
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis acid–base 1,2-addition reactions: synthesis of pyrylium borates from en-ynoate precursors†
Lewis C.
Wilkins
a,
Hugh B.
Hamilton
a,
Benson M.
Kariuki
a,
A. Stephen K.
Hashmi
bc,
Max M.
Hansmann
b and
Rebecca L.
Melen
*a
aSchool of Chemistry, Main Building, Cardiff University, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: melenr@cardiff.ac.uk; Tel: +(44) 2920 879667
bOrganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
cChemistry Department, Faculty of Science, King Abdulaziz University (KAU), Jeddah 21589, Saudi Arabia
First published on 25th September 2015
Abstract
Treatment of methyl (Z)-2-alken-4-ynoates with the strong Lewis acid tris(pentafluorophenyl) borane, B(C6F5)3, yield substituted zwitterionic pyrylium borate species via an intramolecular 6-endo-dig cyclisation reaction.
Some of the first examples of pyrylium salts were reported over 100 years ago through the reaction between acetophenone and sulphuric acid to yield the 2,4,6-triphenyl pyrylium cation.1 While the significance of this compound was not fully appreciated at the time, it has since spawned intense interest in various areas such as a basis for photo-sensitisers,2 redox mediators3 and photo-induced free radical polymerisations.4 Due to these interesting physical properties, pyrylium salts play a fundamental role in materials chemistry.5 Alongside this prominence in materials chemistry, pyrylium compounds are extremely important intermediates in synthetic chemistry and are vital in the generation of complex cyclic compounds with various functionality such as extended macrocyclic arenes6inter alia.7 In particular, these electron deficient aromatic compounds have significance in medicinal and pharmaceutical chemistry for the synthesis of many biologically active drugs and bio-imaging compounds.8
Much of the reason why these pyrylium systems are useful in such applications arise from the 6-membered ring system similar to phenyl and pyridyl derivatives, albeit with reduced aromaticity compared to benzene as a result of the incorporation of the more electronegative oxygen atom.9 The perturbation to the aromaticity caused by the incorporation of an oxycation renders these pyrylium systems susceptible to attack by nucleophiles at the 2-, 4- or 6-positions. This reactivity is of necessity in the formation of aryl-substituted phosphinines,10 pyridinium salts11 and substituted furans.12
The formation of pyrylium salts as intermediates using π-Lewis acidic transition metals, such as gold, has been shown to be a reliable synthetic methodology through π-activation of the alkyne unit of enynone and enynal reagents.13 This proceeds through a trans-oxy-auration step across the alkyne moiety via a 6-endo-dig cyclisation mechanism. This intermediate can then undergo many transformations such as acting as the diene component in [4 + 2] intermolecular cycloaddition reactions with alkynes to yield acylnaphthalene derivatives.14 Our previous studies have shown that the reactions of B(C6F5)3 with organic substrates containing π-bonds together with nucleophilic ketones,15 esters,16 or amides17 result in a range of carboboration and 5-exo-dig 1,2-addition reactions.18 The work discussed here extends our research to en-ynoate substrates in 6-endo-dig cyclisation reactions to generate pyrylium borate species.
The reaction between the strong Lewis acid B(C6F5)3 and functionalised methyl (Z)-2-alken-4-ynoates (1a and 1b) proceeded rapidly at room temperature with an immediate colour change to a dark orange-red colour associated with the formation of the 2-methoxy pyrylium borate products (2a and 2b, Scheme 1). In situ multinuclear NMR studies showed almost quantitative conversion in as little as 1 h, although isolated yields were more modest at 65% (2a) and 64% (2b).
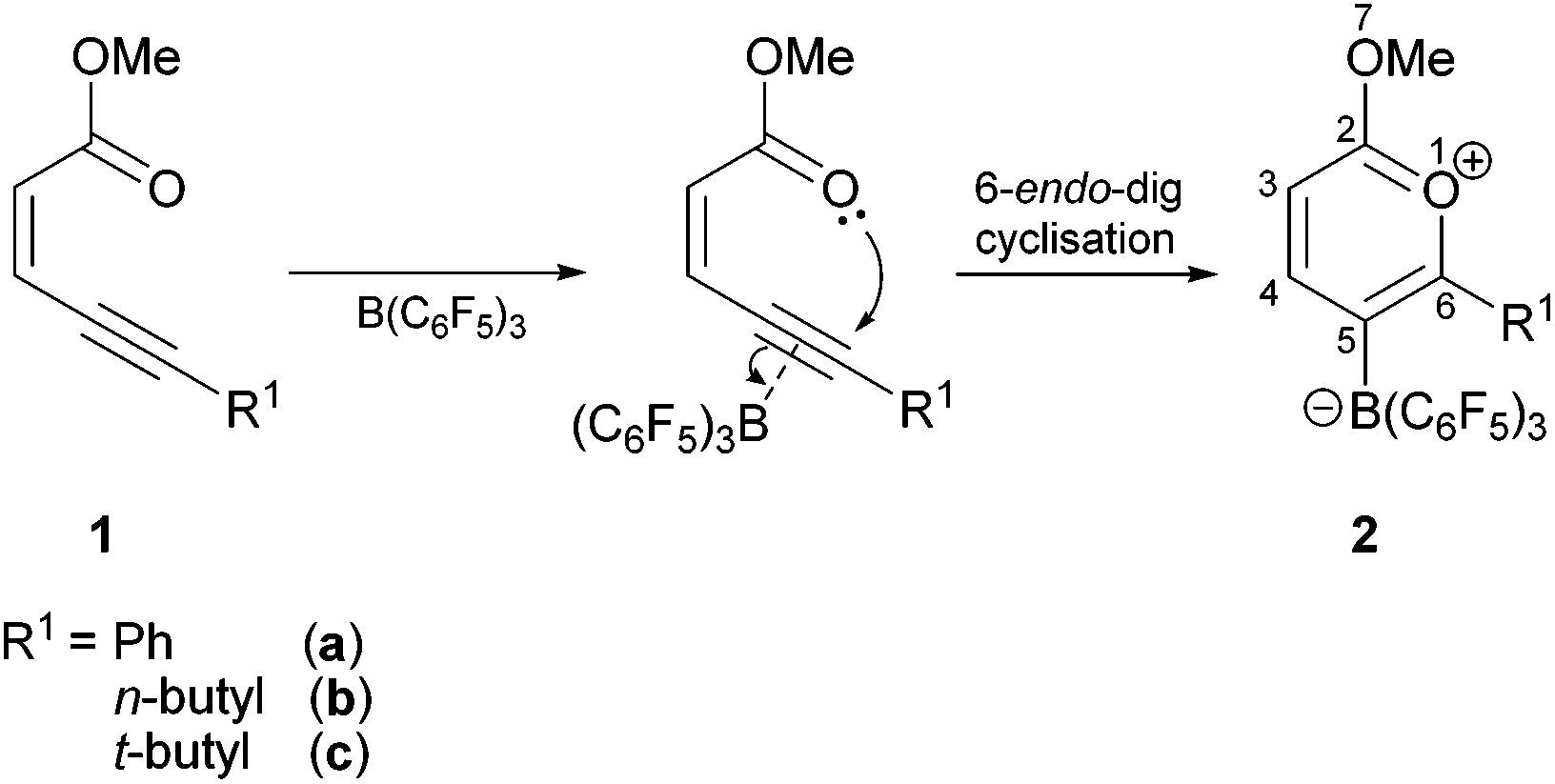 | ||
| Scheme 1 Proposed mechanistic pathway of the reaction between B(C6F5)3 and methyl en-ynoates. | ||
The 1H NMR spectra were diagnostic revealing a prominent resonance of the vinylic proton β to the carbonyl at C(4) at δ = 8.65 and 8.47 ppm for 2a and 2b respectively (cf. δ = 6.37 ppm (1a) and 6.15 ppm (1b)). In addition, the bulky nature of the three C6F5 groups at boron would appear to restrict motion of the adjacent C(6) substituent, leading to two independent proton environments of the CH2 group in the nBu functionality of 2b reflected in the observation of two distinct broad singlets in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at δ = 1.43 and 0.64 ppm. Similarly, the constrained geometry is reflected in separate chemical environments for the F atoms of the C6F5 groups, evident in the 19F NMR spectra of 2b (Fig. 1, bottom) which reveals a total of fifteen resonances for the ortho (δ = −128.2 to −135.0 ppm (6F)), para (δ = −158.9 to −159.9 ppm (3F)) and meta (δ = −162.8 to −165.3 ppm (6F)) fluorine atoms. Heating an NMR sample of 2b in d8-toluene to 80 °C showed no coalescence of the proton environments of the methylene group attached to C(6) in the 1H NMR spectrum. In addition, the 19F NMR showed no discernible coalescence of the fluorine environments on the C6F5 rings suggesting a considerable energy barrier to rotation of the pyrylium cation with respect to the B(C6F5)3 group. The 19F NMR of 2a (Fig. 1, top) exhibits similar behaviour, although the spectra in this case are somewhat simpler with just eight peaks for the ortho (δ = −128.6 (1F), −130.3 (4F) and −131.1 ppm (1F)), para (δ = −159.2 (1F) and −160.0 ppm (2F)) and meta (δ = −163.5 (1F), −164.7 (1F) and −165.4 ppm (4F)) fluorine atoms. 11B NMR spectra lead to sharp resonances at δ = −14.4 and −14.5 ppm for 2a and 2b which has become synonymous with these four coordinate vinyl borate complexes.17,18
:1 ratio at δ = 1.43 and 0.64 ppm. Similarly, the constrained geometry is reflected in separate chemical environments for the F atoms of the C6F5 groups, evident in the 19F NMR spectra of 2b (Fig. 1, bottom) which reveals a total of fifteen resonances for the ortho (δ = −128.2 to −135.0 ppm (6F)), para (δ = −158.9 to −159.9 ppm (3F)) and meta (δ = −162.8 to −165.3 ppm (6F)) fluorine atoms. Heating an NMR sample of 2b in d8-toluene to 80 °C showed no coalescence of the proton environments of the methylene group attached to C(6) in the 1H NMR spectrum. In addition, the 19F NMR showed no discernible coalescence of the fluorine environments on the C6F5 rings suggesting a considerable energy barrier to rotation of the pyrylium cation with respect to the B(C6F5)3 group. The 19F NMR of 2a (Fig. 1, top) exhibits similar behaviour, although the spectra in this case are somewhat simpler with just eight peaks for the ortho (δ = −128.6 (1F), −130.3 (4F) and −131.1 ppm (1F)), para (δ = −159.2 (1F) and −160.0 ppm (2F)) and meta (δ = −163.5 (1F), −164.7 (1F) and −165.4 ppm (4F)) fluorine atoms. 11B NMR spectra lead to sharp resonances at δ = −14.4 and −14.5 ppm for 2a and 2b which has become synonymous with these four coordinate vinyl borate complexes.17,18
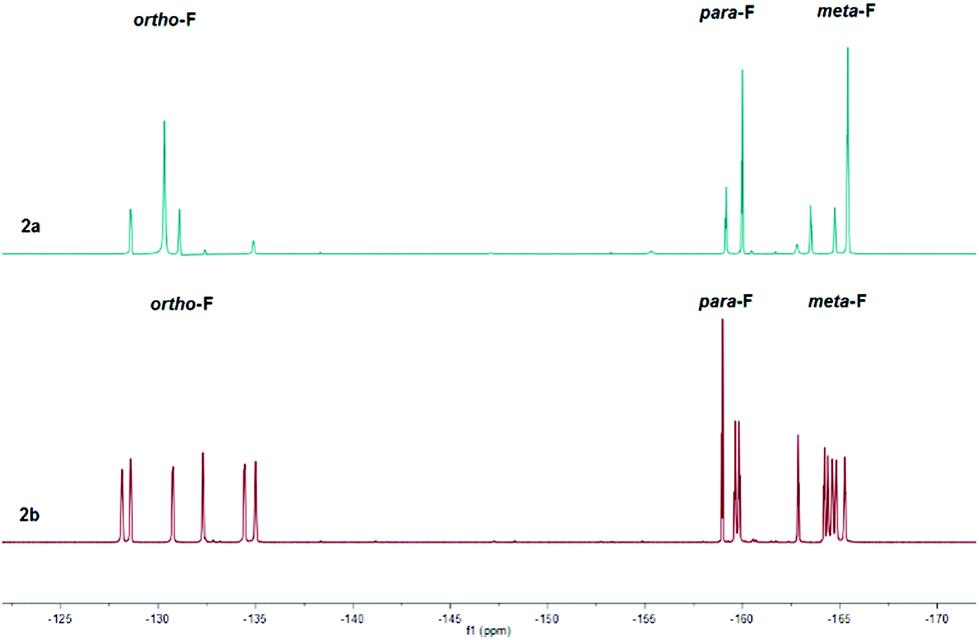 | ||
| Fig. 1 Stacked 19F NMR spectra of 2a and 2b. | ||
Examination of the solid-state single crystal structures of 2 unambiguously confirmed the product of the cyclisation process as the aforementioned pyrylium-borate species (Fig. 2). 2a and 2b both crystallised in the monoclinic P21/n space group with two and one molecule in the asymmetric unit respectively. The phenyl substituent attached to the ring in 2a rotates out of the plane of the pyrylium ring by 66.1(2)° and 58.4(2)°, this loss of planarity also prevents an extended aromatic network with the pyrylium fragment. This is in contrast to that observed previously for dioxolium compounds which contain a cationic 5-membered [C2O2C′]+ ring.16a In these cases, when the C′ carbon is substituted by an aromatic ring then the two rings lie in the same plane offering the possibility for stabilisation of the positive charge through delocalisation.16a Regarding compound 2a, the rotation of the phenyl group out of the plane of the pyrylium ring arises chiefly through reduction of steric congestion. This can be clearly observed in the space-filling models (Fig. 3) of both 2a and 2b. Additionally the reaction of B(C6F5)3 with 1c gave inconclusive results presumably due to the steric congestion that would arise from the neighbouring borate and tBu groups. In 2a the rotation of the C6H5 ring out of the plane of the pyrylium ring allows it to interact with one of the C6F5 rings through a displaced π-stacking interaction with a centroid⋯centroid distance of 3.6421(10) Å leading to an essentially eclipsed geometry in which the C(5)C(6)Ph plane deviates out of the C(5)BC6F5 plane by 0.11(14)°. 2b however, adopts a more conventional staggered geometry whereby the C(6)n-butyl bond is out of C(5)BC6F5 plane by 49.9(2)° which is consistent with the higher symmetry observed for 2a with respect to 2b as detected via NMR spectroscopy. The structures of both 2a and 2b show similar metric parameters for the pyrylium ring. In both cases the C5O heterocycle is essentially planar verified by a maximum deviance from planarity with root mean square deviation values of 0.023 and 0.026 Å (2a) and 0.013 Å (2b). The C–C bond lengths (Table 1) (1.372(3)–1.410(3) Å) are intermediate between those for a typical C–C single bond (ca. 1.53 Å) and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (ca. 1.33 Å) and are more typical for C–C bonds found in more delocalised systems (1.38 Å in benzene).19
C double bond (ca. 1.33 Å) and are more typical for C–C bonds found in more delocalised systems (1.38 Å in benzene).19
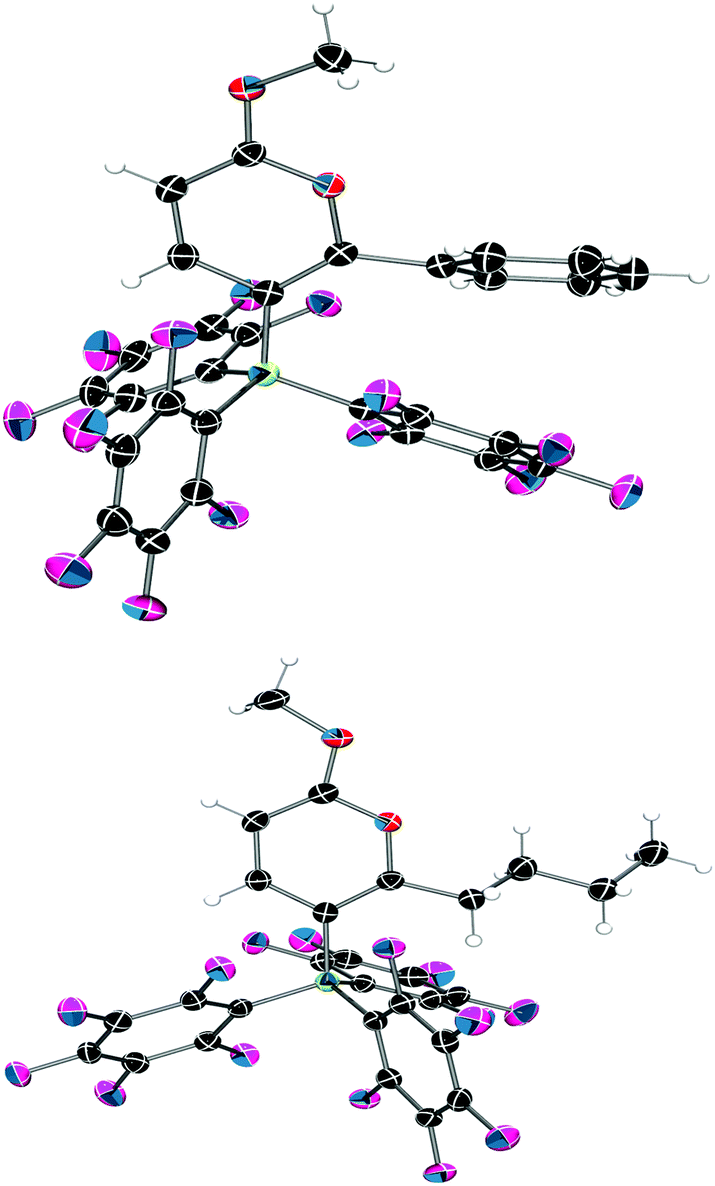 | ||
| Fig. 2 Solid-state structures of 2a (top) and 2b (bottom) with 50% probability ellipsoids. C: black, O: red, B: yellow-green, F: pink, H: white. One of the two crystallographically independent molecules of 2a is shown. | ||
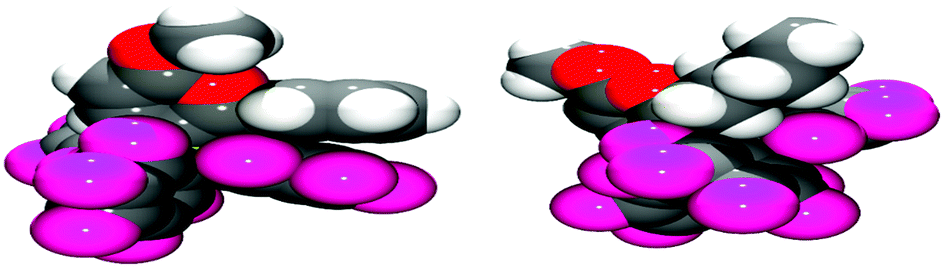 | ||
| Fig. 3 Space-filling representation of structures of 2a (left) and 2b (right). C: black, O: red, B: yellow-green, F: pink, H: white. One of the two crystallographically independent molecules of 2a is shown. | ||
|
|
|||||
|---|---|---|---|---|---|
| Bond | Bond length/Å | ||||
| 2a | 2b | ||||
| Exp. | Calc. | Exp. | Calc. | ||
| C2–C3 | 1.371(3) | 1.388(2) | 1.386 | 1.374(3) | 1.378 |
| C3–C4 | 1.362(3) | 1.360(2) | 1.384 | 1.382(3) | 1.390 |
| C4–C5 | 1.430(2) | 1.435(2) | 1.423 | 1.410(3) | 1.410 |
| C5–C6 | 1.361(2) | 1.365(2) | 1.377 | 1.372(3) | 1.384 |
| C2–O1 | 1.316(2) | 1.304(2) | 1.319 | 1.316(3) | 1.320 |
| C6–O1 | 1.404(2) | 1.397(2) | 1.391 | 1.370(3) | 1.370 |
| C2–O7 | 1.306(2) | 1.303(2) | 1.316 | 1.311(3) | 1.316 |
| C5–B | 1.665(2) | 1.650(2) | 1.671 | 1.657(3) | 1.673 |

The C–O bond lengths associated with the O(1)C(2)O(7) fragment are almost identical within error (1.303(2)–1.316(2) Å) and are intermediate between a formal double and single carbon-oxygen bond (ca. 1.23 and 1.42 Å respectively).19
The nature of the bonding in 2 has been investigated by way of DFT calculations carried out at the B3LYP/6-311G* level (see ESI†). The calculated structure was in good agreement with that observed experimentally with similar geometric parameters for the cationic C5O ring (Table 1) with C–C bond lengths intermediate between single and double bonds characteristic of a delocalised structure. An NBO analysis clearly reveals strong delocalisation around the carbon-containing part of the cationic pyrylium ring but a more localised polar structure in the vicinity of the oxygen atoms. The Wiberg bond indices and NBO charges for 2a (as representative of 2a and 2b) are presented in Fig. 4. The electronegative nature of the oxygen atoms affords strong bond polarisation through the σ-framework reflected in the large partial charges at the carbon atoms bonded to oxygen, as well as the oxygen atoms themselves. This is particularly evident for C(2) bonded to both oxygen atoms and is compensated by oxygen lone pair π-back-donation to the strongly cationic carbon centre (evidenced in the second order perturbation analysis). It is clear from these data and other studies that incorporation of oxygen into the ring leads to a disruption of the aromaticity.9 Nucleus independent chemical shift, NICS(0) calculations at the B3LYP/6-311G*20 level showed that although aromatic, a lower degree of aromaticity is observed when compared to benzene (−8.91 ppm) with 2a and 2b giving NICS(0) values of −3.93 and −4.86 ppm respectively (Fig. 5).
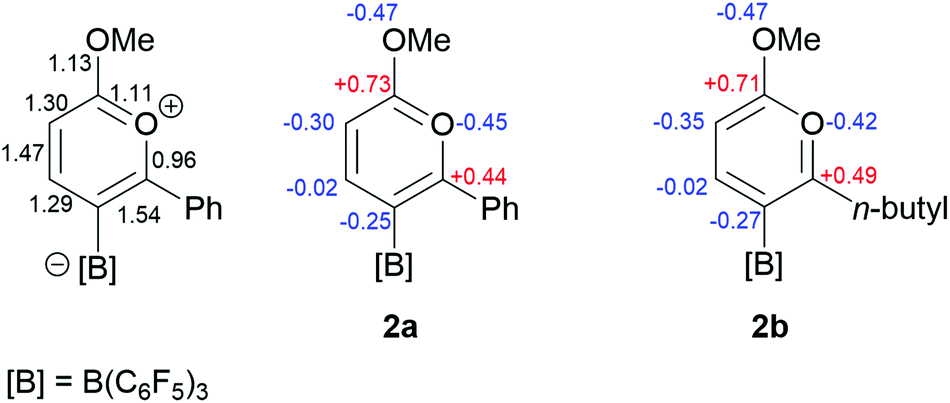 | ||
| Fig. 4 (left) Wiberg bond indices for 2a and (right) NBO charges for the core of the pyrylium cation 2a and 2b. | ||
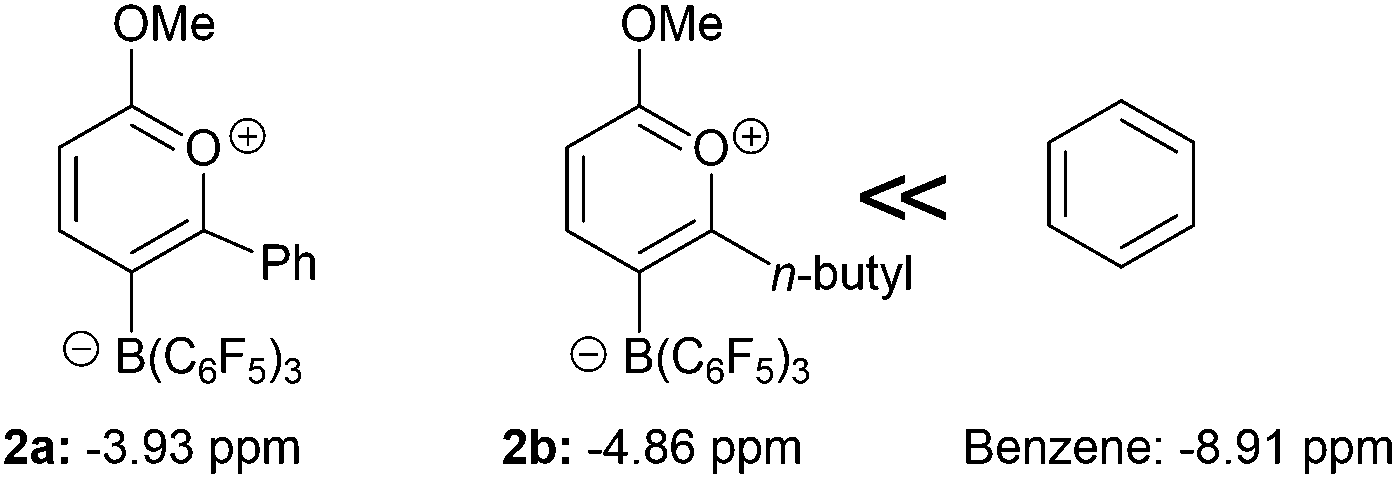 | ||
| Fig. 5 Aromaticity of compounds 2 relative to benzene using nucleus independent chemical shift calculations. | ||
The mechanism for this reaction would appear to follow a similar pathway to that which has been identified in previous work with propargyl esters16 and amides,17i.e. 1,2-addition of the Lewis basic oxygen and the Lewis acid across the alkyne moiety. In the case of both propargyl esters16 and propargyl amides17 a 5-exo-dig cyclisation affords dioxolium and oxazole products respectively. However, in the current case the 6-endo-dig cyclisation pathway appears preferred over formation of the 5-membered ring or indeed 1,1-carboboration which has been observed for alkynes.21 The rapid formation of a stable 6-membered, planar, delocalised 6π Hückel aromatic system in 2 explains the driving force for the 6-endo-dig cyclisation over the 5-exo-dig pathway yielding the less stable furanium derivative.22
In conclusion, this work has shown the ease at which pyrylium borate salts can be prepared in a very clean and fast reaction from methyl (Z)-2-alken-4-ynoate precursors. Importantly, in these systems we can isolate the vinyl borate species as a stable product in the reaction, this is in contrast to that seen in transition metal catalysed systems as the analogous vinyl gold intermediates have been seldom observed or isolated until recent years as the deauration step is typically rapid.23 Further work will probe the breadth of substrate scope and subsequent reactivity of the pyrylium borate products, all of which are ongoing projects in our group. The hydrolysis and/or proto-deboronation of these products will be investigated in the synthesis of pyrones. Indeed, isocoumarins and α-pyrones are key structural units in natural products and many biologically active pharmaceuticals e.g. in HIV protease inhibitors.24 Such systems have been synthesised by similar electrophilic cyclisation reactions using other main group Lewis acids (e.g. using iodine or selenium electrophiles).25 Unlike the cyclisations described above, and those performed by transition metals such as gold, the ability for this transformation to trap the pyrylium species en route to the pyrone product also permits these products to be used for further transformations including reactions with nucleophiles, cycloaddition reactions or cross-coupling of the boronate with organo-halides. Our on-going studies in this area will permit us to develop these types of reagents in the synthesis of useful precursors in a variety of areas of synthetic, biological and materials chemistry.
Acknowledgements
H. B. H. would like to thank the EPSRC-funded Bath/Bristol/Cardiff Catalysis Centre for Doctoral Training (EP/L016443/1). M. M. H. is grateful to the Fonds der Chemischen Industrie for a Chemiefonds scholarship and the Studienstiftung des deutschen Volkes. We thank W. R. Grace for the kind donation of B(C6F5)3. We also thank Dr James Platts for his assistance in NICS calculations as well as Dr Joel Loveridge for his support with NMR spectroscopy.Notes and references
- S. V. Kostanecki and G. Rossbach, Chem. Ber., 1896, 29, 1488 CrossRef.
- A. Amat, A. Arques and M. A. Miranda, Appl. Catal., B, 1999, 23, 205 CrossRef CAS.
- (a) A. Corma, V. Fornes, H. Garcia, M. A. Miranda and M. J. Sabater, J. Am. Chem. Soc., 1994, 116, 9767 CrossRef CAS; (b) S. Liang, L. Zhu and J. K. Hurst, Langmuir, 2012, 28, 12171 CrossRef CAS PubMed.
- M. El-Roz, J. Lalevée, F. Morlet-Savary, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7369 CrossRef CAS.
- (a) F. Garcia, J. M. Garcia, B. Garcia-Acosta, R. Martinez-Manez, F. Sancenon and J. Soto, Chem. Commun., 2005, 2790 RSC; (b) Y. Li, G. Zhang, G. Yang, Y. Guo, C. A. Di, X. Chen, Z. Liu, H. Liu, Z. Xu, W. Xu, H. Fu and D. Zhang, J. Org. Chem., 2013, 78, 2926 CrossRef CAS PubMed.
- S. Höger, S. Rosselli, A.-D. Ramminger and V. Enkelmann, Org. Lett., 2002, 4, 4269 CrossRef PubMed.
- (a) A. R. Katritzky, P. Czerney and J. R. Levell, J. Org. Chem., 1997, 62, 8198 CrossRef CAS PubMed; (b) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications, John Wiley & Sons, 3rd edn, 2013 Search PubMed.
- (a) M. R. Detty and P. B. Merkel, J. Am. Chem. Soc., 1990, 112, 3845 CrossRef CAS; (b) P. Kovacic, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 501 CrossRef CAS PubMed; (c) B. A. Rao, J.-Y. Lee and Y.-A. Son, Supramol. Chem., 2014, 27, 191 CrossRef; (d) M. Shariatgorji, A. Nilsson, P. Källback, O. Karlsson, X. Zhang, P. Svenningsson and P. Andren, J. Am. Soc. Mass Spectrom., 2015, 26, 934 CrossRef CAS PubMed.
- I. V. Omelchenko, O. V. Shishkin, L. Gorb, J. Leszczynski, S. Fias and P. Bultinck, Phys. Chem. Chem. Phys., 2011, 13, 20536 RSC.
- C. Müller, D. Wasserberg, J. J. M. Weemers, E. A. Pidko, S. Hoffmann, M. Lutz, A. L. Spek, S. C. J. Meskers, R. A. J. Janssen, R. A. Van Santen and D. Vogt, Chem. – Eur. J., 2007, 13, 4548 CrossRef PubMed.
- (a) A. R. Katritzky, R. H. Manzo, J. M. Lloyd and R. C. Patel, Angew. Chem., Int. Ed. Engl., 1980, 19, 306 CrossRef; (b) R. Awartani, K. Sakizadeh and B. Gabrielsen, J. Chem. Educ., 1986, 63, 172 CrossRef CAS.
- I. Francesconi, A. Patel and D. W. Boykin, Synthesis, 1999, 61 CrossRef CAS.
- (a) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458 CrossRef CAS PubMed; (b) B. F. Straub, Chem. Commun., 2004, 1726 RSC; (c) S. Zhu, Z. Zhang, X. Huang, H. Jiang and Z. Guo, Chem. – Eur. J., 2013, 19, 4695 CrossRef CAS PubMed.
- N. Asao, K. Takahashi, S. Lee, T. Kasahara and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 12650 CrossRef CAS PubMed.
- R. L. Melen, L. C. Wilkins, B. M. Kariuki, H. Wadepohl, L. H. Gade, A. S. K. Hashmi, D. W. Stephan and M. M. Hansmann, Organometallics, 2015, 34, 4127 CrossRef CAS.
- (a) M. M. Hansmann, R. L. Melen, F. Rominger, A. S. K. Hashmi and D. W. Stephan, Chem. Commun., 2014, 50, 7243 RSC; (b) M. M. Hansmann, R. L. Melen, F. Rominger, A. S. K. Hashmi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 777 CrossRef CAS PubMed.
- R. L. Melen, M. M. Hansmann, A. J. Lough, A. S. K. Hashmi and D. W. Stephan, Chem. – Eur. J., 2013, 19, 11928 CrossRef CAS PubMed.
- R. L. Melen, Chem. Commun., 2014, 50, 1161 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- A. Stanger, J. Org. Chem., 2006, 71, 883 CrossRef CAS PubMed.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839 RSC.
- H. Chen, R. Caughey, R. Liu, M. Mcmills, M. Rupp, W. H. Myers and W. Dean Harman, Tetrahedron, 2000, 56, 2313 CrossRef CAS.
- (a) A. S. K. Hashmi, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247 CrossRef CAS PubMed; (b) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, Adv. Synth. Catal., 2010, 352, 971 CrossRef CAS; (c) A. S. K. Hashmi, A. M. Schuster, S. Gaillard, L. Cavallo, A. Poater and S. P. Nolan, Organometallics, 2011, 30, 6328 CrossRef CAS.
- J. V. N. Vara Prasad, K. S. Para, E. A. Lunney, D. F. Ortwine, J. B. Dunbar, D. Ferguson, P. J. Tummino, D. Hupe and B. D. Tait, J. Am. Chem. Soc., 1994, 116, 6989 CrossRef CAS.
- T. Yao and R. C. Larock, J. Org. Chem., 2003, 68, 5936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, calculations and crystallographic data of all structures. [1417004–1417005]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03340c |
| This journal is © The Royal Society of Chemistry 2016 |