
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide-directed assembly of functional supramolecular polymers for biomedical applications: electroactive molecular tongue-twisters (oligoalanine–oligoaniline–oligoalanine) for electrochemically enhanced drug delivery†
John G.
Hardy
*ab,
Megan N.
Amend
b,
Sydney
Geissler
ab,
Vincent M.
Lynch
c and
Christine E.
Schmidt
*ab
aDepartment of Biomedical Engineering, The University of Texas at Austin, Austin, TX 78712, USA
bJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Biomedical Sciences Building JG-53, P.O. Box 116131, Gainesville, FL 32611-6131, USA. E-mail: johnhardyuk@gmail.com; schmidt@bme.ufl.edu
cDepartment of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA
First published on 8th June 2015
Abstract
We report the preparation and characterization of films of electroactive supramolecular polymers based on non-electroactive oligoalanines and electroactive oligoanilines. Fibroblasts adhered to and proliferated on the films, and the delivery of the clinically relevant anti-inflammatory drug dexamethasone phosphate could be enhanced upon the application of an electrical stimulus.
Technologies that enable precise control of the amount of drugs in the blood stream or specific tissues facilitate the maintenance of the amount of drug within the therapeutic window, and controlling the chronopharmacology of a drug is particularly useful for the treatment of diseases with specific chronobiologies (e.g. cancers, infectious diseases, pain).1–3 Drug delivery systems that respond to chemical (e.g. enzymes, pH) or physical (e.g. electric/magnetic fields, light, pH, temperature) stimuli are potentially applicable for the treatment of such conditions.2,3 Here we report the application of electroactive polymers (EAPs) that allow drug delivery enhanced by the application of an electrical stimulus.
EAPs have interesting electronic and optical properties and are currently investigated for use in electronic4 and biomedical5–8 industries. Polyaniline, polypyrrole and polythiophene derivatives are the EAPs most commonly investigated for biomedical applications.9,10 In recent years block copolymers incorporating electroactive blocks (frequently oligoaniline-based polymers doped with camphorsulfonic acid, CSA, Fig. 1) have been investigated for use as electroactive tissue scaffolds capable of the electrical stimulation of the cells inhabiting them,11 and as materials for electrochemically-triggered drug delivery.12
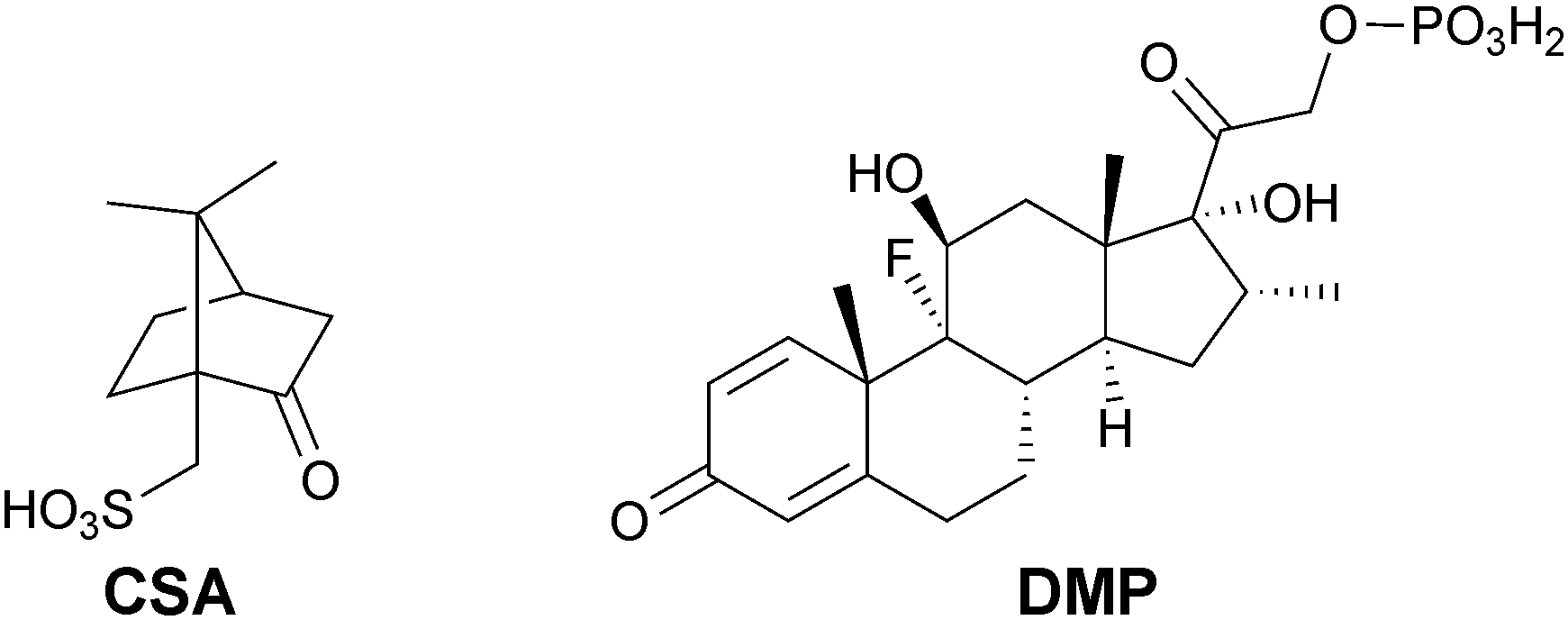 | ||
| Fig. 1 Camphorsulfonic acid (CSA) and dexamethasone phosphate (DMP). | ||
Polyaniline- and polypyrrole-based systems have been used to deliver a variety of drugs, including; adenosine triphosphate, dexamethasone phosphate (DMP, Fig. 1), DNA, dopamine, nerve growth factor and N-methylphenothiazine, as discussed in greater depth in reviews by Garg and co-workers13 and Ndesendo and co-workers.14 Here we report the application of electroactive molecular tongue-twisters (MTTs) based on electroactive oligoanilines and non-electroactive oligoalanines (i.e. oligoalanine–oligoaniline–oligoalanine) for the delivery of dexamethasone phosphate. The MTTs are solution processable and assemble into hierarchically structured supramolecular polymers15–21 because of hydrogen bonding interactions between the oligoalanines in the solid state. Such supramolecular polymers have prospects for the preparation of conformal electroactive coatings for implantable biomaterials.
The electrochemically responsive blocks of oligoaniline were terminated with amines which enabled the initiation of ring-opening polymerization of α-amino acid N-carboxyanhydrides,22 in this case alanine N-carboxyanhydride (Ala-NCA). The oligoanilines (tetraaniline and hexaaniline) were prepared using the methodology described in the literature (Scheme S1, ESI†),23,24 as was Ala-NCA (Scheme S2, ESI†),25 and they were polymerized at room temperature in anhydrous DMF, precipitated in diethyl ether and dried under vacuum (Scheme 1). The hexafluoroisopropanol (HFIP)-soluble fraction was extracted and dried under vacuum, yielding off-white solids that were used without further purification; tetraaniline-based molecular tongue-twister 1 (MTT1, oligoalanine–tetraaniline–oligoalanine) and hexaaniline-based molecular tongue-twister 2 (MTT2, oligoalanine–hexaaniline–oligoalanine), that are depicted in Scheme 1.
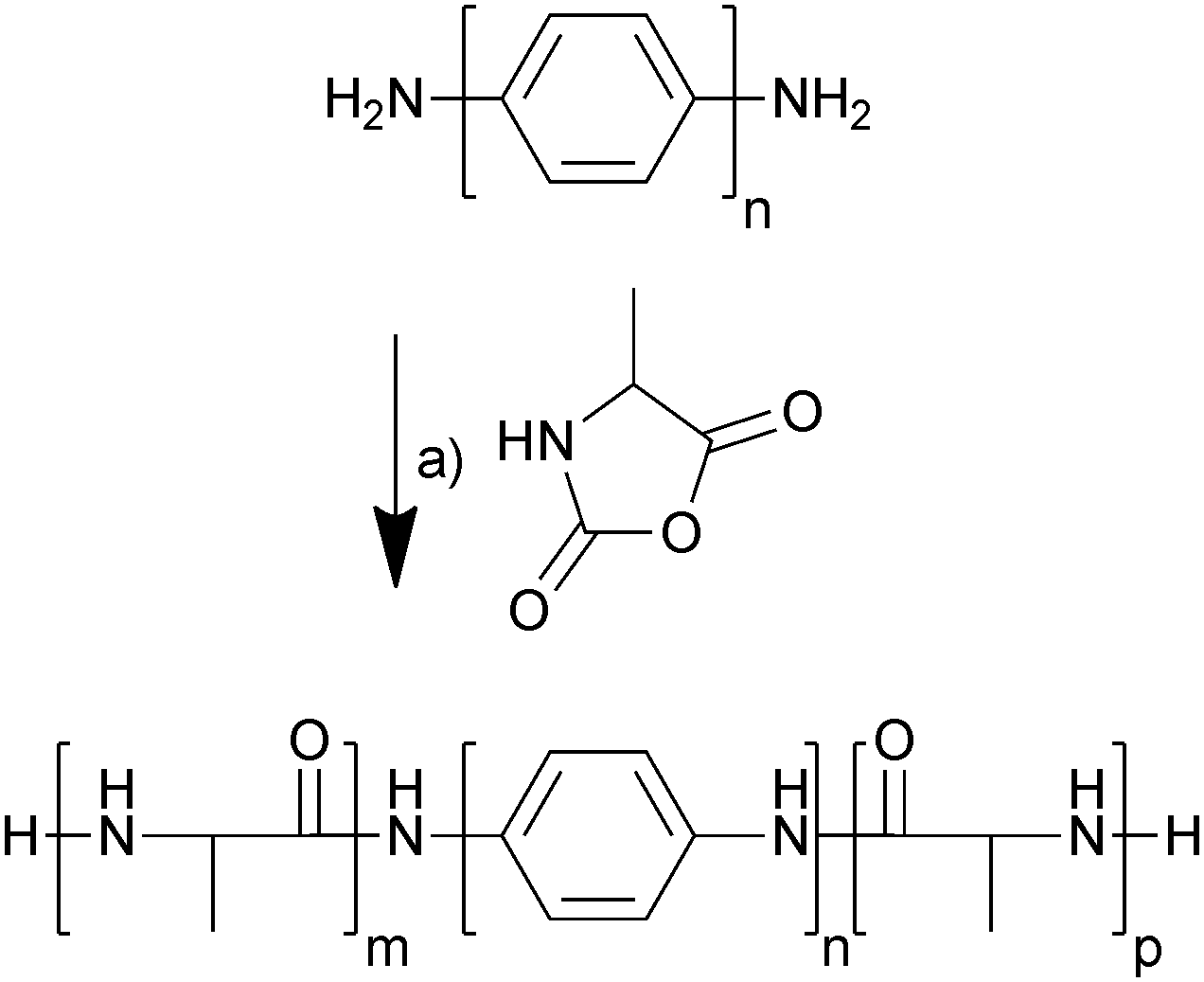 | ||
| Scheme 1 Synthesis of molecular tongue-twisters (oligoalanine–oligoaniline–oligoalanine) from alanine N-carboxyanhydrides and amine-terminated oligoanilines. (a) DMF, Ar, 96 h. For tetraaniline-based MTT1, n = 2, and m + p ≈ 10; whereas for hexaaniline-based MTT2, n = 4, and m + p ≈ 26. | ||
The success of the polymerizations was confirmed spectroscopically. The presence of characteristic peaks in the IR spectra (Fig. S1, ESI†) for amides and oligoanilines (shoulders at ca. 1541 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretches) and 1496 cm−1 (C–C stretching)).26 NMR in deuterated HFIP suggested that MTT1 incorporated ca. 10 alanine units per tetraaniline (in line with the synthesis), whereas MTT2 incorporated 26 alanine units per hexaaniline, suggesting that polymers based on the less soluble hexaaniline required a greater number of alanines for the resulting polymer to be soluble. Yet their low solubility hampered determination of the molecular weight distributions via standard techniques such as GPC/SEC, light scattering or MALDI-TOF mass spectrometry.
N and CC stretches) and 1496 cm−1 (C–C stretching)).26 NMR in deuterated HFIP suggested that MTT1 incorporated ca. 10 alanine units per tetraaniline (in line with the synthesis), whereas MTT2 incorporated 26 alanine units per hexaaniline, suggesting that polymers based on the less soluble hexaaniline required a greater number of alanines for the resulting polymer to be soluble. Yet their low solubility hampered determination of the molecular weight distributions via standard techniques such as GPC/SEC, light scattering or MALDI-TOF mass spectrometry.
Casting dilute solutions of the MTTs in HFIP allowed the preparation of water insoluble films of a few μm in thickness with μm scale roughness (Ra) or root mean square roughness (Rq) as determined by profilometry (Table 1). Undoped films were blue, whereas films doped with CSA (10 wt%) were green.
| R a (μm) | R q (μm) | X c (%) | Conductivity, σ (S cm−1) | |
|---|---|---|---|---|
| a As determined by XRD. b Not applicable. | ||||
| Glass substrate | 0.730 ± 0.153 | 0.905 ± 0.182 | N/Aa,b | N/A |
| MTT1 undoped | 0.760 ± 0.299 | 3.272 ± 6.352 | 68.2a | 1.59 × 10−8 ± 28% |
| MTT1 doped with CSA | 0.760 ± 0.516 | 1.020 ± 0.637 | 30.1a | 2.49 × 10−8 ± 33% |
| MTT2 undoped | 0.700 ± 0.018 | 0.085 ± 0.024 | 0.0a | 7.32 × 10−8 ± 32% |
| MTT2 doped with CSA | 0.475 ± 0.009 | 0.060 ± 0.014 | 0.0a | 8.55 × 10−8 ± 32% |
IR spectra of the films recorded in attenuated total reflectance (ATR) mode revealed subtle differences in the hierarchical supramolecular assembly of the polymers, with evidence of both α-helices and β-sheets in all spectra (Fig. S1, ESI†),27–29 with the α-helices induced by HFIP and the intermolecular β-sheets being partially responsible for the insolubility of the polymers in water (also observed in oligoalanine-rich proteins).30–32 The oligoalanine blocks in undoped films of MTT1 assembled into both α-helices (amide I, 1649 cm−1) and β-sheets (amide I, 1692 cm−1; amide II, 1530 and 1517 cm−1). Doping MTT1 with CSA altered the assembly of the MTTs, yet both α-helices (amide I, 1649 cm−1; amide II, 1543 cm−1) and β-sheets (amide I, 1690 cm−1; amide II, 1530 and 1514 cm−1) were present, although the β-sheet content was clearly reduced. The oligoalanine blocks in undoped films of MTT2 were predominantly amorphous (amide I, 1646 cm−1; amide II, 1533 cm−1) with some β-sheets (amide I, 1629 cm−1; amide II, 1518 cm−1). Doping MTT2 with CSA reduced the β-sheet content (weak amide I, 1690 cm−1; weak amide II, 1530 and 1514 cm−1), and the chains were predominantly amorphous (amide I, 1646 cm−1; amide II, 1535 cm−1).27–29
Wide angle XRD patterns for the films (Fig. S2, ESI†) supported the results from IR spectroscopy, which showed evidence of both α-helices (broad peak at 2θ = 19.4°, d-spacing 4.58 Å) and β-sheets (broad peak at 2θ = 16.7° and 19.9°, d-spacings of 5.31 and 4.46 Å, respectively) also observed in oligoalanine-rich proteins.29 Films composed of MTT1 showed some peaks reported for tetraaniline-based supramolecular polymers (peaks at 2θ = 11.7°, 19.5°, 20.3°, 20.7° and 29.7°, d-spacings of 7.56, 4.55, 4.37, 4.29 and 3.01 Å, respectively),33 and were clearly more crystalline (higher Xc) than those of CSA-doped MTT1 (Fig. S2, ESI† and Table 1) because the presence of the bulky CSA anions interfered with the assembly of the oligoaniline blocks.12,34 Films of MTT2 were amorphous in both the undoped or CSA-doped state.
The conductance of films of the MTTs were measured as previously reported.12 Undoped films of tetraaniline-based MTT1 had a conductivity of ca. 1.6 × 10−8 S cm−1, that was moderately increased by doping with CSA to ca. 2.5 × 10−8 S cm−1 (Table 1). Undoped films of hexaaniline-based MTT2 had a conductivity of ca. 7.3 × 10−8 S cm−1, that was moderately increased by doping with CSA to ca. 8.6 × 10−8 S cm−1 (Table 1). The higher conductivity of MTT2-based films was because of the greater level of conjugation in the backbone of hexaaniline than in tetraaniline. These conductivities are lower than those of mammalian tissues (typically ≥10−4 S cm−1)35–37 that should enable the controlled delivery of a drug upon the application of an electrical potential to the EAPs.
We studied the release profiles of the anti-inflammatory drug DMP from MTT films into phosphate buffered saline (PBS) in the absence or presence of an electrical stimulus using the experimental setup depicted in Fig. S3 (ESI†). Cyclic voltammograms (Fig. S4, ESI†) of the DMP-doped films of MTT1 presented a redox process at ca. 0.26 V (leucoemeraldine to emeraldine 1 transition), whereas those for MTT2 presented a process at 0.42 V (emeraldine 1 to emeraldine 2 transition), confirming the greater conjugation in the backbone of the longer oligomers (potentials are reported vs. a Ag/AgCl reference electrode calibrated with ferrocenemethanol).
We monitored the release of DMP at specific time points spectroscopically, using its characteristic UV absorption at 242 nm. DMP loadings in the films were at 10 wt%. Passive release from films of MTT1 (Fig. 2, grey checked bars) and MTT2 (Fig. 2, black checked bars) was observed, however, release was moderately enhanced by the application of an electrical stimulus (Fig. 2; MTT1 grey bars, MTT2 black bars), typically by 10 to 20% for the first five rounds of stimulation, after which the profiles for MTT1 in the absence/presence of stimulation was observed to be equal which suggests that it is more prone to spontaneous de-doping than the more highly conjugated MTT2. We believe that the release profiles could be improved by tuning both the length of the electroactive oligoaniline, the ratio of electroactive to non-electroactive peptide blocks, and the identity of the non-electroactive peptide (i.e. composition and sequence of amino acids).
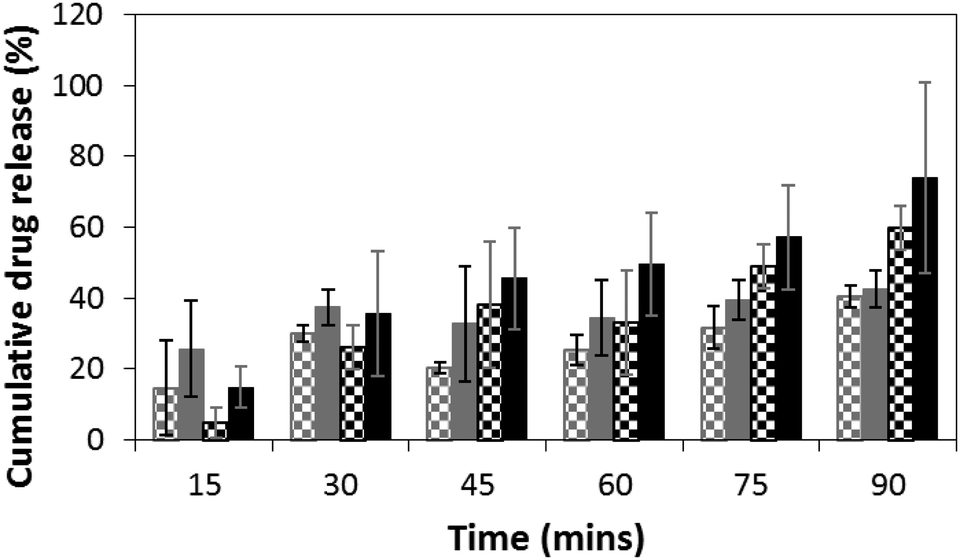 | ||
| Fig. 2 DMP release from films of MTT1 and MTT2 in the absence and presence of electrical stimulation as determined by UV spectroscopy. MTT1 without electrical stimulation checked grey bars; MTT1 with electrical stimulation solid grey bars; MTT2 without electrical stimulation checked black bars; MTT2 with electrical stimulation solid black bars. | ||
Analogous electroactive block copolymers incorporating CSA-doped oligoalanines have been shown to support the adhesion of a variety of cells including fibroblasts, keratinocytes, osteoblasts, PC12 cells and Schwann cells.11 We found that cell viability for the HDFs cultured on the films of MTTs was comparable, and indeed somewhat better than TCP controls (Fig. 3A). However, the adhesion of human dermal fibroblasts (HDFs) to films of undoped or CSA-doped MTT1 was poor by comparison with commercially available Corning Costar® tissue culture plate (TCP) controls as determined using the AlamarBlue® assay (Fig. 3B), and cells had relatively rounded morphologies indicative of poor cell adhesion (Fig. 4A and B), particularly by comparison with TCP controls (Fig. S5, ESI†). Interestingly, HDF adhesion to films of undoped or CSA-doped MTT2 was moderately better, with cells more likely to adopt a spread morphology (Fig. 4C and D). For certain applications, non cell-adhesive biomaterials are interesting candidates, such as for the manufacture of anti-adhesion membranes;38 however, for other applications (e.g. tissue scaffolds) cell adhesion is beneficial. We believe that cell adhesion could be improved by incorporating cell adhesive peptides (e.g. the ubiquitous RGD)10,16,39–41 in the backbone of such EAPs.
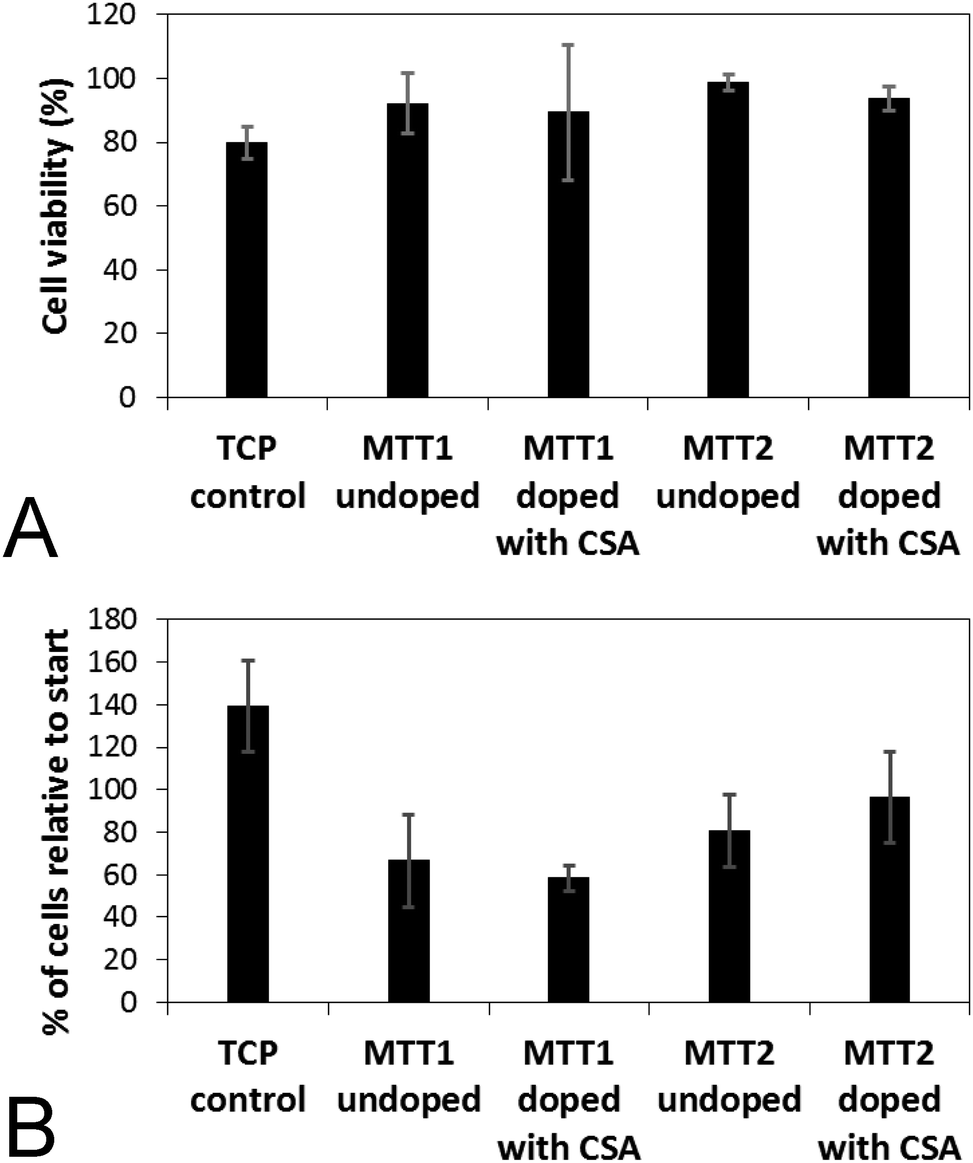 | ||
| Fig. 3 (A) Assessment of the cell viability of human dermal fibroblasts on various surfaces after 2 days in culture as determined using a LIVE/DEAD® Viability/Cytotoxicity Kit. (B) Assessment of cell adhesion on various surfaces after 2 days as determined by the AlamarBlue® assay. | ||
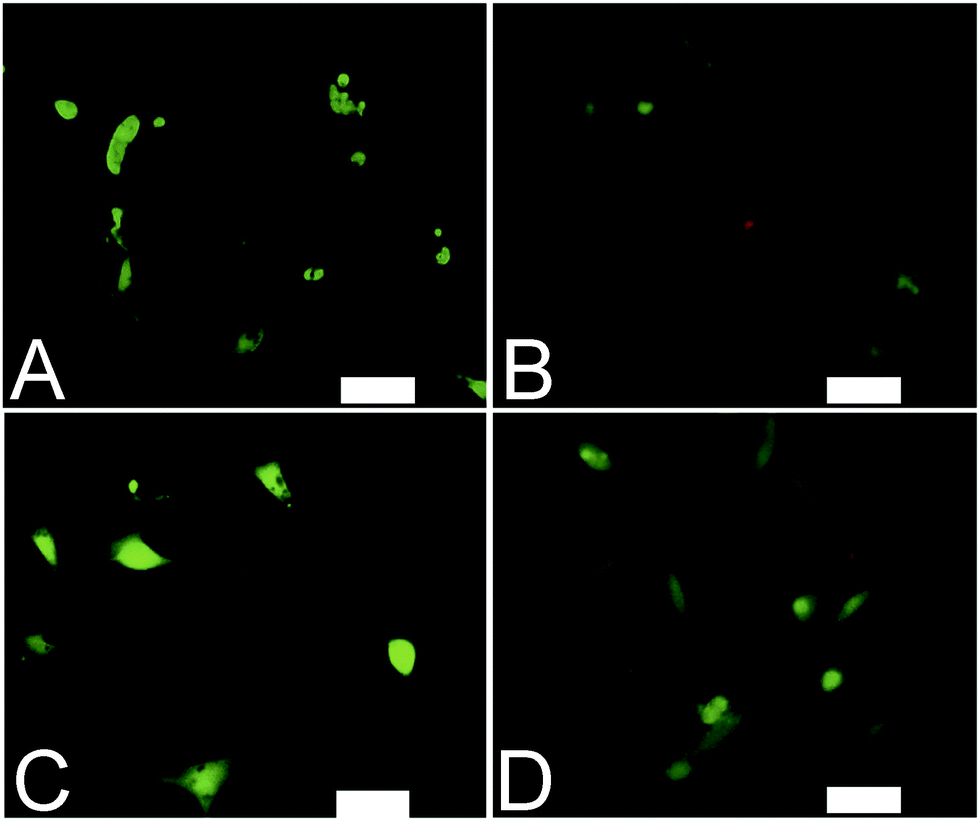 | ||
| Fig. 4 Adhesion of human dermal fibroblasts on various surfaces after 4 days in culture. (A) Undoped MTT1. (B) CSA-doped MTT1. (C) Undoped MTT2. (D) CSA-doped MTT2. Live cells were stained green by calcein and dead cells were stained red by ethidium using a LIVE/DEAD® Viability/Cytotoxicity Kit. Scale bars represent 100 μm. | ||
Conclusions
Herein we described electrically-triggered drug release from electroactive molecular tongue-twisters (i.e. oligoalanine–oligoaniline–oligoalanine). The electroactive ABA block copolymers based on (A) non-electroactive oligoalanines, and (B) electroactive blocks of oligoanilines, represents a novel platform for drug delivery. The physicochemical properties of the films were characterized by various techniques, and we found that hexaaniline-based MTT2 was more conductive and appeared to be more electrochemically stable towards de-doping than tetraaniline-based MTT1. While HDFs cultured on the surfaces of such films adhered weakly, this could be improved by incorporating cell-adhesive moieties.39–41 While imperfect, such EAPs represent valuable lead structures for the development of materials that enable us to deliver drugs with clinically relevant chronopharmacologies, and facilitate the treatment of conditions with specific chronobiologies (e.g. infectious diseases, pain). We believe that it should be possible to rationally control the release profiles and cell–biomaterial interactions by tuning the length of the electroactive oligoaniline, the ratio of electroactive to non-electroactive peptide blocks, and the identity of the non-electroactive peptide (i.e. composition and sequence of amino acids). Specifically, increasing the length of the oligoalanine segments should render them more conductive and electrochemically stable (i.e. less likely to be spontaneously de-doped in biological milieu). The composition and sequence of amino acids in the non-electroactive peptide dictate the charge and hydrophobicity of the materials, which play roles in the self-assembly of the peptides (i.e. hydrogen bonding interactions, electrostatic interactions) and protein deposition on their surfaces which may be important for cell adhesion. Cell–biomaterial interactions could be controlled through the inclusion of cell-adhesive peptides (e.g. RGD, YIGSR, KQAGDV, KHIFSDDSSE, KRSR),39–41 and protease-labile domains (APGL, VRN, or indeed oligoalanines such as those in the backbone of MTT1 and MTT2 that are degraded by elastase),40 potentially facilitate their degradation in vivo. Furthermore, molecules with higher ratios of electroactive to non-electroactive peptide blocks should be more conductive, and the inclusion of charged amino acids may facilitate the inclusion of cationic drugs (antibacterials, antiarrhythmics, etc.).42Acknowledgements
We thank Andrea P. Carranza, YoungIn Jun, Jong M. Kim, David J. Mouser, Min Joo Sohn and Pei-Yun Tseng for early work related to the synthesis of well-defined oligomers of aniline, and David Aguilar Jr. and Harrison J. Matthews for assistance with early work related to the drug delivery studies. We thank the University of Texas at Austin for financial support of Jong M. Kim, David J. Mouser and Min Joo Sohn in the form of Undergraduate Research Fellowships. At the Department of Chemistry at the University of Florida we thank Kari Basso and Louis Searcy for attempts at mass spectrometry analysis. We thank the University of Florida for financial support in the form of startup resources.Notes and references
- A. S. Mandal, N. Biswas, K. M. Karim, A. Guha, S. Chatterjee, M. Behera and K. Kuotsu, J. Controlled Release, 2010, 147, 314 CrossRef CAS PubMed.
- B. Chertok, M. J. Webber, M. D. Succi and R. Langer, Mol. Pharmaceutics, 2013, 10, 3531 CAS.
- M. E. Caldorera-Moore, W. B. Liechty and N. A. Peppas, Acc. Chem. Res., 2011, 44, 1061 CrossRef CAS PubMed.
- P. Leclere, M. Surin, P. Jonkheijm, O. Henze, A. P. H. J. Schenning, F. Biscarini, A. C. Grimsdale, W. J. Feast, E. W. Meijer, K. Mullen, J. L. Bredas and R. Lazzaroni, Eur. Polym. J., 2004, 40, 885 CrossRef CAS PubMed.
- M. Berggren and A. Richter-Dahlfors, Adv. Mater., 2007, 19, 3201 CrossRef CAS PubMed.
- Z. L. Yue, S. E. Moulton, M. Cook, S. O'Leary and G. G. Wallace, Adv. Drug Delivery Rev., 2013, 65, 559 CrossRef CAS PubMed.
- A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701 CrossRef CAS PubMed.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679 CrossRef CAS.
- A. D. Bendrea, L. Cianga and I. Cianga, J. Biomater. Appl., 2011, 26, 3 CrossRef CAS PubMed.
- J. G. Hardy, J. Y. Lee and C. E. Schmidt, Curr. Opin. Biotechnol., 2013, 24, 847 CrossRef CAS PubMed.
- B. L. Guo, L. Glavas and A. C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263 CrossRef CAS PubMed.
- J. G. Hardy, D. J. Mouser, N. Arroyo-Currás, S. Geissler, J. K. Chow, L. Nguy, J. M. Kim and C. E. Schmidt, J. Mater. Chem. B, 2014, 2, 6809 RSC.
- D. Svirskis, J. Travas-Sejdic, A. Rodgers and S. Garg, J. Controlled Release, 2010, 146, 6 CrossRef CAS PubMed.
- V. Pillay, T. S. Tsai, Y. E. Choonara, L. C. du Toit, P. Kumar, G. Modi, D. Naidoo, L. K. Tomar, C. Tyagi and V. M. Ndesendo, J. Biomed. Mater. Res., Part A, 2014, 102, 2039 CrossRef PubMed.
- R. Dong, Y. Zhou, X. Huang, X. Zhu X, Y. Lu and J. Shen, Adv. Mater., 2015, 27, 498 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS PubMed.
- M. Hasegawa and M. Iyoda, Chem. Soc. Rev., 2010, 39, 2420 RSC.
- S. H. Kim and J. R. Parquette, Nanoscale, 2012, 4, 6940 RSC.
- A. J. Hodgson, K. Gilmore, C. Small, G. G. Wallace, I. L. Mackenzie, T. Aoki and N. Ogata, Supramol. Sci., 1994, 1, 77 CrossRef CAS.
- E. K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Börner and P. Bäuerle, Adv. Mater., 2009, 21, 1562 CrossRef CAS PubMed.
- A. Digennaro, H. Wennemers, G. Joshi, S. Schmid, E. Mena-Osteritz and P. Bäuerle, Chem. Commun., 2013, 49, 10929 RSC.
- Z. Zhang, S. Wei and L. Han, Adv. Mater. Res., 2011, 284–286, 2110 CrossRef CAS.
- I. Rozalska, P. Kułyk and I. Kulszewicz-Bajer, New J. Chem., 2004, 28, 1235 RSC.
- I. Kulszewicz-Bajer, I. Rozalska and M. Kurłyek, New J. Chem., 2004, 28, 669 RSC.
- G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koninga and A. Heise, Polym. Chem., 2010, 1, 514 RSC.
- Z. Pang, J. Fu, P. Lv, F. Huang and Q. Wei, Sensors, 2014, 14, 21453 CrossRef CAS PubMed.
- U. Slotta, M. Tammer, F. Kremer, P. Koelsch and T. Scheibel, Supramol. Chem., 2006, 18, 1 CrossRef PubMed.
- F. Paquet-Mercier, T. Lefevre, M. Auger and M. Pezolet, Soft Matter, 2013, 9, 208 RSC.
- O. S. Rabotyagova, P. Cebe and D. L. Kaplan, Macromol. Biosci., 2010, 10, 49 CrossRef CAS PubMed.
- J. G. Hardy and T. R. Scheibel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3957–3963 CrossRef CAS PubMed.
- J. G. Hardy and T. R. Scheibel, Biochem. Soc. Trans., 2009, 37, 677 CrossRef CAS PubMed.
- J. G. Hardy, A. Leal-Egaña and T. R. Scheibel, Macromol. Biosci., 2013, 13, 1431 CrossRef CAS PubMed.
- Z. Shao, Z. Yu, J. Hu, S. Chandrasekaran, D. M. Lindsay, Z. Wei and C. F. J. Faul, J. Mater. Chem., 2012, 22, 16230 RSC.
- Y. Wang, H. D. Tran, L. Liao, X. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365 CrossRef CAS PubMed.
- C. Gabriel, S. Gabriel and E. Corthout, Phys. Med. Biol., 1996, 41, 2231 CrossRef CAS.
- S. Gabriel, R. W. Lau and C. Gabriel, Phys. Med. Biol., 1996, 41, 2251 CrossRef CAS.
- S. Gabriel, R. W. Lau and C. Gabriel, Phys. Med. Biol., 1996, 41, 2271 CrossRef CAS.
- P. H. Zeplin, N. C. Maksimovikj, M. C. Jordan, J. Nickel, G. Lang, A. H. Leimer, L. Römer and T. Scheibel, Adv. Funct. Mater., 2014, 24, 2658 CrossRef CAS PubMed.
- C. Vallejo-Giraldo, A. Kelly and M. J. Biggs, Drug Discovery Today, 2014, 19, 88 CrossRef CAS PubMed.
- H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24, 4353 CrossRef CAS.
- K. G. Sreejalekshmi and P. D. Nair, J. Biomed. Mater. Res., Part A, 2011, 96A, 477 CrossRef CAS PubMed.
- K. I. Umehara, T. Iwatsubo, K. Noguchi, T. Usui and H. Kamimura, Xenobiotica, 2008, 38, 1203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, synthetic schemes, FTIR spectra, XRD spectra, voltammograms, cell adhesion assay data, image of HDFs on TCP control substrate. See DOI: 10.1039/c5tb00106d |
| This journal is © The Royal Society of Chemistry 2015 |