
Molecular docking studies and biological evaluation of chalcone based pyrazolines as tyrosinase inhibitors and potential anticancer agents
Hua-Li Qin*a,
Zhen-Peng Shanga,
Ibrahim Jantanb,
Oya Unsal Tanc,
Muhammad Ajaz Hussaind,
Muhammad Sherd and
Syed Nasir Abbas Bukhari *b
*b
aDepartment of Pharmaceutical Engineering, School of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, 205 Luoshi Road, Wuhan, 430070, P. R. China. E-mail: qinhuali@whut.edu.cn
bDrug and Herbal Research Centre, Faculty of Pharmacy, Universiti Kebangsaan Malaysia, Jalan Raja Muda Abdul Aziz, 50300 Kuala Lumpur, Malaysia. E-mail: snab@ukm.edu.my; Fax: +60326983271; Tel: +601123695295
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Hacettepe University, Ankara 06100, Turkey
dDepartment of Chemistry, University of Sargodha, Sargodha 40100, Pakistan
First published on 18th May 2015
Abstract
Studies on the discovery of tyrosinase enzyme inhibitors and exploration for better cytotoxic agents remain an important line in drug discovery and development. A series of synthetic chalcones and pyrazoline derivatives was evaluated for their inhibitory effects on the diphenolase activity of mushroom tyrosinase. The effects of these compounds on proliferation and microtubule assembly were also evaluated in seven different cancer cell lines. The results revealed that some of the synthetic compounds showed significant inhibitory activity, with four compounds being more potent tyrosinase inhibitors than the reference standard inhibitor kojic acid. Several compounds were toxic to cancer cell lines. Compound 1a was found to possess the highest anticancer activity towards all cell lines with an IC50 in the range of 0.9–2.2 μM. Seven of the compounds showed considerable tubulin polymerization activity at a concentration of 25 μM. Molecular modeling studies of these synthetic compounds were performed to investigate their interactions with the tyrosinase enzyme. The structure–activity relationship (SAR) study using in-silico analysis matched well with the in vitro tumour cell inhibitory activity.
1. Introduction
Melanogenesis is the process of melanin production by melanocytes in the basal layer of the dermis inside the skin and hair follicles.1 Melanin, one of the most extensively distributed pigments is found in animals, fungi and bacteria, and is responsible for pigmentation and other functions including chelation.2 The color of human skin and hair also relies on melanin.1 The role of melanin is to protect the skin from ultraviolet (UV) damage by absorbing the UV sunlight and removing reactive oxygen species (ROS). Several dermatological disorders lead to the accumulation of a disproportionate level of epidermal pigmentation.3 Hyperpigmentary disorders such as actinic and senile lentigines, melasma and post-inflammatory hyperpigmentation are major cosmetic problems for which patients pursue medical advice. These conditions affect the populations with darker skin complexion, particularly Hispanics and Asians with greater severity and frequency.4 Moreover, melanin production-mediated browning is unwanted in mushrooms, vegetables and fresh fruits5,6 as it lessens the commercial worth of the products. Therefore, enzymatic browning in farm-products and hyperpigmentation in human skin are a main distress in cosmetic, pharmaceutical and agricultural fields.7Tyrosinase has also been linked to other neurodegenerative diseases including Parkinson's disease.8 Tyrosinase is associated with three various biochemical processes in insects like defensive encapsulation and melanization of foreign organisms, wound healing and sclerotization of cuticle.9 Tyrosinase inhibitors have become increasingly important because they inhibit the synthesis of melanin.10,11 They play major role in numerous fields such as cosmetic industry,12,13 medications,14 and in the food industry7,15 for their potential to reduce pigmentation. Therefore, the development of effective and safe tyrosinase inhibitors is of great interest in the cosmetic, agricultural and medical industries.
Among the tyrosinase inhibitors, carbonyl based compounds including aurones,16,17 chalcones,18 flavanones19 and pyrazole derivatives20,21 have gained attention owing to their interaction with the hydrophobic protein pocket nearby the binuclear copper active site of tyrosinase.22 Our research group recently demonstrated a series of α, β-unsaturated carbonyl based compounds as prospective tyrosinase inhibitors.23 The inhibition mechanism analysis showed that this kind of compounds, analogous to thiosemicarbazide derivatives and curcumin,24 have potential to complex the two copper ions in the active site of tyrosinase.
Based on this finding, we postulate that the coupling products of properly substituted benzaldehydes with ketones not only complex the binuclear copper active site of tyrosinase but also interact with the hydrophobic enzyme pocket, therefore improving inhibitor-enzyme binding affinity and enhancing inhibitory effects on mushroom tyrosinase. Chemically, chalcones are the coupling products of benzaldehydes with ketones, playing their role as intermediates in the biosynthesis of flavonoids. Chalcones are of great interest because of their broad spectrum of biological activities. Our research group and other researchers have reported the pharmacological potential and biological properties of chalcones in recent review articles.25–28 The anticancer potential of chalcones has triggered extensive efforts to identify novel chalcone-based anticancer drug candidates in oncology.29,30 The cytotoxic activity of chalcones is often linked with their ability of interfering with microtubule formation and tubulin inhibition, which is vital in cellular processes like cell replication, cytoplasmic organelle movement, mitosis and for the regulation of cell shape.31
The promising anticancer and tyrosinase inhibitory potential of chalcones motivated us to evaluate a series of synthetic chalcones and pyrazolines (that were substituted at their N1 position by thioacetamide and arylacetamide groups) for their inhibitory effects on human cancer cell lines and on the diphenolase activity of mushroom tyrosinase. The activity of chalcones in a microtubular polymerization assay was also investigated.
2. Results and discussion
2.1. Chemistry
All the compounds were synthesized as shown in Schemes 1–3 and were recently reported.32 The Claisen–Schmidt condensation reaction was used to synthesize chalcones (1–6) in which equimolar ketones and aldehydes were reacted in the presence of a base. The chalcones (1–6) and thiosemicarbazide were refluxed at alkaline pH to yield pyrazolines (1a–6a). Same reaction was used to synthesize compounds (1b–6b) by refluxing chalcones (1–6) with N-(4-chlorophenyl) semicarbazide in alkaline conditions. All the synthesized compounds were purified by crystallization techniques and column chromatography. A thin layer chromatography (TLC) technique using pre-coated silica plate was employed to investigate the purity of compounds. The successful synthesis of the new compounds was confirmed by a variety of analytical techniques such as MS, 13C NMR, 1H NMR, and elemental analysis.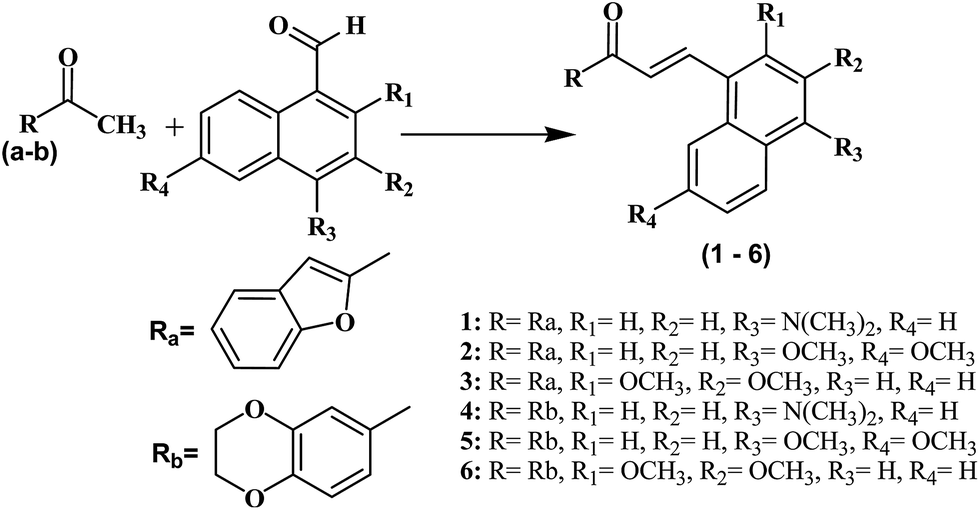 | ||
| Scheme 1 Structures and synthesis scheme of chalcones derivatives (1–6). Reagents and conditions: (i) NaOH, EtOH, room temperature. 49–72% yield. | ||
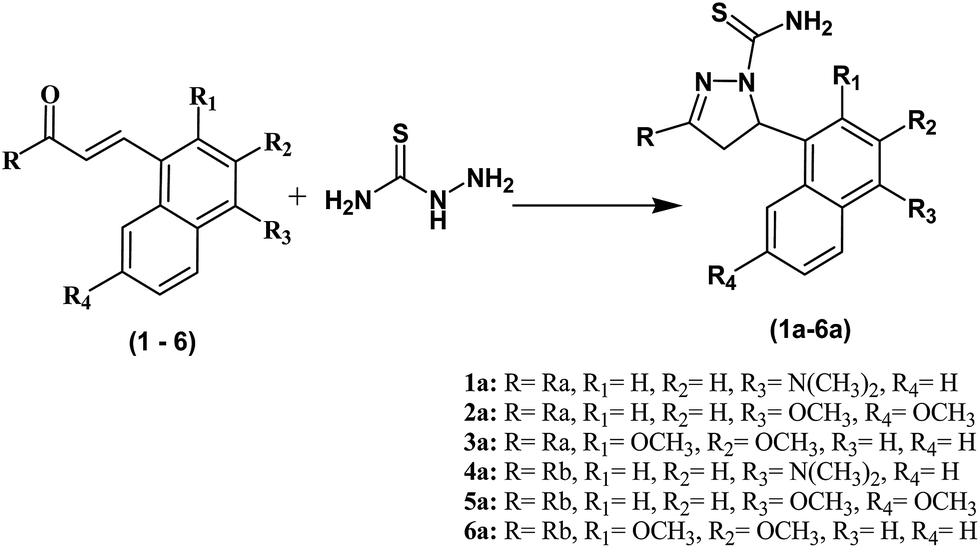 | ||
| Scheme 2 Synthesis of pyrazoline derivatives (1a–6a). Reagents and conditions: (i) NaOH, EtOH, room temperature. 32–51% yield. | ||
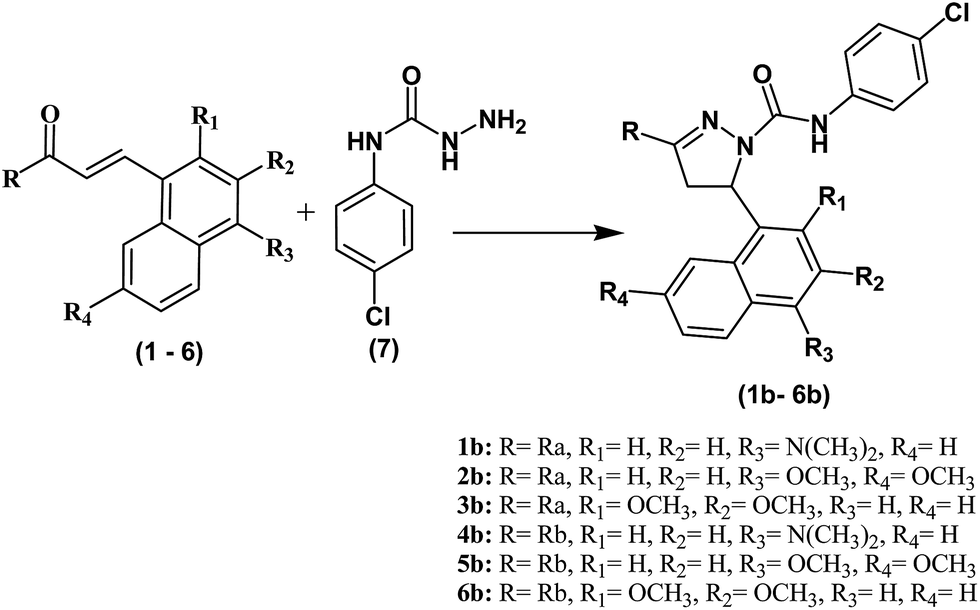 | ||
| Scheme 3 Synthesis of pyrazoline derivatives (1b–6b). Reagents and conditions: (i) NaOH, EtOH, room temperature. 37–61% yield. | ||
2.2. Effects on the diphenolase activity of mushroom tyrosinase
All of the synthetic compounds were subjected to tyrosinase inhibition assay with L-DOPA as substrate, as per method previously reported by our group with some minor alterations.23 Positive control in the assay was kojic acid (5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one) as reported previously.23 The IC50 values of derived pyrazolines and substituted chalcones against tyrosinase are summarized in Table 1, and IC50 values are presented as mean of three experiments.| Compounds | Mean ± SEM | IC50 μM |
|---|---|---|
| 1 | 49.5 ± 1.4 | 21.22 ± 1.25 |
| 2 | 56.2 ± 2.7 | 19.84 ± 2.53 |
| 3 | 52.8 ± 6.2 | 29.66 ± 4.29 |
| 4 | 72.4 ± 1.9 | 14.27 ± 2.21 |
| 5 | 27.4 ± 6.2 | 59.27 ± 2.45 |
| 6 | 12.2 ± 1.8 | 87.24 ± 4.65 |
| 1a | 61.5 ± 4.7 | 12.22 ± 1.65 |
| 2a | 43.4 ± 7.1 | 31.99 ± 4.56 |
| 3a | 45.5 ± 3.9 | 39.08 ± 1.58 |
| 4a | 81.5 ± 2.3 | 8.14 ± 2.61 |
| 5a | 38.8 ± 2.1 | 45.75 ± 3.64 |
| 6a | 35.7 ± 2.9 | 62.42 ± 7.21 |
| 1b | 79.5 ± 3.5 | 5.13 ± 1.12 |
| 2b | 69.2 ± 1.8 | 14.84 ± 1.32 |
| 3b | 61.7 ± 4.4 | 16.22 ± 1.87 |
| 4b | 89.7 ± 3.2 | 4.72 ± 0.65 |
| 5b | 60.0 ± 3.6 | 18.93 ± 1.70 |
| 6b | 52.6 ± 1.5 | 27.46 ± 2.49 |
| Kojic acid | 47.5 ± 5.2 | 12.42 ± 2.55 |
The results revealed that most of the compounds showed potent inhibition on mushroom tyrosinase with IC50 values ranging from 4.72 to 87.24 μM. Particularly, the compounds 1b–6b having 4-chlorophenylsemicarbazide exhibited more potent inhibitory activities than other compounds. Moreover, the compounds 1a, 4a, 1b and 4b showed stronger inhibitory activity than the reference standard inhibitor kojic acid, and 3-(2,3-dihydro-benzo[1,4]dioxin-6-yl)-5-(4-dimethylamino-naphthalen-1-yl)-4,5-dihydro-pyrazole-1-carboxylic acid (4-chloro-phenyl)-amide (4b) demonstrated the most potent tyrosinase inhibitory activity with the IC50 value of 4.72 μM.
The results revealed that as compared to chalcones, pyrazolines showed more potent inhibitory activities. Weak tyrosinase inhibitory activity was exhibited by most of the chalcone analogues. Additionally, the results exhibited that the group of thiosemicarbazide and 4-chlorophenylsemicarbazide played vital role in increasing activities against tyrosinase. These may be associated with the structure of tyrosinase which contained a type-3 copper center with a coupled dinuclear copper active site in the catalytic core. The tyrosinase inhibition by compounds 1a–6a relied on the capability of sulfur atom to chelate with the dinuclear copper in the active site, and tyrosinase would lose its catalyzing capacity after forming the complex.33 An interesting observation was found that compounds containing heterocycles as 4, 4a, and 4b were found more active than compounds bearing same substitution patterns (1, 1a and 1b) but different ring system.
2.3. Cell viability assay
The rule of three (activity-exposure-toxicity), presents the most complicated challenge in the design of drug candidates to the development phase. Absorption, distribution, metabolism and excretion (ADME) studies are extensively used in drug discovery to adjust this balance of properties essential to transform lead compounds into drugs that are both effective and safe for patients.34 Thus, in vitro cell viability assay was carried out using the human mammary gland epithelial cell line (MCF-10A). MCF-10A cells were treated with the synthesized compounds for 96 h and cell viability was measured by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The results are shown according to the percentage of toxicity (Table 2). All the compounds were found to be nontoxic with most of the compounds showing more than 93% cell viability. So, the newly synthesized compounds are biologically safe and could be used as the therapeutic agents for future drug discovery studies.| Comp. | Cell viability % | Antiproliferative activity IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| HT-29 | PC-3 | MCF-7 | H-460 | A-549 | PaCa-2 | Panc-1 | ||
| a HT-29 (colon cancer cell line): PC-3 (prostate cancer cell line): MCF-7 (breast cancer cell line): H-460 (lung cancer cell line): A-549 (epithelial): PaCa-2 (pancreatic carcinoma cell line): Panc-1(pancreas cancer cell line). | ||||||||
| 1 | 97 | 6.7 ± 0.8 | 5.2 ± 1.2 | 7.9 ± 0.2 | 6.9 ± 0.5 | 9.5 ± 2.9 | 9.6 ± 2.7 | 8.5 ± 0.5 |
| 2 | 96 | 24.2 ± 2.1 | 19.7 ± 1.5 | 25.4 ± 0.9 | 24.4 ± 1.2 | 35.7 ± 1.9 | 40.3 ± 6.5 | 31.4 ± 2.7 |
| 3 | 96 | 28.4 ± 1.5 | 27.0 ± 2.7 | 29.6 ± 1.3 | 28.6 ± 1.7 | 42.0 ± 1.5 | 47.6 ± 4.4 | 37.0 ± 4.2 |
| 4 | 98 | 9.3 ± 2.9 | 7.2 ± 2.2 | 10.5 ± 1.7 | 9.5 ± 0.2 | 13.4 ± 1.2 | 14.2 ± 0.4 | 11.9 ± 1.7 |
| 5 | 99 | 17.2 ± 3.4 | 18.4 ± 2.1 | 18.4 ± 1.2 | 17.4 ± 1.2 | 25.2 ± 1.5 | 28.0 ± 0.7 | 22.3 ± 1.3 |
| 6 | 97 | 19.6 ± 1.1 | 17.2 ± 2.4 | 20.8 ± 2.9 | 19.8 ± 2.2 | 28.8 ± 1.4 | 32.2 ± 1.4 | 25.4 ± 2.2 |
| 1a | 92 | 1.3 ± 0.4 | 0.9 ± 0.5 | 2.2 ± 0.3 | 1.4 ± 0.8 | 1.10 ± 0.5 | 0.9 ± 1.8 | 1.2 ± 0.2 |
| 2a | 91 | 4.2 ± 1.4 | 3.2 ± 1.4 | 4.1 ± 2.2 | 2.3 ± 2.1 | 2.0 ± 1.0 | 3.8 ± 2.5 | 1.5 ± 2.1 |
| 3a | 93 | 7.5 ± 1.9 | 8.2 ± 1.7 | 10.4 ± 1.2 | 10.6 ± 0.5 | 9.9 ± 1.6 | 10.1 ± 3.5 | 13.2 ± 1.4 |
| 4a | 92 | 3.2 ± 1.1 | 2.8 ± 1.5 | 4.1 ± 1.5 | 3.3 ± 0.9 | 4.0 ± 0.5 | 3.3 ± 1.2 | 3.7 ± 0.4 |
| 5a | 94 | 10.2 ± 1.7 | 11.2 ± 0.6 | 11.1 ± 2.1 | 10.3 ± 1.2 | 14.5 ± 2.6 | 15.6 ± 1.7 | 12.8 ± 2.2 |
| 6a | 92 | 12.0 ± 2.2 | 17.0 ± 1.0 | 12.9 ± 0.6 | 12.1 ± 1.3 | 17.2 ± 1.1 | 18.7 ± 1.3 | 15.2 ± 2.8 |
| 1b | 95 | 4.7 ± 0.9 | 3.9 ± 1.2 | 5.2 ± 1.6 | 4.6 ± 1.9 | 5.9 ± 0.8 | 5.7 ± 1.2 | 5.3 ± 1.5 |
| 2b | 94 | 12.1 ± 1.2 | 10 ± 1.3 | 12.6 ± 0.5 | 12.0 ± 0.8 | 17.0 ± 1.1 | 18.6 ± 1.8 | 15.1 ± 1.2 |
| 3b | 94 | 12.4 ± 1.5 | 9.0 ± 2.5 | 12.9 ± 2.6 | 12.3 ± 2.0 | 17.5 ± 3.2 | 19.2 ± 2.8 | 15.4 ± 2.2 |
| 4b | 96 | 8.5 ± 0.7 | 5.3 ± 1.3 | 9.0 ± 1.1 | 8.4 ± 1.9 | 11.6 ± 1.2 | 12.3 ± 4.1 | 10.3 ± 1.4 |
| 5b | 95 | 15.9 ± 2.5 | 14.7 ± 2.2 | 16.4 ± 0.8 | 15.8 ± 0.2 | 22.7 ± 2.5 | 25.3 ± 2.3 | 20.0 ± 2.2 |
| 6b | 94 | 16.2 ± 1.5 | 16.5 ± 3.2 | 16.7 ± 0.7 | 16.1 ± 2.8 | 23.2 ± 2.8 | 25.8 ± 2.3 | 20.4 ± 1.4 |
| Flavopiridol | 4.2 ± 1.2 | 5.5 ± 2.1 | 3.1 ± 1.4 | 6.8 ± 1.2 | 5.4 ± 0.8 | 4.9 ± 1.7 | 2.2 ± 0.3 | |
2.4. Effects on human cancer cell lines
The antiproliferative activity of all synthetic compounds on the growth of human pancreas cancer cell line (Panc-1), pancreatic carcinoma cell line (PaCa-2), epithelial cancer cell line (A-549), lung cancer cell line (H-460), breast cancer cell line (MCF-7), colon cancer cell line (HT-29) and prostate cancer cells (PC-3) was determined using the propidium iodide (PI) fluorescence assay. The compound 1a exhibited highest anticancer activity against all cell lines with IC50 in the range of 0.9–2.2 μM. This was narrowly followed by compound 2a (1.5–4.2 μM). The results of these two compounds were comparable against all seven cell lines used for the experiment.All thiosemicarbazide pyrazolines (reaction products of chalcones having different substitutions and thiosemicarbazide) exhibited stronger anticancer growth inhibitory effects on all seven cell lines in comparison to parent chalcones (Table 2). The IC50 of this group of compounds was in the range of 1.3 to 12.0 μM. The other type of pyrazolines (1b–6b) (reaction products of chalcones having various substitutions and 4-chlorophenylsemicarbazide) showed a weaker growth inhibitory effect on cancer cells as compared to thiosemicarbazide pyrazolines (1a–6a).
In general, all synthetic compounds exhibited comparable antiproliferative activity and the average difference in inhibitory activities was not more than 10% against all cancer cell lines. For all compounds, the median inhibition concentration (IC50) was calculated by using GraphPad Prism software (GraphPad Software, San Diego, CA, USA), and an interesting relationship was observed among the compounds in terms of their structural features on the basis of IC50. The compounds (1–3, 1a–3a and 1b–3b) having (type A) 2,3-dihydro-benzofuran moiety were most active as compared to those (4–6, 4a–6a and 4b–6b) possessing dihydro-benzo[1,4]dioxine in place of 2,3-dihydro-benzofuran. Furthermore, it was seen that six compounds (1, 4, 1a, 4a, 1b and 4b) bearing 4-dimethylamino substitution strongly inhibited the growth of cancer cell lines in contrast with other compounds having dimethoxy substitutions.
2.5. Effects on tubulin polymerization
The Fig. 1 summarizes the tubulin polymerization activity of all the synthetic compounds. The synthetic chalcones (1–6) showed no significant influence on tubulin assembly, which proposes a different mechanism for the observed cytotoxicity than tubulin inhibition (Table 2). Conversely, the most active anticancer compounds (1a–6a and 1b–4b) in propidium iodide fluorescence assay exhibited significant inhibition of tubulin assembly. None of the synthetic compounds showed microtubule-stabilizing activity similar to the established antimitotic chemotherapeutic drug docetaxel.35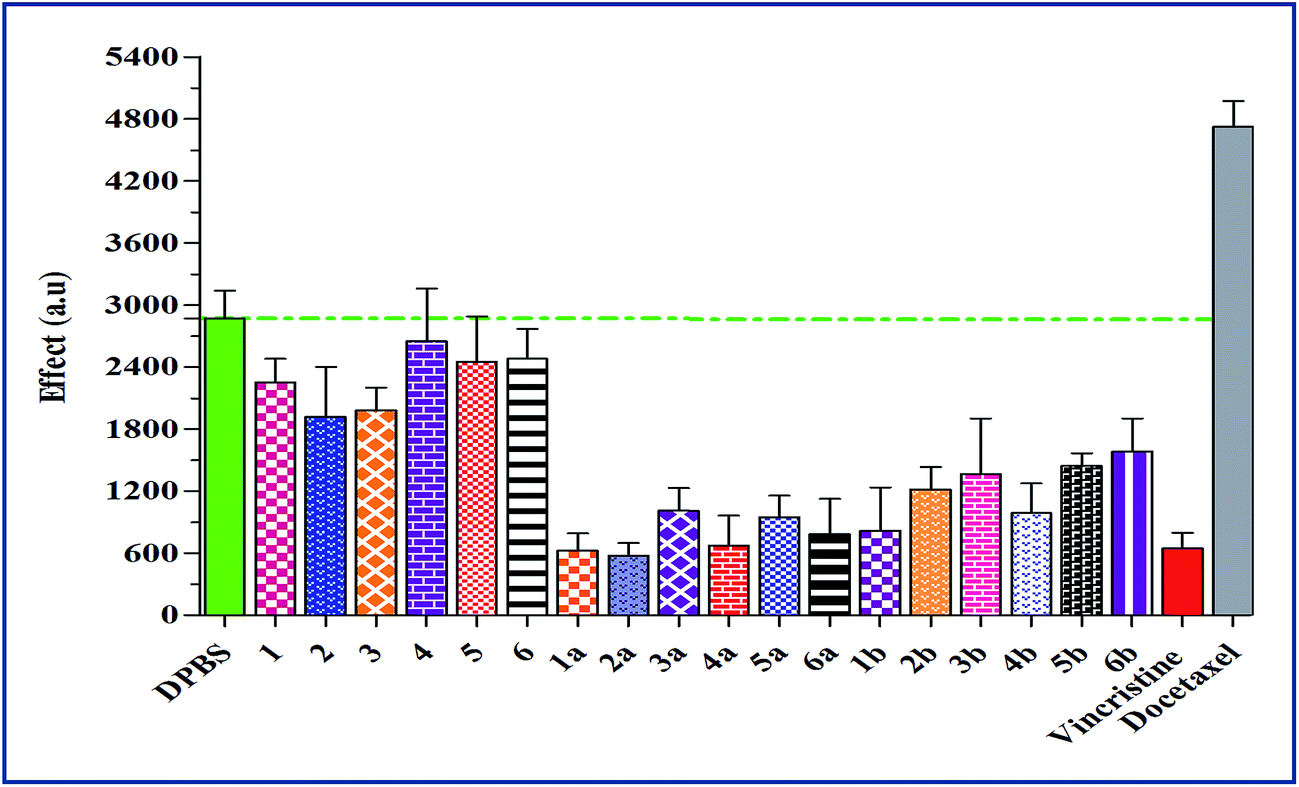 | ||
| Fig. 1 Effect of synthetic compounds on tubulin polymerization activity at concentration of 25 μM. Results are the mean values of three experiments, n = 3. While vincristine and docetaxel (3 μM) were used as reference compounds. All compounds showed the inhibition of tubulin as below the horizontal line indicate tubulin inhibition, only docetaxel is above the horizontal line indicating tubulin stabilization and compounds (4, 5 and 6) close to the horizontal line are considered to be inactive. The y-axis demonstrates tubulin polymerization activity measured at 15 min (arbitrary units) in the growth phase of the tubulin polymerization curve. | ||
2.6. Structure–activity relationship using in silico analysis
The structure–activity relationship is a vital aspect in determining compounds with low bioactivity or unwanted effects. Osiris Property Explorer36 is a knowledge-based activity prediction tool which estimates drug score, drug likeliness and unwanted characteristics like irritant, tumorigenic, mutagenic and reproductive effects of novel compounds on the basis of chemical fragment profiles of already available drugs and non-drugs as reported. From the calculations of Osiris Property Explorer (Table 3), majority of the synthetic compounds have been expected to exhibit strong activity. These findings interrelated well with the anticancer in vitro results for compounds 1a–6a, therefore matching the estimation levels to maximum degree. Low hydrophilicities and hence high log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values cause reduced permeation or absorption. It has been reported that the compounds which have logP value less than 5.0 have a chances of being well-absorbed. Same for the other physicochemical characteristics (Table 2) it can be described that these compounds have prospective to be novel principle compounds in development of anticancer agents. The physicochemical characteristics of majority of the tested compounds were found inside the optimal range (molecular weight <500; TPSA <140 and clogP < 5).37,38 The calculated results of only third series of compounds (1b–6b) synthesized via chemical reaction of chalcones (1–6) with N-(4-chlorophenyl) semicarbazide were not found to be ideal in few aspects.
P values cause reduced permeation or absorption. It has been reported that the compounds which have logP value less than 5.0 have a chances of being well-absorbed. Same for the other physicochemical characteristics (Table 2) it can be described that these compounds have prospective to be novel principle compounds in development of anticancer agents. The physicochemical characteristics of majority of the tested compounds were found inside the optimal range (molecular weight <500; TPSA <140 and clogP < 5).37,38 The calculated results of only third series of compounds (1b–6b) synthesized via chemical reaction of chalcones (1–6) with N-(4-chlorophenyl) semicarbazide were not found to be ideal in few aspects.
| Comp. | Mol. wt | clogP |
Drug-score | TPSA | Drug-likeness | Sa | Mb | Tc | Id | Re |
|---|---|---|---|---|---|---|---|---|---|---|
| a G = green colour indicates drug-conform behaviour; R = red colour indicates undesired effects like mutagenicity. aS (solubility). bM (mutagenic effect). cT (tumorigenic effect). dI (irritant effect). eR (reproductive effect). | ||||||||||
| 1 | 341.0 | 4.90 | 0.14 | 33.45 | 1.9 | −6.67 | R | R | G | G |
| 2 | 358.0 | 4.86 | 0.24 | 48.67 | 3.45 | −6.67 | G | R | G | G |
| 3 | 358.0 | 4.86 | 0.19 | 48.67 | 2.16 | −6.67 | G | R | G | G |
| 4 | 359.0 | 4.37 | 0.09 | 38.77 | −6.26 | −5.66 | R | R | G | G |
| 5 | 376.0 | 4.37 | 0.15 | 53.99 | −4.79 | −5.66 | G | R | G | G |
| 6 | 376.0 | 4.34 | 0.12 | 53.99 | −6.01 | −5.66 | G | R | G | G |
| 1a | 414.0 | 5.19 | 0.09 | 92.31 | 3.13 | −5.54 | R | R | G | R |
| 2a | 431.0 | 5.15 | 0.15 | 107.5 | 4.97 | −5.54 | G | R | G | R |
| 3a | 431.0 | 5.15 | 0.12 | 107.5 | 3.83 | −5.54 | G | R | G | R |
| 4a | 432.0 | 4.63 | 0.06 | 97.63 | −4.52 | −4.87 | R | R | G | R |
| 5a | 449.0 | 4.59 | 0.10 | 112.8 | −2.75 | −4.87 | G | R | G | R |
| 6a | 449.0 | 4.59 | 0.07 | 112.8 | −3.80 | −4.87 | G | R | G | R |
| 1b | 509.0 | 7.01 | 0.08 | 63.3 | 5.08 | −9.31 | R | R | G | G |
| 2b | 525.0 | 6.97 | 0.12 | 78.52 | 6.89 | −9.31 | G | R | G | G |
| 3b | 525.0 | 6.97 | 0.10 | 78.52 | 5.87 | −9.31 | G | R | G | G |
| 4b | 527.0 | 6.45 | 0.04 | 68.62 | −2.61 | −8.64 | R | R | G | G |
| 5b | 544.0 | 6.41 | 0.08 | 83.84 | −0.87 | −8.64 | G | R | G | G |
| 6b | 544.0 | 6.41 | 0.06 | 83.84 | −1.8 | −8.64 | G | R | G | G |
2.7. Molecular docking studies
In order to know the binding modes of the synthesized compounds, molecular docking study of the compounds 4b, 4a, 4, 1b and 1a was carried out against tyrosinase enzyme. Tyrosinase enzyme possesses binuclear copper binding site, and kojic acid and tropolone bind to the entrance of this site.39,40 To discover the probable binding sites in the enzyme, Site Finder application in MOE was used. The area comprising S1 and S2 site was designated as active pocket (Fig. 2). S1 holds binuclear copper binding site while S2 locates nearby the entrance of this site.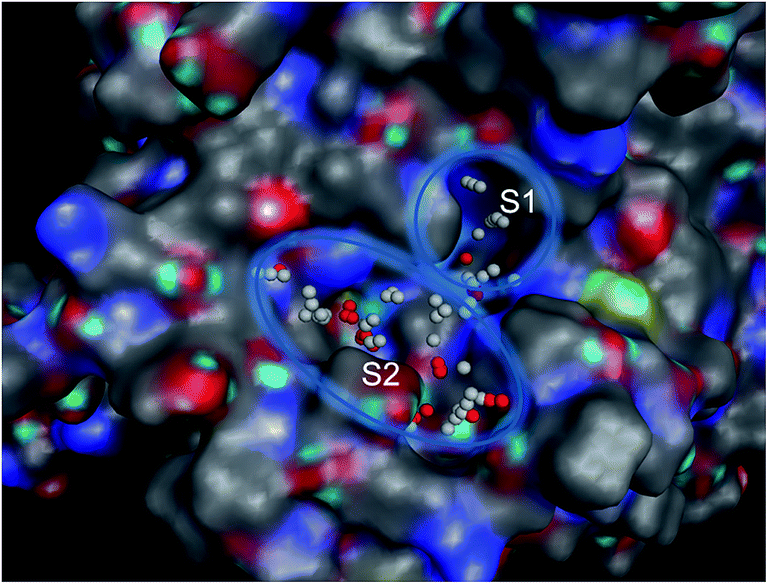 | ||
| Fig. 2 Binuclear copper binding site (S1) and its entrance (S2) region of mushroom tyrosinase enzyme. | ||
The best poses of 4b, 4a, 4, 1b and 1a achieved by docking results were superimposed to equate their orientations (Fig. 3F). It was seen that benzofuran and benzodioxine moieties were fitted well into the cavity formed by His259, His263 and Phe264. Conversely, the naphthalene moieties of these compounds were positioned at the entry of copper binding site (Fig. 3F) and majority of them interacted with His244 and Val283. As the active compounds which were previously synthesized23 also interacted with these residues, it can be assumed that His244 and Val283 could be vital residues for activity. The results of docking for 4b, 4a, 4, 1b and 1a are shown in Fig. 3A–E respectively.
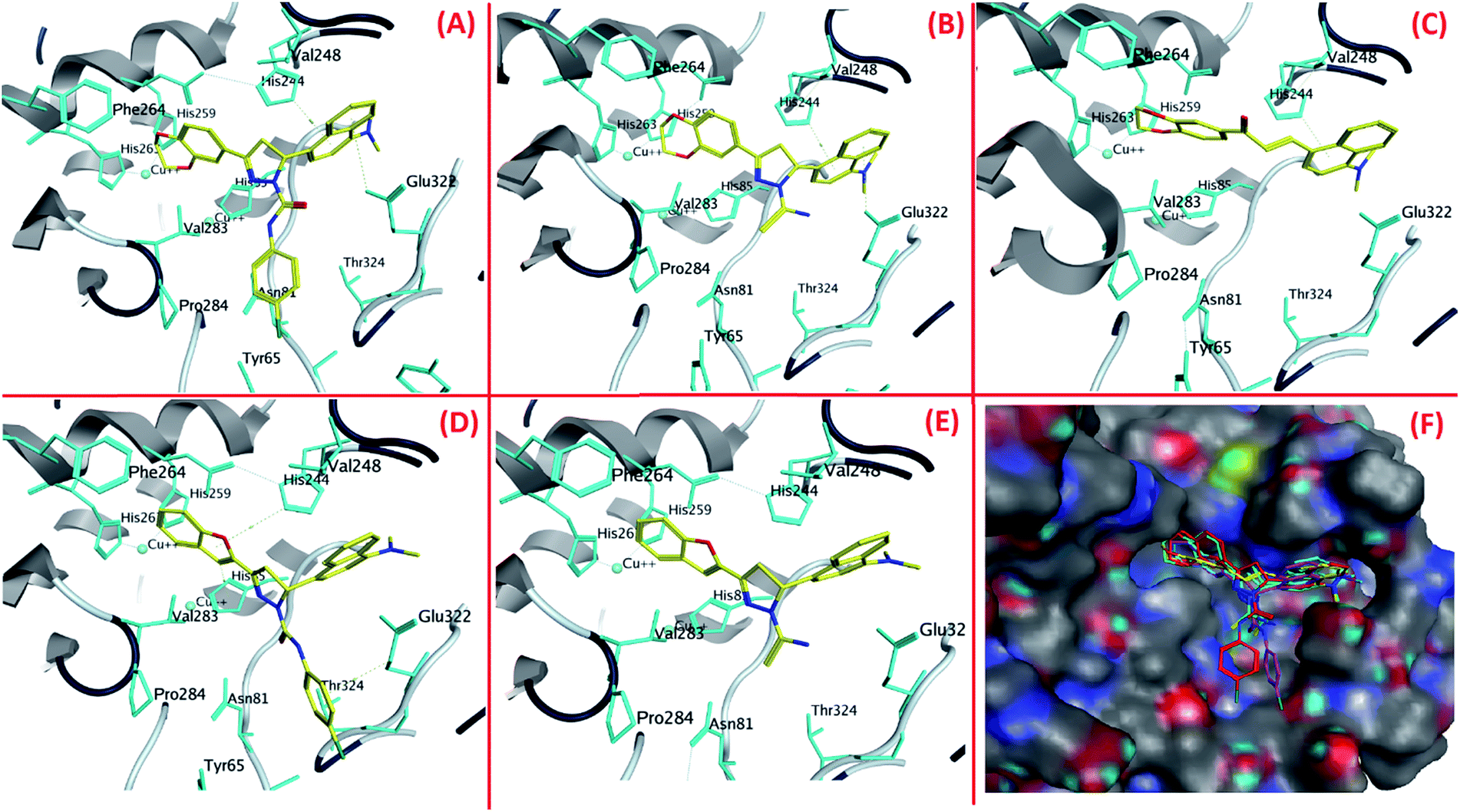 | ||
| Fig. 3 The orientations of (A) 4b, (B) 4a, (C) 4, (D) 1b and (E) 1a in tyrosinase active site and (F) superimposition of the best poses of compound 4b, 4a, 4, 1b and 1a. | ||
3. Methods and materials
3.1. Chemical reagents and instruments
All reagents and chemicals used in this study were purchased from Acros Organics, Merck and Sigma-Aldrich, were of analytical grade, and were used as supplied, unless otherwise specified. A characteristic work-up comprised washing with brine and drying the organic layer with anhydrous magnesium sulfate afore concentration in vacuo. 13C and 1H NMR spectra were recorded on a JEOL ECP spectrometer operating at 500 MHz, with CDCl3 or DMSO-d6 as the solvents and Me4Si as internal standard. High resolution mass spectra (HRMS) were obtained by the electrospray ionization mass spectrometry (ESI-MS) using MicroTOF-Q mass spectrometer (Bruker). Fison EA 1108 elemental analyzer was used to obtain the microanalyses data. Silica gel 60 (230–400 mesh) (Merck) was used to carry out flash column chromatography whereas thin layer chromatography (TLC) was performed on pre-coated silica plates (kiesel gel 60 F254, BDH). Melting points of the compounds were determined on an electrothermal instrument and were uncorrected. The synthesized compounds were envisioned by illumination under ultraviolet (UV) light (254 nm) or by the use of vanillin stain subsequently charring on a hotplate.3.2. Synthesis of chalcone and derived pyrazoline derivatives
The method for the synthesis of chalcones and pyrazolines has been reported previously.32 The key steps of synthesis are shown in the Schemes 1–3. For the synthesis of chalcone derivatives (1–6), respective ketones (10 mmol) were added to a 15 mL solution of the respective aldehydes (10 mmol) in ethanol (10 mmol chalcones (1–6) and 10 mmol thiosemicarbazide/4-chlorophenylsemicarbazide for synthesis of pyrazolines). The mixture of the reaction was made alkaline by the drop wise addition of 50% NaOH solution, and afterwards the reaction mixture was stirred at 27 °C for 2–18 h. TLC monitored the reaction completion. Color changes and appearance of precipitate in the reaction mixture were observed and recorded, as indicators of product formation. Upon completion, the reaction mixture was poured into 50 mL of acidified ice (using 1 mL concentrated HCl). It was extracted with 50 mL ethyl acetate, washed with water (150 mL) followed by drying and concentration in vacuo to produce either solids or oils as the reaction product. The obtained products were further purified by either column chromatography or by recrystallization. The spectral data of the most active tyrosinase inhibitors and anticancer compounds are provided below.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 7.78 (d, J = 8.5 Hz, H), 7.66 (d, J = 7.0 Hz, H), 7.56 (d, J = 7.3 Hz, H), 7.41 (d, J = 6.0 Hz, H), 7.37–7.31 (m, J = 8.0 Hz, 3H), 7.15 (s, H), 7.12 (d, J = 8.0 Hz, H), 6.82 (d, J = 8.5 Hz, H), 6.61 (d, J = 7.5 Hz, H), 5.12 (m, J = 8.4 Hz, 4H), 3.72 (s, 6H); 13C NMR (DMSO-d6) δ: 32.2, 32.8, 82.2, 82.6, 111.3, 118.2, 119.6, 123.5, 124.3, 126.3, 127.2, 128.2, 128.8, 129.1, 129.5, 132.4, 133.4, 137.3, 146.5, 148.8, 152.4, 154.6, 190.2. Mass spectrum (ESI) m/z: 382.62 [M + Na]+; microanalysis calculated for C23H21NO3 (359.42), C, 76.86%; H, 5.89%; N, 3.90%. Found C: 77.12%, H: 5.98%, N: 3.98%.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 8.80 (s, D2O exchangeable, 2H, NH2), 7.78–7.30 (m, J = 8.0 Hz, 10H), 7.19 (s, 1H), 5.52–5.47 (dd, J = 12.0 Hz, 1H), 4.21–4.13 (dd, J = 12.0 Hz, 1H), 3.87 (s, 6H), 3.45–3.29 (dd, J = 17.5 Hz, 1H); mass spectrum (ESI) m/z: 415.62 [M + H]+; microanalysis calculated for C24H22N4OS (414.52), C, 69.54%; H, 5.35%; N, 13.52%; S, 7.74%. Found C: 69.63%, H: 5.29%, N: 13.61%, S: 7.79%.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 8.87 (s, D2O exchangeable, 2H, NH2), 7.89–7.29 (m, J = 8.0 Hz, 8H), 7.16 (s, 1H), 6.92 (d, J = 8.0 Hz, 1H), 5.57–5.50 (dd, J = 11.5 Hz, 1H), 4.22–4.19 (dd, J = 11.5 Hz, 1H), 3.85 (s, 6H), 3.47–3.34 (dd, J = 17.5 Hz, 1H); mass spectrum (ESI) m/z: 432.52 [M + H]+; microanalysis calculated for C24H21N3O3S (431.51), C, 66.80%; H, 4.91%; N, 9.74%; S, 7.43%. Found C: 66.97%, H: 4.98%, N: 9.73%, S: 7.41%.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 8.94 (s, D2O exchangeable, 2H, NH2), 7.82–7.47 (m, J = 6.5 Hz, 8H), 7.12 (s, 1H), 5.42–5.32 (dd, J = 12.0 Hz, 1H), 5.12 (m, J = 8.0 Hz, 4H), 4.22–4.15 (dd, J = 12.0 Hz, 1H), 3.80 (s, 6H), 3.24–3.12 (dd, J = 16.0 Hz, 1H); mass spectrum (ESI) m/z: 455.64 [M + Na]+; microanalysis calculated for C24H24N4O2S (432.54), C, 66.64%; H, 5.59%; N, 12.95%; S, 7.41%. Found C: 66.68%, H: 5.56%, N: 12.99%, S: 7.43%.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 8.91 (s, NH), 7.75–7.17 (m, J = 8.0 Hz, 14H), 7.09 (s, 1H), 5.42–5.34 (dd, J = 12.0 Hz, 1H), 4.18–4.12 (dd, J = 12.0 Hz, 1H), 3.82 (s, 6H), 3.22–3.16 (m, J = 17.5 Hz, 1H); mass spectrum (ESI) m/z: 510.13 [M + H]+; microanalysis calculated for C30H25ClN4O2 (509.00), C, 70.79%; H, 4.95%; N, 11.01%. Found C: 70.83%, H: 4.98%, N: 10.98%.:CH2Cl2, 2:1); 1H NMR (500 MHz, CDCl3) δ: 8.94 (s, NH), 7.80–7.27 (m, J = 8.0 Hz, 12H), 7.15 (s, 1H), 5.36–5.27 (dd, J = 12.5 Hz, 1H), 5.11 (m, J = 8.0 Hz, 4H), 4.28–4.16 (dd, J = 12.5 Hz, 1H), 3.86 (s, 6H), 3.33–3.19 (dd, J = 16.0 Hz, 1H); mass spectrum (ESI) m/z: 528.05 [M + H]+; microanalysis calculated for C30H27ClN4O3 (527.01), C, 68.37%; H, 5.16%; N, 10.63%. Found C: 68.39%, H: 5.14%, N: 10.68%.3.3. Tyrosinase assay
The complete methodology of spectrophotometric assay for tyrosinase was reported previously by our research group.23 To screen the synthesized compounds for the diphenolase inhibitory activity of tyrosinase, L-DOPA was used as a substrate. The positive controls used in the assay were 4-butylresorcinol and kojic acid. DMSO was used to prepare 2% stock solutions of synthesized compounds and were diluted using phosphate buffer (pH = 6.8). The compounds were pre-incubated in 50 mM phosphate buffer (pH = 6.8) with thirty units of mushroom tyrosinase (0.5 mg mL−1) at 25 °C for 10 min. Afterwards, L-DOPA (0.5 mM) was added to the reaction mixture. The reaction was monitored by following the formation of L-DOPA chrome for 1 min, measured by the change in absorbance at 475 nm. The measurements were taken in triplicates for each concentration and average was used for further calculations. The IC50 values were obtained by the interpolation of dose–response curves.3.4. MTT assay
MTT assay was carried out to determine the effect of synthetic compounds on normal (MCF-10A) mammary epithelial cells. The MCF-10A cells were maintained in Dulbecco's modified Eagle's medium/Ham's F-12 medium (1:1) with epidermal growth factor (20 ng mL−1), hydrocortisone (500 ng mL−1), insulin (10 μg mL−1), 2 mM glutamine and 10% foetal calf serum. The cells were passaged every 2–3 days using trypsin ethylenediaminetetraacetic acid (EDTA). MCF-10A cells were seeded at a density of 104 cells per mL in 96-well flat-bottomed plates. After 24 h, the medium was cautiously removed and tested compounds (curcumin analogues) were added in quadruplicate in 200 μL of medium to give a final concentration of not more than 0.1% (v/v) dimethylsulfoxide (DMSO). Afterwards, the cells were kept to grow for 96 h at 37 °C in the incubator. The medium was then removed and fresh medium with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (0.4 mg mL−1) was added in each well for 3 h. All medium was removed and the formazan product produced by live cells was solubilised with 150 μL of DMSO. The plates were vortexed and the absorbance at 540 nm was measured using a microplate reader. The results were expressed as the percentage inhibition of proliferation in comparison to controls containing 0.1% DMSO.
3.5. Antiproliferative activity
To determine the antiproliferative activity, propidium iodide (PI) fluorescence assay was performed on HT-29 (colon cancer cell line), H-460 (lung cancer cell line), HL-60 (promyelocytic leukemia cell line), PC-3 (prostate cancer cell line) and MCF-7 (breast cancer cell line) cell lines.41 Propidium iodide (PI) is a dye which attaches to DNA, provides an accurate and rapid means for calculating total nuclear DNA. The principle behind this assay is that PI is unable to enter an intact cell membrane and the fluorescence signal intensity of the PI is directly related to the quantity of DNA in each cell. Therefore, the cells which have impair membranes or changed permeability profiles are counted as dead cells. The cells for the assay were seeded at 3000–7500 cells per well in 200 μL media in sterile tissue culture grade 96-well plates and allowed to recover for 24 h in humidified 5% CO2 incubator at 37 °C. Afterwards, the compounds (in 0.1% DMSO) were added in triplicate wells at concentrations of 10 μM. 0.1% DMSO alone was used as control. The 96-well plate was then kept in the incubator for 48 h. After the completion of selected treatment time, the medium was aspirated and cells were treated with 25 μL of PI (50 μg mL−1 in water/medium) per well. The 96-well plates containing samples were then frozen at −80 °C for 24 h, thawed and allowed to equilibrate to room temperature. The readings were taken on a fluorometer (Polar-Star BMG Tech), using 530 nm excitation and 620 nm emission wavelength. Finally, the percent cytotoxicity of the compounds was calculated by using following formula:where AC = absorbance reading of control and ATC = absorbance reading of treated cells. A standard drug inhibitor flavopiridol (0.5 μM) was used to compare the results.
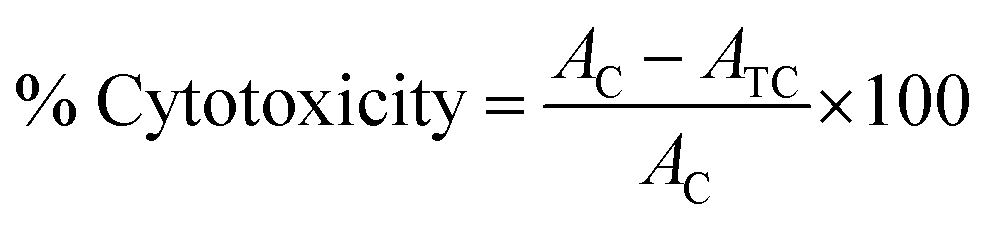
3.6. Tubulin polymerization assay
The effects of synthesized compounds on tubulin polymerization were determined using a commercial kit (Tubulin Polymerization Assay Kit (Porcine tubulin and Fluorescence based), Cytoskeleton Inc., Denver, CO, USA), which utilized fluorescent reporter enhancement (the standard assay protocol was used).42 The fluorescence was measured by using FLUO star OPTIMA and the test substances (dissolved in DMSO) were evaluated at 5 and 25 μM. The experiments were performed in triplicate and the mean values are presented. Vincristine (Apoteket AB, Sweden) and docetaxel (Apoteket AB, Sweden) and were used as positive destabilizing and stabilizing controls, respectively. Both were diluted with PBS and used at a concentration of 3 μM.3.7. Molecular modeling
Molecular modeling studies were performed on MOE (The Molecular Operating Environment) Version 2011.10, software available from Chemical Computing Group Inc., 1010 Sherbrooke Street West, Suite 910, Montreal, Canada H3A2R7, http://www.chemcomp.com. The ligands were built by using the builder tool of the MOE program and were subjected to energy minimization (MMFF94x, gradient: 0.05). The X-ray crystallographic structure of mushroom tyrosinase (PDB: 2Y9W)39 was obtained from the Protein Data Bank. The Structure Preparation process in MOE was used to correct the errors of the protein. Hydrogens were added after correction and partial charges (Gasteiger methodology) were calculated. The possible binding pocket was detected using site finder application. The docking study was performed by default Triangle Matcher placement method. To rank the final poses, GBVI/WSA dG scoring function (which estimates the free energy of binding of the ligand from a given pose) was used. From the top two lowest S scores in the output databases of MOE docking, the ligand–enzyme complex for each ligand was manually selected. The selected complexes were minimized by using LigX utility which holds the receptor atoms far from the ligand fixed whereas permits receptor atoms near the ligand to move.3.8. Statistical analysis
The analysis was carried out by using a complete randomized design and treatments were run in triplicates (n = 3). Statistical analysis was done using analysis of variance (ANOVA) method of general linear model (GLM) procedure using SAS V8 (Cary, NC, USA) for multiple comparisons. P < 0.05 was considered to be statistically significant.4. Conclusion
Among tested chalcone and pyrazoline derivatives, four compounds (1a, 4a, 1b and 4b) exhibited the most potent inhibitory effect on mushroom tyrosinase, even better than the positive control. The compound 4b exhibited highest inhibition of tyrosinase with IC50 4.72 μM, indicating that 4b was a three-fold more effective tyrosinase inhibitor than kojic acid (IC50 12.42 μM). All compounds were evaluated for their anticancer activity and for their effect on microtubule assembly. Several compounds showed cytotoxic activities towards cancer cell lines. Seven of the compounds were discovered to be strong tubulin polymerization inhibitors. The results suggest that these molecules can serve as interesting candidates for the treatment of tyrosinase-related disorders and as leads for the development of new and potent tyrosinase inhibitors and anticancer agents. Nevertheless, from a clinical point of view, this model using mushroom tyrosinase is not adequate for assessing active molecules intended for human use. Hence, more investigation of these potent compounds using human tyrosinase and mechanistic studies for anticancer properties are underway in our laboratory, and the research results will be reported in due course.Notes
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Research Incentive Fund of Universiti Kebangsaan Malaysia Arus Perdana grant (AP-2014-023). HQ is grateful to Wuhan University of technology for financial support.References
- R. A. Spritz and V. J. Hearing Jr, Adv. Hum. Genet., 1994, 22, 1–45 CAS.
- J. M. Menter and I. Willis, Pigm. Cell Res., 1997, 10, 214–217 CrossRef CAS PubMed.
- K. Shankar, K. Godse, S. Aurangabadkar, K. Lahiri, V. Mysore, A. Ganjoo, M. Vedamurty, M. Kohli, J. Sharad, G. Kadhe, P. Ahirrao, V. Narayanan and S. A. Motlekar, Dermatol Ther., 2014, 4, 165–186 CrossRef PubMed.
- A. J. Stratigos and A. D. Katsambas, Am. J. Clin. Dermatol., 2004, 5, 161–168 CrossRef PubMed.
- I. Kubo, I. Kinst-Hori, K. Nihei, F. Soria, M. Takasaki, J. S. Calderon and C. L. Cespedes, Z. Naturforsch. C Bio. Sci., 2003, 58, 719–725 CAS.
- F. Artés, M. Castañer and M. I. Gil, Food Sci. Technol. Int., 1998, 4, 377–389 CrossRef PubMed.
- M. R. Loizzo, R. Tundis and F. Menichini, Compr. Rev. Food Sci. Food Saf., 2012, 11, 378–398 CrossRef CAS PubMed.
- J. Segura-Aguilar, I. Paris, P. Muñoz, E. Ferrari, L. Zecca and F. A. Zucca, J. Neurochem., 2014, 129, 898–915 CrossRef CAS PubMed.
- M. Ashida and P. T. Brey, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 10698–10702 CrossRef CAS.
- M. C. Balcos, S. Y. Kim, H. S. Jeong, H. Y. Yun, K. J. Baek, N. S. Kwon, K. C. Park and D. S. Kim, Acta Pharmacol. Sin., 2014, 35, 489–495 CrossRef CAS PubMed.
- S.-M. Lee, Y.-S. Chen, C.-C. Lin and K.-H. Chen, Int. J. Mol. Sci., 2015, 16, 1495–1508 CrossRef CAS PubMed.
- E. Mendes, M. d. J. Perry and A. P. Francisco, Expert Opin. Drug Discovery, 2014, 9, 533–554 CrossRef CAS PubMed.
- A. Kim and J. Y. Ma, Pharmacogn. Mag., 2014, 10, S463–S471 CrossRef PubMed.
- L. Kolbe, T. Mann, W. Gerwat, J. Batzer, S. Ahlheit, C. Scherner, H. Wenck and F. Stäb, J. Eur. Acad. Dermatol. Venereol., 2013, 27, 19–23 CrossRef CAS PubMed.
- Y.-F. Lin, Y.-H. Hu, H.-T. Lin, X. Liu, Y.-H. Chen, S. Zhang and Q.-X. Chen, J. Agric. Food Chem., 2013, 61, 2889–2895 CrossRef CAS PubMed.
- R. Haudecoeur and A. Boumendjel, Curr. Med. Chem., 2012, 19, 2861–2875 CrossRef CAS.
- I. E. Orhan and M. T. Khan, Curr. Top. Med. Chem., 2014, 14, 1486–1493 CrossRef CAS.
- T.-S. Chang, Int. J. Mol. Sci., 2009, 10, 2440–2475 CrossRef CAS PubMed.
- M. A. Peralta, M. G. Ortega, A. M. Agnese and J. L. Cabrera, J. Nat. Prod., 2011, 74, 158–162 CrossRef CAS PubMed.
- S. S. Gawande, S. C. Warangkar, B. P. Bandgar and C. N. Khobragade, Bioinorg. Med. Chem., 2013, 21, 2772–2777 CrossRef CAS PubMed.
- A. You, J. Zhou, S. Song, G. Zhu, H. Song and W. Yi, Bioinorg. Med. Chem., 2015, 23, 924–931 CrossRef CAS PubMed.
- S. Khatib, O. Nerya, R. Musa, S. Tamir, T. Peter and J. Vaya, J. Med. Chem., 2007, 50, 2676–2681 CrossRef CAS PubMed.
- S. N. A. Bukhari, I. Jantan, O. Unsal Tan, M. Sher, M. Naeem-ul-Hassan and H.-L. Qin, J. Agric. Food Chem., 2014, 62, 5538–5547 CrossRef CAS PubMed.
- J. Liu, W. Yi, Y. Wan, L. Ma and H. Song, Bioinorg. Med. Chem., 2008, 16, 1096–1102 CrossRef CAS PubMed.
- S. N. Bukhari, S. G. Franzblau, I. Jantan and M. Jasamai, Med. Chem., 2013, 9, 897–903 CrossRef CAS.
- S. N. Bukhari, I. Jantan and M. Jasamai, Mini-Rev. Med. Chem., 2013, 13, 87–94 CrossRef CAS.
- S. N. Bukhari, M. Jasamai and I. Jantan, Mini-Rev. Med. Chem., 2012, 12, 1394–1403 CAS.
- M. J. Matos, S. Vazquez-Rodriguez, E. Uriarte and L. Santana, Expert Opin. Ther. Pat., 2015, 25, 351–366 CrossRef CAS PubMed.
- C. Karthikeyan, N. S. Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 97–115 CrossRef CAS.
- H.-M. Wang, L. Zhang, J. Liu, Z.-L. Yang, H.-Y. Zhao, Y. Yang, D. Shen, K. Lu, Z.-C. Fan, Q.-W. Yao, Y.-M. Zhang, Y.-O. Teng and Y. Peng, Eur. J. Med. Chem., 2015, 92, 439–448 CrossRef CAS PubMed.
- S. Ducki, Anti-Cancer Agents Med. Chem., 2009, 9, 336–347 CrossRef CAS.
- S. N. Bukhari, X. Zhang, I. Jantan, H. L. Zhu, M. W. Amjad and V. H. Masand, Chem. Biol. Drug Des., 2015, 85, 729–742 CAS.
- C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183–191 CrossRef CAS PubMed.
- A. E. Nassar, A. M. Kamel and C. Clarimont, Drug Discovery Today, 2004, 9, 1055–1064 CrossRef CAS.
- V. A. de Weger, J. H. Beijnen and J. H. M. Schellens, Anti-Cancer Drugs, 2014, 25, 488–494 CrossRef CAS PubMed.
- http://www.organic-chemistry.org/prog/peo/.
- D. Zhang, A. Templeton, W. Marinaro, A. F. Rumondor, F. Kesisoglou, B. Duersch, K. Thompson, J. Stellabott and M. Kress, in Discovering and Developing Molecules with Optimal Drug-Like Properties, ed. A. C. Templeton, S. R. Byrn, R. J. Haskell and T. E. Prisinzano, Springer, New York, 2015, vol. 15, ch. 14, pp. 469–507 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- W. T. Ismaya, H. J. Rozeboom, A. Weijn, J. J. Mes, F. Fusetti, H. J. Wichers and B. W. Dijkstra, Biochemistry, 2011, 50, 5477–5486 CrossRef CAS PubMed.
- M. Sendovski, M. Kanteev, V. S. Ben-Yosef, N. Adir and A. Fishman, J. Mol. Biol., 2011, 405, 227–237 CrossRef CAS PubMed.
- W. A. Dengler, J. Schulte, D. P. Berger, R. Mertelsmann and H. H. Fiebig, Anti-Cancer Drugs, 1995, 6, 522–532 CrossRef CAS PubMed.
- D. Bonne, C. Heuséle, C. Simon and D. Pantaloni, J. Biol. Chem., 1985, 260, 2819–2825 CAS.
| This journal is © The Royal Society of Chemistry 2015 |