mostly present at neutral pH as the dithiol form, i.e. the productive monoanionic species of type 16a (Fig. 5A) represents less than 1% of the total. In contrast, the monoanionic species for the SeEA analog is significantly more populated (∼7%). Thus, the higher acidity of selenols relative to thiols might contribute to the higher reactivity of the SeEA group by both increasing the concentration of the productive monoanionic species and lowering the barrier of activation for the N,Se-acyl shift. DFT computations aiming at identifying the transition states for the N,Se-acyl shift are in progress and will be reported in due course.
Kinetically controlled ligation using SEA and SeEA latent thio(seleno)ester peptides
The availability of fast and slow reacting thioester functionalities or thioester surrogates can be useful for designing kinetically controlled ligations (KCLs), which involve an NCL reaction between a Cys peptide and a fast reacting thioester in the presence of a slow reacting one. The field has been pioneered by Kent and coworkers who reacted a peptide arylthioester in the presence of a peptide alkylthioester.47 Later on, other KCLs have been developed using O,S-68 or N,S-acyl shift systems.33,38,44,69,70 Here we show that SeEA ligation can be performed in the presence of a SEA moiety thanks to the large difference of reactivity between SeEA and SEA groups towards Cys peptides. In particular, the sequential use of SeEA and SEA ligations enabled to develop the three peptide segment assembly method depicted in Scheme 5. | ||
| Scheme 5 Kinetically controlled ligation using sequential SeEA and SEA ligations. | ||
The reduction of the SeEA peptide segment 10c (or 10d) required the presence of TCEP at the first stage of the process. Here again, Se = TCEP (80 mM) proved to be a useful additive for minimizing the deselenization of the SeEA group by TCEP. Once the SeEA ligation was complete, the addition of the Cys peptide 21 (or 22) trigged the SEA ligation step and the formation of the target polypeptide. The assembly of the model peptide segments 10d, 19 and 22 involved the formation of two Gly–Cys junctions and yielded the 26 amino acids peptide 23d (27% overall after HPLC purification). The method was also applied to the synthesis of the 85 amino acids Kringle 1 domain (K1) of hepatocyte growth factor/scatter factor (HGF/SF).42,71 This example involved the assembly of peptide segments 10c (HGF/SF 125–148), 18 (HGF/SF 149–176) and 21 (HGF/SF 177–209) through the sequential formation of Lys–Cys and Tyr–Cys junctions. Fig. 6 highlights the efficiency of the one-pot process which yielded the K1 polypeptide 23c with 21% overall yield after HPLC purification. The K1 polypeptide produced in this work using the SeEA/SEA KCL method was found to be identical by LC-MS to a sample obtained by another route (see ESI†).42
 | ||
| Fig. 6 Monitoring of the one-pot assembly of K1 polypeptide using HPLC. (A) Few minutes after dissolving peptide segments 10c and 18 (4 mM each). (B) SeEA ligation step after 30 h. The peak marked by an asterisk corresponds to the partial cyclization of peptide 18. (C) SEA ligation step 72 h after the addition of Cys peptide 21. For (A–C), the analysis were run on a C3 Zorbax column with a linear gradient of 0% eluent B (ACN/H2O (4/1 by vol) containing 0.1% TFA) in eluent A (water containing 0.1% TFA) to 100% eluent B in eluent A over 30 min, UV detection at 214 nm. (D) Purified K1 polypeptide. The analysis was run on a C18 Xbridge column using the above conditions. | ||
Synthesis of large SeEA peptides and total synthesis of biotinylated NK1 domain of hepatocyte growth factor
So far, KCL methods were used for accessing proteins composed of less than 100 amino acids. One reason for this is the difficulty in accessing large peptide segments (>50 AA) equipped with a reactive C-terminal thio or selenoester group. Usually, large peptides are prepared by coupling chemoselectively several unprotected peptide segments together. However, such processes are hardly compatible with the presence of highly reactive functional groups such as selenoesters. A potential solution to this problem is to introduce the selenoester moiety after the chain assembly.An appealing alternative is to use a latent selenoester during the assembly of the peptide. With such a strategy, the formation of side-products during the assembly of the latent selenoester is minimized, while the KCL process can be triggered in a subsequent step when needed.
In a previous communication we showed that the SeEA group is compatible with the NCL and SEA ligations in the presence of reducing thiols such as MPAA or DTT.46 We show here that the sequential use of NCL and SEA ligations in a one-pot process permitted the synthesis of the 118 amino acids SeEA peptide 31 as illustrated in Scheme 6 (21% based on 26, one-pot process III). Peptide 31 corresponds to HGF 31–148 and was assembled using MPA thioester peptide 26, SEA peptide 27 and SeEA peptide 30.
 | ||
| Scheme 6 Total synthesis of SeEA peptide 31 by a sequential NCL/SEA ligation process (118 amino acids, one-pot process III) and of NK1-B protein by a SeEA/SEA KCL process (180 amino acids, one-pot process IV). | ||
Peptide 26 was produced using one-pot process I by ligating MPA thioester 24 with SEA peptide 25 at pH 7 in the presence of MPAA, followed by the in situ exchange of the SEA group by MPA (46% for the two steps). SEA peptide 27 was obtained by standard Fmoc SPPS starting from SEA polystyrene resin.32,48 The third segment, i.e. SeEA peptide 30, was produced starting from MPA thioester 29 using one-pot process II. One-pot process II involved first the introduction of the SeEA group by treatment with diselenide 7, metallic selenium and TCEP. The second step allowed the formation of the cyclic diselenide by oxidation with DMSO, while the N-terminal acetoacetyl group was removed in the final step by addition of hydroxylamine.72 The successful preparation of SeEA peptide 31 set the stage for the assembly of the biotinylated NK1 domain of HGF (NK1-B), a protein composed of 180 amino acids (Scheme 6, one-pot process IV). NK1 protein was modified by a C-terminal biotinylated lysine residue to allow the development of binding assays in the future. NK1-B was assembled using the SeEA/SEA KCL method depicted in Scheme 6 (one-pot process IV), which is almost identical to the SeEA/SEA KCL discussed above (see Scheme 5). Gratifyingly, the sequential assembly of SeEA peptide 31, SEA peptide 18 and Cys peptide 33 furnished successfully NK1-B polypeptide (32% based on 31, 3.1% based on 24, Fig. 7), showing the usefulness of S(Se)EA chemistry for accessing challenging proteins.
 | ||
| Fig. 7 Characterization of NK1-B protein. (A) LC analysis of NK1-B. (A) LC trace, eluent A 0.10% formic acid (FA) in water, eluent B 0.10% FA in CH3CN/water: 4/1 by vol. C3 Zorbax 300SB 3.5 μm (4.6 × 150 mm) column, gradient 0–100% B in 30 min (1 mL min−1, detection 215 nm). (B) ESI MS high resolution spectrum. (C) Deconvoluted spectrum and comparison with the theoretical profile. | ||
Conclusions
In conclusion, SeEA peptides are highly stable peptide derivatives which behave as latent selenoesters upon reduction of the cyclic diselenide. They are easily accessible through the chemoselective transamidation of SEA peptides or the selenol–thioester exchange of thioester peptides in water by an excess of bis(2-selenylethyl)amine. This reagent is formed in situ by reduction of 1,2,5-diselenazepane or 1,2,3,6-triselenazocane by TCEP. The presence of Se = TCEP in the exchange mixture minimizes the extrusion of one selenium atom from SeEA diselenide. SeEA peptides react faster than peptide alkylthioesters or SEA peptides with cysteinyl peptides. The large difference of reactivity between SeEA and SEA functionalities enabled designing a one-pot kinetically controlled three peptide segment assembly process working in the N-to-C direction. The method was validated by the total synthesis of hepatocyte growth factor K1 (85 AA) and biotinylated NK1 (NK1-B, 180 AA) domains. NK1-B is by far the largest polypeptide produced by using a KCL process. The synthesis of a latent SeEA selenoester with a size >100 amino acids and the assembly of a 180 amino acids protein by a series of one-pot processes show the great potential of S(Se)EA chemistries for accessing challenging proteins.Acknowledgements
LR and MC thank the Région Nord Pas-de-Calais and the Région Centre councils for a PhD fellowship. YMC thanks the HHMI-Institut Pasteur de Lille program for financial support. We thank the Chemistry Systems Biology (CSB) facility for technical help and advice. JCMM acknowledges the University of Liège for financial support (WG-13/03) and the F.R.S.-FNRS for the access to the HPC facilities (CéCI). We thank Jean-Michel Saliou for HR MS analyses (P3M facility, Institut Pasteur de Lille).Notes and references
- K. Mandal, B. L. Pentelute, D. Bang, Z. P. Gates, V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2012, 51, 1481–1486 CrossRef CAS PubMed.
- R. J. Clark and D. J. Craik, Methods Enzymol., 2012, 503, 57–74 CAS.
- R. J. Clark and D. J. Craik, Biopolymers, 2010, 94, 414–422 CrossRef CAS PubMed.
- G. T. Dolphin, J. Am. Chem. Soc., 2006, 128, 7287–7290 CrossRef CAS PubMed.
- J. A. Camarero, J. Pavel and T. W. Muir, Angew. Chem., Int. Ed., 1998, 37, 347–349 CrossRef CAS.
- J. A. Camarero and T. W. Muir, Chem. Commun., 1997, 1369–1370 RSC.
- C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC.
- K. S. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2011, 49, 9126–9131 CrossRef PubMed.
- K. S. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon and A. Brik, Angew. Chem., Int. Ed., 2011, 50, 6137–6141 CrossRef CAS PubMed.
- B. Fierz, C. Chatterjee, R. K. McGinty, M. Bar-Dagan, D. P. Raleigh and T. W. Muir, Nat. Chem. Biol., 2011, 7, 113–119 CrossRef CAS PubMed.
- E. Boll, H. Drobecq, N. Ollivier, L. Raibaut, R. Desmet, J. Vicogne and O. Melnyk, Chem. Sci., 2014, 5, 2017–2022 RSC.
- E. Boll, H. Drobecq, N. Ollivier, A. Blanpain, L. Raibaut, R. Desmet, J. Vicogne and O. Melnyk, Nat. Protoc., 2015, 10, 269–292 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS PubMed.
- T. Durek and P. F. Alewood, Angew. Chem., Int. Ed., 2011, 50, 12042–12045 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS PubMed.
- A. Ghassemian, X. Vila-Farres, P. F. Alewood and T. Durek, Bioorg. Med. Chem., 2013, 21, 3473–3478 CrossRef CAS PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed.
- F. Nagaike, Y. Onuma, C. Kanazawa, H. Hojo, A. Ueki, Y. Nakahara and Y. Nakahara, Org. Lett., 2006, 8, 4465–4468 CrossRef CAS PubMed.
- H. Hojo, Y. Onuma, Y. Akimoto, Y. Nakahara and Y. Nakahara, Tetrahedron Lett., 2007, 48, 25–28 CrossRef CAS.
- S. Tsuda, A. Shigenaga, K. Bando and A. Otaka, Org. Lett., 2009, 11, 823–826 CrossRef CAS PubMed.
- M. Taichi, X. Hemu, Y. Qiu and J. P. Tam, Org. Lett., 2013, 15, 2620–2623 CrossRef CAS PubMed.
- J.-S. Zheng, S. Tang, Y.-C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475–2484 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier, A. Vallin and O. Melnyk, J. Org. Chem., 2011, 76, 3194–3202 CrossRef CAS PubMed.
- R. B. Martin, S. Lowey, E. L. Elson and J. T. Edsall, J. Am. Chem. Soc., 1959, 81, 5089–5095 CrossRef CAS.
- J. Kang, J. P. Richardson and D. Macmillan, Chem. Commun., 2009, 407–409 RSC.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS PubMed.
- K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto and A. Otaka, ChemBioChem, 2011, 12, 1840–1844 CrossRef CAS PubMed.
- F. Burlina, G. Papageorgiou, C. Morris, P. D. White and J. Offer, Chem. Sci., 2014, 5, 766–770 RSC.
- V. P. Terrier, H. Adihou, M. Arnould, A. F. Delmas and V. Aucagne, Chem. Sci., 2016, 7, 339–345 RSC.
- O. Melnyk and V. Agouridas, Curr. Opin. Chem. Biol., 2014, 22, 137–145 CrossRef CAS PubMed.
- E. Boll, J. Dheur, H. Drobecq and O. Melnyk, Org. Lett., 2012, 14, 2222–2225 CrossRef CAS PubMed.
- L. Raibaut, P. Seeberger and O. Melnyk, Org. Lett., 2013, 15, 5516–5519 CrossRef CAS PubMed.
- R. Desmet, M. Pauzuolis, E. Boll, H. Drobecq, L. Raibaut and O. Melnyk, Org. Lett., 2015, 17, 3354–3357 CrossRef CAS PubMed.
- E. C. Johnson and S. B. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- L. Raibaut, J. Vicogne, B. Leclercq, H. Drobecq, R. Desmet and O. Melnyk, Bioorg. Med. Chem., 2013, 21, 3486–3494 CrossRef CAS PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El-Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- F. Ancot, C. Leroy, G. Muharram, J. Lefebvre, J. Vicogne, A. Lemiere, Z. Kherrouche, B. Foveau, A. Pourtier, O. Melnyk, S. Giordano, A. Chotteau-Lelievre and D. Tulasne, Traffic, 2012, 13, 1261–1272 CrossRef CAS PubMed.
- R. Yang, W. Hou, X. Zhang and C.-F. Liu, Org. Lett., 2012, 14, 374–377 CrossRef CAS PubMed.
- W. Hou, X. Zhang, F. Li and C. F. Liu, Org. Lett., 2011, 13, 386–389 CrossRef CAS PubMed.
- L. Raibaut, H. Drobecq and O. Melnyk, Org. Lett., 2015, 17, 3636–3639 CrossRef CAS PubMed.
- D. Bang, B. L. Pentelute and S. B. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- N. Ollivier, L. Raibaut, A. Blanpain, R. Desmet, J. Dheur, R. Mhidia, E. Boll, H. Drobecq, S. L. Pira and O. Melnyk, J. Pept. Sci., 2014, 20, 92–97 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier and O. Melnyk, Org. Lett., 2011, 13, 1560–1563 CrossRef CAS PubMed.
- F. W. Hoffmann, R. J. Ess, T. C. Simmons and R. S. Hanzel, J. Am. Chem. Soc., 1956, 78, 6414 CrossRef CAS.
- C. Walling and R. Rabinowitz, J. Am. Chem. Soc., 1957, 79, 5326 CrossRef CAS.
- M. D. Gieselman, L. Xie and W. A. van der Donk, Org. Lett., 2001, 3, 1331–1334 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- S. Dery, P. S. Reddy, L. Dery, R. Mousa, R. N. Dardashti and N. Metanis, Chem. Sci., 2015, 6, 6207–6212 RSC.
- L. Henriksen, in The Chemistry of Organic Selenium and Tellurium Compounds, ed. S. Patai, John Wiley & Sons, Chichester, 1987, vol. 2, pp. 393–420 Search PubMed.
- J. Y. C. Chu and D. G. Marsh, J. Org. Chem., 1976, 41, 3204–3205 CrossRef CAS.
- T. Koide, H. Itoh, A. Otaka, H. Yasui, M. Kuroda, N. Esaki, K. Soda and N. Fujii, Chem. Pharm. Bull., 1993, 41, 502–506 CrossRef CAS PubMed.
- N. Ollivier, A. Blanpain, L. Raibaut, H. Drobecq and O. Melnyk, Org. Lett., 2014, 16, 3032–4035 CrossRef PubMed.
- W. Amelung and S. Brodowski, Anal. Chem., 2002, 74, 3239–3246 CrossRef CAS PubMed.
- R. Szostak, J. Aube and M. Szostak, Chem. Commun., 2015, 51, 6395–6398 RSC.
- C. Cox and T. Lectka, Org. Lett., 1999, 1, 749–752 CrossRef CAS PubMed.
- D. G. A. Johansson, G. Wallin, A. Sandberg, B. Macao, J. Aqvist and T. Härd, J. Am. Chem. Soc., 2009, 131, 9475–9477 CrossRef CAS PubMed.
- A. R. Fersht, J. Am. Chem. Soc., 1971, 93, 3504–3515 CrossRef CAS PubMed.
- A. P. Arnold, K. S. Tan and D. L. Rabenstein, Inorg. Chem., 1986, 25, 2433–2437 CrossRef CAS.
- L. Riauba, G. Niaura, O. Eicher-Lorka and E. Butkus, J. Phys. Chem. A, 2006, 110, 13394–13404 CrossRef CAS PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS PubMed.
- M. Meloun and S. Bordovská, Anal. Bioanal. Chem., 2007, 389, 1267–1281 CrossRef CAS PubMed.
- J. S. Zheng, H. K. Cui, G. M. Fang, W. X. Xi and L. Liu, ChemBioChem, 2010, 11, 511–515 CrossRef CAS PubMed.
- H. Ding, A. Shigenaga, K. Sato, K. Morishita and A. Otaka, Org. Lett., 2011, 13, 5588–5591 CrossRef CAS PubMed.
- T. Nakamura, A. Shigenaga, K. Sato, Y. Tsuda, K. Sakamoto and A. Otaka, Chem. Commun., 2014, 50, 58–60 RSC.
- C. Simonneau, L. Berenice, A. Mougel, E. Adriaenssens, C. Paquet, L. Raibaut, N. Ollivier, H. Drobecq, J. Marcoux, S. Cianferani, D. Tulasne, H. de Jonge, O. Melnyk and J. Vicogne, Chem. Sci., 2015, 6, 2110–2121 CAS.
- E. Boll, J. P. Ebran, H. Drobecq, O. El-Mahdi, L. Raibaut, N. Ollivier and O. Melnyk, Org. Lett., 2015, 17, 130–133 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculations, synthetic protocols and characterization for all compounds. See DOI: 10.1039/c5sc03459k |
| ‡ Laurent Raibaut and Marine Cargoët contributed equally to this work. |
| § Note that triselenide 8 was found to decompose partially into diselenide 7 plus selenium upon storage (see ESI†). Therefore, the combined use of diselenide 7 and metallic selenium is a useful alternative to triselenide 8 for accessing SeEA peptides. |
| This journal is © The Royal Society of Chemistry 2016 |