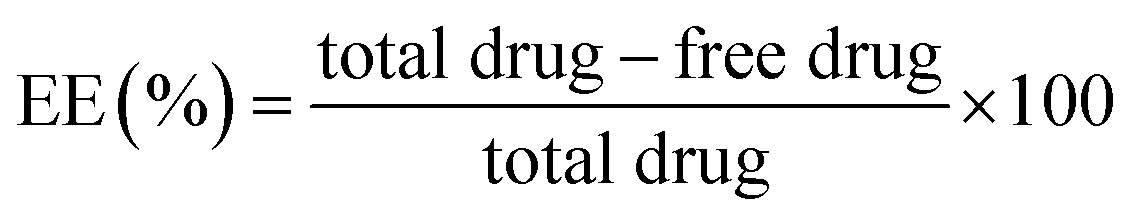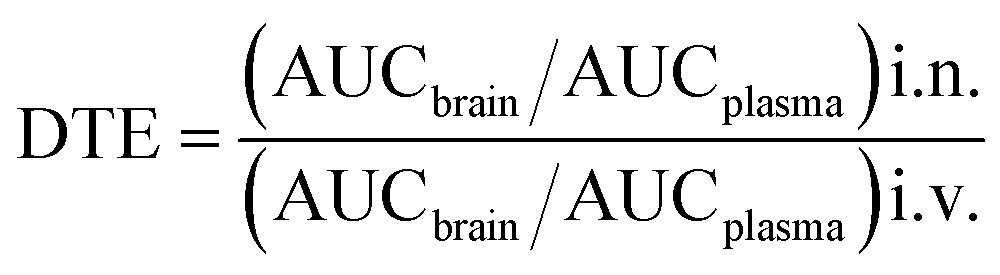DOI:
10.1039/C5RA19793G
(Paper)
RSC Adv., 2016,
6, 2032-2045
Intranasal delivery of asenapine loaded nanostructured lipid carriers: formulation, characterization, pharmacokinetic and behavioural assessment†
Received
24th September 2015
, Accepted 16th December 2015
First published on 4th January 2016
Abstract
The aim of the present research work was to develop asenapine (ASM) loaded nanostructured lipid carriers (ANLC) for the delivery of drugs in the brain by an intranasal route to enhance therapeutic efficacy. A quality by design approach was used for development and optimization of ANLC. A total of five independent variables were selected, in which three were compositions and two were process variables, while particle size and entrapment efficiency were selected as response variables. The final optimized batch was evaluated by various in vitro characterizations as well as in vivo brain and plasma pharmacokinetic studies. Finally, the ANLC was assessed for efficacy and safety profiling for upto three weeks by a behavior model viz. catalepsy, induced locomotor and paw test in Charles Foster rats. The observed particle size, entrapment efficiency and zeta potential of ANLC was found to be 167.30 ± 7.52 nm, 83.50 ± 2.48% and −4.33 ± 1.27 mV, respectively. Surface characterization studies demonstrated a spherical shape with a smooth surface of ANLC which follows the Korsmeyer–Peppas in vitro release kinetic model (r2 = 0.9911, n = 0.53). A brain pharmacokinetic study indicated a significantly higher (p < 0.05) peak drug concentration (Cmax: 74.13 ± 6.73 ng mL−1), area under the drug concentration–time curve (AUC0–24 h: 560.93 ± 27.85 h ng mL−1) and mean residence time (MRT: 7.1 ± 0.13 h) of ANLC compared to ASM in the brain via an intranasal route. The results of behaviour studies of ANLC showed a significant decrease in extra-pyramidal side effects with increasing antipsychotic effect after 1–2 week(s) of treatment. These findings demonstrate that nanostructured lipid carriers could be a new promising drug delivery system for intranasal delivery of asenapine in the treatment of schizophrenia.
1. Introduction
Schizophrenia is a severe chronic debilitating brain disease, afflicting more than 21 million people worldwide, and more prevalent in urban population than rural population albeit without any sexual disposition.1,2 The age of onset is generally between 20 and 35 year and it is characterized by positive, negative symptoms and cognitive dysfunction. Schizophrenia has devastating effects on several aspects of a patient's life. It is amongst the top ten ailments world-wide and on average reduces the patient's life span by ten years. More than 34% of patients demonstrate adherence to therapy problems during the first 4–6 weeks of treatment, with the number rising to 74% within the next 2 years resulting in a significantly high rate of relapse, risk and length of hospitalization.3–6 In treatment of schizophrenia, atypical antipsychotic drugs are more prominently used as compared to typical drugs due to lower incidence of extrapyramidal symptoms, less tardive dyskinesia, less dysphoria and better cognition.7
Asenapine maleate (ASM) is a newer atypical antipsychotic drug and its action is mediated through a combination of antagonist activity at 5-HT2A and D2 receptors.8 It is slightly soluble in water with free base log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P 6.33 and classified as BCS class II drug.9 It is approved for treatment of schizophrenia in adults and as an adjunctive therapy with lithium or valproate for acute treatment of manic or mixed episodes associated with bipolar I disorder. Asenapine is the first antipsychotic drug to be administered sublingually for twice daily (5 mg and 10 mg tablets dosage from are available). However one has to abstain from eating and drinking for 10 minutes after sublingual administration. The bioavailability of ASM was found to be around 35% by sublingual route while it was <2% via oral due to its high gastro-hepatic metabolism.8,10,11 Despite its therapeutic potential in schizophrenia treatment, drawback with current dosage forms of ASM (low bioavailability, drinking and eating restriction, twice a day dosing regimen and extra pyramidal side effect) is still a very challenging task for pharmaceutical researchers. Thus, new formulation strategies have to be adopted to overcome the problem of asenapine delivery.12
P 6.33 and classified as BCS class II drug.9 It is approved for treatment of schizophrenia in adults and as an adjunctive therapy with lithium or valproate for acute treatment of manic or mixed episodes associated with bipolar I disorder. Asenapine is the first antipsychotic drug to be administered sublingually for twice daily (5 mg and 10 mg tablets dosage from are available). However one has to abstain from eating and drinking for 10 minutes after sublingual administration. The bioavailability of ASM was found to be around 35% by sublingual route while it was <2% via oral due to its high gastro-hepatic metabolism.8,10,11 Despite its therapeutic potential in schizophrenia treatment, drawback with current dosage forms of ASM (low bioavailability, drinking and eating restriction, twice a day dosing regimen and extra pyramidal side effect) is still a very challenging task for pharmaceutical researchers. Thus, new formulation strategies have to be adopted to overcome the problem of asenapine delivery.12
In treatment of CNS diseases, it has been reported that intranasal (i.n.) route opens a new possibility of non-invasive delivery of drugs due to high blood flow, porous endothelial membrane, large surface area and avoidance of first pass metabolism.13,14 The nanoparticulate drug delivery has exhibited great potential in movement of drugs across blood brain barriers (BBB) via different transport mechanism. The inherent properties such as nano size, tailored surface, solubility improvement, release modification and multi-functionality facilitate enhancement of bioavailability, efficacy and targetability. The polymeric and/or lipid nanoparticles in size less than 200 nm are widely preferable carriers for brain delivery.15–17 Lipid based nanoparticles shows advantages in brain targeted drug delivery over polymeric nanoparticles due to its rapid uptake by the brain, biocompatibility, biodegradability and less toxicity. The avoidance of organic solvent in production of lipid nanoparticles is one of the unique features associated with them. Nanostructure lipid carriers (NLC) are alternative to the solid lipid nanoparticles (SLN), composed of both solid lipid and liquid lipid. NLC are superior to SLN in respect to higher drug loading, smaller particle size and no drug leakage during storage by lipid polymorphism.18–21
Novel dosage forms are complex and require manipulation of intricate process variables. Thus, its development requires critical process control to obtain desired output. In development of novel dosage form, a crucial issue is to design and optimize a formulation with define therapeutic benefit. Historically, optimization process involved trial and error method in which one variable was changed at a time while keeping others constant. Outcome from these methods overlook the interaction among different factors and may not give optimum values. Hence, regulatory agencies like FDA (USA) and EMA (European Union) have espoused a paradigm shift from trial and error estimation of variables to quality by design (QbD) approach. Response surface methodology (RSM) is one of the QbD based approach used for pharmaceutical product development. Among RSM, Box–Behnken design (BBD) is more cost-effective than other techniques such as central composite design, three-level factorial design and D-optimal design, as it requires lesser experimental runs for optimization of a process at set independent variables.22,23
The aim of this study was to develop asenapine loaded nanostructure lipid carriers (ANLC) employing quality by design principle. The BBD was used to analyze effect of critical parameters of composition and process variables for optimization. ANLC was characterized for particle size, shape, in vitro release, stability and solid state characteristics. Further, in vivo brain and plasma pharmacokinetic studies were performed in Charles Foster (CF) rats. In addition, potential of ANLC was evaluated on animal model for three weeks to access it's therapeutic efficacy and extra pyramidal symptoms.
2. Materials and methods
2.1. Materials
Asenapine maleate (ASM) was a gift sample obtained from Ranbaxy Labs Ltd, Gurgaon, India. Arteether was provided as gift sample from Edelwiss Life Sciences, Chandigarh, India. Levodopa and Carbidopa were purchased from Intas Pharmaceutical Ltd, Ahmedabad, India. Glyceryl monostearate (GMS) was generously donated by Lupin Research Park, Pune, India. Oleic acid (OA) and Tween 80 (polyoxyethylene sorbitan monooleate, T 80) were purchased from SDFCL, Mumbai, India. Dialysis membranes (molecular weight cut-off between 12000 and 14000) were purchased from HiMedia, Mumbai, India. Nanosep Centrifugal filter devices (Omega Membrane, MWCO 100 kDa) were purchased from Pall Life Sciences, Mumbai, India. The water used in all experiments was ultrapure, obtained from a Millipore-DirectQ UV, Millipore, France. The solvents and chemicals used for analysis of drug were HPLC grade. All other chemicals used in the research work were of analytical grade and used as obtained.
2.2. Preparation of nanostructure lipid carriers
Nanostructure lipid carriers (NLC) were prepared by high shear homogenization and sonication method.24 Briefly, specific quantity of glyceryl monostearate (solid lipid), oleic acid (liquid lipid) and asenapine maleate (drug) were mixed and kept in molten state at 70 °C. In another beaker, 50 mL of aqueous phase containing Tween-80 as surfactant was kept at 70 °C on magnetic stirrer (RCT basic, IKA; India). This molten lipid and drug were poured drop wise into aqueous phase under high shear homogenization (Ultra Turrax T25, IKA, India) using S25-10G probe. The resulting suspension was ultrasonicated at 60% amplitude at 0.5 s frequency using probe Ultrasonicator (Hielscher® UP200H, Germany). The final volume of nanosuspension was adjusted to 50 mL and stored at room temperature. The nanosuspension was evaluated for particle size and entrapment efficiency after 24 h of preparation.
2.3. Development of formulation by quality by design
The development of novel dosage form by QbD required in-depth knowledge of product characteristics, source of variability, formulation and manufacturing process variables (including drug substance, excipient and process parameters). This knowledge is then used to implement a flexible and robust manufacturing process that can adapt and produce a consistent product over time. Some of the salient features of QbD include: (a) defining quality target product profile (b) identifying potential critical quality attributes (CQAs) of the drug product (c) determining the critical quality attributes of the drug substance, excipients (d) selecting an appropriate manufacturing process (e) defining a control strategy.25 The quality target product profile (QTPP), critical material attributes (CMA) and critical process parameter (CPP) for asenapine loaded nanostructure lipid carriers have been discussed in detail in the ESI section 2.3.1 and 2.3.2.†
With the exception of few studies26 application of experimental design approach for developing novel drug delivery system emphasizes on optimization of composition variables only.27,28 However, it is already established that process parameters also play a crucial role in novel dosage form. Here, BBD was selected to optimize asenapine loaded nanostructure lipid carriers using Design-Expert software (Version 7.0.0, Stat-Ease Inc., Minneapolis, USA). This design was specifically selected for exploration of complete design space with reduced experimental runs, without aliasing interaction factors.23,29,30 Five independent variables were selected in which three were composition variables and two were process variables. The factors and their levels were chosen on the basis of trial batches and data mining. The variables (A) liquid lipid to solid lipid ratio, (B) drug to solid lipid ratio, (C) aqueous surfactant concentration, (D) homogenization speed and (E) sonication time were selected as independent factors. Particle size (Y1) and entrapment efficiency (Y2) were selected as dependent variables (response). The independent variable and their levels with set constraint for optimization are presented in Table 1.
Table 1 Investigated dependent, independent variables and their levels in Box–Behnken experimental design
| Independent variables |
Levels |
| Low (−1) |
Medium (0) |
High (+1) |
| A = OA/GMS (w/w) |
0.1 |
0.15 |
0.2 |
| B = ASM/GMS (w/w) |
0.1 |
0.15 |
0.2 |
| C = Tween-80 (% w/v) |
0.5 |
1.0 |
1.5 |
| D = homogenization speed, HS (rpm) |
8000 |
12000 |
16000 |
| E = sonication time, ST (min) |
5 |
10 |
15 |
| Dependent variables |
Constraint |
| Y1 = particle size, PS (nm) |
Minimum |
| Y2 = entrapment efficiency, EE (%) |
Maximum |
2.4. Optimization and model validation
A suitable model was selected based on the lack of fit test and model statistic data. The response was fitted to linear, two factor interaction, quadratic and cubic model then evaluated by statistical significance of coefficient, PRESS (predicted residual sum of squares) and r2 values. Based on model, a polynomial equation was generated to describe the effect of factors on response by Design Expert Software. Based on dependent variables constraint, optimized batch was selected by numerical method with maximum desirability factor. This optimized asenapine loaded nanostructure lipid carrier formulation (ANLC) was used for further in vitro and in vivo characterization.
2.5. HPLC analysis
High-performance liquid chromatography (HPLC) analyses for quantification of Asenapine were performed by Waters HPLC 515 having Rheodyne7725i injector fitted with 20 μL loop. The chromatographic separation of asenapine was achieved by reverse phase C18 spherisorb column (5.0 μm ODS 24.6 mm × 250 mm) connected with guard column (5.0 μm ODS, 4.6 mm × 10 mm) at room temperature and detected by photodiode array (PDA) 2998 detector (Waters, USA). The mobile phase consist of acetonitrile and phosphate buffer (1.36 g of KH2PO4 in 1000 mL Millipore water, pH 3.3 adjusted with triethyl amine and o-phosphoric acid) in the ratio of 75:25 and at a flow rate of 1.0 mL min−1. Data was further processed by Empower Pro2 software at λmax 268 nm. The retention time was found to be 4.076 min for asenapine. The method was validated according to the ICH guidelines with respect to system suitability, linearity, limit of quantification and detection, precision, accuracy, robustness, and specificity. The standard calibration curve was linear with linear regression coefficient of (r2) 0.9988 over the range of 10.0–100.0 μg mL−1.31,32
2.6. Determination of particle size, PDI and zeta potential
Particle size was determined by measuring random changes in intensity of light scattered by suspended particles during their Brownian motion. This technique is commonly known as dynamic light scattering (DLS) or photon correlation spectroscopy (PCS). The particle size, polydispersity index (PDI) and zeta potential were determined by particle size analyzer (Delsa Nano C, Beckman Coulter, UK) at 25 °C. Polydispersity index indicates the distribution of particle size of nanoparticles which reveal nature of distribution like monodisperse and polydisperse.33 All studies were performed in triplicates and mean value was considered for data analysis.
2.7. Determination of entrapment efficiency
The entrapment efficiency (EE) was estimated with method described by Vuddanda et al. 2014.34 Accurately measured 500 μL nanosuspension was placed in the upper chamber of Nanosep centrifuge tubes having ultra filter with molecular weight cut-off 100 kDa (Pall Life Sciences, India). Nanosep was centrifuged at 15000 rpm for 30 min using a cooling centrifuge at 4 °C (C-24, Remi). The free amount of asenapine in the filtrate was collected from lower chamber and estimated by HPLC method. The EE was calculated by the following equation:
2.8. In vitro drug release study
In vitro drug release study of ASM and optimized NLC (ANLC) were performed using dialysis bag method. The ASM and ANLC suspensions equitant to 10 mg were filled in pretreated dialysis bag (Dialysis Membrane-135, Molecular weight cut off between 12 and 14 kDa, HiMedia, Mumbai, India) and immersed in 100 mL of phosphate buffer pH 7.4 to mimic biological fluid. The phosphate buffer was magnetically stirred at 100 rpm at 37 °C and 1.0 mL aliquots were withdrawn from release medium at predetermined time for 24 h and replaced with fresh phosphate buffer. The solution was filtered by 0.45 μm syringe filter and concentration of asenapine was measured by HPLC method. With the help of DDsolver software, in vitro drug release data was fitted into various release model like zero order, first order, Higuchi, Korsemeyer–Peppas and Hixson–Crowell to understand the mechanism of drug release from lipid matrix.35,36
2.9. Stability study
Stability study was carried out at 30 ± 2 °C, 65 ± 5% RH for three months. Sealed vials of ANLC suspension was placed in stability chamber. ANLC was analyzed for particle size, zeta potential, entrapment efficiency and in vitro drug release profile comparison (f1: difference factor, f2: similarity factor) at each month.
2.10. Surface characterization
2.10.1. Transmission electron microscopy. The size and morphology of ANLC were observed using a TEM (TECNAI-12) operated at 120 keV. One drop of appropriately diluted nanosuspension was spread on 400 mesh gold coated copper grid. The grid was air dried at room temperature under vacuum for 24 h before observation.
2.10.2. Atomic force microscopy. The external morphology of ANLC was further visualized by scanning probe microscope (NTEGRA Prima, NT-MDT) in semi contact mode. ANLC suspension was diluted 10 times with distilled water and one drop of nanosuspension was placed on the small microscope slide to form a dry film of suspension for observation.
2.11. Solid state characterization
2.11.1. FT-IR. The IR spectra of ASM, GMS and lyophilized ANLC were recorded by Fourier Transform Infrared Spectroscopy (FTIR-8400S, Shimadzu). Sample preparation involved mixing the sample with potassium bromide (KBr) in 1:50 ratio, triturated in glass mortar, pelletized, and finally placed in sample holder. The spectrum was scanned over the wavenumber of 4000–400 cm−1.
2.11.2. Differential scanning calorimetry. The DSC was performed to evaluate any change in drug with respect to melting enthalpy, glass transition temperature and any interactions with excipients. The physical state of ASM, GMS, and lyophilized ANLC were characterized by the differential scanning calorimetry (DSC Q1000, TA instrument). About 2–5 mg of sample was placed in standard aluminium pans and scanned in the range from 5 °C to above the melting point with temperature increment speed of 10 °C min−1 under the dry nitrogen used as effluent gas (flow rate 50 mL min−1).
2.11.3. X-ray diffraction. The physical properties of asenapine in pure form and inside the lipid matrix were measured by X-Ray Powder Diffraction (XRD). X-ray powder scattering measurements were carried out to check the crystallinity of drug in pure and lyophilized ANLC. Study was performed on a Siemens DIFFRACplus 5000 powder diffractometer with CuKα radiation (1.54056 Å). The tube voltage and amperage were set at 40 kV and 40 mA, respectively. Each sample was scanned between 10° and 40° in 2θ with a step size of 0.01° at 1 step per s.37
2.12. In vivo brain and plasma pharmacokinetic study
The experimental protocols were duly approved by Institutional Animal Ethical Committee (Dean/44094/2013-14). In vivo pharmacokinetic study for assessment of availability of ASM in plasma and brain were performed in Male Charles Foster (CF) rats (200–240 g). All rats were kept at normal room temperature in 12 h light/dark cycle with food and water ad libitum. The rats were acclimatized for intranasal delivery with normal saline before one week of experimentation. In two groups for intranasal delivery, required volume was administered in two divided part into each nostril of rat using micropipette. In third group, ASM was delivered through intravenous tail vein injection. All three groups received 1.0 mg kg−1 equivalent asenapine dose of ASM (i.v., i.n.) and ANLC (i.n.). In each group, five animals per time point were sacrificed for collection of their blood and brain. Plasma was separated by centrifuging blood sample at 4000 rpm at 4 °C for 20 min. However, brain samples were taken and homogenized in distilled water using a tissue homogenizer. In processing of brain homogenate and plasma, 100 μL of brain and plasma sample were extracted twice by liquid–liquid extraction procedure using n-hexane with 2% iso-propyl alcohol and arteether (IS) as internal standard. The asenapine quantification in plasma was done by partially validated method in Aekspert ultra LC 100-XL HPLC system equipped with Q-trap 5500 LC-MS/MS (AB Sciex), consisting of flow control valves, vacuum degasser, ekspert 100 pump with ekspert 100-XL autosampler. The Q1/Q3 transitions of m/z 286.1/229.0 and 330.3/267.4 were used to quantify asenapine and IS, respectively. UFLC elution was carried out in isocratic mode with mobile phase consisting of 85:15 (v/v); acetonitrile:ammonium acetate buffer (10 mM, pH 4) at a flow rate of 0.45 mL min−1 with 40 °C oven temperature using a Phenomenex C18 column.38,39 The peak drug concentration (Cmax) and its time (Tmax), area under drug concentration–time curve (AUC) and mean residence time (MRT) of both ASM and ANLC in brain and plasma were calculated using a non-compartmental analysis by Phoenix 64 Software (WinNonlin 6.4, CERTARA). The drug targeting efficiency (DTE) of nanocarriers via intranasal route to brain was calculated according to following equation:40
in above equation, AUCbrain and AUCplasma are areas under drug concentration–time curves for brain and plasma after intranasal and intravenous administration.
2.13. Animal behavioural study
2.13.1. Animal and dose. CF rats (180–220 g) were used for all behaviour studies of ASM and ANLC. The rats were divided into separate groups (five animals per group) and housed in a polypropylene (421 × 290 × 190 mm) cage at normal room temperature in 12 h light/dark cycle. They had free access of food and water. ASM and ANLC equivalent to 1.0 mg kg−1 asenapine were given in all studies via nasal route using micropipette. The group administered with intranasal blank NLC suspension was considered as vehicle control for catalepsy, induced locomotor and paw test. Further, one more group administered with intra-peritoneal L-dopa (10 mg kg−1) and carbidopa (2.5 mg kg−1) was considered as positive control in induced locomotor activity test. In most studies, the animal behaviour models for screening of developed formulation are evaluated on a one day treatment response, which raises concerns about validity of this therapeutic-like behaviour especially in a disease like schizophrenia where pharmacological effect (therapeutic effect or side effect) are usually manifested after 2–3 weeks of treatment. Thus, our experiments were performed for 21 days and observations were recorded on 1st, 7th, 14th and 21st days of treatment in order to account for any inconsistency.41 The data were reported in mean ± SD for each group.
2.13.2. Catalepsy test. This animal test was performed to evaluate the effect of delivery system on extrapyramidal side effects associated with asenapine. Briefly, rat forepaws were placed on horizontal bar fixed at a height of 10 cm above the surface whilst their hind limbs rested on a platform. The amount of time animal remains immobile was calculated. After administration of vehicle control, ASM and ANLC; the time spent in atypical position was recorded after 1 h dosing and mean values were reported.42
2.13.3. Induced locomotor activity test. This behavioural model is based on a hypothesis that increase in locomotor activity is due to an increased dopaminergic activity in the mesolimbic system. Indeed, all antipsychotics have antagonist effects on dopamine agonist induced hyperactivity.43 The locomotor count was determined by Digital Actophotometer (IKON, India). On the day of observation, ASM and ANLC group received the respective formulation followed by administration of intra-peritoneal L-dopa (10 mg kg−1) and carbidopa (2.5 mg kg−1). The locomotor activity was measured for 5 min by placing the animals in Actophotometer 1 h after drug administration.44,45
2.13.4. Paw test. The paw test is model for a prediction of both therapeutic potential as well as extrapyramidal side effects (EPS) associated with any antipsychotic drug. The increase in hindlimb retraction time (HRT) was associated with the antipsychotic potential, whereas the increase in forelimb retraction time (FRT) was associated with the potential to induce EPS. Also, this model has unique feature for differentiating classical antipsychotics which are equipotent in prolonging both the forelimb retraction time (FRT) and hindlimb retraction time (HRT) and atypical antipsychotics which are much more potent in prolonging HRT than FRT.46 The test was performed on a Perspex platform measuring 30 cm × 30 cm, with a height of 20 cm. The top of the platform had two holes of 3.5 cm diameter for the forelimbs and two larger holes of 4.5 cm diameter for hind limbs and a slit for the tail. For both FRT and HRT, the minimum time was set to 1 s and maximum to 60 s. Experiment was performed in triplicate in five minute interval and average FRT and HRT were then calculated for each rat.
3. Results and discussion
3.1. Optimization of NLC and statistical analysis of variables
A total of 46 experimental runs were generated from five factors, three levels Box–Behnken statistical experimental design with 6 centre point. The independent variables and responses are given in Table 3 of the ESI.† The polynomial equations were generated for each response which explained the main effects, interaction effects and quadratic effect of independent variables. The smaller value of PRESS statistic and non significant lack of fit indicates the better model towards data points. A three-dimensional response surface plot was used to study interaction effects of independent variables on the response. A mathematical equation was generated for each response to evaluate the effect of factors. In this equation, a positive and negative value of independent factor represents direct and indirect effect on response.47,48
Table 2 Statistical analysis results of lack of fit test and model summary for particle size and entrapment efficiencya
| Model |
Lack of fit tests |
Model summary statistics |
Remark |
| SS |
df |
MS |
F-Value |
p-Value prob. > F |
SD |
r2 |
Adjusted r2 |
Predicted r2 |
PRESS |
| SS: sum of squares, df: degree of freedom, MS: mean square, SD: standard deviation, PRESS: Predicted Residual Error Sum of Squares; 2FI: two factor interaction, +: PRESS statistic not defined, p-value < 0.05 was consider as statistically significant. |
| Particle size |
| Linear |
10991.06 |
34 |
323.27 |
101.47 |
<0.0001 |
16.80 |
0.8839 |
0.8690 |
0.8462 |
14580.54 |
|
| 2FI |
10889.56 |
24 |
453.73 |
142.42 |
<0.0001 |
19.39 |
0.8849 |
0.8254 |
0.7136 |
27146.81 |
|
| Quadratic |
50.02 |
19 |
2.63 |
0.83 |
0.6578 |
1.66 |
0.9993 |
0.9987 |
0.9975 |
232.59 |
Suggested |
| Cubic |
14.02 |
4 |
3.50 |
1.10 |
0.4478 |
1.82 |
0.9997 |
0.9985 |
− |
+ |
Aliased |
| Pure error |
15.93 |
5 |
3.19 |
− |
− |
− |
− |
− |
− |
− |
|
|
| Entrapment efficiency |
| Linear |
282.71 |
34 |
8.32 |
5.18 |
0.0369 |
2.73 |
0.9601 |
0.9550 |
0.9453 |
398.05 |
|
| 2FI |
189.14 |
24 |
7.88 |
4.91 |
0.0423 |
2.61 |
0.9729 |
0.9589 |
0.9269 |
532.26 |
|
| Quadratic |
55.84 |
19 |
2.94 |
1.83 |
0.2608 |
1.63 |
0.9912 |
0.9839 |
0.9664 |
244.65 |
Suggested |
| Cubic |
0.28 |
4 |
0.070 |
0.043 |
0.9953 |
0.96 |
0.9989 |
0.9944 |
− |
+ |
Aliased |
| Pure error |
8.03 |
5 |
1.61 |
− |
− |
− |
− |
− |
− |
− |
|
Table 3 Optimal predicted and experimental batch with composition and process variables with responses
| Batch |
Composition and process variables |
Response |
| OA/GMS (w/w) |
ASM/GMS (w/w) |
Tween-80 (% w/v) |
HS (rpm) |
ST (minute) |
PS (nm) |
EE (%) |
PDI |
ZP (mV) |
| Predicted |
0.20 |
0.12 |
1.50 |
15948.27 |
5.00 |
167.54 |
83.96% |
— |
— |
| Experimental (ANLC) |
0.20 |
0.10 |
1.50 |
16000.00 |
5.00 |
167.30 ± 7.52 |
83.50 ± 2.48 |
0.261 ± 0.024 |
−4.33 ± 1.27 |
The preliminary diagnostic study for entrapment efficiency revealed the anomalous response of batch NLC 22. It might be unpredictable experimental error. Indeed, this batch was excluded from further statistical analysis and batch optimization. Analysis of lack of fit and model fit summary revealed significant lack of fit for polydispersity index response in all studies model (data not shown). So, this response was not considered for optimization of formulation. Further, only particle size and entrapment efficiency were selected for optimization and their respective lack of fit and model fit summary are presented in Table 2. This table showed that the best-fitted model for dependent variable was quadratic. The non-significant model term was removed from analysis and it was not consider for generation of polynomial response equation of particle size and entrapment efficiency.
3.1.1. Effect on particle size. The particle size (PS) of the prepared batches was in range of 147.78–334.61 nm. The quadratic model was selected for the analysis of variables on particles size based on the lack of fit test and model summary statistics. ANOVA for response surface quadratic model showed that A, B, C, D, E, AD, A2, B2, C2, D2 and E2 were significant model terms. The final mathematical model in terms of coded factors determined by Design-Expert software is shown below:
| PS = +275.85 − 4.54A + 25.20B − 45.01C − 29.34D − 42.71E + 3.16AD − 4.57A2 + 10.05B2 − 20.73C2 − 15.64D2 − 21.90E2 |
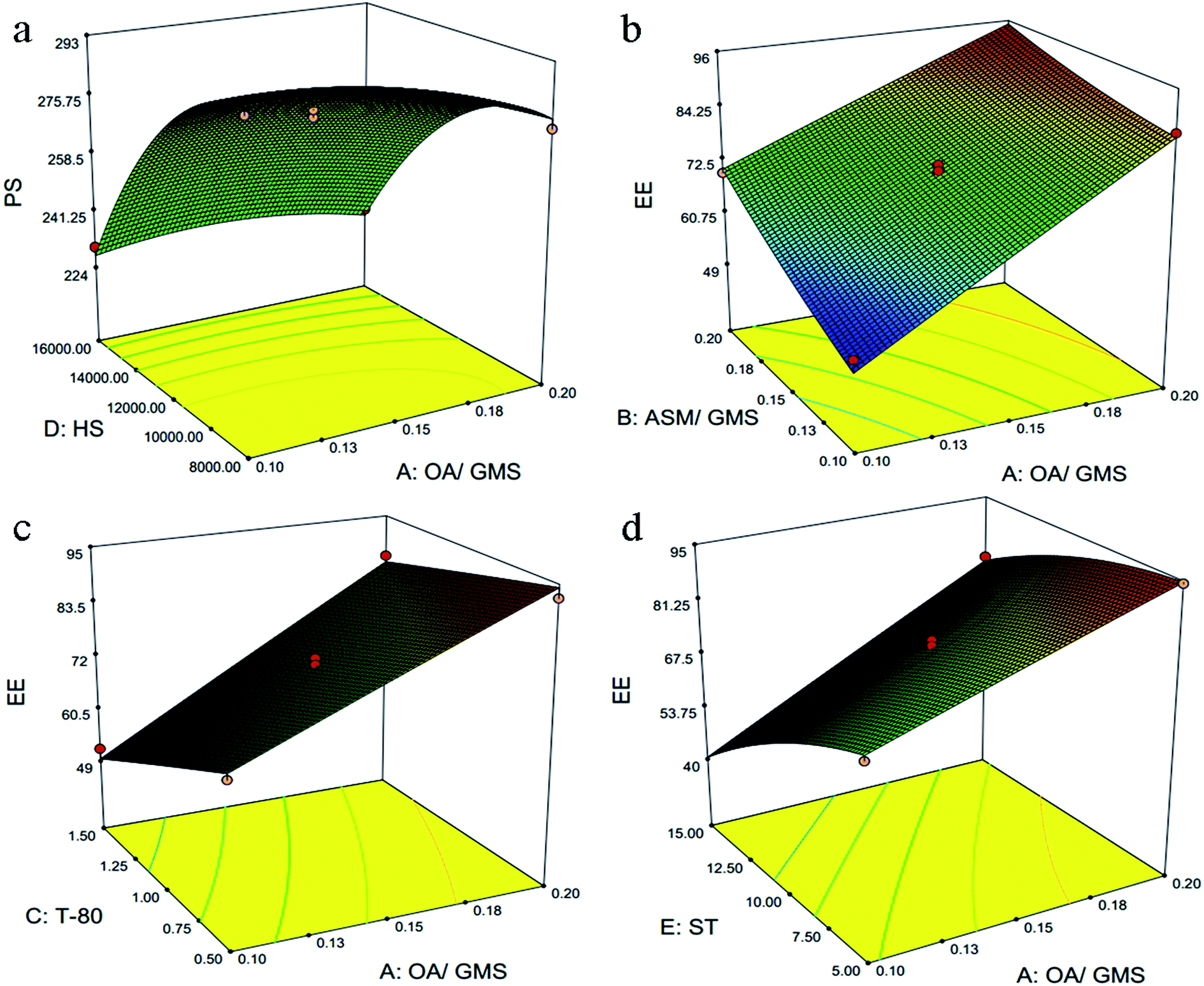
Lower value of PRESS (232.59) and non significant lack of fit (F-value = 0.83, and p value = 0.6578) for quadratic model suggested that this model is fit to describe the effect of independent variables on particle size. Except ASM/GMS ratio (B), which have positive coefficient, all others model terms have negative coefficients. This suggested that particle size increases with increase in ASM/GMS ratio (B). Also, higher coefficient value (45.01) of surfactant concentration (C) suggested it had most significant effect on the particle size followed by E, D, B and A. It shows that stabilization effect of surfactant is critical factor for the preparation of NLC. While in case of the interaction effects between different factors, only OA/GMS ratio (A) and homogenization speed (D) had combined significant effect on particle size. It was visually discerned from 3D response surface plots in Fig. 1a.
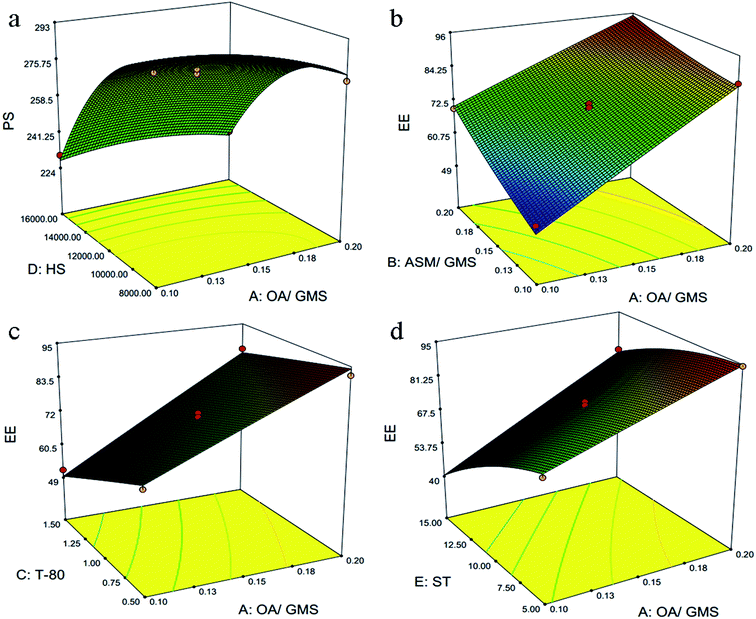 |
| | Fig. 1 Graphical illustrations representing the interaction effect of independent variable on particle size (PS) and entrapment efficiency (EE). In particle size, only one interaction effect (liquid/solid lipid ratio to homogenization speed) showed significant effect (a). Moreover, entrapment efficiency was controlled by three interactions, liquid/solid lipid ratio to drug/solid lipid ratio (b), liquid/solid lipid ratio to surfactant (c) and liquid/solid lipid ratio to sonication time (d), respectively. | |
3.1.2. Effect on entrapment efficiency. The entrapment efficiency (EE) was in between the 51.23–92.56% in developed batches. The lack of fit test and model summary statistics elucidated higher adjusted (0.9839) and predicted (0.9664) r2 values for quadratic model. Thus this model was selected for analysis of independent variables on entrapment efficiency. From the ANOVA, only terms A, B, C, D, E, AB, AC, AE, D2 and E2 were significant. The final mathematical equation with significant model term in coded factors is shown below:
| EE = +74.62 + 15.31A + 7.41B − 7.11C − 4.95D − 11.78E − 2.94AB + 2.35AC + 3.50AE − 1.35D2 − 3.28E2 |
Lower value of PRESS (244.65) and non significant lack of fit (F-value = 1.83, and p value = 0.2608) for quadratic model suggested that this model fit to describe the effect of independent variables on entrapment efficiency. There was increase in entrapment efficiently with increase in the model term A and B. This indicates that higher concentration of liquid lipid and drug is favourable for higher entrapment efficiency. This could be justified by the higher solubility of drug in liquid lipid as compared to solid lipid in selected drug/lipid ratios. Moreover, the surfactant concentration (C), homogenization speed (D) and sonication time (E) have negative coefficient indicating that EE is inversely proportional to these factors. Here, the interaction term AB, AC and AE had the significant effect on the EE and it has been shown in Fig. 1b–d as 3D response surface plot.
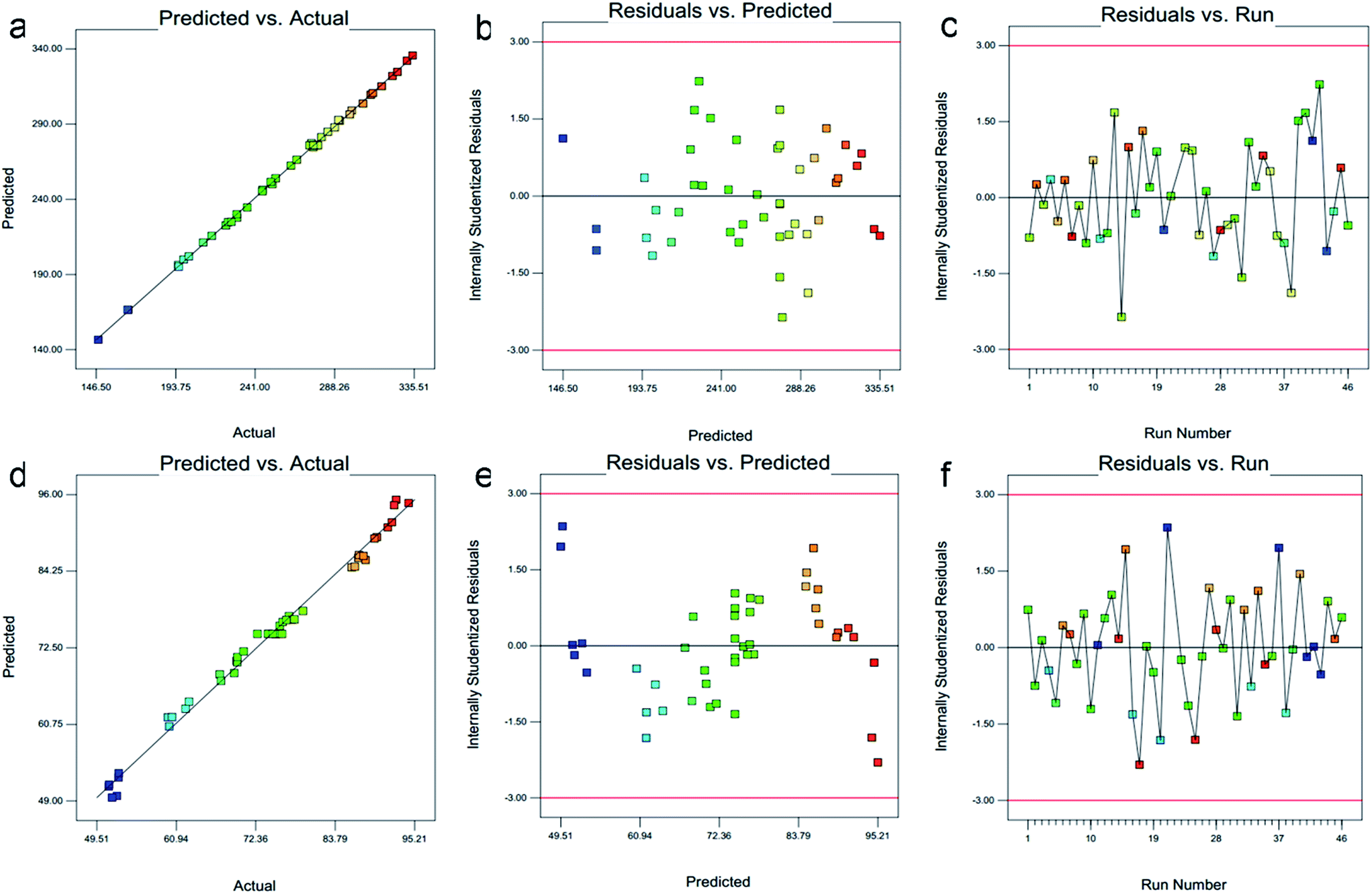
3.1.3. Risk assessment. Risk assessment is a valuable science-based process used in quality risk management that can aid in identifying which material attributes and process parameters potentially have an effect on product CQAs. For preparation of NLC and their set CQPP, particle size and entrapment efficiency were significantly controlled by selected composition and process variables in defined design space. Moreover, optimum value of PDI could be selected from different solutions appeared in numerical optimization. Linearity in predicted vs. actual response and symmetrical distribution pattern in residual vs. predicted, residual vs. run graph for both particle size, entrapment efficiency suggested that model is fit and the possibility of other missing variables which may be determinant of ANLC-CQPP are low (Fig. 2). Apart from the individual factors, only interaction effect of AD had significant effect on particle size. Further, interaction effect of AB, AC and AE had significant effect on EE (Fig. 1).
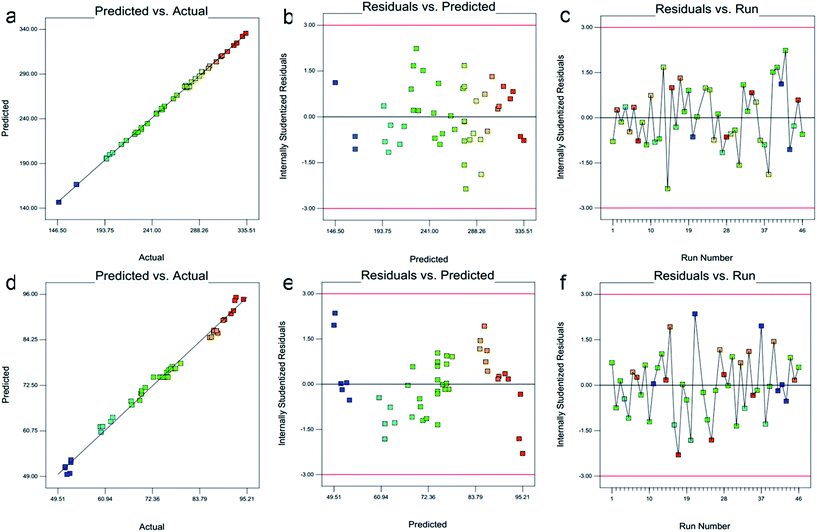 |
| | Fig. 2 Diagnostic illustration graph between predicted vs. actual, residual vs. predicted and residual vs. run for particle size (a–c) and entrapment efficiency (d–f), respectively. All these graph shows well controlled compositions and process variables without any interfering factors. | |
3.1.4. Optimization and validation. The optimization of batch was performed by numerical method with maximum desirability factor. The predicted value of PS and EE were found to be 167.54 nm, 83.96%, respectively at A (0.20 w/w), B (0.12 w/w), C (1.50% w/v), D (15948.27 rpm) and E (5.0 min) (Fig. 3). Therefore, a new batch of NLC with the predicted level of independent variables was prepared to confirm the validity of optimization. The observed response of PS and EE were 167.30 ± 7.52 nm and 83.50 ± 2.48%, respectively at set experimental run condition, A = 0.2 w/w, B = 0.10 w/w, C = 1.5% w/v, D = 16000 rpm and E = 5 min. The predicted values are in good agreement with observed values demonstrating the reliability of this model in predicting a desirable NLC system for asenapine. The nanoparticles of this size range have been found to be suitable for brain targeting as they preferentially accumulate in brain and demonstrate superior clinical efficacy.49,50 These nanoparticles are transported through blood brain barriers by utilizing different mechanism such as adsorptive transcytosis, inhibition of efflux pumps (p-glycoprotein) and passive diffusion from endothelial cells to the brain cell.51,52 So, this optimized batch (ANLC) was selected for in vitro characterization, pharmacokinetic and pharmacological evaluation.
 |
| | Fig. 3 Graphical representation of in vitro release and surface characterization. Drug release profile graph of ASM, ANLC in pH 7.4 phosphate buffer and stability study batch release on day 30, 60 and 90 of ANLC, mean ± SD, n = 3 (a). TEM image of ANLC shows spherical shape particles without any aggregation. Scale bar represents 500 nm (b). AFM image of ANLC demonstrating smooth surface of the particle (measurement scale: 1 μm × 1 μm × 160 nm) (c). | |
3.2. Zeta potential of ANLC
The zeta potential is one of the fundamental parameter to evaluate stability of colloidal system. However, it foremost depends on the chemical nature and interaction between lipid, surfactant and drug. The zeta potential of the optimized asenapine loaded nanostructure lipid carrier (ANLC) was found to be −4.33 ± 1.27 mV. The negative potential and low value of zeta potential justifies acidic nature of lipid matrix and nonionic nature of surfactant, respectively. Normally, colloidal dispersion with zeta potential above the range of −30 to +30 mV is considered as stable. But, this empirical rule does not apply to steric/surface stabilization provided by Tween-80, which continues to stabilize the particles even under low values of zeta potential due to shift in shear plane of particles.
3.3. In vitro drug release study
The in vitro drug release of ASM and ANLC are depicted in Fig. 3a. The ASM produced more than 90% drug release in 12 h and approximate 100% in 24 h indicating creation of perfect sink condition of dissolution media for release study. The release study of freshly prepared optimized ANLC (d0) demonstrated biphasic release pattern, burst release followed by sustained release of drug upto 24 h study. Initial burst release may be attributed to presence of adsorbed free drug or liquid lipid soluble drug on outer surface, which accounts for quick release into the surrounding media. Further, sustained release pattern was contributed by the entrapped drug inside lipid matrix. The release profile of the asenapine from ANLC was fitted into the zero order (r2 = 0.7962), first order (r2 = 0.9328), Higuchi (r2 = 0.9895), Korsmeyer–Peppas (r2 = 0.9911, n = 0.53) and Hixson–Crowell (r2 = 0.9896) release kinetics model. Based on the value of coefficient of correlation, the Korsmeyer–Peppas model was found to be best fit model. It indicates that the drug release follows the anomalous transport (n value between 0.5 and 1.0) of drug i.e. release mechanism is not well known or more than one type of release phenomena could be involved.53
3.4. Stability study
The results of the stability study are presented in Table 4 and Fig. 3a. As shown in table, particle size increases while zeta potential and entrapment efficiency decreases with time. However, the statistical analysis indicated that these changes were not significant (p > 0.05). The release profile of ANLC on day 0, 30, 60 and 90 were compared by difference factor (f1) and similarity factor (f2) considering day 0 release as reference. Generally, f1 values lower than 15 (0–15) and f2 values higher than 50 (50–100) shows similarity of dissolution profile. The observed values of f1 (3.48–6.77) and f2 (75.31–85.95) justified the similarity of release profile of ANLC. These finding indicates that the optimized ANLC were physically stable with no aggregation and similarity in drug release pattern during 90 days.
Table 4 Stability studies results of particle size, entrapment efficiency and in vitro release profile comparisona
| Day |
Particle size (nm) |
Zeta potential (mV) |
Entrapment efficiency (%) |
In vitro release profile comparison |
| f1 |
f2 |
| f1: difference factor; f2: similarity factor, mean ± SD, n = 3. |
| 0 |
167.30 ± 7.52 |
−4.33 ± 1.27 |
83.50 ± 2.48 |
Reference |
Reference |
| 30 |
168.26 ± 7.33 |
−4.51 ± 0.36 |
83.36 ± 3.22 |
3.48 |
85.95 |
| 60 |
172.91 ± 5.21 |
−3.96 ± 1.70 |
82.37 ± 2.81 |
5.72 |
78.34 |
| 90 |
175.21 ± 3.88 |
−3.69 ± 1.19 |
82.04 ± 2.06 |
6.77 |
75.31 |
3.5. Surface characterization
TEM (Fig. 3b) and AFM (Fig. 3c) image of ANLC showed distinct clear spherical particles size (<200 nm), which was close to the results obtained by dynamic light scattering (DLS) method. Moreover, the actual particle size reported by TEM and AFM were found to be less than dynamic light scattering results. It is postulated that these differences were shown by difference in size measurement methodology. The solvent layer attached to particle; called hydrodynamic size is measured in DLS, which is always greater than actual particle size measured by TEM and AFM.
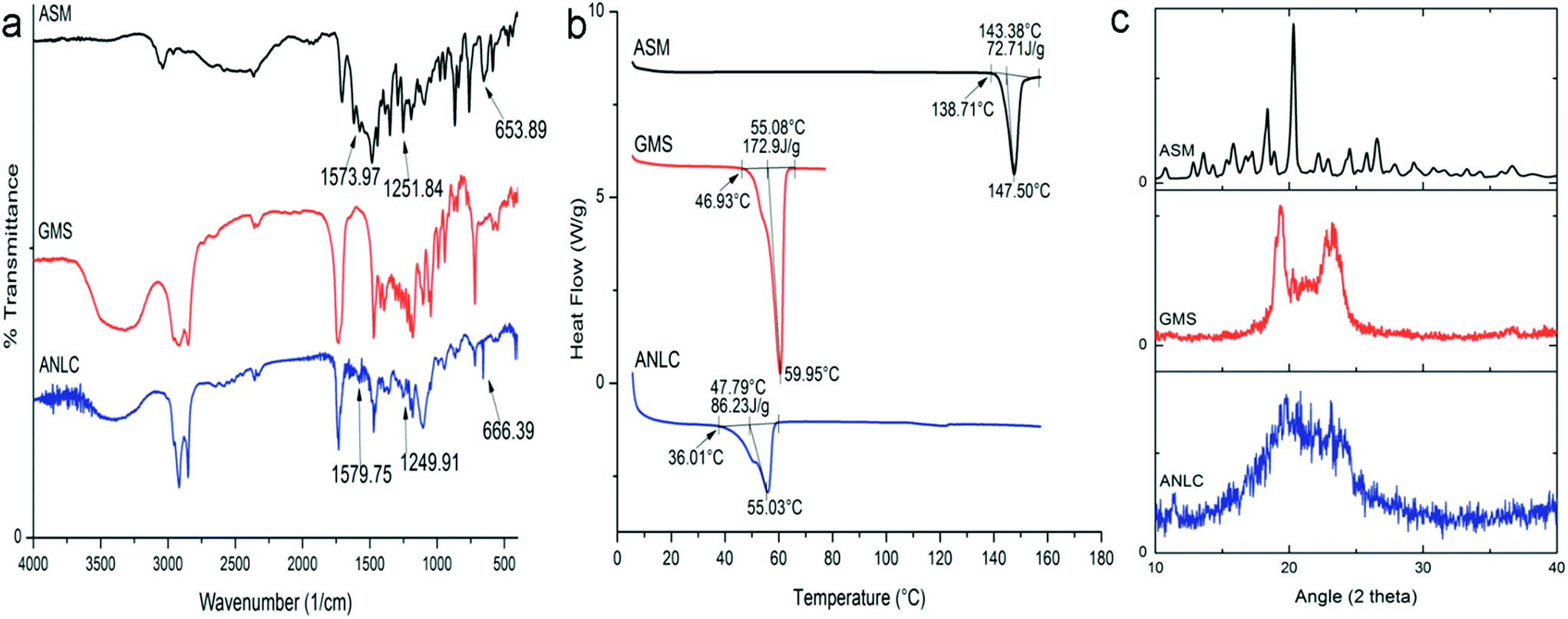
3.6. Solid state characterization
3.6.1. FT-IR. The overlay spectrum of ASM, GMS and ANLC are shown in Fig. 4a. The IR spectrum of asenapine maleate revealed the characteristics absorption bands at 653.89 cm−1 (C–Cl), 1192.67 cm−1 (C–O–C), 1251.84 cm−1 (–N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) , 3° amine), 1573.97 cm−1 (C–C, aromatic ring), 1705.13 cm−1 (–C
, 3° amine), 1573.97 cm−1 (C–C, aromatic ring), 1705.13 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3037.99 cm−1 (–OH). However, all the characteristic peaks of ASM could not be included for interaction study in ANLC owing to similarity in some of functional groups common to GMS, oleic acid and Tween 80. The existence of 653.89 cm−1, 1251.84 cm−1 and 1573.97 cm−1 in ASM and their corresponding bands 666.39 cm−1, 1249.91 cm−1 and 1579.75 cm−1 in ANLC confirms the presence of asenapine maleate in nanostructure lipid carriers indicating no chemical interaction between the drug and lipid matrix. The slight shifting of absorption bands of ASM in ANLC can be attributed to change in molecular environment and intermolecular interactions associated with dispersed drug molecule in lipid excipients.
O), 3037.99 cm−1 (–OH). However, all the characteristic peaks of ASM could not be included for interaction study in ANLC owing to similarity in some of functional groups common to GMS, oleic acid and Tween 80. The existence of 653.89 cm−1, 1251.84 cm−1 and 1573.97 cm−1 in ASM and their corresponding bands 666.39 cm−1, 1249.91 cm−1 and 1579.75 cm−1 in ANLC confirms the presence of asenapine maleate in nanostructure lipid carriers indicating no chemical interaction between the drug and lipid matrix. The slight shifting of absorption bands of ASM in ANLC can be attributed to change in molecular environment and intermolecular interactions associated with dispersed drug molecule in lipid excipients.
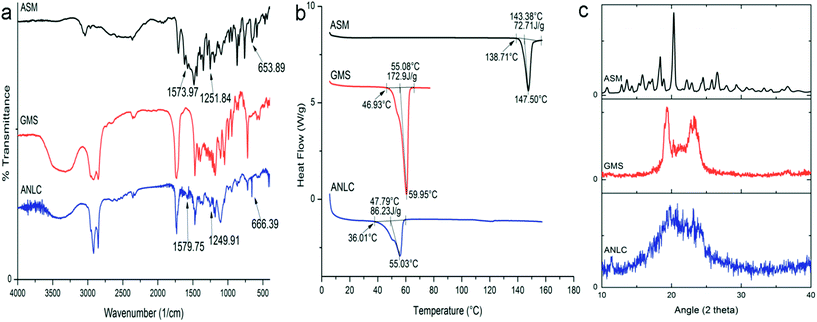 |
| | Fig. 4 FTIR (a), DSC (b) and XRD (c) graphs of ASM, GMS and ANLC. FTIR spectra indicated the presence of asenapine characteristics peaks in ANLC which suggest no interaction between drug and excipient. DSC thermogram suggested crystalline nature of pure drug however it was available in molecular disperse state in ANLC supported by complete disappearance of ASM endothermic peak. XRD spectra of ASM and GMS revealed crystalline nature but significant loss of peaks of the ASM in ANLC suggesting amorphous nature in ANLC. | |
3.6.2. Differential scanning calorimetry. DSC thermogram of ASM, GMS and ANLC are shown in Fig. 4b. A sharp endothermic peak in ASM (Tonset = 138.71 °C, ΔH = 72.71 J g−1) and GMS (Tonset = 46.93 °C, ΔH = 172.9 J g−1), corresponding to its melting point demonstrated crystalline nature of these substances. A broad asymmetric melting peak (Tonset = 36.01 °C, ΔH = 86.23 J g−1) was observed in the thermogram of ANLC. The lack of ASM melting peak in ANLC may be attributed to molecularly dispersed state of asenapine maleate in lipid matrix. The presence of broader endothermic peak below the melting point of GMS in ANLC correlated with possible effects of liquid lipid and surfactant on crystal lattice of GMS. Further, particle size also has pronounced effect on the melting endotherm of lipid in nanosize range according to Gibbs–Thomson equation.54 According to this equation, different lipid nanoparticles melt at different temperatures which results in peak broadening and shift in melting transition at lower temperature as compared to bulk lipid.
3.6.3. X-ray diffraction. XRD spectra of ASM, GMS and ANLC are shown in Fig. 4c. The XRD diffraction pattern of ASM exhibit sharp peaks at 2θ angle 14.4, 16.0, 16.6, 18.3, 19.4, 20.2, 21.8, 23.2, 23.8, 25.0, 26.0 and 26.6 degree which demonstrates crystalline nature of drug. Further, bulk GMS in crystalline form exhibited 2θ angle characteristic peaks at 19.3, 22.7 and 23.3 degree. The diffraction pattern of ANLC resembles GMS with total absence of 2θ characteristic peak of ASM. These patterns of ANLC suggested amorphous nature of asenapine in lipid matrix with significant distortion in crystal lattice of GMS. XRD data for ASM, GMS in bulk and ANLC were good agreement with DSC results.
3.7. In vivo brain and plasma pharmacokinetic study
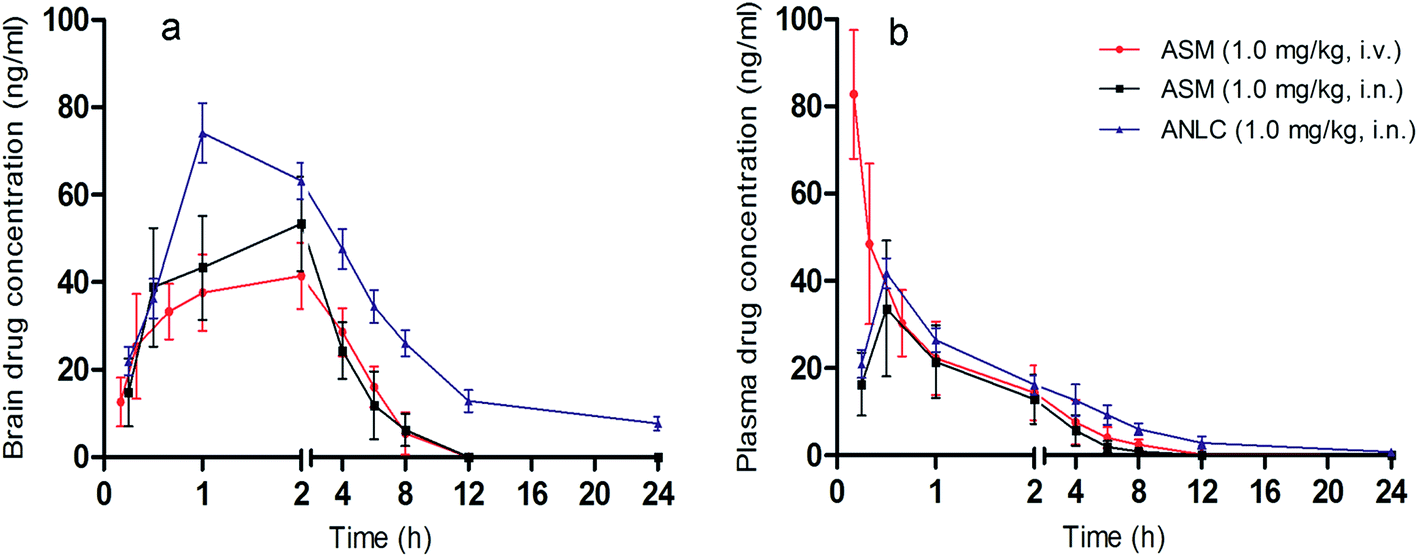
Different pharmacokinetic parameters were evaluated to observe the effect of ANLC on absorption and disposition of asenapine through nasal route. Drug concentration–time profiles in brain and plasma are shown in Fig. 5. The pharmacokinetic parameters obtained from these profiles are presented in Table 5. ANLC showed significantly higher (p < 0.05) Cmax (74.13 ± 6.73 ng mL−1), AUC0–24 h (560.93 ± 27.85 h ng mL−1) and MRT (7.1 ± 0.13 h) in brain compared to pure drug (ASM) when both were administered by i.n. route. All these contributed to 1.34 and 2.68 times higher bioavailability of drug in plasma and brain, respectively after i.n. administration of ANLC. This can be attributed to the nanosize of the carriers as well as presence of oleic acid (liquid lipid) and Tween 80 (surfactant) which leads to enhance the permeation of ANLC across the respiratory epithelium and vessels fenestrate the brain. These finding is well correlated with earlier reports for brain targeting potential of nanoparticles by Tween 80.55,56 After i.n. administration, delivery of the drug to brain has pronged approach, one through olfactory and trigeminal neural pathways and another after permeation from respiratory epithelium to blood and then to brain.57,58 This could be further related with the higher drug targeting efficiency (2.07) of ANLC to brain via i.n. route. In contrast, ASM in i.v. route elicited significantly higher Cmax (82.76 ± 14.78 ng mL−1) in plasma compared to ANLC and ASM via i.n. route owing to instantaneous availability of all free drugs in systemic circulation without absorption phase. Higher AUC0–24 h and MRT of ANLC indicates that drug remains in the brain and blood for longer period of time due to sustained release of drug from lipid matrix. The results obtained from above study suggested the potential of ANLC for brain targeting.
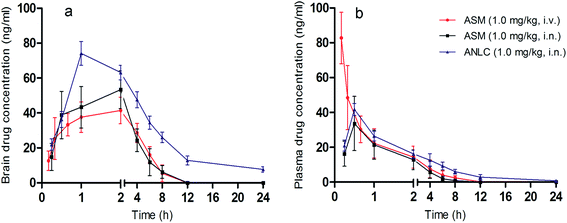 |
| | Fig. 5 In vivo brain uptake and pharmacokinetic study. Brain (a) and plasma (b) drug concentration time profile of ASM (via i.v. and i.n.) and ANLC (via i.n.) route. Mean ± SD, n = 5. | |
Table 5 Pharmacokinetic parameters data of ASM (i.v., i.n.) and ANLC (i.n.) in CF ratsa
| Pharmacokinetic parameters |
Organ |
ASM (i.v.) |
ASM (i.n.) |
ANLC (i.n.) |
| The parameters Cmax, Tmax, AUC0–24 h, and MRT of groups were compared by one way ANOVA followed by Tukey's post hoc test. (p < 0.05) compared to ASM (i.v.). (p < 0.05) compared to ASM (i.n.); mean ± SD, n = 5. |
| Cmax (ng mL−1) |
Brain |
41.46 ± 7.57 |
53.34 ± 9.76 |
74.13 ± 6.73b,c |
| Plasma |
82.76 ± 14.78 |
33.65 ± 15.52b |
41.76 ± 3.47b |
| Tmax (h) |
Brain |
2.0 ± 0 |
2.0 ± 0 |
1.0 ± 0b,c |
| Plasma |
0.17 ± 0 |
0.5 ± 0b |
0.5 ± 0b |
| AUC0–24 h (h ng mL−1) |
Brain |
202.70 ± 35.65 |
209.42 ± 42.48 |
560.93 ± 27.85b,c |
| Plasma |
115.63 ± 25.53 |
68.25 ± 21.34b |
154.46 ± 10.61b,c |
| MRT (h) |
Brain |
3.0 ± 0.21 |
2.8 ± 0.26 |
7.1 ± 0.13b,c |
| Plasma |
2.1 ± 0.28 |
2.0 ± 0.23 |
5.3 ± 0.18b,c |
| Absolute bioavailability (%) |
Brain |
— |
103.31 |
276.72 |
| Plasma |
— |
59.02 |
133.58 |
| DTE |
|
|
1.75 |
2.07 |
3.8. Animal behavioural study
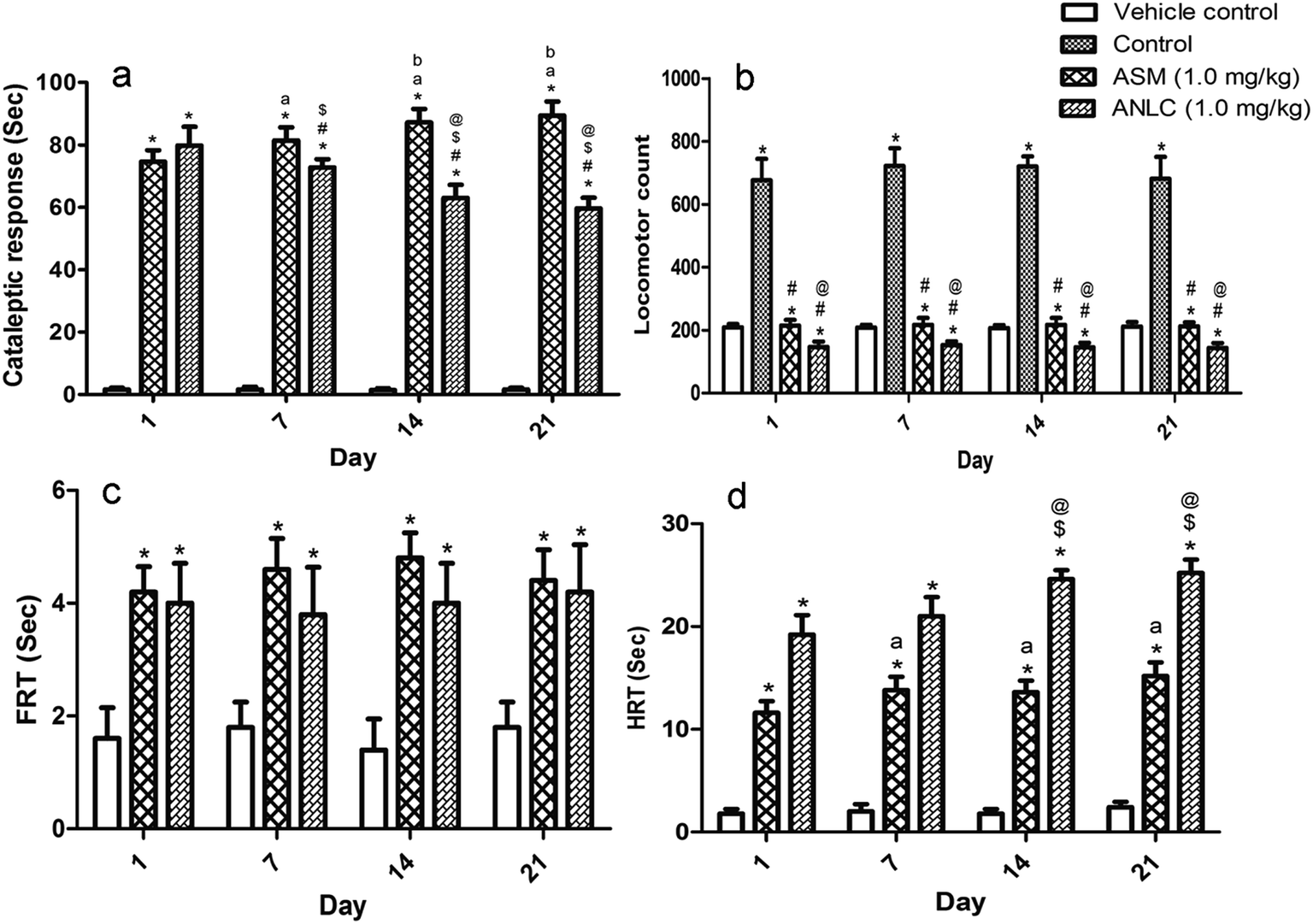
3.8.1. Catalepsy test. The cataleptic study was evaluated upto 21 day to observe effect of formulation on side effect associated with asenapine. Fig. 6a depicts extrapyramidal side effect of ASM and ANLC in normal rats in terms of cataleptic behaviour. Two-way ANOVA revealed that there were significant difference among groups [F (2, 48) = 3152, p < 0.05]. However, there were no significant difference among days [F (3, 48) = 1.087, p > 0.05]. But, there was a significant interaction between group and days [F (6, 48) = 26.23, p < 0.05]. The post hoc test showed that cataleptic response of ASM and ANLC were differed significantly from control group (p < 0.05) on all days. The first day observation showed no significant difference (p > 0.05) in response between ASM and ANLC. However, response exhibited on 7th (p < 0.05), 14th (p < 0.05) and 21st (p < 0.05) days of observation between ASM and ANLC group were significant. When groups were compared on different days, catalepsis was significantly increased in ASM (p < 0.05) and decreased in ANLC (p < 0.05) on 7th, 14th and 21st day in comparison to response of 1st day. In both ASM and ANLC group, no significant difference (p > 0.05) in interday response amongst groups were observed between 14th and 21st day. The reduction in cataleptic response by ANLC can be explained on the basis of sustained drug release, which leads to lesser fluctuation in drug concentration contributing clinical benefits in terms of constant antipsychotic efficacy and reduction of extra-pyramidal side effects. This could justify the superiority of sustained release over immediate release formulation for management of side effect in Schizophrenia.59
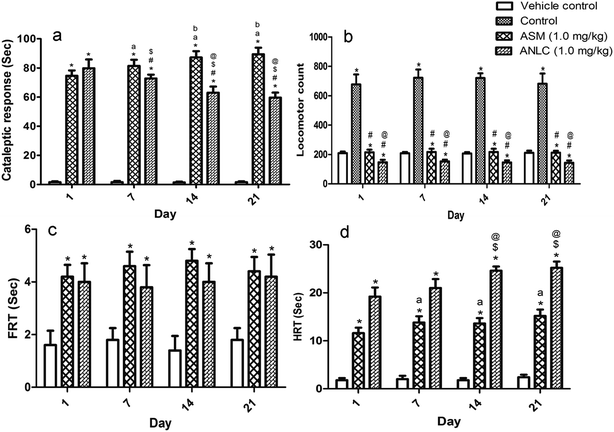 |
| | Fig. 6 Animal behavioural assessment on 1st, 7th, 14th and 21st day of study. (a) Cataleptic response of vehicle control, ASM and ANLC groups. *(p < 0.05) compared to vehicle control; #(p < 0.05) compared to ASM; a,b(p < 0.05) compared to 1st, 7th day response of ASM respectively; $,@(p < 0.05) compared to 1st, 7th day response of ANLC respectively. (b) Locomotor count of vehicle control, control, ASM and ANLC. *(p < 0.05) compared to vehicle control; #(p < 0.05) compared to control; @(p < 0.05) compared to ASM. (c) Paw test response (FRT) of control, ASM and ANLC groups. *(p < 0.05) compared to vehicle control. (d) Paw test response (HRT) of control, ASM and ANLC groups. *(p < 0.05) compared to vehicle control; a(p < 0.05) compared to 1st day response of ASM; $,@(p < 0.05) compared to 1st and 7th day response of ANLC respectively. The statistical calculation was performed by two way ANOVA followed by Bonferroni post hoc test. Mean ± SD, n = 5. | |
3.8.2. Induced locomotor activity test. The results of the locomotors activity test are shown in Fig. 6b. The two way ANOVA demonstrated a significant difference among group [F (3, 64) = 1287, p < 0.05]. Moreover, there were no significant difference among days [F (3, 64) = 0.9212, p > 0.05] and no significant interaction between group and days [F (9, 64) = 0.7116, p > 0.05]. The post hoc test revealed a significant increase in locomotor count in control group as compared to vehicle control and reduction of locomotors count in ASM and ANLC group as compared to control during three week period of behavioural observation. Also significant difference in count were observed between groups treated with ASM and ANLC from day 1 to day 21 (p < 0.05). As shown in results it was stipulated that the antagonistic activity was achieved by both ASM and ANLC, demonstrated the sensitivity of asenapine against D2 receptor. Further, significant count reduction in ANLC as compared to ASM at same dose confirmed that nanostructure lipid carriers had ability to cross the blood brain barrier resulting in higher dose dependent D2 receptor antagonistic activity.60
3.8.3. Paw test. The FRT was defined as the time it took the rat to withdraw one forelimb from front hole. Likewise, the HRT was defined as the time it took the rat to withdraw one hindlimb from rear hole. One hour after drug administration, the four paws of each animal were placed in the holes on the surface of paw test platform. The forelimb retraction time (FRT) and hindlimb retraction time (HRT) of different groups are shown in Fig. 6c and d, respectively. Two way ANOVA of FRT showed that significant difference among groups [F (2, 48) = 126.3, p < 0.05]. However, there were no significant difference among days [F (3, 48) = 0.2879, p > 0.05]. The interaction between group and days [F (6, 48) = 0.7424, p > 0.05] were also not significant. The post hoc analysis indicates significant increase in FRT in ASM and ANLC group when compared to control (p < 0.05). However, no significant difference was found between ASM and ANLC on different day (p > 0.05). The two way ANOVA of HRT demonstrated significant difference in groups [F (2, 48) = 1500, p < 0.05] and days [F (3, 48) = 22.73, p < 0.05]. Also, there was a significant interaction between the group and day [F (6, 48) = 7.373, p < 0.05]. Post hoc analysis of HRT showed that ASM had significant difference in HRT on 7th, 14th and 21st day compared to 1st day (p < 0.05), however, differences were not observed among 7th, 14th and 21st day (p > 0.05). Inter-day analysis of ANLC demonstrated that there was no significant difference between 1st & 7th day (p > 0.05) and 14th & 21st day (p > 0.05) in HRT. Moreover, HRT of both 14th & 21st day were significantly differ to 1st & 7th day (p < 0.05) in ANLC.ASM and ANLC showed HRT greater than corresponding FRT. The significantly higher HRT right from 1st week in case of group treated with ANLC in comparison to corresponding groups treated with ASM indicates that ANLC is better therapeutically. Higher HRT can be correlated to the increased asenapine concentration in brain owing to nanostructure lipid carrier delivery. It shows that developed nanostructure lipid carriers have potential to target brain resulting in higher HRT in ANLC group. It was also observed that HRT increased on subsequent days in groups treated with ASM and ANLC upto 1 and 2 week, respectively. This could be explained on the basis of pharmacological effect of asenapine which takes around 2 weeks to show optimum clinical benefit. The sustained and targeted delivery of asenapine by ANLC can be directly correlated to increase in HRT as compared to ASM during behavioural observation period.
4. Conclusions
The present study was demonstrated the systematic development of ANLC by QbD approach with predetermined properties of formulation ideal for brain delivery. Based on the QTPP, CMA and CPP, the five factors, three levels BBD was selected using GMS, oleic acid, Tween-80 as solid lipid, liquid lipid and surfactant, respectively. The predicted response of optimized batch was in good correlation with observed particle size (167.30 ± 7.52 nm) and entrapment efficiency (83.50 ± 2.48%), which also demonstrated the reliability of this model. The in vitro drug release study revealed anomalous release upto 24 h, which is best fitted with Korsmeyer–Peppas model. Surface and solid state characterization of ANLC demonstrated smooth surface, spherical shape particles; in which asenapine was presented in molecular dispersed state in lipid matrix. Further, brain and plasma pharmacokinetic studies and long term animal behavioural assessment revealed high brain bioavailability, better therapeutic and safety profile of ANLC compared to pure drug via intranasal route. However, biochemical and toxicological studies at pre-clinical level are obligatory for the further evaluation.
Conflict of interest statement
The authors declare no competing financial interest.
Acknowledgements
The authors are thankful to Ranbaxy Laboratories Ltd (Gurgaon, India) for providing asenapine maleate as gift sample. The authors acknowledge the help of Central Instrument Facility Centre of IIT (BHU) Varanasi for AFM studies. The first author is thankful to STINT project for providing support for training on solid state characterization technique and DSC and XRD analysis of samples at Luleå University of Technology, Luleå, Sweden.
References
- W. Rossler, H. J. Salize, J. van Os and A. Riecher-Rossler, Eur. Neuropsychopharmacol., 2005, 15, 399–409 CrossRef PubMed.
- Schizophrenia, WHO, http://www.who.int/mental_health/management/schizophrenia/en/, accessed on April 20, 2015.
- P. J. Weiden and M. Olfson, Schizophr. Bull., 1995, 21, 419–429 CrossRef CAS PubMed.
- T. P. Gilmer, C. R. Dolder, J. P. Lacro, D. P. Folsom, L. Lindamer, P. Garcia and D. V. Jeste, Am. J. Psychiatry, 2004, 161, 692–699 CrossRef PubMed.
- J. A. Lieberman, T. S. Stroup, J. P. McEvoy, M. S. Swartz, R. A. Rosenheck, D. O. Perkins, R. S. Keefe, S. M. Davis, C. E. Davis, B. D. Lebowitz, J. Severe and J. K. Hsiao, N. Engl. J. Med., 2005, 353, 1209–1223 CrossRef CAS PubMed.
- M. Valenstein, F. C. Blow, L. A. Copeland, J. F. McCarthy, J. E. Zeber, L. Gillon, C. R. Bingham and T. Stavenger, Schizophr. Bull., 2004, 30, 255–264 CrossRef PubMed.
- R. Tandon, Current Psychosis and Therapeutics Reports, 2006, 4, 40–49 CrossRef.
- F. I. Tarazi and M. Shahid, Drugs Today, 2009, 45, 865–876 CrossRef PubMed.
- J. A. Bartlett and K. van der Voort Maarschalk, AAPS PharmSciTech, 2012, 13, 1110–1115 CrossRef CAS.
- US Food and Drug Administration, Saphris (Asenapine) Sublingual Tablets, http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022117s017s018s019lbl.pdf, accessed on April 03, 2015.
- S. C. Stoner and H. A. Pace, Clin. Ther., 2012, 34, 1023–1040 CrossRef CAS PubMed.
- L. Citrome, Expert Opin. Drug Saf., 2014, 13, 803–830 CAS.
- S. Patel, S. Chavhan, H. Soni, A. K. Babbar, R. Mathur, A. K. Mishra and K. Sawant, J. Drug Targeting, 2011, 19, 468–474 CrossRef CAS PubMed.
- L. Kozlovskaya and D. Stepensky, J. Controlled Release, 2013, 171, 17–23 CrossRef CAS PubMed.
- L. Kozlovskaya, M. Abou-Kaoud and D. Stepensky, J. Controlled Release, 2014, 189, 133–140 CrossRef CAS PubMed.
- A. Mistry, S. Stolnik and L. Illum, Int. J. Pharm., 2009, 379, 146–157 CrossRef CAS PubMed.
- M. I. Alam, S. Beg, A. Samad, S. Baboota, K. Kohli, J. Ali, A. Ahuja and M. Akbar, Eur. J. Pharm. Sci., 2010, 40, 385–403 CrossRef CAS PubMed.
- R. Tiwari and K. Pathak, Int. J. Pharm., 2011, 415, 232–243 CrossRef CAS PubMed.
- P. Blasi, S. Giovagnoli, A. Schoubben, C. Puglia, F. Bonina, C. Rossi and M. Ricci, Int. J. Pharm., 2011, 419, 287–295 CrossRef CAS PubMed.
- S. Eskandari, J. Varshosaz, M. Minaiyan and M. Tabbakhian, Int. J. Nanomed., 2011, 6, 363–371 CAS.
- M. Patel, E. B. Souto and K. K. Singh, Expert Opin. Drug Delivery, 2013, 10, 889–905 CrossRef CAS PubMed.
- A. Mujtaba, M. Ali and K. Kohli, Int. J. Biol. Macromol., 2014, 69, 420–429 CrossRef CAS PubMed.
- G. Y. Li, M. Zhong, Z. D. Zhou, Y. D. Zhong, P. Ding and Y. Huang, Int. J. Biol. Macromol., 2011, 49, 970–978 CrossRef CAS PubMed.
- A. Garg and S. Singh, Colloids Surf., B, 2011, 87, 280–288 CrossRef CAS PubMed.
- L. X. Yu, Pharm. Res., 2008, 25, 781–791 CrossRef CAS PubMed.
- D. S. Singare, S. Marella, K. Gowthamrajan, G. T. Kulkarni, R. Vooturi and P. S. Rao, Int. J. Pharm., 2010, 402, 213–220 CrossRef CAS PubMed.
- F. Wang, L. Chen, S. Jiang, J. He, X. Zhang, J. Peng, Q. Xu and R. Li, J. Liposome Res., 2014, 24, 171–181 CrossRef CAS PubMed.
- D. Alukda, T. Sturgis and B. B. Youan, J. Pharm. Sci., 2011, 100, 3345–3356 CrossRef CAS PubMed.
- B. Singh, R. Kumar and N. Ahuja, Crit. Rev. Ther. Drug Carrier Syst., 2005, 22, 27–105 CrossRef CAS PubMed.
- B. Singh, M. Dahiya, V. Saharan and N. Ahuja, Crit. Rev. Ther. Drug Carrier Syst., 2005, 22, 215–294 CrossRef CAS PubMed.
- S. K. Singh, P. R. Vuddanda, S. Singh and A. K. Srivastava, BioMed Res. Int., 2013, 2013, 909045 Search PubMed.
- ICH, Q2(R1) – Validation of Analytical Procedures: Text and Methodology, (2005), http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf, accessed on January 15, 2015.
- A. Jain, S. K. Mishra, P. R. Vuddanda, S. K. Singh, R. Singh and S. Singh, Nanomedicine, 2014, 10, 1031–1040 CrossRef CAS PubMed.
- P. R. Vuddanda, A. Mishra, S. K. Singh and S. Singh, Pharm. Dev. Technol., 2014, 1–9, DOI:10.3109/10837450.2014.908302.
- Y. Zhang, M. Huo, J. Zhou, A. Zou, W. Li, C. Yao and S. Xie, AAPS J., 2010, 12, 263–271 CrossRef CAS PubMed.
- M. Fazil, S. Md, S. Haque, M. Kumar, S. Baboota, J. K. Sahni and J. Ali, Eur. J. Pharm. Sci., 2012, 47, 6–15 CrossRef CAS PubMed.
- M. A. Mohammad, A. Alhalaweh and S. P. Velaga, Int. J. Pharm., 2011, 407, 63–71 CrossRef CAS PubMed.
- Y. Singh, M. K. Hidau, J. Krishna and S. K. Singh, Xenobiotica, 2015, 45, 731–740 CrossRef CAS PubMed.
- M. Kumar Hidau, Y. Singh, S. Shahi, P. Mounika and S. Kumar Singh, Curr. Pharm. Anal., 2015, 11, 35–42 CrossRef.
- A. Serralheiro, G. Alves, A. Fortuna and A. Falcão, Eur. J. Pharm. Sci., 2014, 60, 32–39 CrossRef CAS PubMed.
- J. A. Lieberman, F. P. Bymaster, H. Y. Meltzer, A. Y. Deutch, G. E. Duncan, C. E. Marx, J. R. Aprille, D. S. Dwyer, X.-M. Li, S. P. Mahadik, R. S. Duman, J. H. Porter, J. S. Modica-Napolitano, S. S. Newton and J. G. Csernansky, Pharmacol. Rev., 2008, 60, 358–403 CrossRef CAS PubMed.
- O. Franberg, C. Wiker, M. M. Marcus, A. Konradsson, K. Jardemark, B. Schilstrom, M. Shahid, E. H. Wong and T. H. Svensson, Psychopharmacology, 2008, 196, 417–429 CrossRef CAS PubMed.
- M. A. Geyer and B. Ellenbroek, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2003, 27, 1071–1079 CrossRef CAS PubMed.
- H. M. Marston, J. W. Young, F. D. Martin, K. A. Serpa, C. L. Moore, E. H. Wong, L. Gold, L. T. Meltzer, M. R. Azar, M. A. Geyer and M. Shahid, Psychopharmacology, 2009, 206, 699–714 CrossRef CAS PubMed.
- M. Kumar, A. Misra, A. K. Babbar, A. K. Mishra, P. Mishra and K. Pathak, Int. J. Pharm., 2008, 358, 285–291 CrossRef CAS PubMed.
- B. A. Ellenbroek, B. W. Peeters, W. M. Honig and A. R. Cools, Psychopharmacology, 1987, 93, 343–348 CrossRef CAS PubMed.
- S. Chopra, S. K. Motwani, Z. Iqbal, S. Talegaonkar, F. J. Ahmad and R. K. Khar, Eur. J. Pharm. Biopharm., 2007, 67, 120–131 CrossRef CAS PubMed.
- A. Mujtaba, M. Ali and K. Kohli, Chem. Eng. Res. Des., 2014, 92, 156–165 CrossRef CAS.
- S. C. Yang, L. F. Lu, Y. Cai, J. B. Zhu, B. W. Liang and C. Z. Yang, J. Controlled Release, 1999, 59, 299–307 CrossRef CAS PubMed.
- S. Md, R. A. Khan, G. Mustafa, K. Chuttani, S. Baboota, J. K. Sahni and J. Ali, Eur. J. Pharm. Sci., 2013, 48, 393–405 CrossRef CAS PubMed.
- S. Wohlfart, S. Gelperina and J. Kreuter, J. Controlled Release, 2012, 161, 264–273 CrossRef CAS.
- H. L. Wong, X. Y. Wu and R. Bendayan, Adv. Drug Delivery Rev., 2012, 64, 686–700 CrossRef CAS PubMed.
- P. Costa and J. M. S. Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- H. Bunjes and T. Unruh, Adv. Drug Delivery Rev., 2007, 59, 379–402 CrossRef CAS PubMed.
- J. Kreuter, Adv. Drug Delivery Rev., 2001, 47, 65–81 CrossRef CAS PubMed.
- B. Wilson, M. K. Samanta, K. Santhi, K. P. Kumar, N. Paramakrishnan and B. Suresh, Brain Res., 2008, 1200, 159–168 CrossRef CAS PubMed.
- R. G. Thorne, G. J. Pronk, V. Padmanabhan and W. H. Frey 2nd, Neuroscience, 2004, 127, 481–496 CrossRef CAS PubMed.
- S. Haque, S. Md, M. Fazil, M. Kumar, J. K. Sahni, J. Ali and S. Baboota, Carbohydr. Polym., 2012, 89, 72–79 CrossRef CAS.
- C. M. Baldwin and L. J. Scott, CNS Drugs, 2009, 23, 261–269 CrossRef CAS PubMed.
- F. I. Tarazi and J. C. Neill, Expert Opin. Drug Discovery, 2013, 8, 93–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19793g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.