
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen oxide removal over hydrotalcite-derived mixed metal oxides
Magdalena
Jabłońska
and
Regina
Palkovits
*
Chair of Heterogeneous Catalysis and Chemical Technology, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: Palkovits@itmc.rwth-aachen.de
First published on 7th August 2015
Abstract
Rigorous regulations of nitrogen oxide emissions require the development of technologies for their removal from exhaust gases. Implementation of appropriate catalysts can potentially promote NOx (NO, NO2) or N2O removal in shorter reaction time and under milder operation conditions. However, several challenges have to be faced upon trying to address nitrogen oxide pollution with catalytic systems such as sufficient catalytic performance, suitable operational temperatures and catalyst poisoning. The flexible structure of hydrotalcite-like compounds offers the opportunity to introduce various metals into the materials to provide active and selective catalysts for NOx and N2O removal. This minireview summarizes the abatement of nitrogen oxides by using hydrotalcite-derived mixed metal oxides. At first, a brief overview on the general features of hydrotalcite-originated mixed metal oxides and their applications in catalysis is provided. Later on, the application of mixed metal oxides as SCR catalysts with both ammonia (NH3-SCR) and hydrocarbons (HC-SCR) as reducing agents is discussed. An overview of the mixed metal oxides applied as catalysts for NOx storage/reduction (NSR) and further in the simultaneous removal of NOx and soot particles is provided. Additionally, this review discusses mixed metal oxides as efficient catalysts for catalytic decomposition (deN2O) and selective catalytic reduction of N2O (N2O-SCR). Finally, the remaining challenges and future trends are highlighted.
 Magdalena Jabłońska | Magdalena Jabłońska received her PhD (2014) degree from the Faculty of Chemistry Technology of Jagiellonian University in Kraków and Eng. (2013) degree from the Faculty of Energy and Fuels of AGH University of Science and Technology. Since 2014, she has started her postdoctoral work with Professor R. Palkovits at RWTH Aachen, Germany. Her research focuses on environmental catalysis, specifically on diesel after-treatment systems. |
 Regina Palkovits | Regina Palkovits is a full professor of Heterogeneous Catalysis & Chemical Technology at RWTH Aachen University. She graduated with a degree in Chemical Engineering from the Technical University of Dortmund in 2003 and carried out her PhD under the supervision of Prof. Ferdi Schüth at the Max-Planck-Institut für Kohlenforschung until 2006. Afterwards, she joined the group of Prof. Bert Weckhuysen at Utrecht University as a postdoctoral fellow. In 2008, she returned as a group leader to the Max-Planck-Institut für Kohlenforschung and since 2010, she has been a professor at RWTH Aachen University. |
Introduction
The National Emission Ceilings Directive (NECD) recognizes four main air pollutants including nitrogen oxides (NOx), sulphur oxides (SOx), non-methane volatile organic compounds (NMVOc), and ammonia (NH3).1 Nitrogen oxides, NOx = NO + NO2, as major air pollutants bring about a series of environmental issues, including photochemical smog, acid rain and ozone depletion,2,3 as well as global warming caused by N2O.4,5 Above 40% of the total NOx released into the atmosphere within European Union countries comes from mobile sources with major contribution from the use of diesel engines.6,7 Emission of N2O from nitric acid plants, besides its emission from adipic acid installations, is the largest among industrial sources.8 A significant contribution of mobile sources, including diesel engines, to N2O emissions cannot be also neglected.9,10 Several advanced options of NO, NO2 and N2O abatement are available nowadays. The most promising technologies are described in the next chapters.Diesel engines operate under lean-burn conditions with an air-to-fuel ratio of A/F = 20–25. These conditions enable efficient fuel combustion saving 30–35% fuel consumption associated with decreased CO2 emissions.11 However, in the presence of excessive O2, NOx cannot be efficiently reduced over the classical three-way catalyst. However, nitrogen oxide exhaust emissions can be removed via (i) direct decomposition of NOx,12 (ii) selective catalytic reduction of NOx based on urea-SCR (NH3-SCR) or hydrocarbon-SCR (HC-SCR),13,14 (iii) NOx storage/reduction (NSR)15 and simultaneous NOx–soot removal.16 Among the proposed methods for N2O emission abatement, catalytic decomposition (deN2O)4,17 and selective catalytic reduction of N2O (N2O-SCR)18 seem to be the most promising approaches.
A great number of scientific publications relate to hydrotalcite-derived mixed metal oxides as catalysts for the processes mentioned above. Hydrotalcite-like compounds belong to a class of natural and synthetic anionic clays19 and are described with the general formula M1−x2+Mx3+(OH)2(An−)x/n·mH2O, where M2+ is a bivalent metal ion (e.g. Mg2+, Ni2+, Zn2+, and Cu2+), M3+ is a trivalent metal ion (e.g. Al3+, Ga3+, Fe3+, and Cr3+), An− is an interlayer anion (e.g. Cl−, F−, CO32−, Cr2O72−, Mo7O246−, and V10O286−) and x represents the molar fraction of M3+ per total metal, with a value varying in the range of 0.17–0.50.20,21 Synthetic materials are prepared by various methods such as (i) induced hydrolysis,22 (ii) rehydration/reconstruction,23 (iii) sol–gel,24 or (iv) hydrothermal methods.25 However, (v) coprecipitation at low supersaturation and at constant pH 7–10 is the most often applied synthesis procedure, due to its simplicity, repeatability and the associated production of hydrotalcite-like compounds with a high degree of crystallinity.19,26 Detailed information on the structural features of hydrotalcite-like compounds, synthesis methods, and their characterization is available in several comprehensive reviews.19,26–33 Hydrotalcite-like compounds are widely used in catalysis, although they are employed as a precursor of the catalyst more often than they are applied as layered materials themselves.19,32,34–40 In fact, calcined hydrotalcite-like compounds containing copper and/or cobalt have been found to be active and selective catalysts for SOx, NOx and N2O removal.41–44 In particular, Cu(Co)–(Mg)–AlOx materials were used as catalysts for removal of (i) NO and SO2 formed in the regenerator of a fluid catalytic cracking (FCC) unit45,46 and also (ii) other N-containing compounds such as ammonia in NH3-SCO47–49 or dimethylformamide in total oxidation.50 The examples presented above show mainly applications of calcined hydrotalcite-like compounds containing elements with redox behaviour within brucite-like layers (Fig. 1). However, materials prepared by exchange with anionic metal precursors of the desired metal in the interlayer space of the hydrotalcite-like compounds or by deposition of inorganic or organometallic precursors on calcined oxides were also applied.41,47 The variety of preparation methods, together with a broad range of ions, which can be incorporated, makes hydrotalcite-like compounds excellent precursors of high performance catalysts.
 | ||
| Fig. 1 Simplified representation of the main routes leading to the formation of supported metal catalysts from hydrotalcite-like precursors (adapted from ref. 35, copyright with kind permission from Springer Science and Business Media). | ||
Selective catalytic reduction of NOx by NH3
The selective catalytic reduction of NOx by ammonia (NH3-SCR) is the most important and well-established process used to abate NOx from stationary sources, according to the following reactions (eqn (1) and (2)):17,51| 4NO + 4NH3 + O2 → 4N2 + 6H2O | (1) |
| 2NO2 + 4NH3 + O2 → 3N2 + 6H2O | (2) |
This method is also used for removing NOx from diesel exhausts, so-called diesel exhaust fluid (DEF), commonly referred to as AdBlue in Europe.52 In this technique, NOx is continuously reduced by NH3 on commercial V2O5–WO3/TiO2 catalysts suitable for temperatures of 250–400 °C.53,54 On the other side, automotive application of this process calls for high NOx removal over a much wider temperature range up to 600 °C in the cycle of diesel particulate filter regeneration (DPF). Additionally, there is a clear trend to replace V-based catalysts. The major reasons are the narrow operating temperature window of the commercial catalysts, phase transformation of anatase to rutile under the reaction conditions, high activity for oxidation of SO2 to SO3 and vanadium pentoxide toxicity.55 Different types of NH3-SCR catalysts, such as (supported) metal oxides, layered clays, mesoporous silicas and zeolites containing various transition metals, e.g. Fe, V, Cr, Cu, Co and Mn, have been studied in the scientific literature.56–58 Several reviews provide detailed information on the mentioned catalytic systems.2,54,59–61 Among the tested catalysts, layered clays, including hydrotalcite-originated mixed metal oxides with various compositions such as Cu–Mg–Al, Co–Mg–Al, and Mg–Fe, compose a large group.47,48Table 1 summarizes the catalytic performance of mixed metal oxides for the selective reduction of nitric oxide by ammonia. Analysis of these results shows that the catalytic systems consisting of copper and/or iron resulted in high activity in NH3-SCR. In addition to data presented in Table 1, suitable catalytic systems also require a wide temperature window of effective operation. The following description covers a detailed overview of the most interesting systems compared to commercial V2O5–WO3/TiO2 as well as copper-zeolites.
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | NO conversion/% (temperature/°C) | Ref. |
|---|---|---|---|---|
| a Studies were also presented below.64,65 | ||||
| V2O5–WO3/TiO2 | Commercial | 0.75% NO; 0.6% NH3; 3.0% O2; He balance; W/F = 240 g s l−1 | 100 (350) | 41 |
| Cu-BEA (1.0 wt% Cu) | Two-step post-synthesis | 0.25% NO; 0.25% NH3; 2.5% O2; He balance; W/F = 300 g s l−1 | 100 (300) | 62 |
| Impregnation | ||||
| 100 (250) | ||||
Mg–Al (70.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29.3 mol%) :29.3 mol%) |
Coprecipitation (470) | 0.2% NO; 0.2% NH3; 3.0% O2; He balance; W/F = 9 g s l−1 | 71 (450) | 63 |
| Mg–Cu–Al (Mg:Al = 67.9:32.1 mol%; 7.2 wt% Cu) |
79 (400) | |||
| Ce/Mg–Al–Fe (Mg:Fe:Al = 4.0:0.8:1.0 mol%; 4.1 mass% Ce) |
Coprecipitation/reconstruction (550/550) | 0.2% NO; 0.2% NH3; 3.0% O2; He balance; GHSV = 185000 h−1 |
71 (350) | 64 |
| Fe/Mg–Al–Fe (Mg:Fe:Al = 4.0:0.8:1.0 mol%; 34.7 mass% Fe) |
82 (350) | |||
| Fe–PW12–clay (composition not shown; 5.0 wt% Fe) | Hydrothermal–anion exchange/impregnation (500–500/500) | 0.1% NO; 0.1% NH3; 2.0% O2; He balance; W/F = 24 g s l−1 | 40 (450) | 65 |
| Mg–Cu–Fe (2.0:0.5:1.0 mol%) |
Coprecipitation (600) | 0.25% NO; 0.25%; 2.5% O2; He balance; W/F = 150 g s l−1 | 80 (300) | 48 |
| Cu/Mg–Al (Mg:Al = 71.0:29.0 mol%; 10.0 wt% CuO) |
Coprecipitation/impregnation (650/600) | 0.75% NO; 0.6% NH3; 3.0% O2; He balance; W/F = 240 g s l−1 | 80 (300) | 41 |
| Mg–Cu–Al (63.8:7.2:29.0 mol%; 12.5 wt% CuO) |
Coprecipitation (650) | 100 (350) | ||
| Mg–Cu–Al (51.0:20.0:29.0 mol%) |
Coprecipitation (600) | 0.25% NO; 0.25% NH3; 2.5% O2; He balance; W/F = 300 g s l−1 | 95 (250) | 66 |
| Mg–Cu–Co–Al (51.0:10.0:10.0:29.0 mol%) |
85 (250) | |||
Carja and Delahay63 worked on Mg(Cu)–Al–Ox derived from oxovanadate-pillared hydrotalcite-like compounds. The catalysts were prepared by coprecipitation using aqueous solutions of appropriate metal nitrates and NaVO3 as the precipitating agent. Both V2O74− and HV2O73− were indicated to predominate in the aqueous vanadate solution. Calcination of the prepared materials at 470 °C led to the formation of amorphous species with weak and broad maxima revealed by X-ray diffraction. The materials were utilized in catalytic tests in the range of 250–500 °C. Mg–Al–Ox (Mg:Al = 70.7:29.3) facilitated above 70% NO conversion at 450 °C with very low selectivity towards N2O. The presence of copper (7.2 wt%) in the structure lowered the maximum conversion temperature with 79% conversion at 400 °C. The tested material series exhibited promising N2 selectivity. Unfortunately, no stability tests of such materials were provided. Based on BET measurements and microscopic analysis (SEM, TEM), the authors suggested that the mesoporous properties of the materials could enhance transport of the reactants to the active surface sites consequently facilitating the apparent catalytic performance. However, detailed structure–performance correlations are not available hindering interpretation of the obtained results. Studies correlating catalytic performance to physico-chemical properties, e.g. to gain knowledge on the role of copper oxide species in such systems, could pave the way for knowledge-driven catalyst optimization. Unfortunately, most investigations published in this area only focus on catalyst composition–performance correlations of materials tested for NH3-SCR. Other works were also presented below.64,65
Carja et al.64 used a reconstruction method to obtain Fe2O3 or CeO2 deposited on a Mg–Al–Fe–O support (Mg:Al:Fe = 4.0:0.8:1.0). Reconstruction of the structure of calcined iron-substituted hydrotalcite-like compounds occurred by modification with aqueous solutions of appropriate iron or cerium sulphates. After calcination at 550 °C, the prepared materials were used for NH3-SCR. The presence of CeO2 (4.1 mass% of Ce) on the surface of the mixed metal oxides improved the catalytic performance only below 250 °C. Above this temperature, Fe2O3 (34.7 mass% of Fe) doped samples revealed higher conversion. No further information of the effect of iron and cerium oxide doping on the material properties was provided.
Wongkerd et al.65 studied as well iron-modified mixed metal oxides. Deoxycholate- and Keggin-type polyoxometalate-pillared hydrotalcite-like compounds were prepared by applying hydrothermal and anion exchange methods consecutively. In the first step, deoxycholate-pillared hydrotalcite-like precursors were prepared by treatment of aqueous solutions of Mg(OH)2, Al(OH)3, deoxycholic acid and NaOH in an autoclave at 150 °C for 48 h. The obtained product was used to prepare both PW12O40- and SiW12O40-pillared hydrotalcite-like compounds, employing aqueous solutions of the appropriate heteropoly acid and sodium hydroxide. However, detailed elemental compositions of all obtained hydrotalcite-like compounds were not provided. Calcination of the obtained samples in the temperature range of 250–500 °C allowed access to materials with different phase compositions and crystallinity. The impact of phase changes of the catalysts on the activity in NH3-SCR was investigated. Amorphous mixed oxides appeared after calcination at 500 °C and facilitated higher catalytic activity in NH3-SCR compared to samples calcined at lower temperatures. All catalysts revealed NO conversions below 35%. However, N2 selectivity above 99% could be reached in the whole temperature range studied. Further impregnation with 5.0 wt% Fe only slightly enhanced NO conversion while N2 selectivity remained constant. The authors suggest the iron modification to increase the Brönsted acidity of the pillared-clay catalysts. However, no further details related to this point were provided. Polyoxotungstophosphate-pillared hydrotalcite-originated mixed metal oxides doped with iron achieved a maximum NO conversion of 40% at 450 °C. Higher catalytic activity with around 70% conversion of NO at 450 °C could be achieved with calcined Fe-containing hydrotalcite-like compounds prepared by coprecipitation with carbonate as the interlayer anion. For Mg–Fe (Mg:Fe = 2.0:1.0) precursors calcined at 600 °C, X-ray diffraction confirmed the presence of MgO and MgFe2O4.48 Introducing 0.5 mol% copper into the Mg–Fe–O material led to an additional formation of Cu2O and CuO, and significantly improved the catalytic activity under the applied reaction conditions. Around 80% NO conversion was reached at 300 °C, together with a N2 selectivity of above 85% over the whole investigated temperature range. Copper loadings of 0.5 to 1.0 mol% were associated with increased crystallinity of both copper oxide phases (Cu2O and CuO). However, higher copper loadings did not influence catalytic activity, and only slightly lowered the selectivity to N2, which was the main reaction product. The changes in NOx conversion could be correlated with changes in the reducibility of the catalysts. The presence of easily reducible copper oxide species in the Mg–Fe–O system extended the low temperature NOx conversion. On the other hand, it led to lower NOx conversion at higher temperatures due to competition with undesired NH3 oxidation. Increasing copper loadings from 0.5 to 1.0 mol% led to the formation of more aggregated copper oxide species, and thus slightly higher NOx conversion in the high temperature region, i.e. above 350 °C, was recorded. Based on these results, one may conclude that by varying the amount of copper and iron in such catalysts and consequently adjusting the reducibility of the catalysts, it is possible to control the NOx conversion temperature window. Further studies in this direction appear promising.
The influence of the transition metal content was also comprehensively investigated over Cu–(Co)–Mg–Al–Ox obtained by calcination of hydrotalcite-like precursors at 600–650 °C.41,66 Montanari et al.41 studied Cu–Mg–Al–Ox with copper oxide loadings in the range of 4.0–12.5 wt%. Such materials were prepared by coprecipitation followed by calcination at 650 °C. Only poorly crystallized MgO was identified in all samples, without evidence of CuO segregation. As a reference, copper oxide deposited on Mg–Al–Ox (Mg:Al = 71.0:29.0) materials was prepared by impregnation aiming at a loading of 10.0 wt% CuO. Crystalline CuO was clearly evident in this case. Catalytic tests with these materials confirmed complete NO conversion at 350 °C with a broad temperature range of effective operation over a Mg–Cu–Al material with 12.5 wt% copper oxide. Lower contents of copper oxide (4.0 and 8.0 wt%) resulted in significantly lower catalytic performance. Additionally, a catalyst prepared by deposition of copper oxide on a calcined support (Cu/Mg–Al) was compared with mixed metal oxides including copper incorporated within the structure (Mg–Cu–Al). Mg–Cu–Al revealed significantly higher catalytic performance in the studied temperature range of 150–500 °C. Additionally, such catalysts showed no deactivation either after four consecutive cycles or during the stability test at 380 °C for 8 h under the reaction conditions. Again, the obtained NOx conversion could be correlated with the reducibility of the tested catalyst (Fig. 2). All hydrotalcite-derived mixed metal oxides showed a reduction peak with a maximum in the temperature range of 240–300 °C, while the CuO-supported catalysts exhibited a significantly lower reduction temperature, i.e. 200 °C. The authors suggest that highly reducible copper oxide species were responsible for the high catalytic performance in the low temperature range, but also significantly contributed to the side-reaction – ammonia oxidation at high temperatures. Therefore, tuning of the nature of copper oxide species and the right copper content seem to be vital factors in order to obtain highly efficient catalytic systems operating in a wide temperature range.
 | ||
| Fig. 2 Results of temperature-programmed reduction (H2-TPR) performed for Mg–Cu–Al–Ox ((a) 12.5 wt%, (b) 8.0 wt% and (c) 4.0 wt% CuO) and Cu/Mg–Al–Ox ((d) 10.0 wt%) (adapted from ref. 41 with kind permission from Elsevier). | ||
Chmielarz et al.66 carried out similar studies with regard to the catalytic performance of Mg–Cu–Al catalysts with 5.0–20.0 mol% content of copper incorporated into the structure of the precursors. Hydrotalcite-like precursors were prepared by coprecipitation, followed by transformation at 600 °C into only poorly crystallized MgO. Catalytic tests over these materials revealed around 95% NO conversion at 250 °C for catalysts with high concentration of copper (Fig. 3). Unfortunately, the authors reported a very narrow temperature range. Materials with copper contents of only 5.0 or 10.0 mol% exhibited lower activity, with slight changes in selectivity to N2 in the range of 200–350 °C. Besides the content of transition metals, the selected element, e.g. Cu and/or Co, also has strong influence on the catalytic performance.66 The catalytic activity decreases in the following order: Cu > Cu–Co > Co. The highest NO conversion was observed for metal oxides containing copper. Cobalt-based catalysts exhibited poor activity, while an increasing content of Co from 5.0 to 20.0 mol% resulted in lower activity but higher selectivity to N2. The obtained results fully corresponded to H2-TPR analysis of copper-containing materials as well as the cobalt-based sample with 10.0 mol% loading presented in another work of Chmielarz et al.67 In order to determine the interaction of NO and NH3 molecules with the catalyst surface, temperature-programmed methods (TPD, TPSR, and stop flow-TPD) as well as FT-IR were applied. The introduction of copper within brucite-like layers caused the formation of weak nitrogen oxide sorption centres on the catalyst surfaces, which played a crucial role in the catalytic process. The weakly chemisorbed nitrogen oxide was nearly completely transformed into nitrogen in the low temperature region. Moreover, the FT-IR results revealed the formation of thermally stable nitrite and/or nitrate species on the catalyst surface at low temperatures. Such species resulted in N2O formation at higher temperature.66
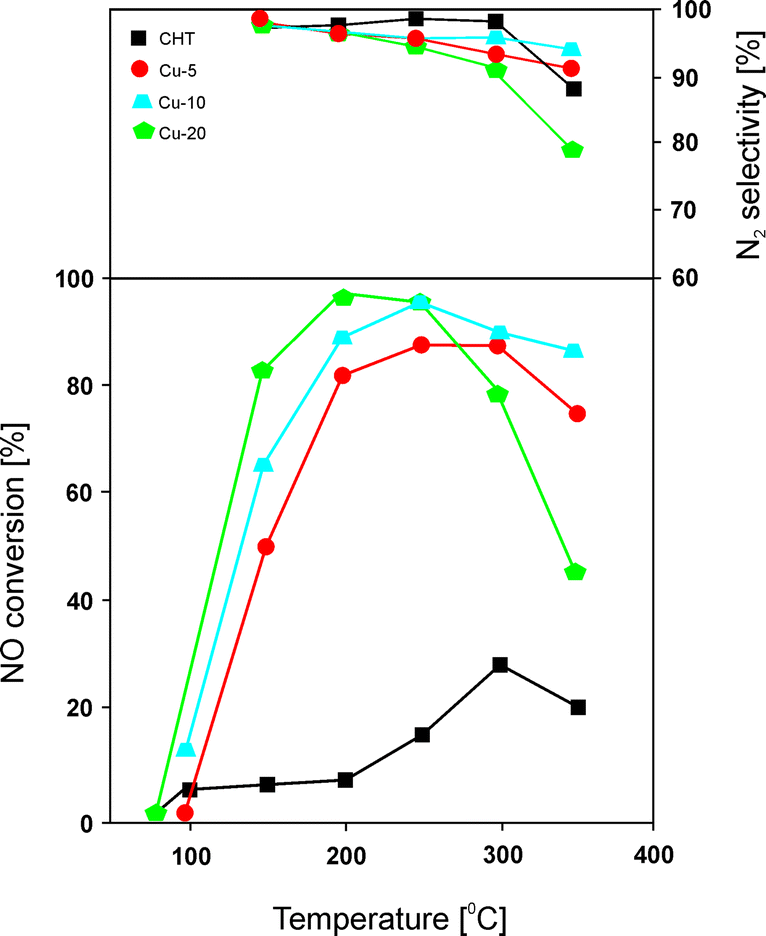 | ||
| Fig. 3 Results of catalytic tests performed for Cu–Mg–Al–Ox. Reaction conditions: 0.25% NO, 0.25% NH3, 2.5% O2, He balance; total flow rate = 40 ml min−1; mass of catalyst = 200 mg (adapted from ref. 66 with kind permission from Elsevier). | ||
In conclusion, a comparison of the catalytic activity of a Mg–Cu–Al–O system with 12.5 wt% CuO to V2O5–WO3/TiO2 as a commercial reference revealed a similar behaviour of both catalysts up to 380 °C. At higher temperatures, the commercial catalyst exhibited superior catalytic performance with 100% NO conversion up to 500 °C. Thus, the advantage of commercial catalysts over mixed metal oxides is the very broad temperature range of effective operation. The selectivity to N2 was slightly better than that for commercial catalysts over the whole temperature range of 100–500 °C.41 Therefore, the results confirmed the strong potential of Mg–Cu–Al–Ox as catalysts in mobile applications, and opened opportunities to improve the catalytic performance of the presented materials. Further studies and stability tests in the presence of typical diesel exhaust gases for this application such as H2O, COx and SOx are still required. Moreover, limited efforts have been made to investigate the material properties or detailed reaction mechanisms over the presented hydrotalcite-originated mixed metal oxides.
Furthermore, a comparison of copper-containing mixed metal oxides together with copper-exchanged zeolites under the same reaction conditions was possible based on the contributions by Chmielarz et al.62,66 Copper-containing BEA catalysts were prepared by a two-step post-synthesis method and conventional wet impregnation. The nature and environment of copper present in the obtained materials were studied by DR UV-vis spectroscopy. Isolated Cu2+ species in pseudo-tetrahedral coordination were present in the materials prepared by the two-step post-synthesis procedure, while different kinds of mononuclear Cu2+ species existed in the wet impregnated sample.62 In comparison, copper in the mixed metal oxides were reported to be present e.g. as mononuclear Cu2+ ions, oligomeric [Cu–O–Cu]n species and bulky CuO crystallites.47 The reducibility of copper oxide species in zeolites was investigated using H2-TPR. The results were in good agreement with the observed catalytic performance. Additionally, it was recognized that the reducibility of metal ions in metal-exchanged zeolites determines the extent of low temperature NO conversion.68 The same species are responsible for ammonia oxidation as the dominating process at higher temperatures. In particular, above 80% NO conversion in the range of 225–525 °C and above 95% selectivity to N2 were achieved over Cu-BEA prepared by impregnation. The data suggest significantly better catalytic performance of copper-zeolites compared to hydrotalcite-derived mixed metal oxides. This conclusion is supported by the results given by Sultana et al.,69 who reported above 80% NO conversion in the range of 250–450 °C and above 97% N2 selectivity in the presence of water vapour and sulphur oxide over (2.4 wt%) Cu-ZSM-5 prepared by an aqueous ion-exchange method. The authors confirmed the primary role of the reducibility of the catalyst compared to their acidity in NH3-SCR. In contrast, a dominant role of acidity was confirmed in the case of HC-SCR.
Selective catalytic reduction of NOx by HCs
The selective catalytic reduction of NOx by hydrocarbons (HC-SCR) is another technology used to remove NOx from the exhaust of lean-burn gasoline engines.53 Attention has been paid to replacing ammonia as a reducing agent with hydrocarbons, due to problems of storage, leakage and transport of liquid ammonia. Ammonia emissions are strictly limited in regulations to 2–10 ppm as a typical NH3 slip threshold limit.70,71 Starting from the work of Iwamoto et al.72 over Cu-ZSM-5 with outstanding activity in HC-SCR at 300 °C, extensive studies have been conducted focusing on Cu-based catalysts for this process.73–75 In view of the considerable research efforts, there is still an open discussion about the roles of copper species coexisting in both the surface and bulk in HC-SCR.76–78Besides copper-based materials, a large number of other types of transition metal ion-exchanged zeolites including Co, Fe, Pt, and Ag79,80 and supported Pt, Pd or Rh14,81 systems have been evaluated. Many hydrotalcite-originated mixed metal oxides revealed promising activity as catalysts for selective reduction of NOx with hydrocarbons.82–84Table 2 summarizes the catalytic performance of mixed metal oxides for the selective reduction of nitric oxide by hydrocarbons. Similar to data collected for NH3-SCR, also in the case of HC-SCR, the maximum conversion of NO was provided for the presented catalytic systems. This representation of the catalytic activity only allows a general overview of the material performance. The most interesting copper-based materials are discussed in more detail.
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | NO conversion/% (temperature/°C) | Ref. |
|---|---|---|---|---|
| Cu-ZSM-5-152 (152/degree of exchange) | Ion-exchange | 0.1% NO; 0.1% C3H6; 10.0% O2; W/F = 300 g s l−1 | 82 (300) | 72 |
| Mg–Al (3.0:1.0 mol%) |
Coprecipitation (800) | 0.1% NO; 0.1% C3H6; 10.0% O2; He balance; W/F = 120 g s l−1 | 16 (600) | 82 |
| Homogeneous precipitation (800) | 40 (550) | |||
| Cu–Ti | Homogeneous precipitation (450) | 0.1% NO; 0.1% C3H6; 10.0% O2; He balance; W/F = 180 g s l−1 | 83 | |
| (2.0:1.0 mol%) |
74 (270) | |||
| (3.0:1.0 mol%) |
76 (260) | |||
| Cu–Al | Coprecipitation (600) | 0.06% NO; 0.06% C3H6; 8.0% O2; He balance; W/F = 150 g s l−1 | 75 | |
| (3.0:7.0 mol%) |
70 (300) | |||
| (4.0:6.0 mol%) |
55 (300) | |||
| Co–Al (0.5:1.0 mol%) |
Coprecipitation (550) | 0.13% NO; 2.0% O2; N2 balance (* with 0.08% C3H8) (** with 0.11% C3H6); GHSV = 11200 h−1 |
*60 (300) | 84 |
| **88 (400) | ||||
| Ni–Al (0.5:1.0 mol%) |
*70 (300) | |||
| **100 (400) | ||||
| Cu–Co–Fe (2.0:8.0:5.0 mol%) |
Coprecipitation (500) | 0.06% NO; 0.06% C3H6; 12.0% O2; 6.0% CO2; 7.0% H2O; He balance; W/F = 72 g s l−1 | 32 (350) | 85 |
| Ni–Al (6.0:2.0 mol%) |
Coprecipitation (450) | 265.0 mbar NO; 265.0 mbar CH4; 27.0 mbar O2; W = 0.25 g | 100 (400) | 86 |
Most catalytic tests were conducted in the presence of propene. In this line, the reaction can be generalized as follows (eqn (3)):
| 2NO + 2C3H6 + 8O2 → N2 + 6H2O + 6CO2 | (3) |
Yuan et al.82 studied the influence of the preparation method of Mg–Al–Ox (Mg:Al = 3.0:1.0) on the acidic properties, and consequently the catalytic activity. For coprecipitation of hydrotalcite-like compounds, aqueous solutions of suitable metal nitrates together with sodium carbonate as a precipitating agent were utilized. Homogeneous precipitation relied on urea as the base retardant. Mixed solutions obtained via homogeneous precipitation were conditioned in different ways: (i) they were transferred into an autoclave and treated at 90 °C for 24 h (hydrothermal method) or (ii) additionally stirred at 90 °C for 24 h before hydrothermal treatment (homogeneous precipitation with hydrothermal treatment). All obtained samples were calcined at 800 °C. MgAl2O4 dominated in the case of samples prepared by homogeneous precipitation and homogeneous precipitation with hydrothermal treatment. Besides the spinel phase, crystalline MgO appeared in hydrothermally treated samples. Mainly magnesium oxide was found in materials obtained by coprecipitation. Activity tests concerning NO reduction by propene revealed 40% NO conversion at 550 °C over the catalysts prepared by homogeneous precipitation. Comparison of the results for all tested materials emphasizes the following order of decreasing catalytic activity despite having comparable composition: homog. precip. > homog. precip. + hydrotherm. treatm. > hydrotherm. treatm. > coprecip. Between 350–650 °C, N2 was the main reaction product. The catalytic activity of the obtained materials was affected by the amount of Lewis acid sites, i.e. higher acidity induced higher catalytic performance (Fig. 4). NH3-TPD measurements revealed significant differences of the desorption profiles. The data confirmed that the preparation methods significantly affected the acidic properties of the calcined catalysts. The catalysts synthesized by homogeneous precipitation were favourable for the formation of the highest quantity of Lewis acid sites among the obtained samples.
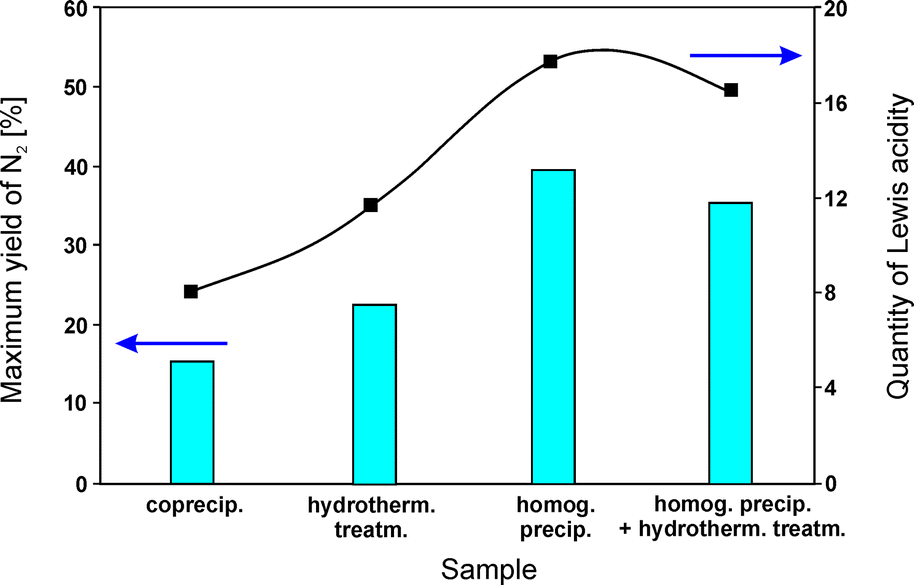 | ||
| Fig. 4 Variations of the quantity of Lewis acidity and catalytic performance of calcined hydrotalcite-like precursors synthesized by different methods (adapted from ref. 82 with kind permission from Elsevier). | ||
Thus, homogeneous precipitation was also applied in further studies over a series of Cu–Ti–Ox materials with Cu:Ti molar ratios of 2.0–5.0:1.0, however, calcined at 450 °C.83 The relatively low temperature treatment did not fully transform the Cu–Ti precursors into mixed metal oxides. In the calcined materials, hydrotalcite-like compounds as well as Cu3TiO4 appeared. For materials with the highest copper loading (Cu:Ti = 4.0–5.0:1.0), the presence of CuO could be confirmed. The superior catalytic performance of Cu–Ti–Ox (Cu:Ti = 3.0:1.0, 43.0 wt% Cu) (Fig. 5) in NO reduction with C3H6 was mainly ascribed to the presence of crystalline Cu3TiO4. Additionally, stability tests over 720 min under the reaction conditions revealed no deactivation of this catalyst or the other obtained samples. The authors concluded that in HC-SCR, surface copper species were more active than bulk copper species. In particular, H2-TPR analysis indicated that the reduction temperature of surface Cu2+ corresponded to the optimal reaction temperature for high catalytic activity. Another factor that influenced the catalytic performance was the quantity of Lewis acid sites. Again, a linear correlation between the quantity of Lewis acidity and the catalytic performance was reported. NO conversion over Cu3Ti1 reached 76% at 260 °C with around 75–85% N2 selectivity in the temperature range of 260–290 °C. Therefore, the material presents a very promising catalytic system for low temperature HC-SCR. The pathway of C3H6-SCR over the obtained catalysts was investigated using FT-IR under the reaction conditions. Based on these results, the authors concluded that the adsorbed nitrates (products of NO oxidation by O2) reacted with acetate and formate (products of partial oxidation of propene) to yield N2, CO and H2O. The nitrates, acetate and formate were found to be essential intermediates for SCR of NO by C3H6. A higher concentration of acetate on Cu3Ti1 could be correlated with its higher catalytic performance.
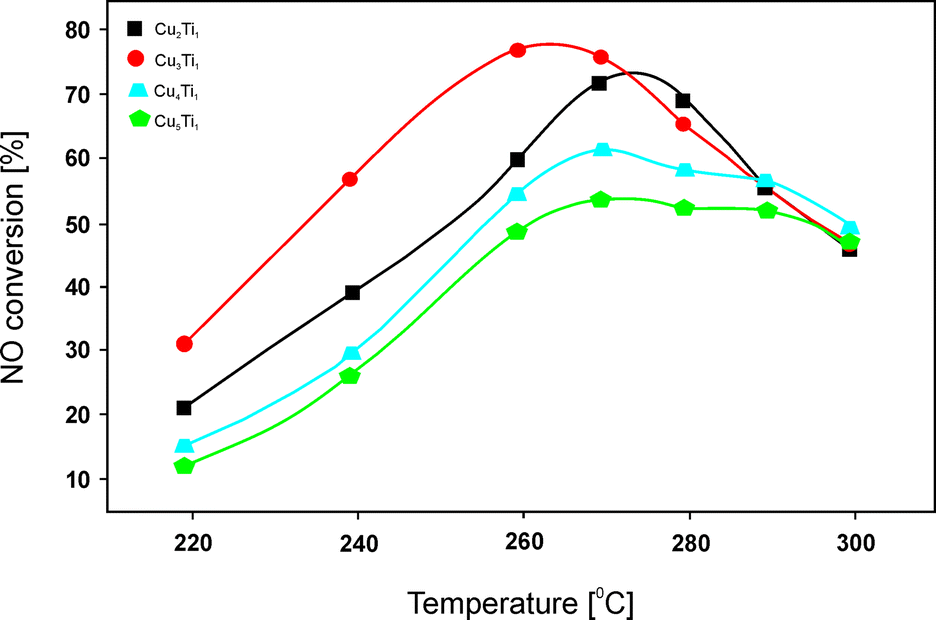 | ||
| Fig. 5 Results of catalytic tests performed for Cu–Ti–Ox. Reaction conditions: 0.1% NO, 0.1% C3H6, 1.0% O2, He balance; total flow rate = 100 ml min−1; mass of catalyst = 300 mg (adapted from ref. 83 with kind permission from Elsevier). | ||
Concerning other mixed metal oxides, Kumar et al.75 studied materials with various ratios of Cu:Al in the range of 1.0–7.0:3.0–9.0. Mg–Al–Ox was prepared by coprecipitation using aqueous solutions of the appropriate metal nitrates together with Na2CO3 and NaOH, followed by subsequent calcination at 600 °C. Amorphous structures with weak and broad reflections of γ-Al2O3 were identified in all calcined samples. Only in the case of high copper loadings (Cu:Al = 7.0:3.0, 41.7 wt% Cu), additional CuO appeared. Formation of surface CuAl2O4 for all samples was also proven using X-ray photoelectron spectroscopy. Interestingly, for Cu–Ti catalysts, high copper loadings of above 40.0 wt% resulted in the most efficient catalysts,83 while for Cu–Al samples, lower copper concentration facilitated superior catalytic performance. Among all tested materials, NO reduction with propene was highly efficient over Cu–Al–Ox with a Cu:Al molar ratio of 3.0:7.0 (13.7 wt% Cu) and reached 70% at 300 °C. Unfortunately, the catalyst only exhibited high activity in a limited temperature range. Information on the stability of this system was not included. For higher copper contents, NO conversion decreased, which was in line with the decreasing intensity of copper in CuAl2O4. For a Cu:Al molar ratio of 7.0:3.0, around 30% conversion (300 °C) was achieved, resembling the lowest NO conversion obtained for the series of tested materials. Therefore, CuAl2O4 was suggested to be a vital factor for obtaining high efficiency in C3H6-SCR. Such a crucial role could not be identified for highly dispersed surface and bulk CuO.
All studies mentioned above utilized C3H6 as the reducing agent of NO. The effect of other reducing agents in HC-SCR was examined by Tret'yakov et al.84 Co–Al–Ox and Ni–Al–Ox with different compositions (M:Al = 0.5–3.0:1.0) were prepared by coprecipitation followed by calcination at 550 °C. Co3O4 and NiO as crystalline phases were found only in samples containing cobalt and nickel, respectively. The materials were investigated in HC-SCR applying C3H6, C3H8 as well as n-C10H22. Among the tested reducing agents, propene proved to be the most efficient one for NO reduction, while materials with a Ni(Co):Al molar ratio of 0.5:1.0 achieved the highest catalytic activity. In particular, Ni–Al–Ox and Co–Al–Ox reached a NO conversion of 100 and 88% at 400 °C, respectively. Unfortunately, below this temperature, both catalysts only exhibited low NO conversions. The high catalytic activity of such samples was suggested to be related to high dispersion of Ni- or Co-containing mixed oxides resulting in a large fraction of active surface sites. The small crystallite size in the case of nickel-containing samples, i.e. below 4 nm, compared to that of cobalt-based samples (5 nm) was associated with the higher catalytic performance between these two samples. These studies present ideal starting points using hydrotalcite-derived mixed metal oxides, but do not provide detailed structure–performance correlations. Incorporating both elements within brucite-like sheets controlling the metal particle size could enhance the catalyst activity. Additionally, the stability of these systems and the detailed reaction pathways of HC-SCR require investigations facilitating knowledge-driven design of the catalysts.
Further studies compared C3H6 and C8H18 as reducing agents in NO reduction over Cu–Co–Fe materials as catalysts.85 The material precursors with different molar ratios (Cu:Co:Fe = 1.0–2.0:1.0–8.0:1.0–5.0) were prepared by coprecipitation and transformed into mixed metal oxides at 500–800 °C. After calcination at 500 °C, the materials possessed crystalline CuFe2O4 and/or CoFe2O4 phases. For calcination temperatures up to 800 °C, crystallinity increased further and Co3O4 and CuO were formed, respectively. Utilizing catalysts with a Cu:Co:Fe molar ratio of 1.0:3.0:2.0 and comparing propene and octane as reductants, superior catalytic activity in the presence of propene could be confirmed. Additionally, catalytic tests revealed higher NO conversion for materials calcined at 500 rather than 800 °C. Among all combinations, materials with the highest concentrations of transition metals (Cu:Co:Fe = 2.0:8.0:5.0) also achieved the highest NO conversion up to 32% at 350 °C. Other materials only reached activity below 20%. Although these data seem to point to relatively low activity, one has to note that these catalytic experiments were conducted in the presence of 7.0 wt% H2O. Unfortunately, catalytic data for these materials under water-free conditions were not provided. Additionally, the presented physicochemical characterization, including XRD, BET, IR and SEM analyses, was not discussed with regard to structure–performance correlations. Consequently, crucial factors influencing the catalytic activity remain unclear. Open challenges exist to explore the role of transition metals in such systems.
Other reductants such as methane were applied to NO reduction catalysed by Mg–Fe–Ox and Ni–Al–Ox with a M2+:M3+ molar ratio of 6.0:2.0.86,87 The materials were prepared by coprecipitation, followed by calcination at 450–500 °C, resulting in the formation of mixed metal oxides with the structural features of poorly crystallized MgO. These catalysts enabled complete NO removal at 400 °C. In comparison, pure NiO at this temperature facilitated only 22% NO removal under the same experimental conditions.86 Unfortunately, the presented studies only report catalytic data for one reaction temperature hampering a direct comparison to other literature systems.
Overall, the most discussed catalytic systems achieved only limited activity in a narrow operating temperature window. Among the tested materials, Cu–Ti–Ox appears promising. Concerning structure–performance correlations of Cu–Ti materials, a linear correlation of activity and the quantity of Lewis acidity of the obtained mixed metal oxides could be identified.76 Similar correlations were observed for Mg–Al–Ox.83 On the other side, the role of Brönsted acid sites remains unclear. However, some studies indicate that both Lewis and Brönsted acid sites are essential for NO reduction with hydrocarbons.88 In view of the complexity of HC-SCR with different catalysts, reductants and reaction conditions, and considering the limited number of studies, there has been no generally accepted role of copper oxide species in copper-containing hydrotalcite-derived mixed metal oxides. Therefore, further studies covering these points are certainly necessary. Moreover, optimization of such materials together with missing stability tests under the reaction conditions, with other components of waste gases, such as H2O, COx, SOx, etc., and further also under real diesel engine conditions is essential for application-oriented material development.
NOx storage/reduction
Another promising technology for NOx removal from diesel exhausts relates to NOx storage/reduction (NSR). This process is based on sequential lean–rich changes in the diesel engine. Under lean conditions (with an oxygen excess), NOx is stored on NSR catalysts as surface NO2− and/or NO3− species. Subsequently, by changing from lean to rich conditions (excess of fuel), nitrites and/or nitrates are catalytically reduced and/or decomposed to N2.15 The NSR mechanism is generally assumed to take place in five steps:89,90A) during lean-burn cycles: (i) NO oxidation to NO2, (ii) NOx adsorption as nitrites and/or nitrates on the basic adsorption sites of the catalysts, and (iii) reductant feed, and
B) during rich-burn cycles: (iv) NOx release from the catalyst, and (v) NOx reduction to N2.
Each step is critical for efficient operation. However, NO-to-NO2 oxidation is known to be an important step for NOx storage since the adsorption of NO2 is more facile than that of NO in the catalysts.90
The first generation of NSR catalysts introduced by Toyota was based on Pt–BaO/Al2O3.91 These catalysts are also referred to as lean NOx trap (LNT) or NOx adsorber catalysts (NACs) and present good activity with 0.58 mmol g−1 NO storage and 97% mean NO conversion at 300 °C.42,92 However, there are some challenges related to limited resistance to sulphur poisoning, thermal degradation, as well as formation of carbon deposits on the catalysts.91,93 In this line, sulphur dioxide-tolerant and stable NSR catalysts with comparable or higher storage/reduction performance in the process are needed. Different types of materials have been tested so far mainly consisting of a NOx storage component based on alkali or alkaline earth metals (e.g. Ca, Sr, Ba, K, and Na) and a catalytic redox component such as transition/noble metals (e.g. Cu, Co, Pt, Rh, and Pd). Both components are usually dispersed on high surface area supports (e.g. Al2O3, Al2O3–CeO2, and Al2O3–SiO2).94–96 Several reviews summarize the tested materials and their properties and provide insights concerning reaction mechanisms in the presence of such catalysts.11,89–91,97,98 Hydrotalcite-originated mixed metal oxides have been widely used as an alternative to traditional NSR catalysts due to their high NOx storage capability and good SOx tolerance, which present a critical point in the development of efficient NSR catalysts.99,100 Recently, reviews focusing on this catalyst type were published by Yu et al.101 and Jabłońska et al.102 The first article concerns mainly the activity and reaction mechanism of selected materials obtained and studied by the authors, while the second review refers only to a selected set of catalyst formulations and their activity in NSR. Therefore, we herein discuss a broad range of catalysts, their preparation and structure–performance correlations with major emphasis on the recent literature not covered in previous reviews yet. Tables 3 and 4 summarize the NOx storage capacity or mean NOx conversion of mixed metal oxides in lean/rich cycles at a given temperature. The presented systems mainly combine redox and basic sites. Indeed, redox sites are necessary to oxidize NO to NO2 and basic sites are necessary to store NO2 in the form of nitrates. Materials containing cobalt and/or calcium as well as ruthenium seem to achieve reasonable NOx storage capacity. Among the materials tested under rich/lean cycles, cobalt-containing materials doped with vanadium or palladium also seem to be possible alternatives to commercial Pt–BaO/Al2O3. On the other side, such systems are not free from drawbacks, e.g. the toxicity of cobalt.
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | NOx storage/mmol g−1 (temperature/°C) | Ref. |
|---|---|---|---|---|
| Pt–BaO/Al2O3 (1.0 wt% Pt, 20.0 wt% Ba) | Impregnation/impregnation (500/500) | 0.1% NO; 3.0% O2; He balance; saturation; W/F = 36 g s l−1 | 0.58 (300) | 92 |
| Pd/Mg–Al (7.0:3.0 mol%; 1.34 wt% Pd) |
Commercial/impregnation (600/500) | 0.05% NO; 0.05% N2; 5.0% O2; He balance; 30 min; W/F = 180 g s l−1 | 0.06 (300) | 103 |
| 0.079% NO; 8.0% O2; N2 balance; 30 min; W/F = 96 g s l−1 | ||||
| Mg–Ru–Al (90.0:1.0:29.0 mol%) |
Coprecipitation (600) | 0.22 (350) | 104 | |
| Mg–Al (3.0:1.0 mol%) |
Coprecipitation (800) | 0.13% NO; 0.01% NO2; 8.0% O2; N2 balance; 30 min; W/F = 120 g s l−1 | 0.09 (300) | 105 |
| Mg–Co–Al (1.0:2.0:1.0 mol%) |
0.20 (300) | |||
| Mg–Co–Al (1.5:1.5:1.0 mol%) |
Coprecipitation (800) | 0.08% NO; 8.0% O2; N2 balance; 30 min; W/F = 120 g s l−1 | 0.07 (300) | 106 |
| Mg–Co–Al–Ti (1.5:1.5:0.9:0.1 mol%) |
0.04 (300) | |||
| Ca–Co–Al (2.0:1.0:1.0 mol%) |
Coprecipitation (800) | 30 min 60 min 60 min 0.08% NO; 8.0% O2; He balance; W/F = 120 g s l−1 | 0.43 (300) | 101, 107 |
| 0.60 (300) | ||||
| Ca–Co–Al–La (2.0:1.0:0.9:0.1 mol%) |
0.63 (300) |
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | Mean NOx conversion/% (temperature/°C) | Ref. |
|---|---|---|---|---|
| Pt–Ba/Al2O3 (1.0 wt% Pt, 15.0 wt% Ba) | Impregnation/impregnation (500/500) | Lean (120 s): 5.0% O2; 10.8% CO2; 0.0954% NO; He balance | 97 (300) | 42 |
| Rich (6 s): 3.3% CO; 1.1% H2; 6000 ppm C3H6; 5.0% O2; 10.8% CO2; 0.0954% NO; He balance; GHSV = 20000 h−1 |
||||
| Pt/Mg–Al (0.33:1.0 mol%) (3.0:1.0 mol%) (1.0 wt% Pt) |
Coprecipitation/impregnation (600/500) | Lean (190 s): 8.0% O2; 0.055% NO; N2 balance | 53 (400) | 108 |
| Rich (110 s): 0.08% C3H6; 0.055% NO; N2 balance; W/F = 45 g s l−1 | ||||
| 81 (400) | ||||
| Pt/Mg–Al (3.0:1.0 mol%; 1.0 wt% Pt) |
Commercial/impregnation (650/550) | Lean (120 s): 5.0% O2; 10.8% CO2; 0.1% NO; He balance | 68 (300) | 109 |
| Rich (6 s): 3.3% CO; 1.1% H2; 0.6% C3H6; 10.8% CO2; 0.1% NO; He balance; GHSV = 20000 h−1 |
||||
| Pt–Cu/Mg–Al (1.0 wt% Pt, 1.0 wt% Cu) (1.0 wt% Pt, 4.0 wt% Cu) (2.3:1.0 mol%) |
Commercial/impregnation (650/550) | Lean (120 s): 5.0% O2; 10.8% CO2; 0.1% NO; He balance | 92 (197) | 100 |
| 97 (300) | ||||
| Rich (6 s): 3.3% CO; 1.1% H2; 0.6% C3H6; 10.8% CO2; 0.0954% NO; He balance; GHSV = 20000 h−1 |
||||
| Pt–Cu/Mg–Al (66.0:34.0 mol%; 1.0 wt% Pt, 4.0 wt% Cu) |
Commercial/impregnation (650/550) | Lean (120 s): 5.0% O2; 10.8% CO2; 0.0954% NO; He balance | 95 (300) | 42 |
| Rich (6 s): 3.3% CO; 1.1% H2; 0.6% C3H6; 5.0% O2; 10.8% CO2; 0.0954% NO; He balance; GHSV = 20000 h−1 |
99 | |||
| Mg–Co–Al (69.0:16.0:15.0 mol%) |
Coprecipitation (650) | Lean (120 s): 13.0% O2; 0.053% NO; 0.005% C3H8; N2 balance | 30 (300) | 110 |
| V/Mg–Co–Al (1.0 wt% V) | Coprecipitation/impregnation (650/550) | Rich (60 s): 8.0% O2; 0.053% NO; 0.07% C3H8; N2 balance (*with 0.006% SO2; 8.0% H2O); W/F = 92 g s l−1 | 99 (300) *80 (300) 99 (300) *65 (300) 70 (300) | |
| Ru/Mg–Co–Al (1.0 wt% Ru) | ||||
| Pt/Mg–Co–Al (1.0 wt% Pt) (69.0:16.0:15.0 mol%) |
||||
| Mg–Zn–Al–Fe (0.75:0.01:0.18:0.06 mol%) |
Coprecipitation (550) | Lean (120 s): 13.0% O2; 0.053% NO; 0.005% C3H8; N2 balance | 80 (450) | 111 |
| Pd/K/Mg–Zn–Al–Fe (0.75:0.01:0.18:0.06 mol%; 1.0 wt% Pd, 1.0 wt% K) |
Coprecipitation/impregnation (550) (H2-red., 450) | Rich (60 s): 0.053% NO; 0.005% C3H8; N2 balance; W/F = 92 g s l−1 | 95 (450) | |
| Pd/K/Mg–Co–Al (0.70:0.15:0.15 mol%; 1.0 wt% Pd, 1.0 wt% K) |
95 (450) |
Mg–Al–Ox was utilized as a support due to its basicity. Precursors for such applications were mainly prepared by coprecipitation followed by calcination, which only led to the formation of poorly crystallized MgO. The NOx storage properties of Mg–Al–Ox varied depending on the Mg:Al ratio; however, only low activities in NOx capture were found. For example, catalysts with a Mg:Al molar ratio of 3.0:1.0 achieved an uptake of 0.09 mmol g−1 NOx at 300 °C, which was related to the lack of redox components in the system.105 Deposition of transition metals on the (calcined) supports and/or incorporating them into the structure of hydrotalcite-like compounds enabled a significantly higher activity. Yu et al.105 studied the influence of different concentrations of cobalt (0.5–3.0 mol%) incorporated into Mg–Al hydrotalcite-like compounds. Calcination at 800 °C led to MgO as well as spinel (Co2AlO4, CoAl2O4 and/or Co3O4) formation for cobalt loadings above 1.0 mol%. Only small amounts of MgO were found in the samples with a Mg:Co:Al molar ratio of 1.0:2.0:1.0, revealing the highest storage capacity reaching 0.20 mmol g−1 NOx at 300 °C, which was more than two times higher compared to that of undoped Mg–Al–Ox. The authors correlated the results with possible redox reactions involving cobalt ions, and thus leading to more NOx adsorbed (eqn (4) and (5)):
| NO + Co3+ − O2− → Co2+ − NO2− | (4) |
| NO2 + Co3+ − O2− → Co2+ − NO3− | (5) |
Further, a migration process of NO2− and NO3− took place from Co to adjacent Mg–Al–Ox to form relatively stable nitrites and/or nitrates. Varying the amounts of magnesium and cobalt in such catalysts and consequently adjusting the adsorption and storage properties of the catalysts could lead to generation of optimum adsorbent catalysts for NSR. Based on the obtained results, further studies appear worthwhile. It should be stressed that in this study, besides NO, NO2 also existed in the feed. A great number of studies have shown that NSR catalysts store NO2 more efficiently than NO.112–114 Thus, the majority of NOx coming out of the lean-burn engine is in the form of NO. Therefore, the oxidation of NO over diesel oxidation catalysts (DOCs) is a very important step in the storage process. In the catalytic tests, the presence of only NO in the feed possibly caused the lower values of NOx storage capacity over Mg–Co–Al and Mg–Co–Al–Ti with similar chemical and/or phase compositions (0.07 and 0.04 mmol g−1 NO at 300 °C, respectively). Additionally, the relatively low NOx storage capacity obtained over Mg–Co–Al–Ti–Ox was reported to be caused by destabilization of adsorbed NOx on the catalyst surface after incorporation of titanium.106 Replacing Mg with Ca improved the NOx storage properties and was related to the enhanced alkalinity of the catalytic system. As a result, Ca–Co–Al–Ox with different compositions (Ca:Co = 0.0–3.0:0.0–3.0) was prepared. Co2AlO4, CoAl2O4 and/or Co3O4 were detected in the most active samples. In particular, the catalyst with a Ca:Co:Al molar ratio of 2.0:1.0:1.0 stored 0.43 and 0.60 mmol g−1 NO at 300 °C within 30 and 60 min, respectively. Incorporating small quantities of La (0.1 mol%) improved the catalytic activity further. Consequently, a Ca–Co–Al–La–Ox catalyst captured 0.63 mmol g−1 NO within 60 min.101,107 In this system, NOx was first oxidized to nitrites/nitrates on the catalytic redox sites (CoOx), and then spilled over to the storage region (CaO). Lanthanum was reported to possibly mediate between them.107 Interestingly, no further activation occurred for partial substitution of calcium in Ca–Co–Al–La samples with barium or strontium. According to the authors, the results indicated that the basicity of the oxide catalysts was not the only factor that determined the NOx storage capability. Other possible explanations were related to incomplete incorporation of barium or strontium into the hydrotalcite-like sheets.101,107 However, no physicochemical characterization was carried out to support this hypothesis.
Besides the above mentioned non-noble metal NSR catalysts, several investigations focused on Pd,103 Pt115 or Ru104 addition. Li et al.104 studied a hydrotalcite-like compound with ruthenium introduced into the structure. The obtained Mg–Ru–Al (Mg:Ru:Al = 90.0:1.0:29.0) precursor was prepared by coprecipitation and transformed at 600 °C into MgO together with a spinel phase (Mg(Al,Ru)2O4). A reasonably high NOx storage capability was found at 250–400 °C with a maximum value of about 0.22 mmol g−1 NO at 350 °C. Based on the NOx adsorption–desorption profiles as well as in situ FTIR studies, they proposed that NOx oxidation takes place starting from O2 adsorption on the surface of Ru, followed by its dissociation to O (eqn (6) and (7)):
| Ru + O2(g) → Ru − O2 | (6) |
| Ru − O2 + Ru → 2Ru − O | (7) |
In the next steps, gaseous NO could react with surface O to form nitrites/nitrates, which then migrate to the basic sites adjacent to the Ru species. Therefore, the adsorption sites on Ru were regenerated (eqn (8) and (9)):
| Ru − O + 2NO(g) + O2− → Ru + 2NO2− | (8) |
| Ru − O + NO2− → Ru + NO3− | (9) |
Additionally, surface O on Ru could also oxidize gaseous NO to gaseous NO2 on the catalyst surface (eqn (10)):
| Ru − O + NO(g) → Ru + NO2(g) | (10) |
The formed NO2 could then be trapped on the basic sites available in the catalysts – probably non-adjacent to Ru species, forming nitrites/nitrates, and the other NO2 species went to the outlet (eqn (11)):
| 2NO2(g) + O2− → NO2− + NO3− | (11) |
This mechanism was proposed for a Ru–Mg–Al–O system; however, it seems that it is valid also in the case of other combinations of noble metals, such as Pd.111
The high NOx storage capacity of Mg–Ru–Al was correlated with the high dispersion of ruthenium species in the catalyst, which provides more active sites to convert NO to nitrites/nitrates, i.e. the major route to store NOx on the catalyst.
Probably, high dispersion was not achieved after deposition of platinum onto Mg–Al–Ox. Therefore, Pt/Mg–Al with a Mg:Al molar ratio of 7.0:3.0 revealed poorer NOx storage (0.06 mmol g−1 NO at 300 °C) compared to the Ru-based system.103 However, these materials facilitated high mean NOx conversion at 300–400 °C.100,108
Cheng et al.108 examined the effect of the Mg:Al molar ratio in the range of 0.33–3.0:1.0 on the catalytic performance of Pt/Mg–Al–Ox. In particular, Pt/Mg–Al (Mg:Al = 3.0:1.0) and Pt/MgO as a reference showed superior NOx conversions with 81 and 94% at 400 °C, respectively. This behaviour could be attributed to the higher basicity in comparison to that of mixed metal oxides with lower Mg:Al molar ratios. Similar trends for temperatures above 200 °C were observed for Pt/Mg–Al (Mg:Al = 1.3–3.0:1.0).109 Increasing activity with an increase in magnesium loading was also found for the catalysts after aging at 800 °C for 24 h with application of steam or SO2-pretreatment. On the contrary, the Pt/MgO reference catalyst lost its activity after hydrothermal treatment. Thus, the obtained results revealed the basic role of hydrotalcite-like precursors in the preparation of efficient NSR catalysts.
Pt/Mg–Al catalysts were prepared by impregnation of calcined Mg–Al hydrotalcite-like compounds (synthesized or commercial) with a solution of an appropriate platinum precursor. Calcination below 600 °C led to the formation of only poorly crystallized MgO. After hydrothermal treatment, MgAl2O4 and crystalline Pt appeared. These phases did not appear after introducing copper into the Pt/Mg–Al–O system. This observation was attributed to potential formation of surface CuAl2O4 and Pt–Cu alloy, which prevented sintering.11 Combinations of Pt–Cu supported on Mg–Al–Ox with different molar ratios of Mg:Al were comprehensively studied by Fornasari et al.42,99,100 For example, Mg–Al–Ox (Mg:Al = 2.3:1.0) obtained by calcination at 650 °C was impregnated with copper and subsequently with platinum salt solutions. Two different loadings of Cu (1.0 or 4.0 wt%) were used to keep the platinum loading constant (1.0 wt%). Pt–(1.0 wt%)Cu/Mg–Al–Ox enabled a maximum NOx conversion of 92% at 197 °C, while 97% mean NOx conversion at 300 °C was achieved over the material containing 4.0 wt% copper.100 Pt–(4.0 wt%)Cu/Mg–Al with a higher Mg:Al molar ratio of 66.0:34.0 showed comparable catalytic activity (95% mean NOx conversion at 300 °C).42,99 After aging at 800 °C for 24 h with steam and SO2-pretreatment, the mean NOx conversion decreased to 77% (400 °C) and 89% (300 °C). However, the overall NOx conversion remained superior to that observed at the same temperatures for a Pt–BaO/Al2O3 reference system. Higher NOx conversion was also reported over the tested mixed metal oxides compared to the reference system during 50 min stability tests in the presence of 0.002% SO2 in the feed. During the test, NOx conversion over Pt–Cu/Mg–Al–Ox decreased by about 5%. Unfortunately, there is still a lack of detailed mechanistic understanding of processes occurring under the reaction conditions and in the presence of sulphur oxide. Further studies in this direction covering these promising catalytic systems are certainly necessary.
Besides catalysts containing platinum–copper, Palomares et al.110 studied also Pt–Co as well as other combinations (Pd–Co, V–Co, and Ru–Co). Cobalt was introduced into the Mg–Al hydrotalcite-like structure during coprecipitation. Different compositions of precursors were utilized (Mg:Co:Al = 64.0:6.0:30.0, 69.0:11.0:20.0 or 69.0:16.0:15.0). After calcination at 650 °C, the obtained materials exhibited a structure of poorly crystallized MgO. Catalytic investigations showed around 30% mean NOx conversion at 300 °C for the last composition presented above. An increasing content of cobalt caused improved redox properties, thus NO was more easily oxidized to nitrites/nitrates (eqn (4) and (5)), which further moved to adjacent Mg–Al–Ox to be stored. Impregnation with Pt, Pd, V or Ru further improved the redox properties and consequently the catalytic performance of the materials. Fig. 6 presents some results of these studies. Both V- and Ru-doped samples facilitated near 100% mean NOx conversion at 300 °C. Both catalysts presented also higher activity at lower temperatures as well as in the presence of 0.006% SO2 and 8.0% H2O compared to Pt–BaO/Al2O3 as a reference. Additionally, stability test for 4 h over the vanadium-containing material in the presence of both water vapour and sulphur oxide revealed a stable NOx conversion of 70%. The authors explained the results by a low SOx capture rate of the magnesium oxide sites together with high resistance to poisoning of vanadium. A detailed analysis of the nature and stability of the active sites in the catalysts was not provided.
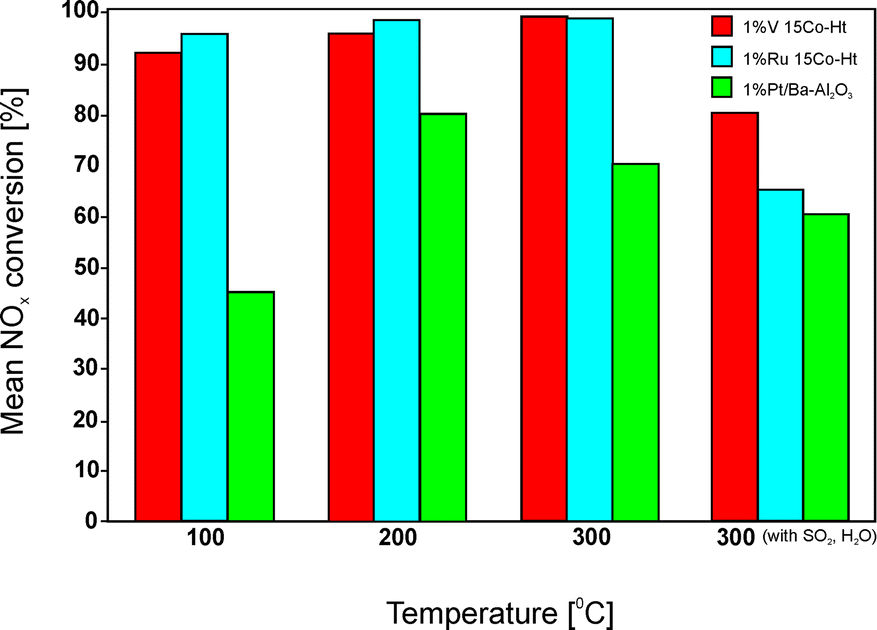 | ||
| Fig. 6 Results of catalytic tests performed for Mg–Co–Al–Ox. Reaction conditions: lean (120 s): 13.0% O2, 0.053% NO, 0.005% C3H8; rich (60 s): 8.0% O2, 0.053% NO, 0.07% C3H8, N2 balance; total flow rate = 650 ml min−1; mass of catalyst = 1000 mg (adapted from ref. 110 with kind permission from Elsevier). | ||
Despite these promising results, further studies on mixed metal oxides mainly focused on problems related to the toxicity of cobalt.116 Consequently, Fe- and/or Zn-containing systems found increasing attention. Catalytic systems based on Mg–Zn–Al–Fe–Ox (Mg:Zn:Al:Fe = 0.75:0.01:0.18:0.06) were studied and their catalytic performance for NSR was compared to that of Mg–Co–Al–Ox (Mg:Co:Al = 0.70:0.15:0.15).114 Both precursors were prepared by coprecipitation and transformed into mixed metal oxides at 550 °C. Mg–Zn–Al–Fe–Ox achieved around 20% lower mean NO conversion at 450 °C compared to Mg–Co–Al–Ox. Modification of both samples with Pd (redox sites necessary to oxidize NO to NO2) and K (basic sites necessary to store such NO2 in the form of nitrates) resulted in significant activation. In this line, both modified catalysts reached around 95% mean NO conversion at 450 °C. Additional stability tests of the iron-containing catalyst in the presence of 2.0% H2O or after aging at 700 °C for 5 h with steam revealed excellent stability. No significant deactivation occurred. However, hydrothermal treatment caused an increased crystallinity of MgO and a spinel phase (MgAl2O4) was formed. The high activity and stability of Mg–Zn–Al–Fe–Ox were related to the synergistic interaction between iron and zinc, which favoured a redox cycle in the reaction (Fig. 7). Furthermore, the presence of potassium and the high dispersion of palladium sites, measured using CO chemisorption, resulted in a shift of the H2-TPR profile to lower temperatures indicating improved redox properties.
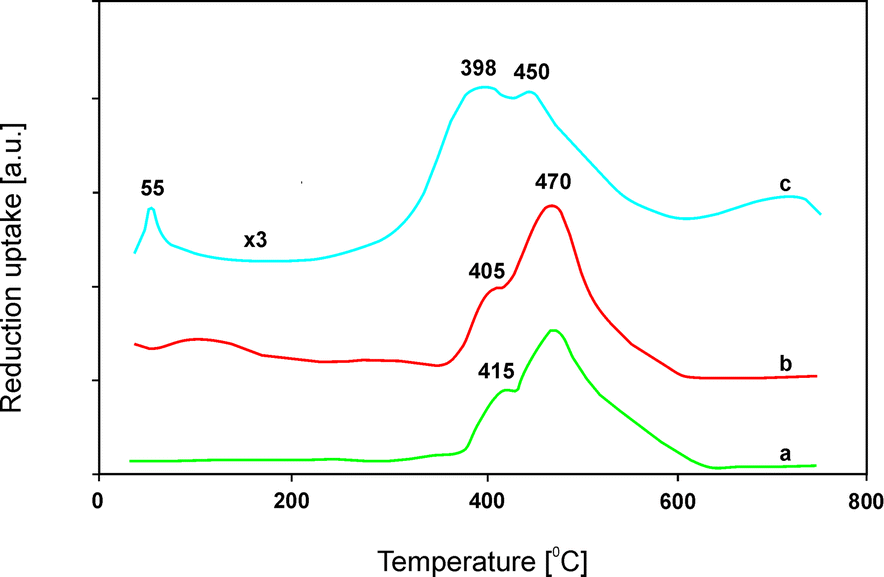 | ||
| Fig. 7 Results of temperature-programmed reduction (H2-TPR) performed for Mg–Fe–Al–Ox (a), Mg–Zn–Al–Fe–Ox (b) and Pd/K/Mg–Zn–Al–Fe–Ox (c) (adapted from ref. 111 with kind permission from Elsevier). | ||
Despite promising catalytic activity, poor activity at temperatures below 400 °C presents a major drawback. Therefore, further optimization of such catalysts is necessary. Nevertheless, analysis of the NSR behaviour of the presented catalysts as well as other materials reviewed in this chapter indicates that mixed metal oxides are realistic options for high performance NOx storage/reduction catalysts. Several systems exhibit excellent resistance to SO2 and higher thermal stability than the reference Pt–BaO/Al2O3. Future studies should address catalyst optimization in the presence of typical exhaust gases such as soot, COx or SOx and subsequently tests under real diesel engine conditions in order to assess the potential of these materials for NSR.
Simultaneous catalytic removal of NOx and diesel soot particulates
Soot is another problematic emission for diesel engines. Diesel particulate filters (DPFs) can trap over 90% of soot particulates from the combustion chamber, and subsequently burn them at temperatures around 600 °C to regenerate the DPF.117 Regeneration of the DPF at lower temperatures can be achieved by applying catalysts. A first technical solution, called continuous regenerating trap (CRT), was proposed by Johnson Matthey.118 The mechanism involves oxidation of NO by a catalyst to NO2, which reacts further with trapped soot to form CO2 and NO. The released NO is oxidized again to NO2. The main drawback of this technology relates to low NOx removal efficiency due to NO2 slip.119 Therefore, another solution for simultaneous catalytic removal of NOx and soot, called the diesel particulate–NOx reduction (DPNR) system, was proposed by Toyota.120 DPNR catalysts work under lean–rich cycle conditions. Under lean conditions, NOx in the exhaust is stored as nitrites and/or nitrates, while under rich conditions, the stored NOx is reduced to N2 by soot, HCs and CO.121 The development of highly efficient and robust catalysts possessing low-temperature activity (200–400 °C) for both NOx removal and soot oxidation is a key point of this technology. Yoshida et al.122 first proposed K–Ce–Mn and K–Ce–Cu catalytic systems for simultaneous NOx–soot removal. Then, supported metal oxides,121,123 zeolites,124 perovskites,125,126 and spinels,127 including Mn, Fe, Ni, Cu, Ag, Pt, K, and Ba, have been studied and most of them showed promising activities in the simultaneous removal of NOx and soot. Also, hydrotalcite-derived mixed metal oxides exhibit high potential in this process.128,129 Recently, Yang et al.16 published a review on hydrotalcite-derived mixed metal oxides. They classified the catalysts according to their compositions (e.g. binary, ternary hydrotalcite-derived catalysts, etc.) and described their NSC and activity in selective NOx reduction and soot combustion. Table 5 summarizes the catalytic performance of mixed metal oxides for NOx storage and removal. The loose contact mixtures (soot and catalyst gently mixed) compared to tight contact mixtures (soot and catalyst mixed in a mortar) are believed to resemble more closely the contact between soot and the catalyst in a real DPF.133 However, tight contact systems showed more pronounced differences of catalytic activity and were preferably applied in scientific studies. Table 5 presents the NOx storage capacity achieved over soot/catalyst mixtures (loose or tight contact) and NOx reduction by the soot reductant at a given temperature.| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | NOx storage/reduction (temperature/°C) | Ref. | |
|---|---|---|---|---|---|
| /mmol g−1 | /% | ||||
| Pt–BaO/Al2O3 (2.0 wt% Pt, 20.0 wt% BaO) | Impregnation/impregnation (700) | 0.03% NO; 10.0% O2; N2 balance | 123 | ||
| Catalyst; W/F = 72 g s l−1 soot/catalyst mixture, loose contact; W/F = 84 g s l−1 | 0.50 (300) | — | |||
| 0.43 (300) | — | ||||
| Mg–Al (2.8:1.0 mol%) |
Coprecipitation (800) | 0.075% NO; 10.0% O2; N2 balance soot/catalyst mixture, tight contact; GHSV = 48000 h−1 |
0.37 (100–272) | 7 (278–700) | 128 |
| Mg–Mn–Al (2.3:0.5:1.0 mol%) (0.5:1.7:1.0 mol%) |
0.66 (100–404) | 20 (327–614) | |||
| 0.09 (100–202) | 7 (263–618) | ||||
| Mg–Mn–Al (1.5:1.5:1.0 mol%) |
Coprecipitation (800) | 0.075% NO; 10.0% O2; N2 balance soot/catalyst mixture, tight contact; GHSV = 48000 h−1 |
0.27 (100–336) | 13 (308–700) | 129 |
| K/Mg–Mn–Al (7.5 wt% K) (20.0 wt% K) (1.5:1.5:1.0 mol%) |
Coprecipitation/impregnation (800/800) | 0.59 (100–422) | 27 (280–605) | ||
| 0.92 (100–560) | 9 (280–700) | ||||
| Mg–Co–Al (0.5:2.5:1.0 mol%) |
Coprecipitation (600) | 0.04% NO; 10.0% O2; N2 balance soot/catalyst mixture, tight contact; W/F = 30 g s l−1 | 0.21 (100–335) | 9 (284–573) | 130 |
| K/Mg–Co–Al (4.5 wt% K) (4.5 wt% K) (0.5:2.5:1.0 mol%) |
Coprecipitation/impregnation (600/500) (500/500) | ||||
| 0.32 (100–348) | 32 (336–700) | ||||
| 0.40 (100–341) | 30 (330–700) | ||||
| Mg–Al–La (3.0:0.9:0.1 mol%) |
Coprecipitation (800) | 0.1% NO; 5.0% O2; He balance catalyst (*0.135% NO; 5.0% O2; He balance soot/catalyst mixture, tight contact); W/F = 30 g s l−1 | 0.24 (100–584) | — | 131 |
| Mg–Cu–Al (1.0:2.0:1.0 mol%) |
0.14 (100–442) | — | |||
| Mg–Cu–Al–La (1.0:2.0:0.9:0.1 mol%) |
0.35 (100–430) | *51 (400–450) | |||
| Mg–Co–Al (0.5:2.5:1.0 mol%) |
Coprecipitation (600) | 0.04% NO; 10.0% O2; N2 balance soot/catalyst mixture, tight contact; W = 0.1 g | 0.80 (350) | — | 132 |
| K/Mg–Co–Al (4.5 wt% K) (10.0 wt% K) (0.5:2.5:1.0 mol%) |
Coprecipitation/impregnation (600/600) | ||||
| 1.73 (350) | — | ||||
| 2.04 (350) | — | ||||
Li et al.128 examined the effect of different Mg:Mn molar ratios of 0.0–2.8:0.0–1.8, as a result of variable oxidation states of manganese (+2, +3, and +4), on the catalytic performance of Mg–Mn–Al–Ox. The materials were prepared by coprecipitation using aqueous solutions of magnesium and aluminum nitrates together with manganese acetate. Na2CO3 and NaOH were used as precipitating agents. The obtained hydrotalcite-like compounds were subsequently calcined at 800 °C. Mn-free samples possessed crystalline structures of MgO and MgAl2O4, while an increasing Mn content was associated with the presence of Mg2MnO4 and/or MnAl2O4. Both Mn-containing spinels enhanced the NOx storage performance. Mg–Mn–Al–Ox with Mg:Mn:Al molar ratios of 2.3:0.5:1.0 and 2.0:0.9:1.0 revealed the highest NOx uptake among all tested samples (0.66 and 0.50 mmol g−1 NO, respectively) and NO reduction (20 and 24%, respectively). Introduction of higher amounts of manganese into the Mg–Mn–Al systems (Mg:Mn molar ratio above 1.6:1.3) resulted in crystalline Mn3O4 and Mn2O3 formation. However, the presence of these two phases seemed to decrease the catalytic activity of the materials. In particular, a NO uptake of 0.09 mmol g−1 with 7% NO reduction was achieved over Mg–Mn–Al–Ox with a Mg:Mn:Al molar ratio of 0.5:1.7:1.0. Therefore, the presence of both Mn4+ and Mn2+ in the samples was found to be essential in order to achieve efficient NOx storage and reduction, respectively. Moreover, based on the presented data, the major role of the transition metal could be identified. Obviously, Mn-free samples caused lower NOx uptake compared to materials with higher loadings of manganese (Mg:Mn = 1.6:1.3 and above such a molar ratio).
While the roles of both MgO and MgAl2O4 were not stressed in the work of Li et al.,128 their promoting effect in the hydrotalcite-derived systems was recognized and described in the scientific literature. Studies aimed at efficient NOx storage below 300 °C assumed poorly crystallized MgO as the NOx storage site in Mg–Al–Ox together with a small amount of noble metals.11,99,103 MgAl2O4 essentially enhanced the high temperature performance. For example, Kwak et al.134 reported relatively low NOx uptake below 300 °C over Pt–BaO/MgAl2O4 compared to commercially applied Pt–BaO/Al2O3. At 350 °C, the NOx uptake over MgAl2O4-supported catalysts was twice as high over the alumina-based ones. Similarly, Takahashi et al.135 reported that a Pt–K/MgAl2O4 catalyst exhibited better NOx uptake properties at 600 °C than Pt–K/Al2O3, which was explained by the enhanced basicity arising from the MgAl2O4 spinel structure.
Further studies of Li et al.129,132 focused on potassium-promoted Mg–Mn(Co)–Al–Ox. Potassium nitrate served as a precursor of K. Mixed metal oxides containing manganese were prepared by coprecipitation followed by calcination at 800 °C. Impregnation with potassium (1.5–20.0 wt%) resulted in K2Mn4O8 formation besides MnAl2O4 and/or Mg2MnO4. Materials of this composition facilitated 0.59 mmol g−1 NO uptake and a maximum NOx reduction of 27% over K/Mg–Mn–Al–O containing 7.5 wt% K. The authors explained the catalytic performance based on the strong interaction between K and Mn species on the carrier facilitating the formation of reactive oxygen species in the new K–Mn–O phase. K2Mn4O8 could more easily oxidize NO to NO2 and store NOx in the form of nitrates, which were subsequently reduced by soot. Interestingly, such a positive effect of K2Mn4O8 on the simultaneous NOx–soot removal has been already reported earlier.136 For higher potassium contents, KNO3 appeared resulting in high NOx storage ability at the cost of poor NO reduction.129
Mixed metal oxides containing cobalt with a Mg:Co:Al molar ratio of 2.5:0.5:1.0 were prepared by coprecipitation using aqueous solutions of the appropriate metal nitrates, followed by calcination at 600 °C.132 Co–Mg–Al–Ox consisted mainly of Co3O4 and/or CoAl2O4, while for materials promoted by potassium (4.5–10.0 wt%), K2O was segregated additionally. With regard to the effect of potassium loading on the catalyst performance, 4.5 wt% K proved to provide the highest activity gain. Even higher potassium loadings resulted in an only limited further increase of NOx storage capacities. Therefore, advanced studies using 4.5 wt% potassium focused on the influence of calcination temperature of hydrotalcite-like precursors (500, 600, 700 or 800 °C).130 Co3O4 and/or CoAl2O4 were identified in the samples calcined at 500 and 600 °C, while calcination at 700 and 800 °C led to the formation of CoAl2O4 or CoAl2O4-like spinels. Mg–Co–Al–Ox was impregnated with 4.5 wt% potassium followed by calcination at 500 °C. A K2O phase appeared for the samples calcined above 600 °C. A K (4.5 wt%)/Mg–Co–Al catalyst calcined at 600 °C achieved the maximum performance for simultaneous soot–NOx removal with a NO uptake of 0.32 mmol g−1 and 32% NO reduction. These results were attributed to the high surface K/Co atomic ratio of the catalyst established by XPS analysis. The strong interaction between surface K and Co induced the formation of reactive oxygen species in the new K–Co–O phase. Active oxygen species could easily react with soot on the catalyst surface. In addition, active oxygen species could facilitate oxidation of NO to NO2, stored as nitrates on K species, followed by their reduction by soot.
Wang et al.131 incorporated rare earth elements such as lanthanum into Mg–(Cu)–Al hydrotalcite-like structures. Mg–(Cu)–Al–(La) precursors were prepared by coprecipitation of an aqueous solution of suitable metal nitrates, sodium carbonate and sodium hydroxide. After calcination at 800 °C, Mg–Al–La–Ox revealed the presence of La2O3 and La10Al4O21, while for Mg–Cu–Al–La–Ox, CuLaO3 together with CuO and MgAl2O4 was identified. In general, samples containing lanthanum exhibited higher NOx adsorption capacities compared to La-free references. La2O3 is a basic oxide, and, accordingly, is able to store NOx. The presence of copper enhanced the redox properties of the materials and facilitated NO oxidation to NO2. A maximum storage capacity of 0.35 mmol g−1 NO together with 51% NO removal could be reached over Mg–Cu–Al–La–Ox. Based on in situ FTIR measurements of the catalyst, a reaction scheme responsible for the high catalytic performance in the simultaneous removal of soot and NOx was proposed as follows (eqn (12)–(14)):
| 2NO + O2 → 2NO2 | (12) |
| 2NO2 + C → CO2 + 2NO | (13) |
| 2NO2 + C → 2CO2 + N2 | (14) |
In contrast to Mg–Cu–Al–La–Ox, Ce-based catalysts doped with cobalt or iron reached significantly lower conversions.137,138 In particular, Fe–Ce–Ox (Fe:Ce = 1.0:9.0) only achieved around 10% NO conversion.138
Summarizing the current state of the art with regard to the simultaneous removal of NOx and diesel soot particulates, the relatively low NOx storage capacities of most materials have to be mentioned. Nevertheless, hydrotalcite-derived mixed metal oxides exhibit significant potential for application in this area. A further advance in this field will clearly depend on a comprehensive understanding of the mechanisms involved in NOx storage and soot removal and potential synergies between these processes.
Catalytic decomposition of N2O
The catalytic decomposition of N2O (deN2O) belongs to the best available technologies for N2O abatement from nitric acid production, which has been recognized as one of the biggest industrial sources of N2O emissions, according to the reaction (eqn (15)):| 2N2O → 2N2 + O2 | (15) |
The possible positions of the catalytic N2O converters were proposed in the installation for nitric acid production: (i) behind the Pt–Rh catalyst for ammonia oxidation, inside the ammonia burner (high-temperature N2O decomposition, 800–950 °C) and (ii) at the end of the pipe (downstream of the absorption column) (low-temperature N2O decomposition, 250–450 °C).4 These different locations of the catalytic N2O converters and consequently different reaction conditions require various catalysts for such processes. The first option calls for extreme thermal stability of the catalyst. For this reason, an interesting alternative limiting N2O emission in such installations seems to be a low temperature process. Therefore, studies frequently focus on the development of catalysts with high activity at temperatures below 450–500 °C. Several catalytic systems have so far been tested for N2O catalytic decomposition in the scientific literature. Especially, Co3O4-based spinels modified with K, Cs or Zn139,140 or zeolites modified with Co, Cu or Fe141,142 as well as supported Rh or Ag catalysts143,144 found attention. Recent reviews summarize the broad collection of tested catalytic systems under different reaction conditions.17,145 Herein, we will only highlight the potential of hydrotalcite-originated materials. Starting from the work of Kannan et al.,146,147 several hydrotalcite-originated mixed metal oxides with different combinations of Cu, Co, Ni, Mn and/or Rh, Pd were applied as catalysts for the decomposition of N2O.148–153Table 6 gives an overview of the catalytic performance of deN2O over selected mixed metal oxides mainly summarizing the maximum N2O conversion. Besides investigations of N2O decomposition in ideal N2O-inert gas feeds, experiments in the presence of other components of exhaust gases, such O2, H2O and NO (the product of ammonia oxidation in nitric acid plants), were carried out. N2O decomposition is most efficiently catalysed by Co, Mn modified with K or Rh as implied by the published results. Analysis of the presented data shows that the process of N2O decomposition is very sensitive to the kind and loading of the catalytically active metal, its form, dispersion, etc. Further details of the selected material system are addressed in the following section.
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | N2O conversion/% (temperature/°C) | Ref. |
|---|---|---|---|---|
| Co3O4 | Commercial | 5.0% N2O; He balance | 100 (550) | 140 |
| (*with 1.0% H2O); GHSV = 7000 h−1 | *100 (600) | |||
| Mg–Al (2.2:1.0 mol%) |
Coprecipitation (500) | 0.0985% N2O; He balance (*with 2.5% O2; 2.0% H2O); W/F = 60 g s l−1 | 1 (450) *1 (450) | 147, 148149 |
| Co–Al (3.0:1.0 mol%) |
84 (450) *25 (450) | |||
| Mg–Co–Al (0.94:2.0:1.0 mol%) |
100 (450) *88 (450) | |||
| Co–Rh–Al (Co:Al = 3.0:1.0 mol%; 0.7 wt% Rh) |
100 (300) *100 (450) | |||
| Mg–Co–Al Mg–Cu–Al (61.0:10.0:29.0 mol%) |
Coprecipitation (600) | 0.5% N2O; 4.5% O2; He balance; W/F = 120 g s l−1 | 90 (650) 100 (650) | 154 |
| Mg–Cu–Co–Al | Coprecipitation (800) | 100 (500) | ||
| K/Mg–Cu–Co–Al (0.9 wt% K) (51.0:10.0:10.0:29.0 mol%) |
Coprecipitation/impregnation (800/600) | 100 (500) | ||
| Ni–Al (4.0:2.0 mol%) |
Coprecipitation (450–500) | 0.1% N2O; He balance; W/F = 60 g s l−1 | 79 (450) | 155, 156 |
| Ni–Al | Coprecipitation (400) | 2.0% N2O; Ar balance (*with 4.0% O2; 8.8% H2O); W/F = 429 g s l−1 | 89 (450) *38 (500) | 157 |
| K/Ni–Al (K:Ni = 0.1:1.0 mol%; Ni:Al = 4.1:1.0 mol%) |
Coprecipitation/impregnation (400/400) | 100 (450) *100 (500) | ||
| Co–Mn–Al (4.0:1.0:1.0 mol%) Mg–Co–Al (2.0:2.0:2.0 mol%) |
Coprecipitation (500) | 0.1% N2O; He balance; W/F = 60 g s l−1 (*with 5.0% O2; 2.0% H2O; W/F = 180 g s l−1) | 82-97 (450) *45 (450) *2 (450) | 158, 159 158 |
| Co–Rh–Al (Co:Al = 3.0:1.0 mol%) Mg–Co–Rh–Al (Mg:Co:Al = 1.0:3.0:1.0 mol%) (0.7 wt% Rh) |
Coprecipitation (450) | 1.0 mbar N2O; He balance (*with 30.0 mbar O2; 0.125 mbar SO2); W/F = 30 g s l−1 | 100 (325) *0 (325) 100 (325) *95 (325) | 160 |
| Co–Rh–Al (75.0:0.5:24.5 mol%) Mg–Rh–Al (71.0:1.0:28.0 mol%) |
Coprecipitation (650) (H2-reduction, 750) | 20.0% N2O; He balance (*with 2.6% H2O); GHSV = 50000 h−1 |
55 (450) *38 (450) 100 (450) *100 (450) | 161 |
| Mg–Rh–Pd–Al (70.0:0.5:1.0:28.5 mol%) |
*95 (450) | |||
| Mg–Rh–La–Al (70.0:0.5:1.0:28.5 mol%) |
*89 (450) | |||
| Co–Al | Coprecipitation (500) | 0.05% N2O; He balance (*with 4.0% O2; 2.6% H2O); W/F = 120 g s l−1 | 89 (450) | 162 |
| K/Co–Al (K:Co = 0.04:1.0 mol%; Co:Al = 3.0:1.0 mol%) |
Coprecipitation/impregnation (500) | 100 (350) *100 (450) | ||
| (700–800) | 100 (300) *92 (360) | |||
| K/Co–Mn–Al (4.0:1.0:1.0 mol%; 0.9 wt% K) |
Coprecipitation/resuspension (500) | 0.1% N2O; He balance; GHSV = 40380 h−1 (*with 5.0% O2; 4.0% H2O) (**with 5.0% O2; 0.17% NO; 0.17% NO2; GHSV = 13460 h−1) |
100 (450) *25 (450) *55 (450) | 44 |
| K/Co–Mn–Al (1.8 wt% K) | Coprecipitation (500) | 0.1% N2O; He balance; W/F = 60 g s l−1 (*with 5.0% O2; 0.9% H2O; 0.005% NO; W/F = 180 g s l−1) | 100 (450) *90 (450) | 163 |
| Na/Co–Mn–Al (1.4 wt% Na) (4.0:1.0:1.0 mol%) |
Coprecipitation/impregnation (500/500) | 100 (450) | ||
| K/Ce–Co (Ce:Co = 0.21:1.0 mol%; 0.7 mol% K) |
Coprecipitation (400) | 0.1% N2O; Ar balance; W/F = 200 g s l−1 | 96 (350) | 164 |
| Co–Al Na/Co–Al (1.5 wt% Na) (3.0:1.0 mol%) |
Coprecipitation (300) Coprecipitation/impregnation (300/300) | 2.0% N2O; Ar balance; W/F = 429 g s l−1 | 82 (450) 100 (450) | 165 |
| Co–Rh–Al Na/Co–Rh–Al (0.75 wt% Na) (2.5:0.05:1.0 mol%) |
Coprecipitation (450) Coprecipitation/impregnation (450) | 0.03% N2O; He balance (*with 4.6% O2; 0.3% H2O; 0.0125% N2; 0.015% NO; 0.3% CO2); W/F = 250 g s l−1 | 100 (320) *100 (450) *100 (430) | 150 |
| Co–Al | 15.0% N2O; 2.0% H2O; He balance GHSV = 18000 h−1 |
166 | ||
| (3.4 wt% Na) | Coprecipitation (500) | 94 (475) | ||
| (1.0 wt% Na) (2.2:1.0 mol%) |
Coprecipitation/impregnation (500) | 94 (475) | ||
| K/Co–Mn–Al (1.1 wt% K) Cs/Co–Mn–Al (3.4 wt% Cs) (4.0:1.0:1.0 mol%) |
Coprecipitation/impregnation (500/500) | 0.1% N2O; He balance; W/F = 180 g s l−1 (*with 5.0% O2; 3.0% H2O) (**with 5.0% O2; 3.0% H2O; 0.02% NO) | 100 (450) *75 (450) **38 (450) 100 (450) *95 (450) **65 (450) | 159 |
Mg–Al–Ox revealed poor activity,149 and even changes of the Mg:Al molar ratio did not influence the catalytic activity.161 Introducing metals with redox properties, such as Ni, Cu or Co, significantly improved the catalytic activity in N2O decomposition. Kannan et al.146–149 utilized Co(Ni,Cu,Mg)–Al–Ox. The precursors were prepared by coprecipitation using aqueous solutions of the appropriate metal nitrates together with Na2CO3 and NaOH as precipitating agents. Armor et al.148 claimed that N2O conversion was maximized over the catalysts calcined at temperatures ranging from 450 to 500 °C. Calcination at 500 °C led to the formation of mixed metal oxides. In particular, Co3O4 and/or CoAl2O4 were identified in Co–Al–Ox. Catalytic tests over the obtained materials revealed the following order of transition metals with decreasing activity in N2O decomposition: Co ≥ Ni > Cu. Regarding cobalt- and nickel-containing materials, the difference in activity was explained based on their redox behaviour. The strength of oxygen bonding in cobalt oxide was suggested to be weaker than that in the corresponding Ni-analogue. Unfortunately, no H2-TPR analysis was provided and the reason for the lower activity of copper-based materials remained unclear.
Further catalytic tests with different Co:Al molar ratios of 1.1–3.55:1.0 confirmed the highest activity over Co:Al = 3.0:1.0. The obtained results indicated increasing activity with increasing Co2+ concentration on the catalyst surface as determined by X-ray photoelectron spectroscopy. As a result, the Co3Al1 catalyst exhibited a maximum N2O conversion of 84% at the reaction temperature of 450 °C. However, addition of 2.0% H2O and 2.5% O2 to the feed reduced the conversion to 25%, thereby limiting the commercial use of this catalyst.
Studies of Pérez-Ramírez et al.160 proved a positive influence on the activity for N2O decomposition over materials with higher Co:Al molar ratios around 1.0–3.0:1.0. On the contrary, Chang et al.167 implied from their results that the catalytic activity decreases with increasing cobalt concentration based on the ratio of Co:Al = 1.0–3.0:1.0. Up to 92% N2O conversion at 450 °C was achieved over Co:Al = 1.0:1.0. 10.0% O2 in the feed stream reduced the N2O conversion to around 85%. Further, the presence of Mg in the Co-containing catalysts considerably influenced the catalytic behaviour mainly due to stabilization of Co2+.160 Therefore, higher N2O conversion with and without O2 (or 2.5% O2 and 2.0% H2O) in the feed was achieved over Mg–Co–Al–Ox (Mg:Co:Al = 0.94:2.0:1.0, 450 °C, 0.1 g of catalyst and 100 ml min−1 total flow rate) and reached near 100% (or 88%).149 In contrast, Obalová et al.158 reported 80% N2O conversion over Mg–Co–Al–Ox with a Mg:Co:Al molar ratio of 2.0:2.0:2.0 (450 °C, 0.3 g of catalyst and 100 ml min−1 total flow rate).
Other studies of Chmielarz et al.154 emphasized even higher activity for samples containing copper than those containing cobalt in Mg–M–Al–Ox systems. For materials prepared by coprecipitation followed by calcination at 600 °C, the catalytic activity over transition metals proceeded as follows: Cu > Co > Fe > Ni. Studies focusing on optimization of chemical (Cu and/or Co, 10.0–15.0 mol%) and phase composition (calcination at 600–800 °C) confirmed maximum activity within the studied series for Mg–Cu–Co–Al–Ox with a Mg:Cu:Co:Al molar ratio of 51.0:10.0:10.0:29.0 calcined at 800 °C. Such relatively high temperature resulted in the formation of highly crystallized MgO and spinels (MgAl2O4, CuAl2O4, CoAl2O4 and/or Co3O4). The obtained results were explained based on the synergistic interaction between copper and cobalt oxides species as well as cobalt present in the form of spinels. It should also be stressed that the obtained results are fully consistent with the positive effect of copper on Co3O4 in N2O decomposition.168 Nevertheless, the stability of the evaluated Mg–Cu–Co–Al–Ox systems under ideal and real reaction conditions requires further investigations.
Kannan and Swamy146,149 reported the comparable activity of Ni–Al–Ox and catalysts containing cobalt. On the other side, catalysts containing additional Mg in the structure were found to be significantly less active catalysts compared to analogous Mg–Co(Cu)–Al–Ox.154 These findings were in agreement with the studies conducted by Obalová et al.155,156 The substitution of Ni with Mg resulted in significantly decreased catalytic activity: Ni:Al (4.0:2.0) > Ni:Mg:Al (3.0:1.0:2.0 > 2.0:2.0:2.0) > Mg:Al (4.0:2.0). A maximum N2O conversion of 79% was achieved at 450 °C over Ni–Al–Ox (Ni:Al = 4.0:2.0). Upon calcination at 450–500 °C, a Ni–Al precursor formed crystalline NiO. Additionally, the ternary mixed metal oxides included MgO, which was inactive in deN2O. However, according to Pérez-Ramírez et al.,160 the presence of magnesium in Ni–Al–O and Co–Al–O catalysts prevented deactivation of the catalysts due to preferential adsorption of SOx on MgO. Consequently, the active sites remained available for N2O reduction.
Integrating both Ni and Mn into one system(Ni–Mn–Ox, Mg–Ni–Mn–Ox) resulted in a low N2O conversion of 20% at 450 °C. (Ni,Mg)6MnO8 present in both samples could be responsible for the low activity.155 Partial substitution of Mn with Al in the series of Co–Mn resulted in higher catalytic activity.169 Obalová et al.44,158,159,169 investigated numerous combinations of Co–Mn–(Al)–Ox and found promising activity in the system with a Co:Mn:Al molar ratio of 4.0:1.0:1.0. The material precursors were prepared by coprecipitation and transformed into CoAl2O4 and/or Co3O4 at 500 °C. They provided a comprehensive study of this system in a N2O–He feed as well as in the presence of H2O, O2 and NO. The N2O conversion reached 82–97% at 450 °C. However, the conversion decreased in the presence of other components besides O2, e.g. 52 and 45% N2O conversions were achieved after passing 2.0% H2O or H2O together with 5.0% O2 over the catalyst, respectively. The activity loss caused by water vapour proved to be reversible.158 Although the authors did not comment on the reversibility of the deactivation caused by O2, both H2O and/or O2 possibly competed with N2O for the same adsorption sites. Considering structure–performance correlation, it was found that the highly efficient Co4Mn1Al1 catalyst contained an optimum surface amount of Co2+/Co3+ and Mn3+/Mn4+ molar ratios determined to be 1.13 and 2.27 by XPS analysis. Another crucial factor established from H2-TPR measurements (Fig. 8) was the optimum amount of components reducible in the temperature range of 350–450 °C – the same range in which deN2O proceeded.
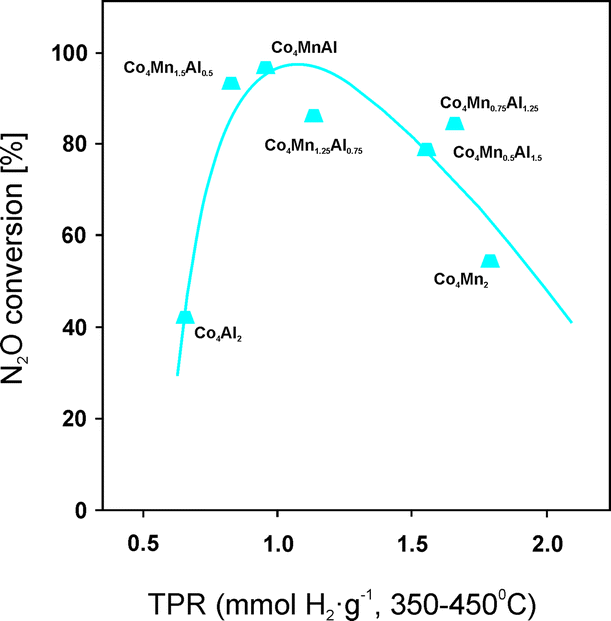 | ||
| Fig. 8 Dependence of N2O conversion on the amount of reducible components in the interval of 350–450 °C. Reaction conditions: 0.1% N2O, He balance; total flow rate = 100 ml min−1; mass of catalyst = 100 mg; temperature = 450 °C (adapted from ref. 169 with kind permission from Elsevier). | ||
Excellent conversion of N2O was achieved over Co–Rh–Al–Ox facilitating 100% conversion of N2O at a process temperature of 300–350 °C.147,149,170 Consequently, such systems present highly active catalysts at low temperatures leading to several studies covering these materials. The material precursors were prepared by coprecipitation followed by calcination at 450–500 °C. Due to the low Rh concentration, only spinels – Co3O4 and/or CoAl2O4 – were detected.149,160 Kannan et al.147–149 studied the effect of rhodium loading (0.3–1.0 wt%) on the catalytic activity and reported an optimal amount of 0.7 wt% Rh for Co–Rh–Al–Ox (Co:Al = 3.0:1.0). They found significant deactivation in the presence of 2.0% H2O and 2.5% O2 below 400 °C and no inhibition above 450 °C. Long-term stability tests were not carried out. Additionally, these studies mainly focused on the correlation of catalyst composition and catalytic activity rather than the explanation of the impact of the rhodium oxidation state, its particle size and dispersion. Nevertheless, several papers indicated the dispersion of rhodium as one of the crucial parameters influencing catalytic performance.171–173 Besides rhodium dispersion, particle size was also found to play a vital role in the high catalytic performance. For example, studies of Parres-Esclapez et al.174 over rhodium supported on γ-Al2O3, MgAl2O4 and SrAl2O3 revealed that the smaller the rhodium particle size, the higher the catalytic activity. Great research efforts have been made in order to determine which rhodium species in the catalytic decomposition of N2O are the most active. However, a final conclusion could not be drawn yet. Available literature data in this field indicate that the actual active species of rhodium depend on the catalyst composition.171,175
Alini et al.161 addressed these points. They studied H2-reduced (5.0% H2, 750 °C) Co–Rh–Al–Ox (Co:Rh:Al = 75.0:0.5:24.5) and Mg–Rh–Al–Ox (Mg:Rh:Al = 71.0:0.5:28.5, 71.0:1.0:28.0 or 80.0:1.0:19.0) at 450 °C, and reported that homogeneously dispersed Rh0 with an average particle size in the range of 1.0–3.0 nm was the active species for N2O decomposition. With 2.6% water vapour in the feed, the activity of the Co-based catalysts decreased from 55 to 38%, while the samples containing magnesium still enabled complete conversion. Interestingly, varying the Mg:Al molar ratios in the absence of H2O did not cause significant changes in the catalytic activity. In contrast, catalysts with a higher Mg:Al ratio (Mg:Rh:Al = 80.0:1.0:19.0) deactivated faster in wet long-term stability tests (after 400 h) compared to Mg:Rh:Al with a molar ratio of 71.0:1.0:28.0. This effect was explained by reoxidation of metallic rhodium. However, this deactivation in the presence of water vapour proved to be completely reversible, gaining full catalytic activity back by further heating the catalysts in reducing atmosphere.
Other reduced materials such as Mg–Rh–Pd–Al–Ox (Mg:Rh:Pd:Al = 70.0:0.5:1.0:28.5 or 70.5:0.5:0.5:28.5) as well as Mg–Rh–La–Al (71.0:1.0:5.0:23.0) showed lower activity in the presence of water vapour compared to Mg–Rh–Al–Ox. These lower N2O conversions, which were nevertheless still higher than that for Co–Rh–Al–Ox, were assigned to the possible segregation of PdO during calcination for the samples containing palladium. Segregated PdO was suggested to inhibit the synergistic interaction between both metallic rhodium and palladium. For La-containing materials, the formation of inactive La2CO5, which could also hinder the interaction of metallic rhodium and Mg–Al–Ox, was proposed to explain the low catalytic activity. Pérez-Ramírez et al.160 additionally confirmed a promoting effect of magnesium on the structure of Co–Rh–Al–Ox. They investigated the effect of 0.125 bar SO2 and 30.0 bar O2 in the feed for Co–Al–Ox (Co:Al = 3.0:1.0, 0.7 wt% Rh) and Mg–Co–Rh–Al–Ox (Mg:Co:Al = 1.0:3.0:1.0, 0.7 wt% Rh). For Co–Al–Ox at 450 °C, the conversion did not change after introducing SO2 and O2 into the feed. However, at temperatures around 325 °C, the catalyst was completely deactivated. After removal of SO2 and O2 from the feed, the activity did not return to its previous level. Additionally, the catalyst exhibited a significant drop of conversion from 80 to 60% during the first 10 h of the stability test at 300 °C. The presence of magnesium in Co–Rh–Al–O improved its stability in the presence of sulphur dioxide and oxygen and reduced deactivation in the long-term tests (100 h).
Although changes in the Mg:Al molar ratio and therefore the basicity of the oxide matrix did not cause significant differences in the activity for N2O decomposition of the mixed metal oxides,161 doping with some alkaline metals, such as Na, Li, K and Cs, was found to significantly increase the catalytic activity.44,163 In particular, the promoting effect of potassium on N2O decomposition was proven to be remarkable. Therefore, studies over various K-doped metal oxide catalysts were widely reported.154,162,163 However, in most cases, the presence of H2O, O2, and/or NOx (NO and NO2) inhibited N2O conversion.155,157,176 Note that such effects depended on the catalyst used as well as the feed composition. Deposition of small amounts of alkaline metals using an impregnation method followed by calcination did not change the structure of the support used. For example, Wu et al.157 studied Ni–Al–Ox (Ni:Al = 4.1:1.0) impregnated with potassium with a K:Ni molar ratio in the range of 0.05–0.2:1.0. The catalysts with a K:Ni molar ratio of 0.1:1.0 and calcined at 300–400 °C were the most active among the tested series. Full conversion of N2O over these catalysts could be reached at 450 °C. Materials with the same composition but calcined at 500 °C revealed lower N2O conversion by about 8%. The catalytic activity decreased with increasing K loading of the samples. Catalytic tests in the presence of 8.8% H2O and/or 4.0% O2 revealed that the presence of oxygen induced moderate inhibition on N2O decomposition over Ni–Al–Ox and K/Ni–Al–Ox (Ni:Al = 4.1:1.0, K:Ni = 0.1:1.0). Although the mixture of H2O and O2 significantly inhibited N2O decomposition, full conversion of N2O was still reached at 500 °C over the catalysts containing potassium, while N2O conversion over undoped Ni–Al–Ox was only 38%. The lower catalytic activity in the presence of H2O and/or O2 was most likely due to competitive adsorption of both components on the same active sites responsible for N2O decomposition. The same effect was observed over other hydrotalcite-derived mixed metal oxides for N2O decomposition, e.g. over K-promoted Co–Al–Ox (Co:Al = 3.0:1.0, K/Co = 0.02–0.12:1.0) studied by Cheng et al.162
The relationship between catalytic performance and content of potassium was further studied by Obalová et al.44,177 Deposition of potassium (0.0–3.0 wt%) on Co–Mn–Al–Ox (Co:Mn:Al = 4.0:1.0:1.0) was carried out by re-suspension of hydrotalcite-like precursors in aqueous solutions of potassium nitrate. After calcination at 500 °C, the samples with 2.7–3.0 wt% K contained spinels as well as KxMnO2. Modification of the Co–Mn–Al–O system with different amounts of potassium significantly changed the catalytic activity. Maximum activity in N2O decomposition could be observed for samples doped with both 0.9 and 1.6 wt% K. All modified samples were tested for N2O decomposition under various feed compositions, including 4.0% H2O, 5.0% O2, 0.17% NO and 0.1% NO2. The presence of H2O and O2 in the feed decreased the catalyst activity substantially. However, the activity could be completely recovered when both components were eliminated from the feed. Also, the presence of NOx influenced N2O decomposition. The catalysts with 0.9 wt% potassium were the most active ones for feeds containing not only N2O but also oxygen and nitrogen oxides.44 Another series of Co–Mn–Al–Ox doped with potassium was obtained by impregnation of (i) calcined hydrotalcite-like compounds or (ii) washed precipitates with an aqueous solution of KNO3.163 Among the tested samples, K (1.8 wt%)/Co–Mn–Al–Ox (Co:Mn:Al = 4.0:1.0:1.0) prepared by doping with potassium salt solution right after coprecipitation exhibited promising activity. Even in the presence of 0.9% H2O, 5.0% O2 and 0.005% NO, the catalyst reached 90% conversion at 450 °C. Catalytic tests of this material in the presence of 4.0% H2O and 5.0% O2 revealed only a 25% decrease in N2O conversion during the stability test for 6 h. In comparison, no loss in N2O conversion was observed for a N2O–He feed and a time-on-stream of 360 h. The excellent performance of this system confirms its great potential as an industrially relevant catalyst for N2O abatement.
Besides studies over deposited potassium, Xue et al.164 investigated the effect of residual potassium remaining after synthesis. Ce–Co–Ox (Ce:Co = 0.21:1.0) was prepared from appropriate metal nitrates with KOH as the precipitating agent, followed by calcination at 400 °C. Co3O4 and CeO2 were present in the material and a cooperative effect caused by their interaction was proposed. For around 0.7 mol% potassium in the sample, a downshift in the catalytic activity of around 50 °C compared to that of the catalyst without residual K occurred. Potassium-doped catalysts achieved around 96% conversion at 350 °C. However, no catalytic or stability tests in the presence of other components (e.g. H2O, O2 and/or NOx) were reported. Future studies should focus on real gas compositions to elucidate the full potential of the presented system. The positive effect of K was stronger compared to those of similar contents of Na (Fig. 9). In fact, the positive effect decreased in this sequence as follows: Cs > Rb > K > Na > undoped > Li.159 Also, studies concerning residual amounts or additionally added sodium were carried out. The influence of residual Na, which remained in trace amounts after washing step was provided by Farris et al.166 Both very low (below 0.16 wt%) and very high (above 15.0 wt%) residual sodium contents caused low N2O conversions of 50% and lower. An optimum sodium content in the range of 3.0–6.0 wt% in Co–Al–Ox was supposed to promote the decomposition of N2O, facilitating over 80% conversion at 475 °C. Doping with 1.0–2.0 wt% Na resulted in comparable results. In this line, deposition of residual sodium using impregnation followed by calcination appears to be more efficient to promote the decomposition of N2O.150 The location of sodium within the material remains unclear. Evidence of sodium species being incorporated into the structure of mixed metal oxides is not yet available. The optimum sodium loading is dependent on the mixed metal oxides used. For example, Xu et al.165 found 1.5 wt% sodium as the optimum loading ensuring higher activity compared to that over undoped Co–Al–Ox. A similar value (1.4 wt%) was reported for Na/Co–Mn–Al–Ox as one of the most active catalysts.163 Pérez-Ramírez et al.150 utilized Co–Rh–Al–Ox with different sodium contents ranging from 0.01 to 5.0 wt%. An optimum loading of the catalyst, which led to higher conversion compared to the undoped support, was established to be 0.75 wt%.
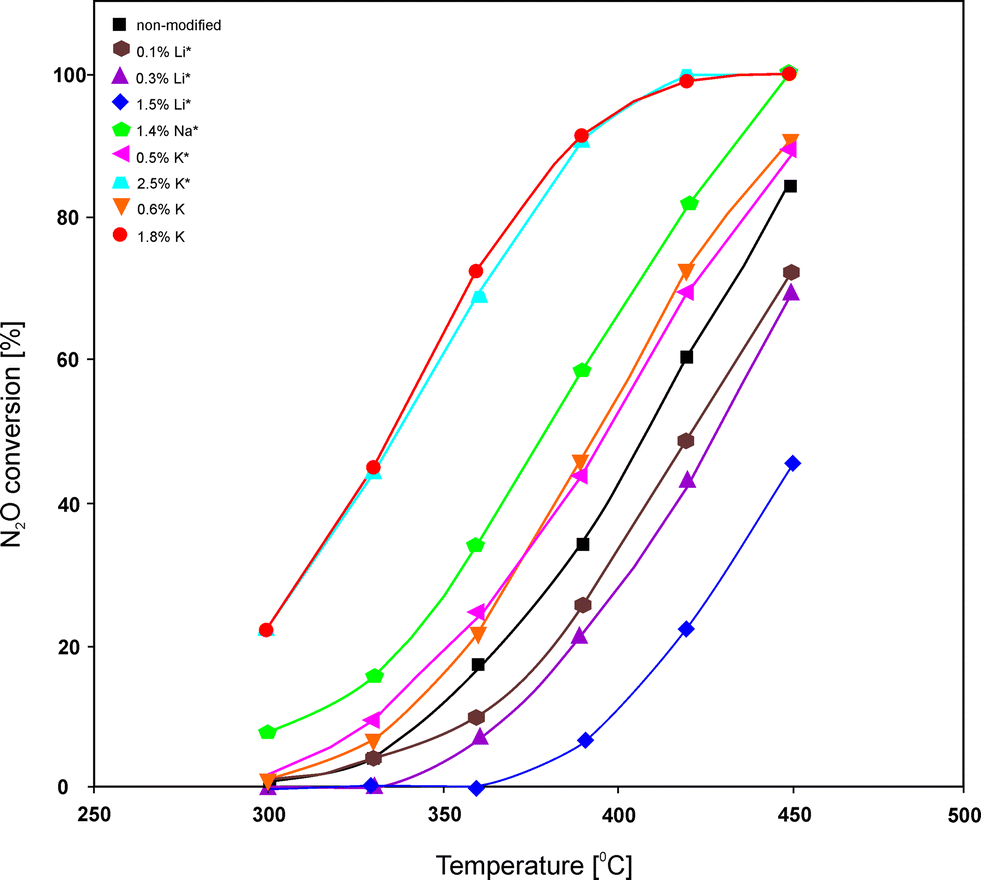 | ||
| Fig. 9 Results of catalytic tests performed for Co–Mn–Al–Ox modified with Li, Na or K; alkali metals were added after coprecipitation of the precursors or by impregnation of calcined precursors (samples labelled as *). Reaction conditions: 0.1% N2O, He balance; total flow rate = 100 ml min−1; mass of catalyst = 100 mg (adapted from ref. 163 with kind permission from Elsevier). | ||
Different explanations of the activating effect of alkali metals deposited on mixed metal oxides or incorporated within their structure were provided.44,157,162 A broad range of studies by Obalová et al.44,159 covering different mixed metal oxides modified with several alkali promoters revealed that the promoting effect of alkali metals was associated with their ionization potential, the charge transfer to the catalyst and a decrease in the binding energies of all catalyst components. The sequence of the promoting effect of alkali metals was explained in terms of charge donation from the alkali metal cations to surface oxygen and further to cobalt and manganese in Co–Mg–Al–Ox.159 Only for the system modified with Li, the N2O conversion of Co–Mg–Al–O decreased, a result well in line with the data published by Obalová et al.163,178
In conclusion, hydrotalcite-derived mixed metal oxides appear to be efficient catalysts for N2O decomposition even under simulated exhaust gas composition containing H2O, O2, SO2 and/or NOx.146–148,158,163 Therefore, this type of material can be considered as a serious candidate for the removal of N2O under industrially relevant conditions. Nevertheless, there is still space for improvement of the activity and stability of the most suitable catalytic systems such as Co–Rh–Al–Ox or K/Co–(Mn)–Al–Ox. Finally, catalytic tests under close to real HNO3 plant conditions are required.
Selective catalytic reduction of N2O
Studies related to the catalytic decomposition of N2O are mainly motivated by the abatement of waste gases of nitric acid production. Nevertheless, this method could also be used for N2O abatement of other waste gases.37,179 Another possibility concerns the selective reduction of N2O by carbon monoxide, ammonia or hydrocarbons. According to the generalized reaction mechanism, the mentioned reductants facilitate the removal of surface oxygen species generated during the decomposition of adsorbed N2O on the surface active site (*) as follows (eqn (16)–(19)):153| N2O + * ↔ N2O * | (16) |
| N2O * → N2 + O * | (17) |
| 2O * ↔ O2 + 2 * | (18) |
| N2O + O * → N2 + O2 + * | (19) |
Thus, they decrease the reaction operation temperature. Data for the catalytic performance of mixed metal oxides for such applications are still limited in the scientific literature. Table 7 summarizes the relevant results. The maximum N2O conversion achieved in deN2O was compared with the one obtained in N2O-SCR.
| Catalyst code | Preparation method (calcination temperature/°C) | Reaction conditions | N2O conversion/% (temperature/°C) | Ref. |
|---|---|---|---|---|
| Co–Mn–Al (4.0:1.0:1.0 mol%) |
Coprecipitation (500) | 0.1% N2O; He balance (*with 0.15% CO) (**with 0.15% CO; 20.0% O2); W/F = 60 g s l−1 | 89 (450) *100 (350) **52 (450) | 180 |
| Co–Rh–Al (1.0:0.2:1.0 mol%) |
Coprecipitation (500) | 1.25% N2O (*with 1.75% CO) (**with 1.75% CO; 0.5% O2); GHSV = 30000 h−1 |
100 (350) *100 (200) **100 (400) | 170, 181 |
| Co–Rh–Ce–Al (1.0:0.2:0.01:1.0 mol%) |
100 (400) *100 (200) **80 (500) | 181 | ||
| Co–Pd–Al (1.0:0.1:1.0 mol%) |
Coprecipitation (500) | 1.25% N2O; GHSV = 30000 h−1 (*2.5% N2O; 1.0% NO; 5.0% CO; **2.5% N2O; 2.0% NO; 3.5% CO; GHSV = 20000 h−1) |
100 (450) *100 (200) **100 (350) | 170 |
| Mg–Fe (70.0:30.0 mol%) |
Coprecipitation (500) | 0.1% N2O (*with 0.1% NH3); W/F = 460 g s l−1 | 80 (500) *100 (450) | 182 |
| Mg–Al–Fe (50.0:45.0:5.0 mol%) |
10 (500) *100 (500) | |||
| Rh/Mg–Al (7.0:1.5 mol%; 1.6 wt% Rh) |
Commercial/impregnation (650/600) | 0.05% N2O; 0.1% C3H6; 5.0% O2; He balance; W/F = 120 g s l−1 | 100 (600) | 183 |
| Mg–Fe (38.1:25.5 wt%) |
Coprecipitation (650) | 0.8% N2O (*with 0.8% C8H10); W/F = 60 g s l−1 | 16 (550) *80 (450) | 184 |
| Mg–Cr (36.6:26.7 wt%) |
*40 (450) |
Co–Mn–Al–O with a Co:Mn:Al molar ratio of 4.0:1.0:1.0, which revealed high potential in the decomposition of N2O, was additionally tested in the selective reduction of nitrous oxide with carbon monoxide.180 The presence of CO significantly increased the N2O conversion. 100% N2O conversion was achieved at 350 °C in the presence of CO, while only 89% conversion was reached at 450 °C for direct N2O decomposition. Adding 20.0% O2 into the feed and maintaining this temperature level, the conversion decreased to below 10%. The significant activity loss in the presence of O2 could be related to competitive adsorption on the active sites responsible for the high catalytic activity.
Decreased N2O conversion in the presence of both carbon monoxide and oxygen was also reported by Chang et al.181 over Co–Rh–(Pd)–(Ce)–Al–Ox. The catalysts were prepared by coprecipitation and transformed at 500 °C into Co3O4 and/or CoAl2O4. N2O was converted at around 200 °C over all catalysts without oxygen in the feed. Instead, in the presence of both 1.75% CO and 0.5% O2, total conversion required 400 °C for Co–Rh–Al–Ox (Co:Rh:Al = 1.0:0.2:1.0). When cerium was incorporated, the catalytic activity significantly decreased. A similar effect was observed after substituting rhodium with palladium in the Co–Rh–Al–O system. On the other side, introducing 0.01% NO into the feed containing nitrous oxide and carbon monoxide did not cause significant inhibition of the tested catalysts. Chang et al.170 additionally examined the effect of NO on the reduction of N2O with CO over Co–Pd–Al–Ox (Co:Pd:Al = 1.0:0.1:1.0). Catalytic tests with different compositions (N2O:NO:CO = 1.5–2.5%:1.0–3.0%:3.5–7.0%) confirmed that N2O conversion could only be improved when excess amounts of carbon monoxide were introduced into the feed. Both of the discussed studies did not present long-term stability tests, but mainly correlated catalyst composition and catalytic activity in various feeds. Further investigations appear necessary aiming at a better understanding of the role of the metals introduced into the presented systems. Mixed metal oxides also exhibited noticeable efficiency in the selective catalytic reduction of nitrous oxide with NH3 and with various other hydrocarbons as reducing agents.182,183 Vulic et al.182 investigated N2O reduction with ammonia over Mg–(Al)Fe–Ox (5.0 or 30.0 mol% Fe). The precursors were prepared by coprecipitation followed by calcination at 500 °C and formation of poorly crystallized MgO. Samples containing a low amount of iron (5.0 mol%), independent of the Mg:Al molar ratios (Mg:Al:Fe = 85.0–30.0:10.0–65.0:5.0), showed N2O conversions of around 10% in the decomposition of nitrous oxide between 450 and 500 °C. Alini et al.161 draw the same conclusion for Mg–Rh–Al–Ox with different Mg:Al molar ratios of 80.0–71.0:19.0–28.0 and a constant loading of rhodium (1.0 mol%). Increasing the iron content up to 30.0 mol% in Mg–Fe–Ox significantly improved the conversion to 80% at 500 °C. However, the same catalysts enabled complete N2O conversion at 450 °C in the presence of ammonia. The catalytic activity also increased for other tested materials, confirming the influence of the redox properties on the catalytic behaviour. More easily reduced Fe3+ species caused lower catalytic activity.
Christoforou et al.183 studied N2O reduction in the presence of 0.1% C3H6 and 5.0% O2. A calcined commercial Mg–Al (Mg:Al = 7.0:1.5) hydrotalcite-like precursor was used as a support for 1.6 wt% rhodium. This material facilitated full N2O conversion at temperatures as high as 600 °C. A comparative study covered also Al2O3, SiO2, ZrO2 and TiO2 as supports. No detailed physicochemical characterization of the materials was provided; however, the authors argued that the differences in activity could be due to variation of rhodium dispersion on the different support materials. Indeed, higher dispersion of the metal could be achieved by its incorporation within brucite-like sheets, instead of its deposition on the calcined hydrotalcite-like compounds. This methodology could certainly improve the results presented herein.
High temperatures for N2O reduction with 0.8% C8H10 (ethylbenzene) were also found by Kuśtrowski et al.184 over a series of Mg–Fe–Cr–Ox catalysts (0.0–25.5 wt% Fe and 0.0–26.7 wt% Cr). The precursors were prepared by coprecipitation and subsequently transformed into mixed metal oxides at 650 °C. Besides poorly crystallized MgO, spinels (MgCr2O4 and/or MgFe2O4) were also detected in all samples. Iron was found to be a more active component than chromium. The catalytic tests revealed high activity only for samples containing the highest amount of Fe, i.e. Fe3+ in the tetrahedral sites of the spinel structure, which was determined by Mössbauer spectroscopy. In particular, Mg–Fe–Ox with 25.5 wt% Fe enabled 80% N2O conversion at 450 °C. At 550 °C, N2O conversion exceeded 92% over all tested catalysts.
As stated above, the selective catalytic reduction of N2O with carbon monoxide, ammonia or hydrocarbons seems to be another attractive route to eliminate nitrous oxide. However, few studies address N2O-SCR over mixed metal oxides. Further investigations concerning the major factors governing the activity and selectivity as well as the influence of water and oxygen in the feed stream appear to be necessary.
Conclusions
Selective catalytic reduction of NOx based on urea-SCR (NH3-SCR) or hydrocarbon-SCR (HC-SCR), NOx storage/reduction (NSR) and simultaneous NOx–soot removal are the predominant methods dedicated to diesel exhaust gas aftertreatment. N2O emission abatement through catalytic decomposition (deN2O) is well established for nitric or adipic acid plants. However, appropriate reductants also facilitate the selective catalytic reduction of N2O (N2O-SCR). Therefore, efficient N2O removal at much lower temperatures becomes possible. This seems to be an opportunity for removal of N2O generated as a by-product in the different stages of diesel after-treatment systems. The diesel oxidation catalyst (DOC), selective catalytic reduction (SCR) and/or selective catalytic oxidation (SCO) can contribute to N2O formation depending on the (i) operating temperatures, (ii) catalysts used and (iii) exhaust gas conditions as well as (iv) after-treatment system configuration. Thus, subsequent reduction of N2O over suitable catalysts is indispensable to meet future regulations.Hydrotalcite-derived mixed metal oxides are one of the classes of materials tested for the reduction of nitrogen oxides from both stationary and mobile sources. The present minireview summarizes the relationship between the different active phases and their catalytic performance. Although, a large number of chemical and/or phase formulations have been reported in order to develop active, selective and stable low-temperature catalysts, major challenges remain. One of the main challenges concerns the design of catalysts able to cope with a broad operating temperature window with high catalytic performance. This point is of special importance considering the broad temperature range of exhaust gases emitted by diesel engines, which can even increase up to 600 °C. Furthermore, the majority of the studies have been carried out under ideal conditions. Only a limited number of investigations considered gas mixtures which mimic real exhaust gases. Overall, hydrotalcites have proven their high potential in several systems for nitrogen oxide removal, e.g. NSR and SCR or deNO2/N2O-SCR and SCR). Bridging fundamental and applied research, further studies with structured, e.g. monolithic catalysts and suitable gas mixtures, appear highly relevant aiming at the development of tailored high-performance catalysts.
Acknowledgements
This work was funded by the Excellence Initiative of the German Federal and State Governments in the frame of the Center for Automotive Catalytic Systems Aachen (ACA) at RWTH Aachen University.References
- NEC Direcctive Status Report 2013 Online, http://www.eea.europa.eu/publications/nec-directive-status-report-2013 (accessed January 2015).
- V. I. Pârvulescu, P. Grange and B. Delmon, Catal. Today, 1998, 46, 233 CrossRef.
- X. Tang, S. Madronich, T. Wallington and D. Calamari, J. Photochem. Photobiol., B, 1998, 46, 83 CrossRef CAS.
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117 CrossRef.
- M. J. Scott, R. D. Sands, N. J. Rosenberg and R. C. Izaurralde, NATO ASI Ser., Ser. I, 2002, 12, 105 Search PubMed.
- F. Klingstedt, K. Arve, K. Eränen and D. Y. Murzin, Acc. Chem. Res., 2006, 39, 273 CrossRef CAS PubMed.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, Cascade Biocatal., 2013, 3, 413 CAS.
- L. Yan, X. Zhang, T. Ren, H. Zhang, X. Wang and J. Suo, Chem. Commun., 2002, 860 RSC.
- K. H. Becker, J. C. Lörzer, R. Kurtenbach, P. Wiesen, T. E. Jensen and T. J. Wallington, Environ. Sci. Technol., 1999, 33, 4134 CrossRef CAS.
- B. Guan, R. Zhan, H. Lin and Z. Huang, Appl. Therm. Eng., 2014, 66, 395 CrossRef CAS.
- G. Centi, G. E. Arena and S. Perathoner, J. Catal., 2003, 216, 443 CrossRef CAS.
- N. Imanaka and T. Masui, Appl. Catal., A, 2012, 431–432, 1 CrossRef CAS.
- Z. M. Liu and S. I. Woo, Catal. Rev.: Sci. Eng., 2006, 48, 43 CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283 CrossRef CAS.
- N. Takahashi, H. Shinjoh, T. Iijima, T. Suzuki, K. Yamazaki, K. Yokota, H. Suzuki, N. Miyoshi, S. Matsumoto, T. Tanizawa, T. Tanaka, S. Tateishi and K. Kasahara, Catal. Today, 1996, 27, 63 CrossRef CAS.
- R. Yang, Y. Gao, J. Wang and Q. Wang, Dalton Trans., 2014, 43, 10317 RSC.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25 CrossRef CAS.
- C. Pophal, T. Yogo, K. Tanabe and K. Segawa, Catal. Lett., 1997, 44, 271 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- M. Jabłońska, L. Chmielarz, A. Węgrzyn, K. Guzik, Z. Piwowarska, S. Witkowski, R. I. Walton, P. W. Dunne and F. Kovanda, J. Therm. Anal. Calorim., 2013, 114, 731 CrossRef.
- P. Kuśtrowski, A. Węgrzyn, L. Chmielarz, A. Bronkowska, A. Rafalska-Łasocha and R. Dziembaj, J. Therm. Anal. Calorim., 2004, 77, 243 CrossRef.
- N. Gutmann and B. Mueller, J. Solid State Chem., 1996, 122, 214 CrossRef CAS.
- F. Kooli, V. Rives and M. A. Ulibarri, Inorg. Chem., 1995, 34, 5114 CrossRef CAS.
- T. Lopez, P. Bosh, E. Ramos, R. Gomez, O. Novaro, D. Acosta and F. Figueras, Langmuir, 1996, 12, 189 CrossRef CAS.
- J. M. Oh, S. H. Hwang and J. H. Choy, Solid State Ionics, 2002, 151, 285 CrossRef CAS.
- J. He, M. Wei, B. Li, Y. Kang, D. G. Evans and X. Duan, Struct. Bonding, 2005, 119, 89 CrossRef.
- S. Miyata, Clays Clay Miner., 1980, 28, 50 CAS.
- D. G. Evans and R. C. T. Slade, Struct. Bonding, 2006, 119, 1 CAS.
- V. Rives and M. A. Ulibarri, Coord. Chem. Rev., 1999, 181, 61 CrossRef.
- U. Costantino, M. Nocchetti, M. Sisani and R. Vivani, Z. Kristallogr., 2009, 224, 273 CrossRef CAS.
- U. Costantino, V. Ambrogi, M. Nocchetti and L. Perioli, Microporous Mesoporous Mater., 2008, 107, 149 CrossRef CAS.
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026 RSC.
- P. Nalawade, B. Aware, V. J. Kadam and R. S. Hirlekar, J. Sci. Ind. Res., 2009, 68, 267 CAS.
- Z. P. Xu, J. Hang, M. O. Adebajo, H. Zhang and C. H. Zhou, Appl. Clay Sci., 2011, 53, 139 CrossRef CAS.
- D. Ticht and B. Coq, CATTECH, 2003, 7, 206 CrossRef.
- A. Vaccari, Catal. Today, 1998, 41, 53 CrossRef CAS.
- S. Kannan, Catal. Surv. Asia, 2006, 10, 117 CrossRef CAS.
- K. H. Goh, T. T. Lim and Z. Dong, Water Res., 2008, 42, 1343 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, Microporous Mesoporous Mater., 2008, 107, 3 CrossRef CAS.
- B. F. Sels, D. E. de Vos and P. A. Jacobs, Catal. Rev.: Sci. Eng., 2001, 43, 443 CAS.
- B. Montanari, A. Vaccari, M. Gazzano, P. Käβner, H. Papp, J. Pasel, R. Dziembaj, W. Makowski and T. Łojewski, Appl. Catal., B, 1997, 13, 205 CrossRef CAS.
- G. Centi, G. Fornasari, C. Gobbi, M. Livi, F. Trifirò and A. Vaccari, Catal. Today, 2002, 73, 287 CrossRef CAS.
- A. Corma, A. E. Palomares, F. Rey and F. Marquéz, J. Catal., 1997, 170, 140 CrossRef CAS.
- L. Obalová, K. Karásková, K. Jirátová and F. Kovanda, Appl. Catal., B, 2009, 90, 132 CrossRef.
- A. Corma, A. E. Palomares, F. Rey and F. Márquez, J. Catal., 1997, 170, 140 CrossRef CAS.
- A. E. Palomares, C. Franch, A. Ribera and G. Abellán, Catal. Today, 2012, 191, 47 CrossRef CAS.
- L. Chmielarz, M. Jabłońska, A. Strumiński, Z. Piwowarska, A. Węgrzyn, S. Witkowski and M. Michalik, Appl. Catal., B, 2013, 130, 152 CrossRef.
- L. Chmielarz, A. Węgrzyn, M. Wojciechowska, S. Witkowski and M. Michalik, Catal. Lett., 2011, 141, 1345 CrossRef CAS.
- M. Jabłońska, Selective Ammonia Oxidation into Nitrogen and Water Vapour, LAP Lambert Academic Publishing, 1st edn, 2014 Search PubMed.
- J. Haber, K. Bahranowski, J. Janas, R. Janik, T. Machej, L. Matachowski, A. Michalik, H. Sadowska and E. M. Serwicka, Stud. Surf. Sci. Catal., 2000, 130, 599 CrossRef.
- P. Forzatti, Appl. Catal., A, 2001, 222, 221 CrossRef CAS.
- C. Enderle, G. Vent and M. Paule, SAE Technical Paper 2008-01-1182, 2008 Search PubMed.
- H. Bosch and F. Janssen, Catal. Today, 1988, 2, 369 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1 CrossRef CAS.
- R. M. Heck, Catal. Today, 1999, 53, 519 CrossRef CAS.
- X. L. Tang, J. M. Hao, W. G. Xu and J. H. Li, Catal. Commun., 2007, 8, 329 CrossRef CAS.
- R. Q. Long and R. T. Yang, Catal. Lett., 2001, 74, 201 CrossRef CAS.
- L. Chmielarz, A. Kowalczyk, M. Wojciechowska, P. Boroń, B. Dudek and M. Michalik, Chem. Pap., 2014, 68, 1219 CrossRef CAS.
- T. Grzybek, Catal. Today, 2007, 119, 125 CrossRef CAS.
- Y. Feng, S. Liu, Ch. Chen, H. Zhao and Y. Xu, Appl. Mech. Mater., 2013, 320, 629 CrossRef.
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147 CrossRef CAS.
- P. Boroń, L. Chmielarz and S. Dzwigaj, Appl. Catal., B, 2015, 168, 377 CrossRef.
- G. Carja and G. Delahay, Appl. Catal., B, 2004, 47, 59 CrossRef CAS.
- G. Carja, S. Dranca, E. Husanu and I. Volf, Environ. Eng. Manage. J., 2009, 8, 553 CAS.
- T. Wongkerd, A. Luengnaruemitchai and S. Jitkarnka, Appl. Catal., B, 2008, 78, 101 CrossRef CAS.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha, D. Majda and R. Dziembaj, Appl. Catal., B, 2002, 35, 195 CrossRef CAS.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha and R. Dziembaj, Appl. Catal., B, 2005, 58, 235 CrossRef CAS.
- K. Arakawa, S. Matsuda and H. Kimoshita, Appl. Surf. Sci., 1997, 121–122, 382 CrossRef.
- A. Sultana, M. Sasaki, K. Suzuki and H. Hamada, Catal. Commun., 2013, 41, 21 CrossRef CAS.
- Air Pollution Control Technology Fact Sheet Online, http://www.epa.gov/ttncatc1/dir1/fscr.pdf, (accessed January 2015).
- Regulation (EC) no 595/2009 of the European Parliament and of the Council of 18 June 2009.
- M. Iwamoto, N. Mizuno and H. Yahiro, Stud. Surf. Sci. Catal., 1993, 75, 1285 CrossRef CAS.
- V. Houel, D. James, P. Millington, S. Pollington, S. Poulston, R. Rajaram and R. Torbati, J. Catal., 2005, 230, 150 CrossRef CAS.
- J. H. Kwak, H. Y. Zhu, J. H. Lee, C. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758 RSC.
- P. A. Kumar, M. P. Reddy, L. K. Ju, B. Hyun-Sook and H. H. Phil, J. Mol. Catal. A: Chem., 2008, 291, 66 CrossRef CAS.
- J. H. Kwak, H. Y. Zhu, J. H. Lee, C. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758 RSC.
- P. A. Kumar, M. P. Redy, L. K. Ju, B. Hyuan-Sook and H. H. Phil, J. Mol. Catal. A: Chem., 2008, 291, 66 CrossRef CAS.
- L. C. Wang, Q. Liu, M. Chen, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Phys. Chem. C, 2007, 111, 16549 CAS.
- K. A. Bethke, M. C. Kung, B. Yang, M. Shah, D. Alt, C. Li and H. H. Kung, Catal. Today, 1995, 26, 169 CrossRef CAS.
- M. Huuhtanen, T. Maunula and R. L. Keiski, Stud. Surf. Sci. Catal., 2005, 158, 1867 CrossRef.
- H. Hamada, Y. Kintaichi, M. Inaba, M. Tabata, T. Yoshinari and H. Tscuchisa, Catal. Today, 1996, 29, 53 CrossRef CAS.
- D. Yuan, X. Li, Q. Zhao, J. Zhao, S. Liu and M. Tadé, Appl. Catal., A, 2013, 451, 176 CrossRef CAS.
- D. Yuan, X. Li, Q. Zhao, J. Zhao, M. Tadé and S. Liu, J. Catal., 2014, 309, 268 CrossRef CAS.
- V. F. Tret’yakov, A. G. Zakirova, A. A. Spozhakina, M. V. Gabrovska, R. Edreva-Kardzhieva and L. A. Petrov, Catal. Ind., 2010, 2, 62 CrossRef.
- H. B. Liu, Z. Y. Huang, L. J. Li, J. T. Huang, C. H. Zhi, H. C. Li, B. G. Wu and Y. Z. Wu, Adv. Mater. Res., 2014, 1033–1034, 1058 CrossRef CAS.
- R. S. Mulukutla and C. Detellier, J. Mater. Sci. Lett., 1997, 16, 752 CrossRef CAS.
- R. S. Mulukutla and C. Detellier, J. Mater. Sci. Lett., 1996, 15, 797 CrossRef CAS.
- N. Li, A. Wang, M. Zheng, X. Wang, R. Cheng and T. Zhang, J. Catal., 2004, 225, 307 CrossRef CAS.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks, Catal. Rev.: Sci. Eng., 2004, 46, 163 Search PubMed.
- L. Lietti, P. Forzatti, I. Nova and E. Tronconi, J. Catal., 2001, 204, 175 CrossRef CAS.
- T. Nakatsuji, R. Yasukawa, K. Tabata, K. Ueda and M. Niwa, Appl. Catal., B, 1999, 21, 121 CrossRef CAS.
- T. J. Toops, D. B. Smith, W. S. Epling, J. E. Parks and W. P. Partridge, Appl. Catal., B, 2005, 58, 255 CrossRef CAS.
- K. Yamazaki, N. Takahashi, H. Shinjoh and M. Sugiura, Appl. Catal., B, 2004, 53, 1 CrossRef CAS.
- E. C. Corbos, X. Courtois, N. Bion, P. Marecot and D. Duprez, Appl. Catal., B, 2008, 80, 62 CrossRef CAS.
- G. Liu and P. X. Gao, Catal. Sci. Technol., 2011, 1, 552 CAS.
- S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054 CrossRef CAS PubMed.
- P. Forzatti, L. Castoldi, L. Lietti, I. Nova and E. Tronconi, Stud. Surf. Sci. Catal., 2007, 171, 175 CrossRef CAS.
- M. S. Brogan, A. D. Clark, M. J. Spencer and R. J. Brisley, SAE Techn. Pap., 1998 Search PubMed.
- G. Fornasari, F. Trifiró, A. Vaccari, F. Prinetto, G. Ghiotti and G. Centi, Catal. Today, 2002, 75, 421 CrossRef CAS.
- F. Basile, G. Fornasari, M. Livi, F. Trini, F. Trifiró and A. Vaccari, Top. Catal., 2004, 30–31, 223 CrossRef.
- J. J. Yu, J. Cheng, Ch. Y. Ma, H. L. Wang, L. D. Li, Z. P. Hao and Z. P. Xu, J. Colloid Interface Sci., 2009, 333, 423 CrossRef CAS PubMed.
- M. Jabłońska, A. E. Palomares, A. Węgrzyn and L. Chmielarz, Acta Geodyn. Geomater., 2014, 11, 175 Search PubMed.
- B. A. Silletti, R. T. Adams, S. M. Sogmon, A. Nikolopoulos, J. J. Spivey and H. H. Lamb, Catal. Today, 2006, 114, 64 CrossRef CAS.
- L. D. Li, J. J. Yu, Z. P. Hao and Z. P. Xu, J. Phys. Chem. C, 2007, 111, 10552 CAS.
- J. J. Yu, Z. Jiang, L. Zhu, Z. P. Hao and Z. P. Xu, J. Phys. Chem. B, 2006, 110, 4291 CrossRef CAS PubMed.
- J. Cheng, X. Wang, C. H. Ma and Z. Hao, J. Environ. Sci., 2012, 24, 488 CrossRef CAS.
- J. J. Yu, X. P. Wang, L. D. Li, Z. P. Hao and Z. P. Xu, Adv. Funct. Mater., 2007, 17, 3598 CrossRef CAS.
- H. Cheng, G. Chen, S. Wang, D. Wu, Y. Zhang and H. Li, Korean J. Chem. Eng., 2004, 21, 595 CrossRef CAS.
- G. Fornasari, R. Glöckler, M. Livi and A. Vaccari, Appl. Clay Sci., 2005, 29, 258 CrossRef CAS.
- A. E. Palomares, A. Uzcategui and A. Corma, Catal. Today, 2008, 137, 261 CrossRef CAS.
- M. Jabłońska, A. E. Palomares and L. Chmielarz, Chem. Eng. J., 2013, 213, 273 CrossRef.
- Y. Su, Mechanistic Investigation of Nitrogen Oxide Storage and Reduction Catalyst, 1st edn., ProQuest, 2007 Search PubMed.
- X. He, M. Meng, J. He, Z. Zou, Z. Li and Z. Jiang, Catal. Commun., 2010, 12, 165 CrossRef CAS.
- Z. Li, M. Meng, F. Dai, T. Hu, Y. Xie and J. Zhang, Fuel, 2012, 93, 606 CrossRef CAS.
- S. F. Kang, Z. Jiang and Z. P. Hao, Wuli Huaxue Xuebao, 2005, 31, 278 Search PubMed.
- M. De Boeck, M. Kirsch-Volders and D. Lison, Mutat. Res., 2003, 533, 135 CrossRef CAS PubMed.
- A. P. Walker, Top. Catal., 2004, 28, 165 CrossRef CAS.
- R. Allansson, P. G. Blakeman, B. J. Copper, H. Hess, P. J. Silcock and A. P. Walker, SAE Technical Paper 2002-01-0428, 2002 Search PubMed.
- A. Setiabudi, M. Makkee and J. A. Moulijn, Top. Catal., 2002, 30, 305 Search PubMed.
- J. Suzuki and S. Matsumoto, Top. Catal., 2004, 28, 171 CrossRef CAS.
- R. Matarrese, L. Castoldi, L. Lietti and P. Forzatti, Top. Catal., 2009, 52, 2041 CrossRef CAS.
- K. Yoshida, S. Makino, S. Sumiya, G. Muramatsu and R. Helferich, SAE Tech. Pap. Ser., 1989 Search PubMed.
- J. Klein, I. Fechete, V. Bresset, F. Garin and V. Tschamber, Catal. Today, 2012, 189, 60 CrossRef CAS.
- Y. Teroka, K. Kanada, H. Furukawa, I. Moriguchi and S. Kagawa, Chem. Lett., 2001, 30, 604 CrossRef.
- Z. Liu, Z. Hao, Y. Guo and Y. Zhuang, J. Environ. Sci., 2002, 14, 289 CAS.
- Y. Teraoka, K. Nakano, S. Kagawa and W. F. Shangguan, Appl. Catal., B, 1995, 5, L181 CrossRef CAS.
- W. F. Shangguan, Y. Teraoka and S. Kagawa, Appl. Catal., B, 1998, 16, 149 CrossRef CAS.
- Q. Li, M. Meng, H. Xian, N. Tsubaki, X. Li, Y. Xie, T. Hu and J. Zhang, Environ. Sci. Technol., 2010, 44, 4747 CrossRef CAS PubMed.
- Q. Li, M. Meng, F. Dai, Y. Zha, Y. Xie, T. Hu and J. Zhang, Chem. Eng. J., 2012, 184, 106 CrossRef CAS.
- Q. Li, M. Meng, N. Tsubaki, X. Li, Z. Li, Y. Xie, T. Hu and J. Zhang, Appl. Catal., B, 2009, 91, 406 CrossRef CAS.
- Z. Wang, X. Yan, X. Bi, L. Wang, Z. Zhang, Z. Jiang, T. Xiao, A. Umar and Q. Wang, Mater. Res. Bull., 2014, 51, 119 CrossRef CAS.
- Q. Li, M. Meng, Z. Q. Zou, X. G. Li and Y. Q. Zha, J. Hazard. Mater., 2009, 161, 366 CrossRef CAS PubMed.
- B. A. A. L. van Setten, J. M. Schouten, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2000, 28, 253 CrossRef CAS.
- J. H. Kwak, D. H. Kim, J. Szanyi, S. J. Cho and Ch. H. F. Peden, Top. Catal., 2012, 55, 70 CrossRef CAS.
- N. Takahashi, S. Matsunaga, T. Tanaka, H. Sobukawa and H. Shihnjoh, Appl. Catal., B, 2007, 77, 73 CrossRef CAS.
- Y. Teraoka, K. Kanada and S. Kagawa, Appl. Catal., B, 2001, 34, 73 CrossRef CAS.
- F. Dai, M. Meng, Y. Zha, Z. Li, T. Hu, Y. Xie and J. Zhang, Fuel Process. Technol., 2012, 104, 43 CrossRef CAS.
- Q. Shen, G. Lu, Ch. Du, Y. Guo, Y. Wang, Y. Guo and X. Gong, Chem. Eng. J., 2013, 218, 164 CrossRef CAS.
- M. Inger, M. Saramok, M. Wilk, P. Stelmachowski, F. Zasada, G. Maniak, W. Piskorz, A. Adamski, A. Kotarba, Z. Sojka and P. Granger, Przemysł Chemiczny, 2009, 88, 1307 CAS.
- P. Stelmachowski, G. Maniak, A. Kotarba and Z. Sojka, Catal. Commun., 2009, 10, 1062 CrossRef CAS.
- J. A. Z. Pieterse, G. Mul, I. Melian-Cabrera and R. W. van den Brink, Catal. Lett., 2005, 99, 41 CrossRef CAS.
- M. Rutkowska, L. Chmielarz, M. Jabłońska, C. J. Van Oers and P. Cool, J. Porous Mater., 2014, 21, 91 CrossRef CAS.
- J. Oi, A. Obuchi, G. R. Bamwenda, A. Ogata, H. Yagita, S. Kushiyama and K. Mizuno, Appl. Catal., B, 1997, 12, 277 CrossRef CAS.
- T. N. Angelidis and V. Tzitzios, Ind. Eng. Chem. Res., 2003, 42, 2996 CrossRef CAS.
- P. Taniou, Z. Ziaka and S. Vasileiadis, American Transactions on Engineering & Applied Sciences, 2013, 2, 149 Search PubMed.
- S. Kannan and C. S. Swamy, Appl. Catal., B, 1994, 3, 109 CrossRef CAS.
- C. S. Swamy, S. Kannan, Y. Li, J. N. Armor and T. A. Braymer, US Pat., 5407652 A, 1995 Search PubMed.
- J. N. Armor, T. A. Braymer, T. S. Farris, Y. Li, F. P. Petrocelli, E. L. Weist, S. Kannan and C. S. Swamy, Appl. Catal., B, 1996, 7, 397 CrossRef CAS.
- S. Kannan, Appl. Clay Sci., 1998, 13, 347 CrossRef CAS.
- J. Pérez-Ramírez, J. M. García-Cortés, F. Kapteijn, M. J. Illán-Gómez, A. Ribera, C. Salinas-Martínez de Lecea and J. A. Moulijn, Appl. Catal., B, 2000, 25, 191 CrossRef.
- L. Oi, A. Obuchi, A. Ogata, G. R. Bamwenda, R. Tanaka, T. Hibino and S. Kushiyama, Appl. Catal., B, 1997, 13, 197 CrossRef.
- J. Pérez-Ramírez, F. Kapteijn and J. A. Moulijn, Catal. Lett., 1999, 60, 133 CrossRef.
- L. Obalová and V. Fíla, Appl. Catal., B, 2007, 70, 353 CrossRef.
- L. Chmielarz, M. Rutkowska, P. Kuśtrowski, M. Drozdek, Z. Piwowarska, B. Dudek, R. Dziembaj and M. Michalik, J. Therm. Anal. Calorim., 2011, 105, 161 CrossRef CAS.
- L. Obalová, K. Jirátová, F. Kovanda, M. Valášáková, J. Balabánová and K. Pacultová, J. Mol. Catal. A: Chem., 2006, 248, 210 CrossRef.
- L. Obalová, M. Valášáková, F. Kovanda, Z. Lacný and K. Kolinová, Chem. Pap., 2004, 58, 33 Search PubMed.
- H. Wu, Z. Qian, X. Xu and X. Xu, Ranliao Huaxue Xuebao, 2011, 39, 115 CAS.
- L. Obalová, K. Jirátova, F. Kovanda, K. Pacultová, Z. Lacný and Z. Mikulová, Appl. Catal., B, 2005, 60, 289 CrossRef.
- L. Obalová, K. Karásková, A. Wach, P. Kuśtrowski, K. Mamulová-Kutláková, S. Michalik and K. Jirátová, Appl. Catal., A, 2013, 462–463, 227 CrossRef.
- J. Pérez-Ramírez, J. Overeijnder, F. Kapteijn and J. A. Moulijn, Appl. Catal., B, 1999, 23, 59 CrossRef.
- S. Alini, F. Basile, A. Bologna, T. Montanari and A. Vaccari, Stud. Surf. Sci. Catal., 2002, 143, 131 CrossRef CAS.
- H. Cheng, Y. Huang, A. Wang, L. Li, X. Wang and T. Zhang, Appl. Catal., B, 2009, 89, 391 CrossRef CAS.
- K. Karásková, L. Obalová, K. Jirátová and F. Kovanda, Chem. Eng. J., 2010, 160, 480 CrossRef.
- L. Xue, C. B. Zhang, H. He and Y. Teraoka, Catal. Today, 2007, 126, 449 CrossRef CAS.
- X. Xu, X. Xu, G. Zhang and X. Niu, Ranliao Huaxue Xuebao, 2009, 37, 595 CAS.
- T. S. Farris, Y. Li, J. N. Armor and T. A. Braymer, US Pat., 5472677 A, 1995 Search PubMed.
- K. S. Chang, H. Song, Y. S. Park and J. W. Woo, Appl. Catal., A, 2004, 273, 223 CrossRef CAS.
- T. Franken and R. Palkovits, Appl. Catal., B, 2015, 176–177, 298 CrossRef CAS.
- L. Obalová, K. Pacultová, J. Balabánová, K. Jirátová, Z. Bastl, M. Valášáková, Z. Lacný and F. Kovanda, Catal. Today, 2007, 119, 233 CrossRef.
- K. S. Chang and X. Peng, J. Ind. Eng. Chem., 2010, 16, 455 CrossRef CAS.
- J. Haber, M. Nattich and T. Machej, Appl. Catal., B, 2008, 77, 278 CrossRef CAS.
- J. Haber, T. Machej, J. Janas and M. Nattich, Catal. Today, 2004, 90, 15 CrossRef CAS.
- K. Yuzaki, T. Yarimizu, K. Aoyagi, S. I. Ito and K. Kunimori, Catal. Today, 1998, 45, 129 CrossRef CAS.
- S. Parres-Esclapez, F. E. López-Suárez, A. Bueno-López, M. J. Illán-Gómez, B. Ura and J. Trawczyński, Top. Catal., 2009, 52, 1832 CrossRef CAS.
- K. Doi, Y. Y. Wu, R. Takeda, A. Matsunami, N. Arai, T. Tagawa and S. Goto, Appl. Catal., B, 2001, 35, 43 CrossRef CAS.
- L. Obalová, F. Kovanda, K. Jirátová, K. Pacultova and Z. Lacný, Collect. Czech. Chem. Commun., 2008, 73, 1045 CrossRef.
- L. Obalová, G. Maniak, K. Karásková, F. Kovanda and A. Kotarba, Catal. Commun., 2011, 12, 1055 CrossRef.
- K. Karásková, L. Obalová and F. Kovanda, Catal. Today, 2011, 176, 208 CrossRef.
- A. Shimizu, K. Tanaka and M. Fujimori, Chemosphere: Global Change Sci., 2000, 2, 425 CrossRef CAS.
- K. Paculová, L. Obalová, F. Kovanda and K. Jirátová, Catal. Today, 2008, 137, 385 CrossRef.
- K. S. Chang, H. J. Lee, Y. S. Park and J. W. Woo, Appl. Catal., A, 2006, 309, 129 CrossRef CAS.
- T. J. Vulic, A. F. K. Reitzmann and K. Lázár, Chem. Eng. J., 2012, 207–208, 913 CrossRef CAS.
- S. C. Christoforou, E. A. Efthimiadis and I. A. Vasalos, Catal. Lett., 2002, 79, 137 CrossRef CAS.
- P. Kuśtrowski, L. Chmielarz, A. Rafalska-Łasocha, B. Dudek, A. Patek-Janczyk and R. Dziembaj, Catal. Commun., 2006, 7, 1047 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |