
Injectable hydrogels based on poly(ethylene glycol) and derivatives as functional biomaterials
Emilia Bakaic
,
Niels M. B. Smeets
and
Todd Hoare
*
Department of Chemical Engineering, McMaster University, 1280 Main St. W, Hamilton, Ontario L8S 4L7, Canada. E-mail: hoaretr@mcmaster.ca
First published on 13th April 2015
Abstract
Hydrogels based on poly(ethylene glycol) (PEG) and derivatives have attracted significant interest in recent years given their capacity to be well-tolerated in vivo in the context of drug delivery and tissue engineering applications. Injectable, in situ-gelling analogues of such hydrogels offer the additional advantages of being easy and non-invasive to administer via the injection of low-viscosity precursor polymer solutions, expanding their scope of potential applications. In this highlight, we first review the design criteria associated with the rational design of in situ-gelling hydrogels for in vivo applications. We then discuss recent progress in the design of injectable PEG hydrogels, specifically highlighting our ongoing work on PEG-analogue hydrogels based on poly(oligoethylene glycol methacrylate) for targeted biomedical applications.
 Emilia Bakaic | Emilia Bakaic is a Ph.D. student in the Department of Chemical Engineering at McMaster University, working in the laboratory of Prof. Todd Hoare. She received her Bachelor's degree in Biomedical Sciences from the University of Waterloo in 2010. Her research focuses on the design of injectable polymeric hydrogel systems for various biomedical applications, including tissue scaffold engineering and drug delivery. |
 Niels M. B. Smeets | Niels M. B. Smeets is a Senior Scientist at EcoSynthetix Inc. in Burlington, Ontario, Canada and was formerly a post-doctoral fellow in Prof. Todd Hoare's laboratory at McMaster University. He received a Ph.D. in polymer chemistry and polymer reaction engineering from Eindhoven University of Technology (the Netherlands) in 2009. His research interests include synthetic polymer design, polymeric materials for biomedical applications, and biomaterials. Smeets has published over 35 journal articles and has 3 pending patent applications. |
 Todd Hoare | Todd Hoare is an Associate Professor and Distinguished Engineering Fellow in the Department of Chemical Engineering at McMaster University. He received his Ph.D. in Chemical Engineering from McMaster in 2006 and joined the faculty after post-doctoral fellowship at the Massachusetts Institute of Technology. Hoare has published 3 book chapters and over 50 journal articles, has one granted and 11 pending patent applications, and won the John Charles Polanyi Prize in Chemistry from the Government of Ontario recognizing his early career accomplishments. |
1 Introduction
Hydrogels are water-swollen polymeric networks capable of absorbing large quantities of water and biological fluids while maintaining a distinct three-dimensional structure.1 The high water content of hydrogels, combined with the structural similarity of hydrogels to natural extracellular matrix (ECM),2 makes hydrogels attractive synthetic materials for biomedical devices including drug release depots,3–5 cell delivery scaffolds,6–8 functional tissues,9–11 or wound dressings.12 The cross-linked, three-dimensional structure, while key to the ultimate applications of hydrogels, also severely limits the ability of hydrogels to be delivered inside the body by minimally invasive routes (i.e. non-surgical procedures such as injections). As a result, significant focus has shifted to the design of “injectable” or “in situ gelling” hydrogels that form after injection in vivo.13,14 The hydrogel precursor solution can be introduced in a minimally invasive manner directly to the area of interest, significantly reducing patient discomfort, risk of infection, recovery time, and treatment cost. Once injected, hydrogel formation occurs through either physical cross-linking, typically exploiting the physiological conditions in the target tissue, or by chemical cross-linking, exploiting click and/or dynamic covalent bond chemistry.15–18 For many applications, the use of chemical cross-linking is preferred to improve the mechanical properties of the soft material, preserve the integrity of the hydrogel over an externally-programmed period of time (based on the type and density of cross-links present), and enhance the chemical flexibility of hydrogel in terms of its ability to be easily functionalized with physical or biological cues that improve its function and/or compatibility with the surrounding tissue.19In this highlight, the relevant design criteria for injectable hydrogels will be briefly outlined and illustrated by reviewing a number of examples from the current state-of-the-art in injectable PEG hydrogels. The highlight will be concluded with a summary of our recent work on injectable PEG-analogue poly(oligoethylene glycol methacrylate) (POEGMA) hydrogels to illustrate the potential of these biomaterials as a potential substitute for conventional PEG-based biomaterials.
2 Design criteria for covalently cross-linked injectable hydrogels
The clinical lifetime of an injectable hydrogel can be divided into three separate stages: (i) pre-gelation (precursor polymers in solution), (ii) therapeutic window (after injection and gelation) and (iii) degradation (hydrogel degradation products). Rational design of injectable hydrogels thus requires careful consideration of the synthetic and biological aspects of the precursor polymers, resulting hydrogel, as well as the hydrogel degradation products. The key design criteria will be discussed accordingly, as schematically depicted in Scheme 1.14,16,18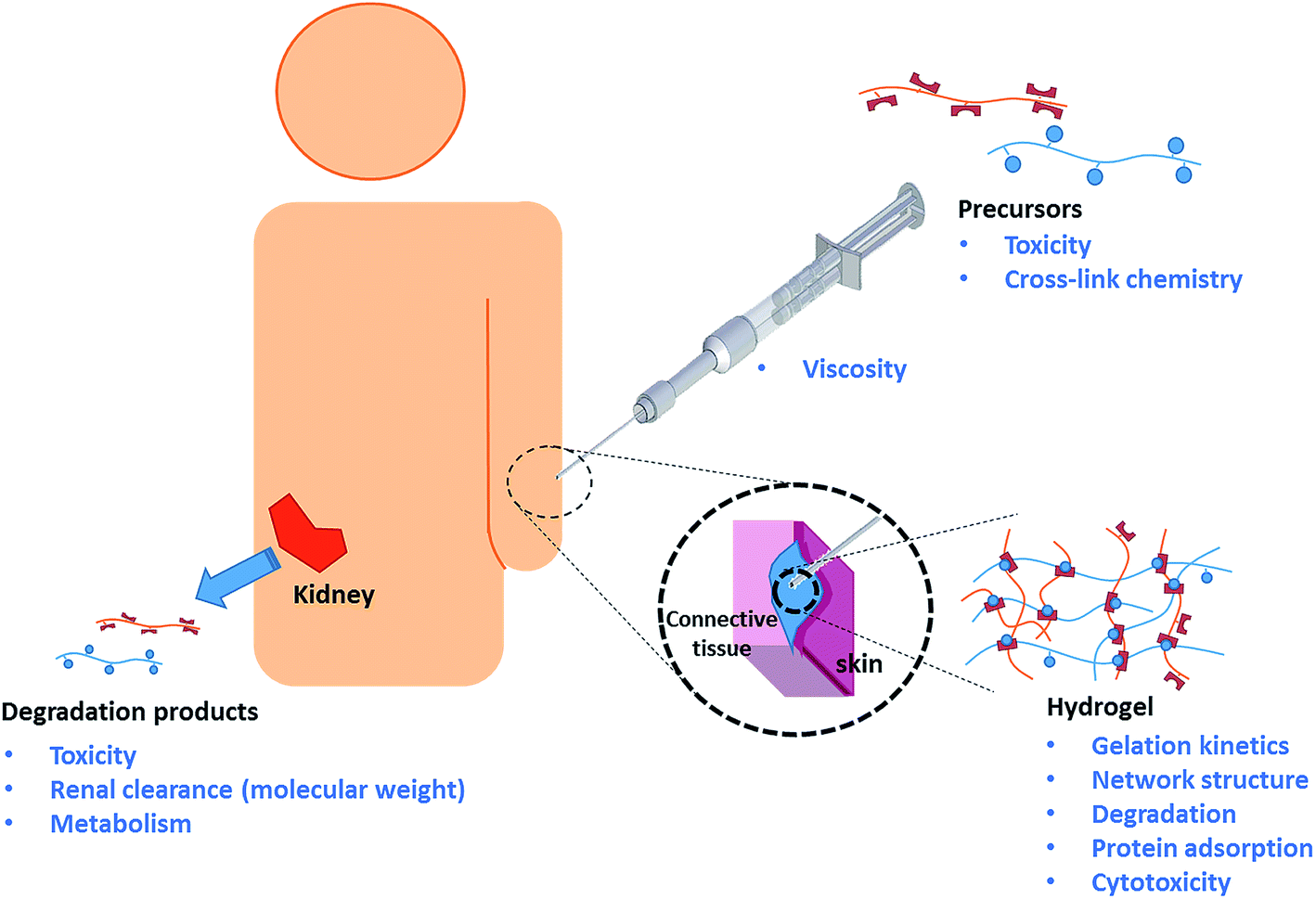 | ||
| Scheme 1 Schematic representation of the various design criteria for injectable hydrogels. | ||
Precursor polymers
Hydrogel precursor polymers can be based on naturally occurring polymers (e.g. collagen,20 fibrin,21,22 hyaluronic acid,23 alginate,24 agarose,25 or dextran26) or synthetic polymers (e.g. poly(N-isopropylacrylamide) (PNIPAAm),27,28 polyacrylamide (PAAm),29 poly(hydroxyethyl methacrylate) (PHEMA), poly(vinylpyrrolidone) (PVP), poly(ethylene glycol) (PEG) or other water-soluble polymers;4,11,30). Although most natural polymers can be directly cross-linked by small bifunctional molecules such as epichlorohydrin or glyoxal,31 a large variety of chemistries has been developed to functionalize these polymers with cross-linkable functional groups and thus avoid the need for often toxic small molecule cross-linkers.32 Synthetic polymers can be functionalized directly through the use of functional comonomers and/or via post-polymerization modification. Regardless of the chemical nature of the precursor, the precursor polymer needs to be stable, non-cytotoxic, and not elicit a significant inflammatory response from the patient's immune system upon injection. This latter point is especially challenging in the context of covalently cross-linked injectable hydrogels since chemically-driven gelation requires that the hydrogel precursors are functionalized with reactive functional groups that are not necessarily bioorthogonal33 (e.g. amine, aldehyde, or thiol groups that are also present in, or cross-reactive with, polypeptides including enzymes, membrane proteins, etc.) However, it must be emphasized that the toxicity of functional polymeric precursors is typically low compared to low molecular weight chemicals with the same functionalities. For example, low molecular weight vinylsulfones (used in Michael-type addition cross-linking with thiols) demonstrate significant toxicity, but polymer-bound vinyl sulfones cannot enter cells and thus do not undergo toxic reactions with glutathione and DNA.34,35 Similarly, the use of low molecular weight glutaraldehyde for gelation has been reported to induce toxicity,36,37 but Hoffmann and co-workers noted no apparent cytotoxic effects associated with chitosan–glutaraldehyde hydrogels cultivated with ATDC5 and chrondrocytes up to 14 days.38 In our experience, both natural (oxidized dextran or carboxymethyl cellulose)39,40 as well as synthetic aldehyde-functionalized precursors do not show any significant toxicity either in vitro (via an MTT assay on 3T3 mouse fibroblasts at concentrations up to 2000 μg mL−1)40,41 or in vivo (via subcutaneous injection in BALB/c mice).41,42The precursor polymers are likely to be stored for a considerable amount of time prior to clinical use and thus need to demonstrate high stability in aqueous solution. Furthermore, as administration occurs through a needle, the precursor solutions need to be of sufficiently low viscosity, or at least possess sufficient shear-thinning properties, to allow for easy injection. For routine applications, the maximum needle size for injections is 25G;43 however, many ophthalmic or cosmetic applications require as small as a 33G needle sizes. This requirement makes both molecular weight control and polymer architecture very important in the design of a scaffold.
Hydrogels
For successful in vivo application, gelation (i.e. cross-linking of the precursor polymers) must occur at physiologically relevant conditions (i.e. 5.5 ≤ pH ≤ 7.5 and T = 37 ± 2 °C) relatively quickly (i.e. seconds to minutes) following injection. The emergence of click chemistries has introduced a variety of cross-linking approaches that are advantageous to the design of injectable hydrogels,44 most notably copper-catalyzed alkyne azide reactions (CuAAC),45 strain promoted azide-alkyne click cycloadditions (SPACC),46 thiol-Michael additions,47 Diels–Alder cycloaddition,48 disulfide formation,49,50 oxime chemistry,51 and Schiff base52 formation. While not all of these cross-link chemistries are strictly bioorthogonal, in vitro and in vivo toxicity of such chemistries can be minimized by controlling gelation kinetics, as fast gelation kinetics reduce the amount of time that the non-bioorthogonal functional groups are exposed to native tissue prior to consumption via cross-linking. Gelation kinetics can be controlled by varying precursor polymer concentration, the number of reactive groups per chain, and/or the molecular weight of the precursor polymers.53–57 Relatively fast gelation kinetics are typically beneficial in an injectable system to minimize the diffusion of precursors from the site of injection, which may cause adverse effects in neighboring tissues and/or prevent hydrogel formation entirely if the dilution is too significant. However, it is important minimize the formation of structural and mechanical defects in the hydrogel network, which directly influences the diffusivity of therapeutics, nutrients, oxygen and (toxic) waste products. Controlling the cross-link density of hydrogels allows for the design of injectable hydrogels that mimic a broad range of tissue mechanics ranging from soft brain tissue (0.2–1 kPa) to relatively stiff cartilage or pre-calcified bone tissue (20–60 kPa).58 However, in most in situ gelling systems, cross-link density and gelation kinetics are inextricably linked (as both are governed by the degree of precursor polymer functionalization), representing a challenge in rational hydrogel design of highly cross-linked gels that can still be delivered by injection or weaker gels that need to be gelled quickly.Aside from certain applications (e.g. dermal fillers),59 injectable hydrogels must be either biodegradable60,61 or bio-erodible62 to avoid surgical removal after its therapeutic lifetime has expired. In tissue engineering applications, controlled degradation is even more important as a functional aspect of the hydrogel that provides temporal cues to regulate cell proliferation and migration within the hydrogel matrix.63 Whereas injectable hydrogels based on natural polymers are intrinsically degradable as a result of enzymatic activity in vivo, the degradation of synthetic precursor hydrogels must be programmed into the hydrogel design by using reversible chemistries that both cross-link the hydrogel and provide a hydrolyzable or oxidative link that facilitates degradation over time. The majority of biorthogonal cross-link chemistries listed earlier are reversible and therefore perfectly suited for the synthesis of injectable hydrogels.
Similar to the precursor polymers, the hydrogel itself must not elicit an inflammatory response once formed in vivo.64 The ability of the hydrogel to “mask” itself from the host's immune system by repelling protein adsorption is crucial to its success both short and long term. Protein adsorption is generally recognized as the first step of an inflammatory cascade that recruits inflammatory macrophages to the site of injury, initiates the formation of granulation tissue and (eventually, at least for slow or non-degrading materials) creates a fibrotic capsule around the material.65,66 This “walling-off” is particularly problematic for hydrogels designed for drug release or tissue regeneration applications as it inhibits the effective diffusion of drugs, nutrients, and/or waste products between the hydrogel and surrounding tissue.67 Although hydrogels are less prone to protein adsorption relative to most biomaterials due to their hydrophilic and highly hydrated nature, the choice of polymeric components in the hydrogel is crucial to further reduce protein adsorption. PEG is still considered the gold-standard polymer to minimize protein adsorption, with PEG derivatives e.g. poly(oligoethylene glycol) (meth(acrylate)),68,69 natural polymers,70 polyoxazolines,71 and zwitterionic polymers72 also demonstrating favorable and, in some cases, superior protein repellency properties.
Degradation products
Hydrogel degradation can occur via direct degradation of the polymer backbone and/or degradation of the cross-links between the individual polymer chains. Degradation of the polymer backbone may proceed via an oxidative,50 hydrolytic,28,73 and/or enzymatic mechanism.74 The rate of degradation depends on the chemical structure of the polymer and the cross-link density, which govern both the thermodynamics of the bond stability as well as the kinetics of diffusion of the bond-breaking stimulus (e.g. water, enzymes, etc.) to the degradation site. Most biopolymers degrade via one or more of these mechanisms, resulting in non-toxic degradation products that can be metabolized by the body. Examples of synthetic polymers that contain either hydrolytically or enzymatically cleavable fragments in the polymer backbone have been reported75 but require elaborate synthesis. Degradation of the cross-links76,77 is therefore the commonly used clearance mechanism for synthetic hydrogels. Consequently, the degradation products of the hydrogel are structurally similar or in some cases identical to the original precursor polymers. Since the carbon–carbon backbone synthetic precursors cannot be metabolized by the body, the molecular weight of such precursor polymers needs to be carefully controlled to prevent bioaccumulation and ensure renal clearance following degradation. Typically, a molecular weight cut-off of 20 × 103 – 60 × 103 g mol−1 has been reported as the kidney clearance limit.78,164Recent progress in the design of self-immolative polymers which degrade systematically following an external stimulus to remove a capping agent should also be noted as a potentially promising strategy for driving hydrogel degradation.79,80 By controlling the stability of the capping material, programmed or even on-demand timed degradation may be achievable using this type of strategy. While current chemistries for the design of self-immolative polymers are not directly amenable to in situ gelation and some concerns persist regarding the ultimate toxicity of the degradation products resulting from many of these proposed mechanisms,2 this strategy may be increasingly important moving forward as the chemistry of self-immolative polymers continues to develop.
3 Current state-of-the-art of injectable poly(ethylene glycol)-based hydrogels
Polyethylene glycol (PEG) is a synthetic polymer that has found wide-spread application in the design of biomaterials due to its demonstrated non-cytotoxic, non-immunogenic and protein repellent properties that effectively mask it from the host's immune system.81 Hydroxyl-terminated di- and multifunctional PEG polymers can readily be functionalized with a variety of desired functional groups (e.g. aminoxy,51 azide,82,83 thiol,84 vinyl sulfone,85 or maleimide,48) and cross-linked into PEG hydrogels using a step-growth polymerization approach,4 resulting in highly organized PEG hydrogel networks.86 These PEG hydrogels have been demonstrated to provide excellent synthetic matrices for the controlled release of therapeutics4,30 as well as tissue engineering applications, particularly when coupled with functional biomacromolecules such as the cell adhesive peptide RGD to enable PEG to effectively support cell growth.87–89 The following discussion will highlight specific cross-linking chemistries that have been reported to generate injectable PEG-based hydrogels.Azide-alkyne cycloadditions
The cycloaddition of an azide with a terminal alkyne has received considerable interest for the design of hydrogels due the high selectivity and bioorthogonality of this cross-linking chemistry. The copper(I)-catalyzed alkyne-azide (CuAAC) click reaction90 is of particular interest as it proceeds under physiological conditions. Ossipov and co-workers91 reported the first azide-alkyne cross-linked hydrogel for a poly(vinyl alcohol) (PVA) system. A number of PEG-based azide-alkyne cross-linked hydrogels have since been reported. For example, Adzima and co-workers92 reported a PEG hydrogel based on 4-arm PEG azide and a bifunctional PEG alkyne. This group demonstrated temporal and spatial control over the CuAAC click reaction by using standard photolithographic techniques to photochemically reduce Cu(II) to Cu(I). This technique offers the ability to selectively pattern hydrogels to e.g. promote cell adhesion and could drastically reduce the amount of Cu(I) required for the synthesis of functional and well-defined PEG hydrogels. The reduction of the Cu(I) concentration is particularly relevant as the associated cytotoxicity of copper ions and reactive oxygen species generated by copper ions in vivo may pose a limitation towards clinical application.19 Alternately, PEG-based hydrogels have been prepared based on mixtures of PEG with other functional entities.92–94 For example, Tan and co-workers94 developed a PEG hydrogel based on PEG-alkyne and an azido-functionalized α-cyclodextrin (αCD) that can cross-link through azide-alkyne chemistry as well as the formation of PEG–αCD inclusion complexes, providing cooperative control over both cross-link density and degradation mechanism/time (Fig. 1).95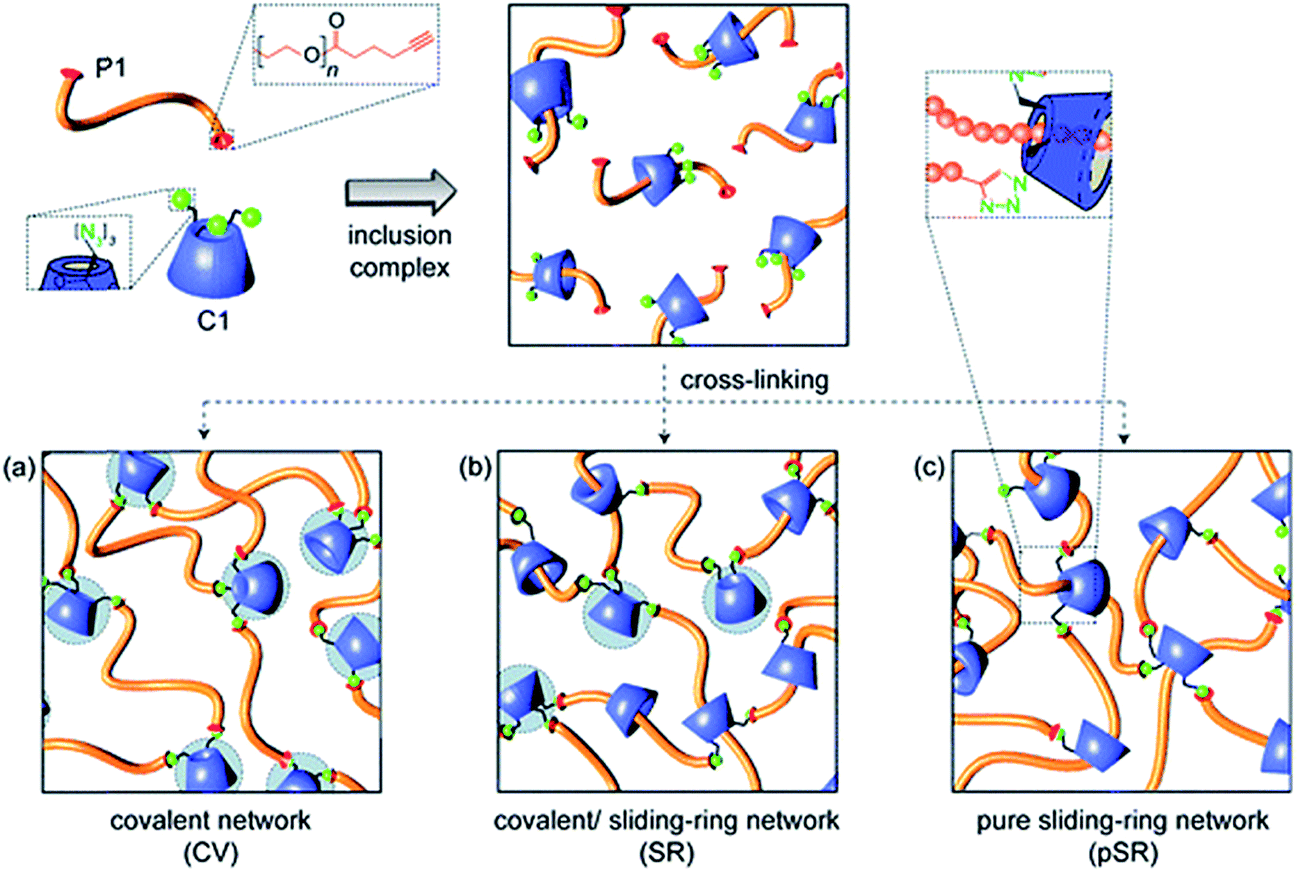 | ||
| Fig. 1 Schematic representation of the formation of α-cyclodextrin (αCD)–polyethylene glycol(PEG) hydrogels through the formation of alkyne-azide chemistry (CV), inclusion complexes (pSR) and a combination of both (SR). Reproduced from ref. 95 with permission from RSC Publishing. | ||
Strain-promoted alkyne-azide cycloaddition (SPAAC)96 has been reported as a promising alternative to CuAAC, as cross-linking can occur under physiological conditions in the absence of Cu(I). Examples of SPAAC cross-linked PEG hydrogels have been reported by Anseth's group,83,97 which demonstrated cross-linking of a 4-arm PEG tetraazide and a bis(cyclooctyne)-peptide. Zheng and co-workers98 reported an injectable PEG hydrogel based on 4-dibenzocyclooctynol (DIBO) functionalized difunctional PEG and a 3-arm glycerol exytholate triazide. These hydrogels formed within 5 minutes with a modulus of ∼0.8 kPa and were capable of sustaining hMSC cell viability for 24 hours in vitro. Furthermore, the polymer precursors did not exhibit significant cytotoxicity at relevant concentrations for hydrogel preparation.98 The high specificity, fast cross-linking kinetics, and excellent bioorthogonality will drive continuing interest in SPAAC, although the currently cumbersome synthesis of the cyclooctynes does at least now limit the broad applicability of this chemistry.
Michael additions
The 1,4-addition of a nucleophile to the β position of an α,β-unsaturated carbonyl compound such as an aldehyde or ketone, commonly referred to as a Michael-type addition, is a widely investigated approach for injectable hydrogel preparation. This cross-link chemistry occurs spontaneously under physiological conditions, especially when thiols are used as the nucleophiles,99,100 and the formed thioether bonds are relatively weak and can be reversible.101–105 Different α,β-unsaturated electrophiles have been used in conjunction with thiols for the preparation of injectable hydrogels.106–110 Although acrylates are the most commonly used electrophiles for Michael additions, maleimides48,105,111 and vinyl sulfones85,112–116 offer higher rates of cross-linking and have attracted more recent attention for this purpose. García and co-workers117 demonstrated the advantages of these alternative electrophiles in preparing functional PEG hydrogels by cross-linking a thiol-containing adhesive peptide (GRGDSPC) with 4-arm PEG containing acrylate (PEG-4-AC), vinyl sulfone (PEG-4-VS), or maleimide (PEG-4-MAL) functional groups. In the presence of 4 mM triethanolamine (TEA), gelation kinetics depended considerably on the choice of Michael acceptor, ranging from 1–5 min for PEG-4-MAL, 30–60 min for PEG-4-VS, and 60 min for PEG-4-AC. Furthermore, the maleimide-based hydrogels required two orders of magnitude less TEA to gel, a significant attribute considering the known cytotoxic effects of TEA on endothelial cells, cells in the ovarian follicles, and pancreatic islet cells.118 These hydrogels can also readily be functionalized using a dithiol protease-cleavable RGD peptide to create an in vivo degradable network that promoted the spreading of encapsulated C2C12 murine myoblasts.119 Successful use of this hydrogel formulation was demonstrated in vivo via injection onto the pericardium of an excised rat heart (Fig. 2).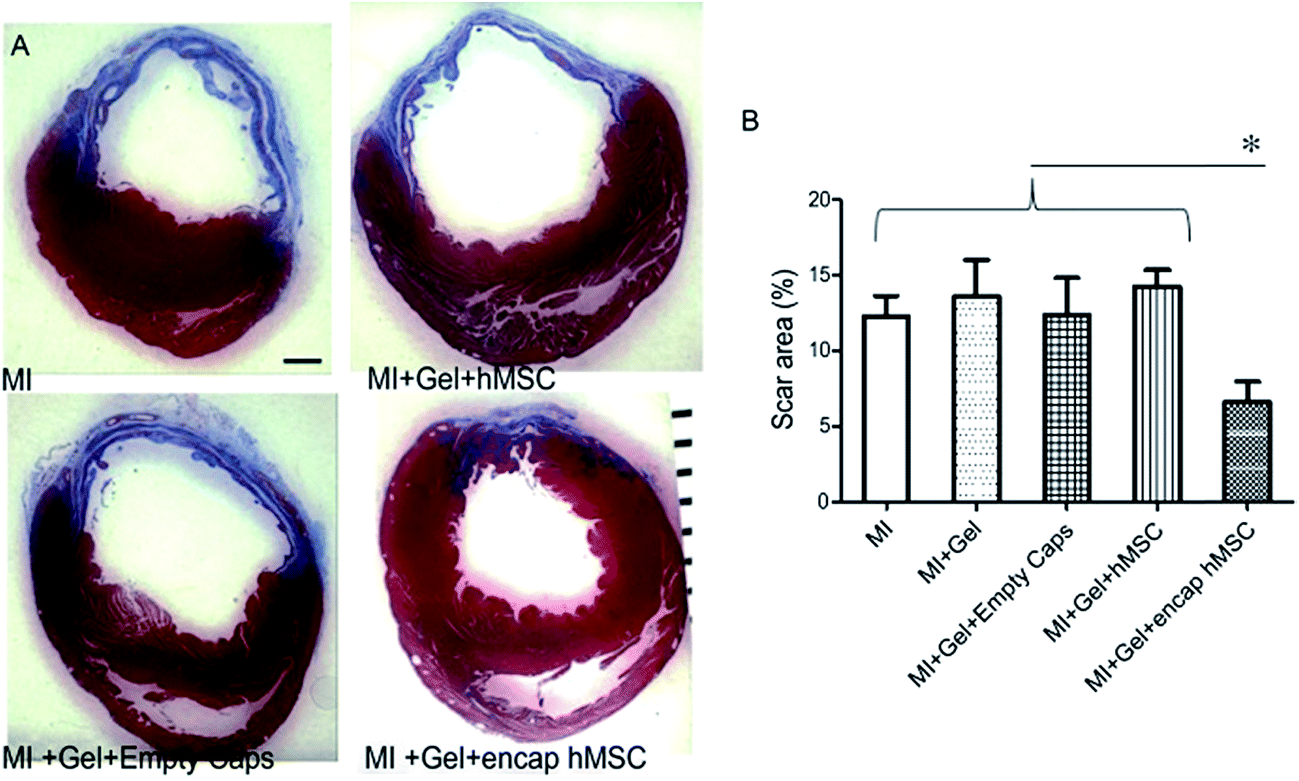 | ||
| Fig. 2 Treatment of hearts using human mesenchymal stem cells (hMSC) encapsulated in a 4-arm PEG maleimide-GRGDSPC dithiol injectable hydrogel showed reduced myocardial scarring after 28 days. (A) Representative sections of infarcted hearts stained with Masson's Trichrome and treated with encapsulated hMSCs or control gels. Blue indicates fibrotic scar. ×15, scale bar = 1 mm. (B) Animals treated with encapsulated hMSCs showed reduced scar area (7 ± 1%; n = 6) at 28 days compared to control treated hearts (MI: 12 ± 1%, n = 8; MI + gel: 14 ± 2%, n = 7; MI + gel + hMSC: 14 ± 1%, n = 7; MI + gel + empty caps: 12 ± 2%, n = 5). Data represent mean ± SEM. *P < 0.05. MI indicates myocardial infarction. Reproduced from ref. 119 with permission from the American Heart Association. | ||
Michael addition cross-linked hydrogels have also been reported in the context of drug delivery applications, with Yu et al. cross-linking tetramaleimide and tetrathiol PEG to form transparent PEG hydrogels that quickly gelled under physiological conditions for the ocular delivery of Avastin®.120 In vitro cytotoxicity indicated that the developed PEG hydrogel had no apparent cytotoxicity on L-929 fibroblast cells after 7 days of incubation; the cytotoxicity of the precursors was not determined despite concerns regarding the potential cross-reactivity and oxidation of thiols in vivo which persists as a potential limitation of thiol Michael additions. Prolonged Avastin® release was shown in vitro from these hydrogels for a period up to 14 days.120
Disulfide formation
Disulfide bonds, formed via the oxidation of two thiol groups, play an essential structural and chemical role in protein folding and assembly. Thiols are protonated and unreactive in their reduced states (such as in the cytoplasm), but form S–S bonds in the presence of an oxidizing environment such as in the endoplasmic reticulum and extracellular space. Disulfide cross-linking is of significant interest for the design of injectable hydrogels as thiol-functionalized precursors readily cross-link in the presence of oxidizing agents such as oxygen121 but then degrade in the presence of reducing agents such as glutathione. Alternately, disulfide rearrangements (commonly found in nature for the formation of disulfide bonds in proteins) can be exploited for the formation of hydrogels and/or to promote hydrogel degradation. PEG-based disulfide cross-linked injectable hydrogels have been reported by Choh and co-workers,122 prepared by coextruding pyridyl disulfide functionalized hyaluronic acid (HA-PD) with a PEG dithiol. Mixing of these precursors at physiological conditions results in macroscopic gelation within 4–5 minutes. Fibroblasts, endothelial cells (HUVECs), and adult stem cells (pMSCs) were successfully encapsulated in vitro and shown to proliferate over several days, even in the presence of unreacted thiols and the low-molecular-weight pyridine-2-thione cross-linking by-product.Diels–Alder cycloaddition
The addition of a conjugated diene to an activated double bond (dienophile) is referred to as a Diels–Alder cycloaddition. The most commonly used Diels–Alder reaction for cross-linking hydrogels involves the reaction between maleimide and furan functionalized precursors.123–125 Diels–Alder reactions occur spontaneously under physiological conditions and are highly selective and relatively biorthogonal, aside from free thiol groups present at relatively low concentration in proteins.126 Although Diels–Alder adducts are considered dynamic covalent bonds, current reversibility of formed adduct requires elevated temperatures beyond physiological temperature.127 However, hydrogel degradation can be promoted under physiological conditions if the diffusion of hydrogel degradation products shifts the equilibrium towards the reverse Diels–Alder reaction. An illustrative example of the use of Diels–Alder chemistry for PEG hydrogels was recently reported by the groups of Bowman and Anseth.48 The hydrogel network was formed using thiol-Michael chemistry, cross-linking a 4-arm PEG maleimide with a 4-arm PEG thiol. The maleimide functional groups are cross-reactive in Diels–Alder chemistry and thus provide a tethering site for a furan-modified RGD peptide (see Fig. 3). By exploiting the reversible nature of the Diels–Alder bond and le Châtelier's principle, the peptide could be release from the hydrogel with a release rate that is simply controlled by the excess of unreacted maleimide groups in the formed hydrogel network. For example, doubling the amount of maleimide groups present in the hydrogel decreased the amount of peptide released from approximately 60% to about 40%. Consequently, this example elegantly demonstrates how two different cross-link chemistries (i.e. thiol-Michael and Diels–Alder) can be used to independently control the rate of hydrogel degradation as well as release.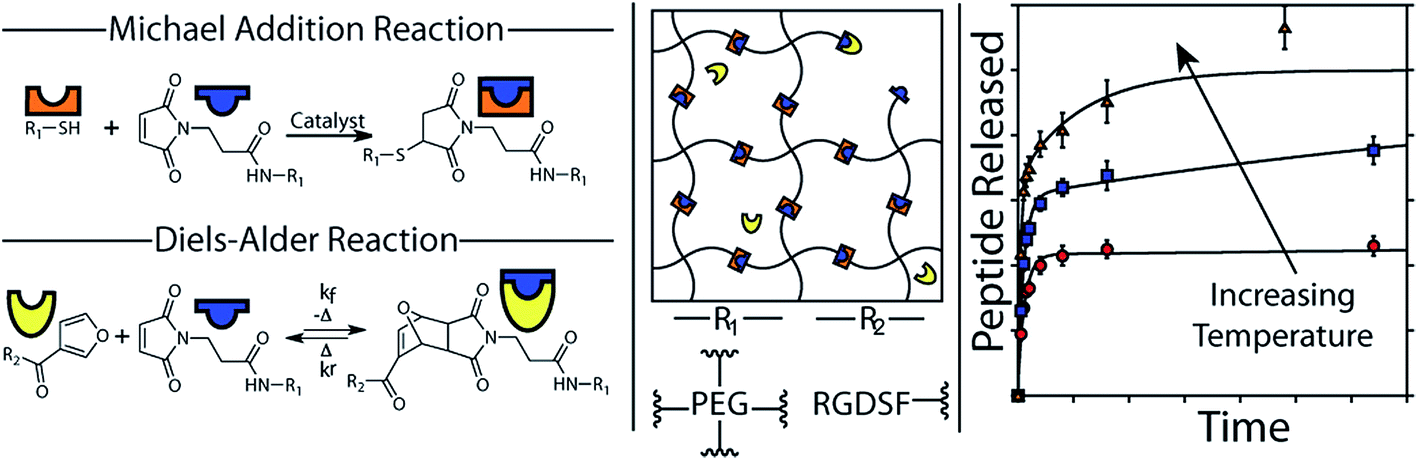 | ||
| Fig. 3 The formation of PEG hydrogels using thiol-Michael addition and the functionalization with a releasable fluorescent RGD peptide using Diels–Alder chemistry. A schematic representation of the hydrogel network and the release kinetics as a function of the temperature are also shown. Reproduced from ref. 48 with permission from ACS publishing. | ||
Schiff base formation
The nucleophilic addition of a nitrogen from an amine or hydrazide (or similar derivatives) to the carbonyl group of a ketone or aldehyde is referred to as a Schiff base or imine bond.128 Schiff-bases are considered dynamic covalent bonds with the advantages of fast and tunable gelation kinetics129 in the absence of a catalyst and complete reversibility. Conventional Schiff base formation between a carbonyl and a primary amine offers only limited hydrolytic stability and thus results in relatively fast hydrogel degradation.128 Increased hydrolytic stability is achievable by increasing the nucleophilicity of the amine derivative, with the majority of the Schiff base cross-linked hydrogels reported have been based on hydrazone bond formation between a hydrazide and an aldehyde.28,73,130–133 An example of a PEG-based Schiff base hydrogel was reported by Saito and co-workers52 for the delivery of doxorubicin. A doxorubicin-loaded hydrogel was prepared by conjugating doxorubicin to a low molecular weight PEG dialdehyde (5 kDa) using Schiff base chemistry, followed by mixing with a solution of a high molecular weight poly(vinylamine) (PVam) (Fig. 4). The precursor gelled quickly upon mixing and the resulting hydrogel showed sufficient hydrolytic stability at physiological pH for the targeted application. Prolonged doxorubicin was demonstrated up to 100 hours.52 However, the functional groups used in Schiff base formation (in particular aldehydes) are cross-reactive with functional groups on biomacromolecules (e.g. amines), such that care must be taken in designing the precursor polymers and gelation times in order to avoid potential toxicity.37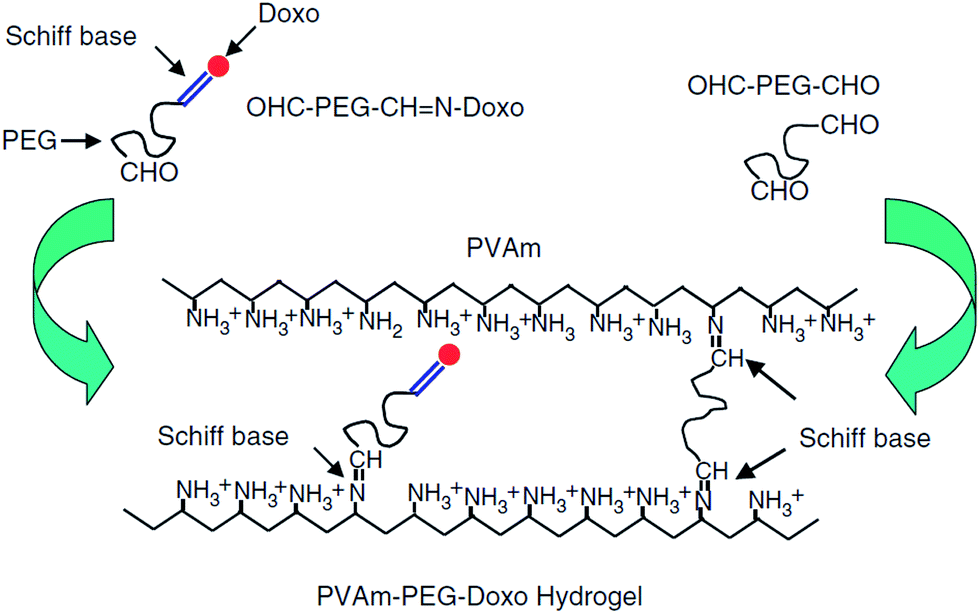 | ||
| Fig. 4 Schematic representation of the formation of poly(vinylamine) (PVAm)–poly(ethylene glycol) (PEG)–doxorubicin(doxo) hydrogels using Schiff base chemistry. Reproduced from ref. 52 with permission from SAGE journals. | ||
Oxime chemistry
Oxime bonds form rapidly between an aldehyde or ketone and a hydroxylamine under physiological conditions and offer enhanced hydrolytic stability relative to Schiff base cross-linking strategies at the cost of requiring an acid catalyst in order to proceed at an appreciable rate. Grover and co-workers51 have used oxime chemistry to synthesize PEG hydrogels through the reaction of an 8-arm aminooxy PEG with glutaraldehyde (Fig. 5). The mechanical properties, gelation kinetics, and water swelling ratios could be tuned according to the weight fraction of precursor polymers and the pH, and hydrogels modified with RGD successfully supported hMSC cell encapsulation. The in vivo potential of oxime cross-linked hydrogels formed between a 4-arm aminooxy PEG and a 4-arm PEG ketone was subsequently evaluated in myocardial tissue by introducing the hydrogel using a catheter.134 While in vitro gelation rates ranged from 30 minutes at pH 4 to >50 hours at pH 7.4, in vivo gelation was observed within 20 minutes independent of the pH of the injection solution (pH 6.0–7.4). This effect can be attributed to the more complex biological environment in vivo relative to in vitro experiments.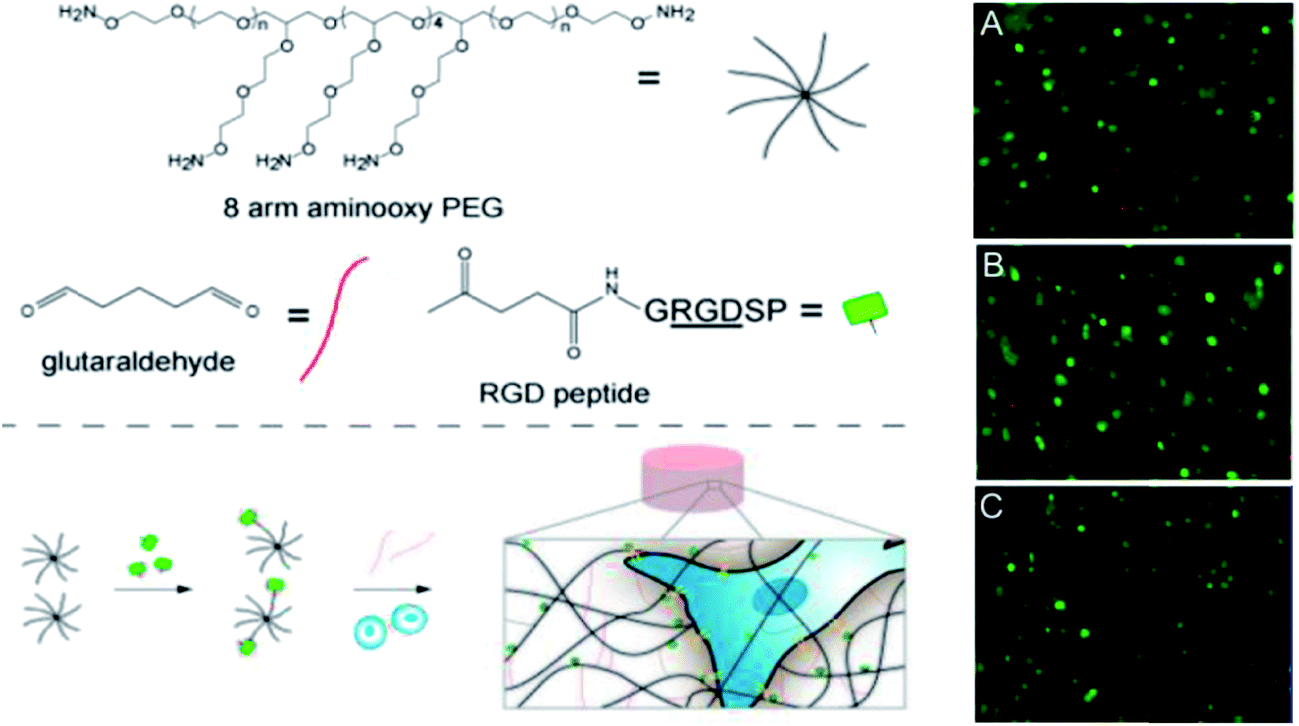 | ||
| Fig. 5 Synthesis and encapsulation of MSCs within RGD-functionalized oxime-cross-linked PEG hydrogels. Reproduced from ref. 51 with permission from the American Chemical Society. | ||
Despite the obvious advantages of using PEG as a precursor polymer for the preparation of injectable hydrogels (specifically, its controlled molecular weight and protein repellent properties), PEG also suffers from a number of limitations. As PEG can only be selectively modified at the α,ω-hydroxyl chain ends, relatively expensive multi-arm PEG precursors must be used as precursor polymers to promote network formation at lower PEG concentrations. Furthermore, incorporation of specific functionalities (e.g. RGD peptide or other biomolecules) effectively competes with cross-linking since these functional tethers need to be incorporated at the same reactive sites; in this sense, the number of cross-links and functional tethers is inherently limited by the number of chain ends on the PEG starting materials, significantly limiting the mechanical strength of the resulting hydrogel. Consequently, we have started exploring injectable PEG analogue polymers that can be synthesized from free-radical polymerization as a potential system to improve upon PEG-based hydrogel designs.
4 Poly(oligoethylene glycol methacrylate)-based hydrogels
In order to address challenges with injectable PEG hydrogels and PEG more broadly as a biomaterial, poly(oligoethylene glycol methacrylate) (POEGMA) is now being investigated as an alternative to PEG.68,135 Unlike PEG, which is typically prepared via ring-opening polymerization and thus cannot readily be co-polymerized with other functional comonomers, POEGMA is synthesized from conventional (or controlled/living) free-radical polymerization,136–138 offering excellent control over polymer composition, functionality, architecture and molecular weight distribution with a range of functional monomers to impart desired functionalities to the polymers. POEGMA can also be made thermoresponsive via the statistical copolymerization of oligo(ethylene glycol) methacrylate (OEGMA) monomers with varying lengths of ethylene oxide (EO) side chains (n).138 For example, statistical copolymers of diethylene glycol methacrylate (M(EO)2MA, n = 2) and OEGMA with an average molecular weight of 475 g mol−1 (OEGMA475, n = 8–9) display a cloud point in water according to a linear correlation between the cloud points of the respective PM(EO)2MA (∼23 °C) and POEGMA475 (∼90 °C) homopolymers.138 The reversible temperature transition shows little hysteresis139 when compared to other thermoresponsive synthetic polymers such as N-isopropylacrylamide (NIPAAm) due to the lack of a hydrogen-bond donor.140 This property is of potential interest both for the design of “smart”, environmentally-responsive biomaterials for on-demand drug delivery, triggerable cell adhesion, etc. as well as more generally as PEG-based materials with significantly improved bioadhesion in the absence of functional peptide tags. At the same time, POEGMA has been demonstrated to display similar bio-inert (“stealth”)141,142 and non-cytotoxic properties143 when compared to PEG. Consequently, we and others have considered POEGMA a promising platform for the synthesis of PEG-analogue hydrogels with improved control over the chemical, physical, and biological properties of the hydrogels.A number of POEGMA-based hydrogels have been reported to-date,144–148 with the majority prepared via conventional free radical polymerization in the presence of a di(meth)acrylate cross-linker to promote gelation. However, with the exception of a 4-arm PEG-b-POEGMA polymer reported by Fechler, which undergoes physical gelation at the physiological temperature of 37 °C,149 none of these previously hydrogels are either injectable or degradable in vivo, which severely limits their potential clinical application.
In response, we have done extensive work in our laboratory on the design of injectable hydrogels based on dynamic covalent chemistry,127,150 with a particular focus on Schiff-base chemistry between aldehyde and hydrazide functional groups (see Scheme 2). Use of this cross-linking chemistry for injectable hydrogels was first proposed by Bulpitt and Aeschlimann151 and has been further developed by Langer's group73,130 and others91,131,152,153 to a broad range of biopolymer, synthetic and composite hydrogel systems. Our choice of hydrazone chemistry for the preparation of injectable hydrogels was driven by the fast gelation kinetics and hydrolytic reversibility of the hydrazone bond. Hydrazide and aldehyde functional groups are also readily introduced onto a range of synthetic and natural polymers using facile and few step copolymerization and post-polymerization modification strategies. The desired physiochemical and biological hydrogel properties can consequently be “dialed-up” by selecting and modulating different hydrogel precursors. Furthermore, hydrazide and aldehyde functionalized precursors provide sufficient bio-orthogonality (even in the presence of potential Schiff-base formation between the pendant aldehydes and available amines on native proteins) to in our experience avoid causing any significant adverse reactions in vitro and in vivo.41,42 We attribute this result to the high reversibility of the aldehyde-amine Schiff base pair (i.e. a hydrazone bond will form preferentially to minimize free energy), the fast gelation kinetics which rapidly consume a large fraction of the free aldehyde groups before proteins can diffuse into the hydrogel, and the high local concentration of hydrazides used during injection which ensures the majority of aldehyde groups are reacted following gel formation.
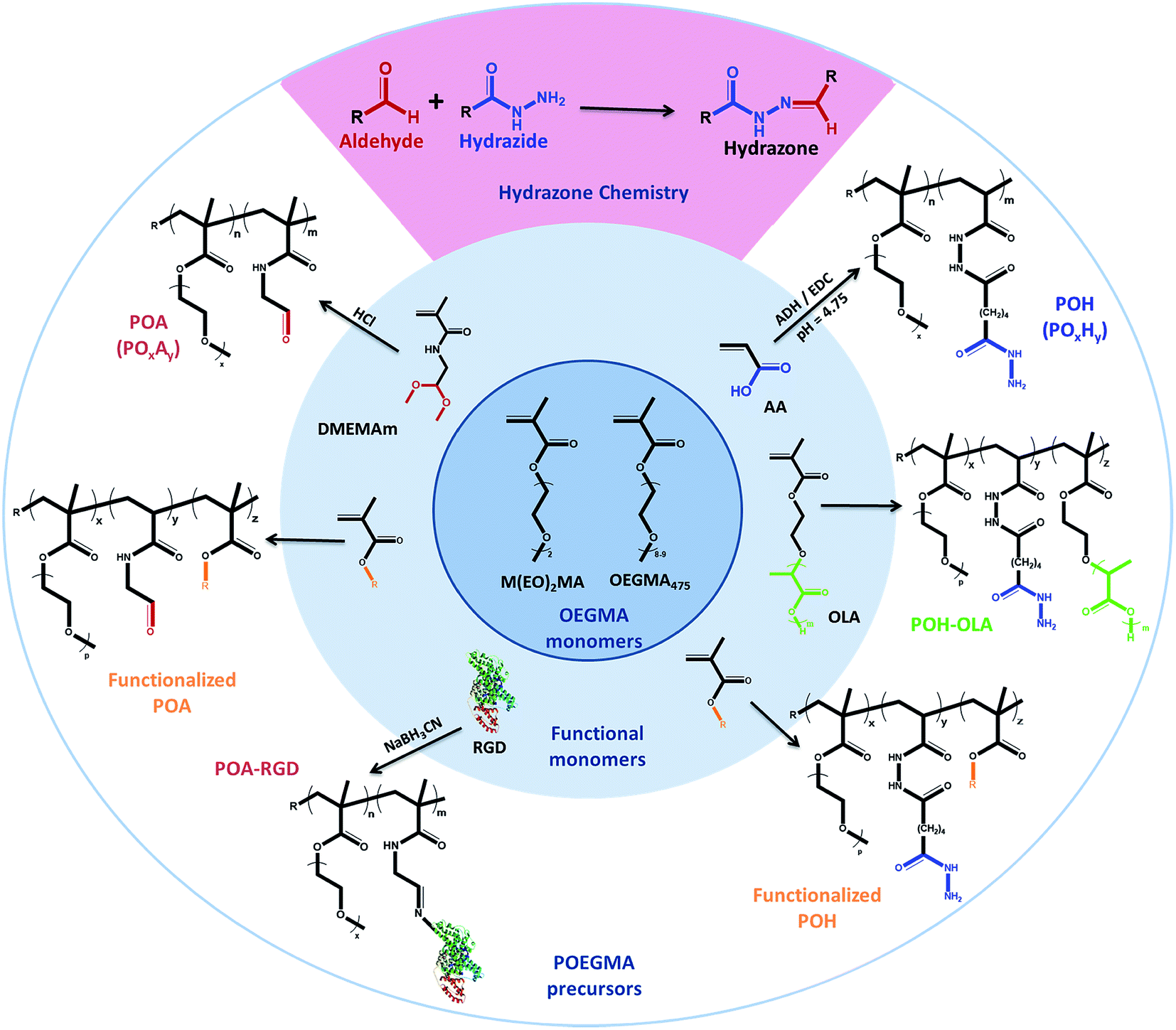 | ||
| Scheme 2 Library of hydrazide and aldehyde-functionalized poly(oligoethylene glycol methacrylate) (POEGMA) hydrogel precursors prepared for designing hydrogels. | ||
Hydrazide-functionalized POEGMA precursors (POH) are synthesized from conventional free-radical copolymerization of M(EO)2MA, OEGMA475 and acrylic acid (AA) in the presence of thioglycolic acid (TGA) to limit the molecular weight below the kidney cut-off, followed by post-polymerization modification using the carbodiimide-mediated coupling of adipic acid dihydrazide (ADH).41 The aldehyde-functionalized POEGMA precursors (POA) are synthesized from the copolymerization of M(EO)2MA, OEGMA475 and N-2,2-diethoxyethyl methacrylamide (DMEMAm) in the presence of TGA, followed by the acid-catalyzed deprotection of the acetal to the aldehyde (see Scheme 2).41 The aldehyde and hydrazide reactive precursors are labeled as POxAy and POxHy respectively, where x denotes the mole percentage of OEGMA475 among the OEGMA monomers used and y denotes the overall mole percentage of DMEMAm or AA in the synthesis recipe. We have exploited the synthetic versatility of the POEGMA platform to synthesize precursor polymers that differ in (i) OEGMA composition (0 ≤ x ≤ 100, and thus phase transition temperature),42 (ii) reactive group content (20 ≤ y ≤ 40, and thus theoretical cross-link density),41 or (iii) the presence of specific functional moieties (e.g. hydrophobic oligo(lactic acid) (OLA)154 or RGD cell-signaling peptide to direct internal domain formation, cell adhesion, etc.)41 (Scheme 2). Since the POH and POA polymers represent both the hydrogel precursors as well as degradation products,41 the design of these polymers with a Mn below the renal clearance-limit of ∼32 kDa should facilitate elimination of the precursors via the kidneys following hydrogel degradation.
POEGMA hydrogels (labeled as POx, where x indicates the mole fraction OEGMA475 among the OEGMA monomers) are generally prepared by co-extruding POH and POA precursor solutions of equal concentration (most typically 150 mg mL−1 = 13 w/w%) and comparable degree of functionality (y = 30 mol%). Despite this similarity in precursor concentration and reactive group content, hydrogel properties vary considerably as a function of the precursor composition (x) (Fig. 6). The gelation time, defined as the time point at which the hydrogel visually no longer flows on the time scale of test tube rotation (5 s) and cross-link density decrease with increasing mole fractions of OEGMA475 (n = 8–9); for example, PO0H30 and PO0A30 gel within 5 s and form a stiff hydrogel (G′ ∼ 10 kPa) whereas PO100H30 and PO100A30 require 20 min to gel and form a relatively weaker hydrogel (G′ ∼ 0.8 kPa). We hypothesize this trend is attributable to the steric hindrance of the longer PEG side chains in OEGMA475 (n = 8–9) that reduces the availability of the reactive aldehyde and hydrazide groups for cross-linking. This difference in gelation kinetics also has a direct impact on the underlying morphology of the hydrogel network formed. A small-angle neutron scattering (SANS) study on these hydrogels revealed that the fast gelling PO0 precursors form an inhomogeneous hydrogel network with a small mesh size, whereas the slow gelling PO100 precursors form a much more homogeneous network with a large mesh size.155
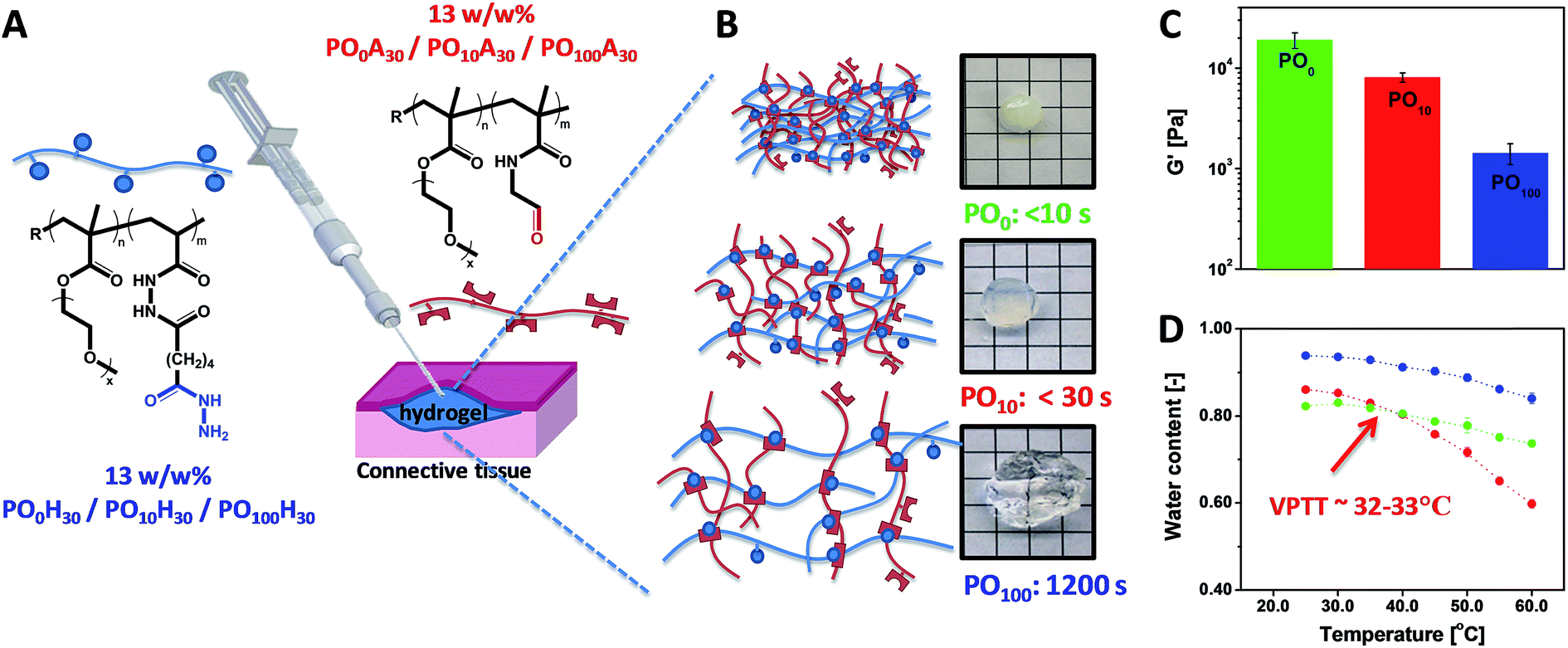 | ||
| Fig. 6 (A) Preparation of POEGMA hydrogels using a double-barrel syringe. (B) Physical appearance and gelation time for PO0, PO10 and PO100 hydrogels. (C) Hydrogel mechanics as measured by the shear storage modulus (G′). (D) Thermoresponsive properties of PO0, PO10 and PO100 hydrogels. Data reproduced from ref. 42 with permission from Elsevier. | ||
The volume phase transition temperature (VPTT) of POEGMA hydrogels aligns well with the cloud point (or lower-critical solution temperature, LCST) reported for analogous statistical POEGMA copolymers of similar M(EO)2MA-OEGMA475 composition.138 Although the cloud points of the POH and POA precursors are well over 50 °C in phosphate-buffered saline (PBS) due to the hydrophilicity of the hydrazide and aldehyde functional groups (key to avoiding local precipitation and subsequent inflammation as well as to further promote clearance of the precursor polymers after the gel has degraded),42 once cross-linked the formed hydrazone bond does not significantly alter the VPTT of the POEGMA hydrogels versus what would be predicted from a statistical copolymer of the two OEGMA comonomers used.42 Consequently, we have been able to prepare POEGMA hydrogels that are completely collapsed at 23 °C (PO0), display a VPTT at ∼33 °C (PO10, comparable to PNIPAAm), or do not display a VPTT up to 60 °C (PO100, comparable to PEG) (Fig. 3D). These hydrogels show remarkable differences in swelling kinetics at physiological temperature due to these different temperature dependencies; PO0 and PO10 hydrogels prepared at 13 w/w% in PBS (water content = 87.0%) de-swell at 37 °C to plateau water contents of 80 w/w% and 78 w/w%, respectively while PO100 hydrogels prepared at the same precursor concentration swell to a plateau water content of 91 w/w% at the same temperature.
Our modular approach to designing hydrogels allows for (i) the incorporation of building blocks with specific functionalities that cannot be attained using synthetic polymers using simple mixing (e.g. superparamagnetic iron oxide nanoparticles)40 and (ii) facile tuning of the hydrogel properties by mixing hydrogel precursors with different properties, avoiding the often trial-and-error optimization procedures conventionally used to synthesize hydrogels with targeted physical propeties.156 Building on this concept, we have exploited our library of POA and POH precursors to prepare “mixed precursor” POEGMA hydrogels that demonstrate this principle, using 2 POA and 2 POH precursors with different x values (i.e. different LCST values) as the precursor units; the high and low transition temperature precursor polymer pairs are mixed at different ratios to tune the gel properties.157 Important macroscopic physiochemical properties such as the gelation time, equilibrium water content, and hydrogel elasticity of these “mixed precursor” hydrogels can be predicted according to the simple rule of mixtures based on the corresponding properties of “single precursor” POEGMA hydrogels (i.e. hydrogels prepared using only 1 aldehyde and 1 hydrazide precursor of the same x) (Fig. 7). Consequently, on a macroscopic scale, desired hydrogel properties can be “dialed-up” simply by mixing different precursor polymers, avoiding the laborious synthesis and characterization required to optimize a hydrogel using trial-and-error synthesis. On a microscopic scale, however, “mixed precursor” hydrogels are less homogeneous than “single precursor” hydrogels and contain large inhomogeneous domains that cause an opaque appearance as well as alter the interactions between the hydrogel with its environment (e.g. protein binding, cell adhesion, drug uptake).155
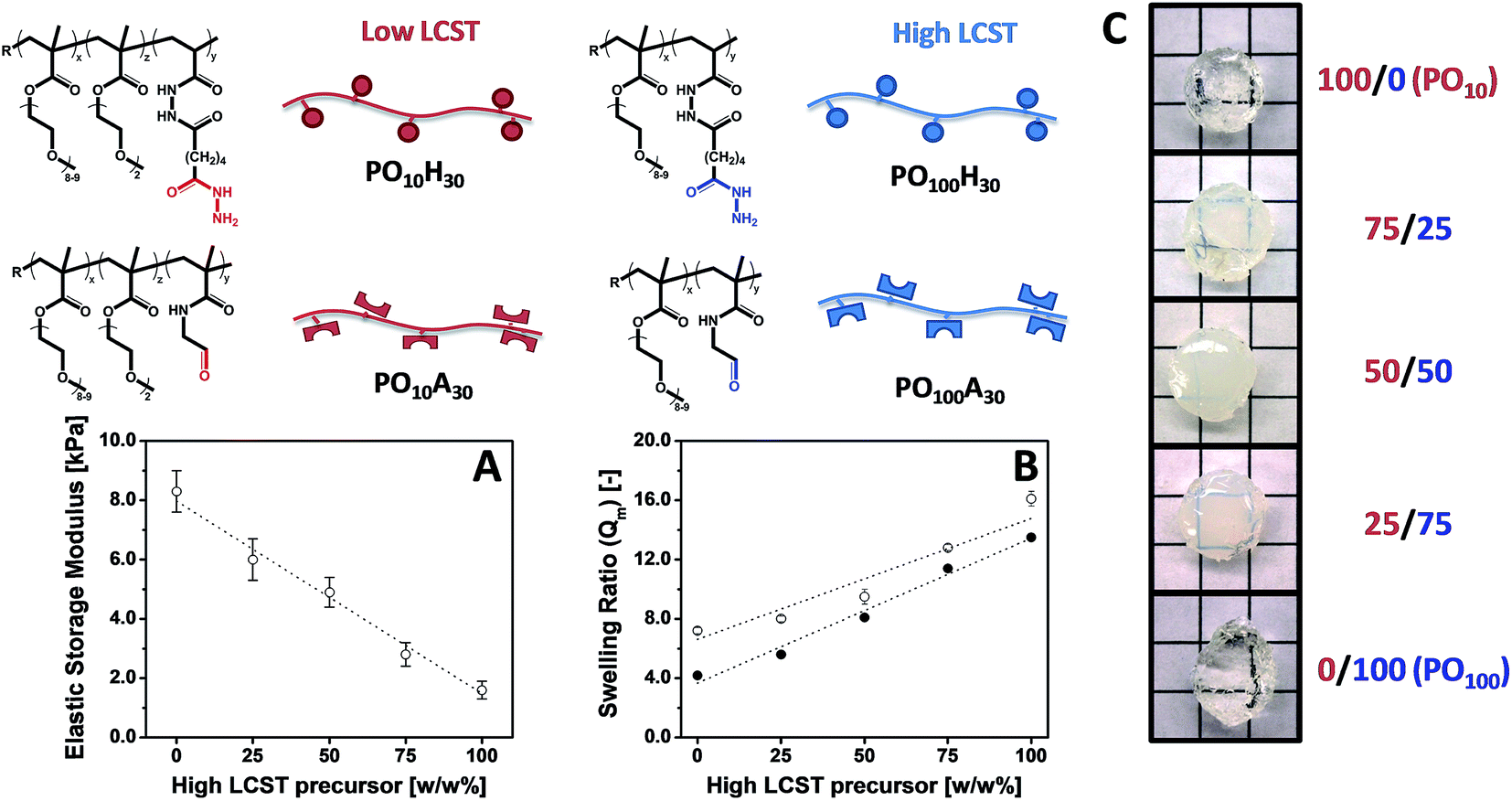 | ||
| Fig. 7 Tuning hydrogel properties by mixing POEGMA precursors of different LCST. (A) Elastic storage modulus (G′), (B) mass-based swelling ratio (Qm) and (C) physical appearance of POEGMA hydrogels containing 100/0, 75/25, 50/50, 25/75 and 0/100 w/w% PO10H30/PO100H30 and PO10A30/PO100A30. Data reproduced from ref. 155 and 157 with permission from Wiley-VCH and ACS. | ||
POEGMA hydrogels with associative hydrophobic domains have also been prepared by synthesizing a PO100H30 precursor polymer containing a functional oligo(lactic acid) methacrylate macromonomer (OLAm, where m represents the lactic acid chain length).154 Co-extrusion with PO100A30 results in the formation of POEGMA hydrogels that are both chemically (via formation of hydrazone bonds) and physically (via self-association of hydrophobic OLA chains) cross-linked, with both the chemical and physical cross-links degradable over time via hydrolysis. Hydrogel swelling, degradation and elasticity can be independently tuned using POH–OLA precursors according to the OLA chain length (m = 4, 8, or 16) and concentration (z = 10, 20, or 30 mol%). SANS analysis of the hydrogel microstructure revealed the presence of hydrophobic associative domains as well as the effect of the competition between physical and chemical cross-linking on the mesh size, which can be directly tuned to create hydrogels with targeted properties and well-defined nanodomains.
Like PEG hydrogels, POEGMA hydrogels hold significant promise in biomedical applications like controlled release and cell scaffolding. Similar to conventional PEG hydrogels, unmodified POEGMA hydrogels show little affinity for hydrophobic therapeutics and generally show significant burst-release and a short therapeutic release window. In an attempt to improve the affinity of PEG-based hydrogels for hydrophobic therapeutics, we explored the release of fluorescein isothiocyanate-labeled bovine serum albumin (FITC-BSA) from “mixed precursor” as well as POEGMA–OLA hydrogels. The inhomogeneous microscopic network structure of “mixed precursor” hydrogels offered somewhat improved control over FITC-BSA burst release but did not significantly extend the therapeutic release window versus conventional, more homogeneous “single precursor” hydrogels. In comparison, the incorporation of oligo(lactic acid) (OLA) pendant side chains into POH precursors resulted in significantly lower burst release as well as moderate control over the duration of the following sustained release.154 However, further optimization of these PO–OLA hydrogels is required to attain release profiles that are clinically relevant.
In a biological setting, POEGMA hydrogels show similar non-cytotoxicity and bio-inert properties to conventional PEG hydrogels, even in the case of thermoresponsive hydrogels with lower VPTT values (Fig. 8). None of the POH or POA precursors showed any decrease in cell viability up to 2000 μg mL−1 in MTT assays on 3T3 mouse fibroblasts,42 and the PEG-analogue PO100 hydrogels exhibited very low adsorption of bovine serum albumin (BSA) and fibrinogen (Fib) on par or better than reported PEG-based materials.158–162 Interestingly, while POEGMA hydrogels above their VPTT also demonstrated extremely low protein adsorption relative to most biomaterials, they were still capable of supporting moderate adhesion of 3T3 mouse fibroblasts.42 Furthermore, the thermoresponsivity of the lower VPTT POEGMA hydrogels can be exploited to turn cell adhesion to the hydrogels effectively on and off; for example, lowering the temperature of a 3T3 fibroblast-adhered PO0 hydrogel to 4 °C (T < VPTT) for just 15 minutes results in significant (∼25%) delamination of cells from the gels for cell recovery without the need to use harsher enzymatic methods such as trypsinization (Fig. 8B).42 Consequently, POEGMA-based hydrogels can be applied in tissue engineering applications not readily served by conventional PEG hydrogels.
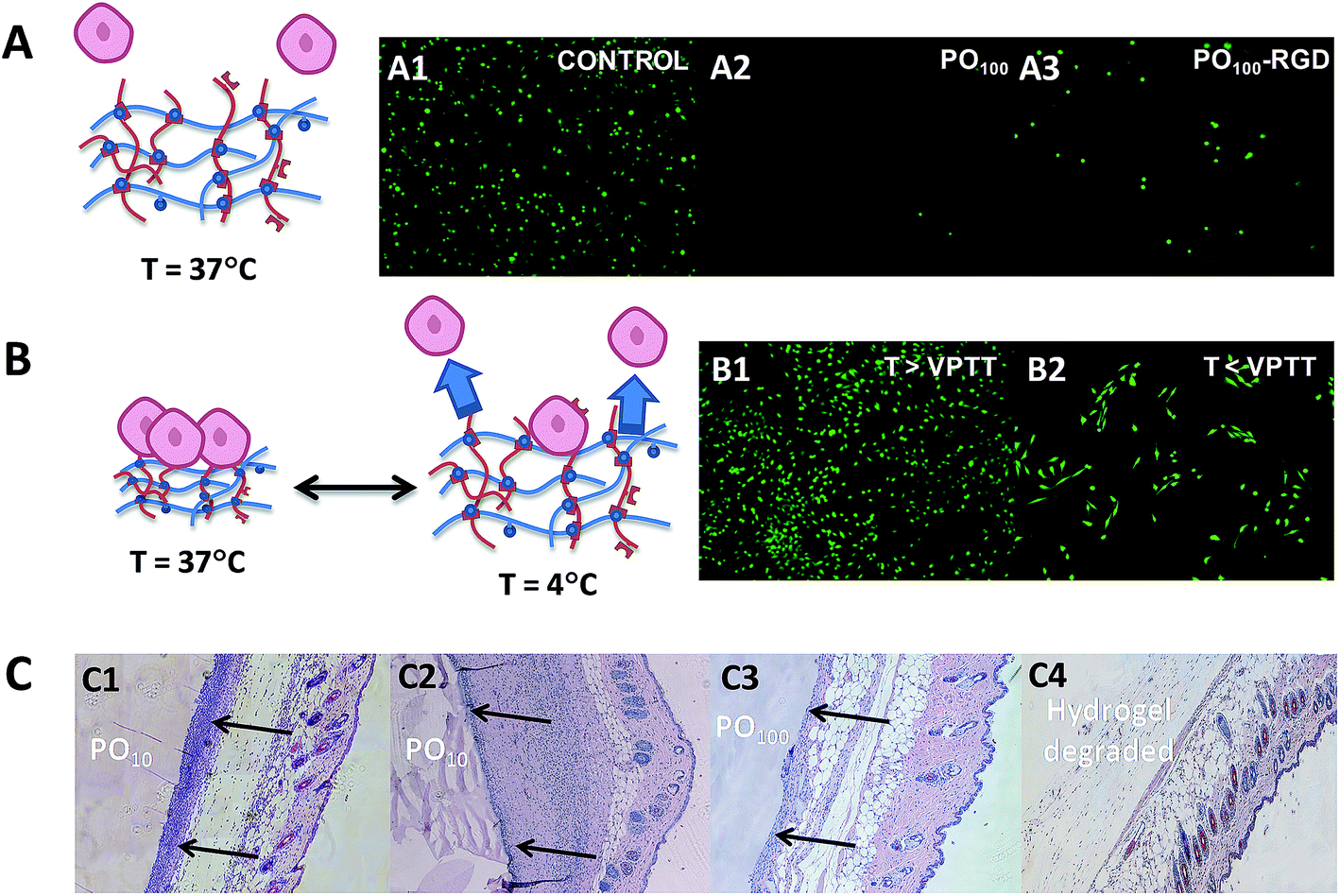 | ||
| Fig. 8 Overview of the biological properties of POEGMA hydrogels. (A) Bio-repellent properties of unmodified POEGMA hydrogels (A2) and controlled adhesion of cells using RGD-functionalized POEGMA hydrogels (A3) relative to a tissue culture polystyrene control (A1). (B) Switching cell adhesion “on” above the VPTT (B1) and “off” below the VPTT (B2). (C) Histological sections following subcutaneous injection following acute (C1 and C3) and chronic (C2 and C4) time points. Complete degradation of a PO100 hydrogel is shown in C4. Images reproduced from ref. 41 and 42 with permission from the Royal Society of Chemistry and Elsevier. | ||
Alternately, analogous to the several examples of PEG-based hydrogels cited previously,48,89,134,163 POA precursors can be also functionalized with RGD to prepare POEGMA hydrogels that selectively promote cell adhesion.41 Subcutaneous injection of these soft materials caused a relatively mild acute inflammatory response (2 days) and little to no chronic inflammation (30 days) (Fig. 8C). Furthermore, in vivo experiments showed that the POEGMA hydrogels could fully degraded under physiological conditions without any obvious signs of local tissue toxicity.42
The results to date demonstrate the potential of POEGMA-based injectable hydrogels for biomedical applications such as controlled release and cell encapsulation. The physiochemical properties of the hydrogels (governed by the network structure) can be easily controlled by the choice of OEGMA and functional monomers during the precursor polymer synthesis. The biological properties of these hydrogels are generally similar to PEG, provided that the VPTT is above physiological temperature. However, the presence of a VPTT also extends the range of applications that can be considered for POEGMA-based hydrogel materials, and the ease of POEGMA modification (particularly relative to PEG) opens up possibilities for specifically tuning these materials to targeted biomedical applications. We are currently exploring the biocompatibility of these POEGMA-based hydrogels in various biological environments as well as the more detailed cell responses (in terms of cell spreading and differentiation) to the various types of hydrogel structures accessible using this approach to assess the applicability of these materials for both in vivo, clinical applications as well as in vitro cell culture-based applications.
5 Summary and perspectives
PEG and PEG-analogue materials hold tremendous potential for the design of both in vivo scaffolds for drug delivery or tissue engineering as well as in vitro for cell culturing applications. Injectable analogues of such hydrogels offer even more promise both for effective 3D scaffolding of cells in these hydrogels by simple mixing as well as non-invasive administration in vivo, reducing the complexity of hydrogel use in the body as well as opening up potential uses of hydrogels in areas generally disfavored for routine surgical procedures if they are avoidable (e.g. the eye). Newer methods to incorporate multiple functional tethers (both physical and biological) on PEG-based hydrogels to direct cell responses in more precise and, in some cases, externally triggered ways suggest the potential use of PEG-based hydrogels in next-generation tissue engineering approaches. However, in our view, the significant synthetic challenges associated with functionalizing PEG, particularly in a manner that is independent of the cross-link density of the ultimate PEG-based hydrogel network, make PEG analogues such as POEGMA of increasing significance along the path of synthetically replicating (to the extent possible) natural cell microenvironments. To this end, an improved understanding of the similarities and differences between the well-established tissue responses and clearance mechanisms of PEG and those of POEGMA is essential to establish whether or not the synthetic versatility offered by POEGMA-based hydrogels can truly be exploited in a broad range of potential biomedical applications.Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Research Council (NSERC) and 20/20: NSERC Ophthalmic Materials Research Network for funding the POEGMA research reviewed in this work.References
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biopolymer-Based Hydrogels as Scaffolds for Tissue Engineering Applications: A Review, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- Z. Li and J. Guan, Thermosensitive Hydrogels for Drug Delivery, Expert Opin. Drug Delivery, 2011, 8, 991–1007 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- T. Hoare and D. S. Kohane, Hydrogels in Drug Delivery: Progress and Challenges, Polymer, 2008, 49, 1993–2007 CrossRef CAS PubMed.
- R. M. A. Hernández, G. Orive, A. Murua and J. L. Pedraz, Microcapsules and Microcarriers for in Situ Cell Delivery, Adv. Drug Delivery Rev., 2010, 62, 711–730 CrossRef PubMed.
- X.-J. Jiang, T. Wang, X.-Y. Li, D.-Q. Wu, Z.-B. Zheng, J.-F. Zhang, J.-L. Chen, B. Peng, H. Jiang and C. Huang, et al., Injection of a Novel Synthetic Hydrogel Preserves Left Ventricle Function after Myocardial Infarction, J. Biomed. Mater. Res., Part A, 2009, 90, 472–477 CrossRef PubMed.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. S. Kumar, Polymeric Scaffolds in Tissue Engineering Application: A Review, Int. J. Polym. Sci., 2011, 2011, 1–19 CrossRef.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Synthetic Matrix Metalloproteinase-Sensitive Hydrogels for the Conduction of Tissue Regeneration: Engineering Cell-Invasion Characteristics, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- J. Zhu, Bioactive Modification of Poly(ethylene Glycol) Hydrogels for Tissue Engineering, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- J. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, Wound Healing Dressings and Drug Delivery Systems: A Review, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- H. Tan and K. G. Marra, Injectable, Biodegradable Hydrogels for Tissue Engineering Applications, Materials, 2010, 3, 1746–1767 CrossRef CAS PubMed.
- Y. Li, J. Rodrigues and H. Tomás, Injectable and Biodegradable Hydrogels: Gelation, Biodegradation and Biomedical Applications, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- S. R. Van Tomme, G. Storm and W. E. Hennink, In Situ Gelling Hydrogels for Pharmaceutical and Biomedical Applications, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- L. Yu and J. Ding, Injectable Hydrogels as Unique Biomedical Materials, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- W. E. Hennink and C. F. van Nostrum, Novel Crosslinking Methods to Design Hydrogels, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef PubMed.
- M. Patenaude, N. M. B. Smeets and T. Hoare, Designing Injectable, Covalently Cross-Linked Hydrogels for Biomedical Applications, Macromol. Rapid Commun., 2014, 35, 598–617 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Click Hydrogels, Microgels and Nanogels: Emerging Platforms for Drug Delivery and Tissue Engineering, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- B. K. Chan, C. C. Wippich, C. J. Wu, P. M. Sivasankar and G. Schmidt, Robust and Semi-Interpenetrating Hydrogels from Poly(ethylene Glycol) and Collagen for Elastomeric Tissue Scaffolds, Macromol. Biosci., 2012, 12, 1490–1501 CrossRef CAS PubMed.
- D. C. S. Pedroso, A. Tellechea, L. Moura, I. Fidalgo-Carvalho, J. Duarte, E. Carvalho and L. Ferreira, Improved Survival, Vascular Differentiation and Wound Healing Potential of Stem Cells Co-Cultured with Endothelial Cells, PLoS One, 2011, 6, e16114 CAS.
- A. F. G. Godier-Furnémont, T. P. Martens, M. S. Koeckert, L. Wan and J. Parks, Composite Scaffold Provides a Cell Delivery Platform for Cardiovascular Repair, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7974–7979 CrossRef PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Diels-Alder Click Cross-Linked Hyaluronic Acid Hydrogels for Tissue Engineering, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- H. R. Moyer, R. C. Kinney, K. A. Singh, J. K. Williams, Z. Schwartz and B. D. Boyan, Alginate Microencapsulation Technology for the Percutaneous Delivery of Adipose-Derived Stem Cells, Ann. Plast. Surg., 2010, 65, 497–503 CrossRef CAS PubMed.
- M. S. Shoichet, R. H. Li, M. L. White and S. R. Winn, Stability of Hydrogels Used in Cell Encapsulation: An In Vitro Comparison of Alginate and Agarose, Biotechnol. Bioeng., 1996, 50, 374–381 CrossRef CAS.
- Y. Liu and M. B. Chan-Park, A Biomimetic Hydrogel Based on Methacrylated Dextran-Graft-Lysine and Gelatin for 3D Smooth Muscle Cell Culture, Biomaterials, 2010, 31, 1158–1170 CrossRef CAS PubMed.
- M. A. Cooperstein and H. E. Canavan, Biological Cell Detachment from poly(N-Isopropyl Acrylamide) and Its Applications, Langmuir, 2010, 26, 7695–7707 CrossRef CAS PubMed.
- M. Patenaude and T. Hoare, Injectable, Degradable Thermoresponsive Poly( N -Isopropylacrylamide) Hydrogels, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS.
- S.-W. Lee, K. E. Tettey, I. L. Kim, J. A. Burdick and D. Lee, Controlling the Cell-Adhesion Properties of Poly(acrylic acid)/Polyacrylamide Hydrogen-Bonded Multilayers, Macromolecules, 2012, 45, 6120–6126 CrossRef CAS.
- N. A. Peppas, K. B. Keys, M. Torres-Lugo and A. M. Lowman, Poly(ethylene Glycol)-Containing Hydrogels in Drug Delivery, J. Controlled Release, 1999, 62, 81–87 CrossRef CAS.
- H. Ismail, M. Irani and Z. Ahmad, Starch-Based Hydrogels: Present Status and Applications, Int. J. Polym. Mater., 2013, 62, 411–420 CrossRef CAS.
- J. F. Robyt, Essentials of Carbohydrate Chemistry, ed. C. R. Chantor, Springer New York, New York, NY, 1998 Search PubMed.
- J. M. Baskin and C. R. Bertozzi, Bioorthogonal Click Chemistry: Covalent Labeling in Living Systems, QSAR Comb. Sci., 2007, 26, 1211–1219 CAS.
- K. L. Dearfield, K. Harrington-Brock, C. L. Doerr, J. R. Rabinowitz and M. M. Moore, Genotoxicity in Mouse Lymphoma Cells of Chemicals Capable of Michael Addition, Mutagenesis, 1991, 6, 519–525 CrossRef CAS.
- A. B. Pratt, F. E. Weber, H. G. Schmoekel, R. Müller and J. A. Hubbell, Synthetic Extracellular Matrices for in Situ Tissue Engineering, Biotechnol. Bioeng., 2004, 86, 27–36 CrossRef CAS PubMed.
- C. R. Lee, A. J. Grodzinsky and M. Spector, The Effects of Cross-Linking of Collagen-Glycosaminoglycan Scaffolds on Compressive Stiffness, Chondrocyte-Mediated Contraction, Proliferation and Biosynthesis, Biomaterials, 2001, 22, 3145–3154 CrossRef CAS.
- L. L. Huang-Lee, D. T. Cheung and M. E. Nimni, Biochemical Changes and Cytotoxicity Associated with the Degradation of Polymeric Glutaraldehyde Derived Crosslinks, J. Biomed. Mater. Res., 1990, 24, 1185–1201 CrossRef CAS PubMed.
- B. Hoffmann, D. Seitz, A. Mencke, A. Kokott and G. Ziegler, Glutaraldehyde and Oxidised Dextran as Crosslinker Reagents for Chitosan-Based Scaffolds for Cartilage Tissue Engineering, J. Mater. Sci.: Mater. Med., 2009, 20, 1495–1503 CrossRef CAS PubMed.
- D. Sivakumaran, D. Maitland and T. Hoare, Injectable Microgel-Hydrogel Composites for Prolonged Small- Molecule Drug Delivery, Biomacromolecules, 2011, 12, 4112–4120 CrossRef CAS PubMed.
- S. B. Campbell, M. Patenaude and T. Hoare, Injectable Superparamagnets: Highly Elastic and Degradable poly(N-Isopropylacrylamide)-Superparamagnetic Iron Oxide Nanoparticle (SPION) Composite Hydrogels, Biomacromolecules, 2013, 14, 644–653 CrossRef CAS PubMed.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Injectable and Tunable Poly(ethylene Glycol) Analogue Hydrogels Based on Poly(oligoethylene Glycol Methacrylate), Chem. Commun., 2014, 50, 3306–3309 RSC.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Injectable Poly(oligoethylene Glycol Methacrylate)-Based Hydrogels with Tunable Phase Transition Behaviours: Physicochemical and Biological Responses, Acta Biomater., 2014, 10, 4143–4155 CrossRef CAS PubMed.
- M. K. Nguyen and D. S. Lee, Injectable Biodegradable Hydrogels, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, Bioorthogonal Click Chemistry: An Indispensable Tool to Create, ACS Macro Lett., 2013, 2, 5–9 CrossRef CAS PubMed.
- C. M. Nimmo and M. S. Shoichet, Regenerative Biomaterials That “Click”: Simple, Aqueous-Based Protocols for Hydrogel Synthesis, Surface Immobilization, and 3D Patterning, Bioconjugate Chem., 2011, 22, 2199–2209 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank and K. Kingsbury, et al., Synthesis of Well-Defined Hydrogel Networks Using Click Chemistry, Chem. Commun., 2006, 2774–2776 RSC.
- Y. Fu and W. J. Kao, In Situ Forming Poly(ethylene Glycol)-Based Hydrogels via Thiol-Maleimide Michael-Type Addition, J. Biomed. Mater. Res., Part A, 2011, 98, 201–211 CrossRef PubMed.
- K. C. Koehler, K. S. Anseth and C. N. Bowman, Diels-Alder Mediated Controlled Release from a Poly(ethylene Glycol) Based Hydrogel, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS PubMed.
- D. A. Ossipov, S. Piskounova, O. P. Varghese and J. Hilborn, Functionalization of Hyaluronic Acid with Chemoselective Groups via a Disulfide-Based Protection Strategy for In Situ Formation of Mechanically Stable Hydrogels, Biomacromolecules, 2010, 11, 2247–2254 CrossRef CAS PubMed.
- X. Z. Shu, Y. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Disulfide Cross-Linked Hyaluronan Hydrogels, Biomacromolecules, 2002, 3, 1304–1311 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biocompatible Hydrogels by Oxime Click Chemistry, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- H. Saito, A. S. Hoffman and H. I. Ogawa, Delivery of Doxorubicin from Biodegradable PEG Hydrogels Having Schiff Base Linkages, J. Bioact. Compat. Polym., 2007, 22, 589–601 CrossRef CAS PubMed.
- H. J. Naghash, O. Okay and H. Yildirim, Gel Formation in Free-Radical Crosslinking Copolymerization, J. Appl. Polym. Sci., 1995, 56, 477–483 CrossRef CAS PubMed.
- O. Okay, H. J. Naghash and I. Capek, Free-Radical Crosslinking Copolymerization: Effect of Cyclization on Diffusion-Controlled Termination at Low Conversion, Polymer, 1995, 36, 2413–2419 CrossRef CAS.
- H. J. Naghash, O. Okay and Y. Yagci, Gel Formation by Chain-Crosslinking Photopolymerization of Methyl Methacrylate and Ethylene Glycol Dimethacrylate, Polymer, 1997, 38, 1187–1196 CrossRef CAS.
- M. Keskinel and O. Okay, Effects of Cyclization and Electrostatic Interactions on the Termination Rate of Macroradicals in Free-Radical Crosslinking Copolymerization, Polym. Bull., 1998, 40, 491–498 CrossRef CAS.
- O. Okay, M. Kurz, K. Lutz and W. Funke, Cyclization and Reduced Pendant Vinyl Group Reactivity during the Free-Radical Crosslinking Polymerization of 1,4-Divinylbenzene, Macromolecules, 1995, 28, 2728–2737 CrossRef CAS.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Growth Factors, Matrices, and Forces Combine and Control Stem Cells, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- J. Kablik, G. D. Monheit, L. Yu, G. Chang and J. Gershkovich, Comparative Physical Properties of Hyaluronic Acid Dermal Fillers, Dermatol. Surg., 2009, 35, 302–312 CrossRef CAS PubMed.
- K. S. Anseth, A. T. Metters, S. J. Bryant, P. J. Martens, J. H. Elisseeff and C. N. Bowman, In Situ Forming Degradable Networks and Their Application in Tissue Engineering and Drug Delivery, J. Controlled Release, 2002, 78, 199–209 CrossRef CAS.
- G. D. Nicodemus and S. J. Bryant, Cell Encapsulation in Biodegradable Hydrogels for Tissue Engineering Applications, Tissue Eng., Part B, 2008, 14, 149–165 CrossRef CAS PubMed.
- A. Sawhney, C. Pathak and J. Hubbell, Bioerodible Hydrogels Based on Photopolymerized Poly(ethylene Glycol)-Co-Poly(alpha-Hydroxy Acid) Diacrylate Macromers, Macromolecules, 1993, 26, 581–587 CrossRef CAS.
- D. Seliktar, Designing Cell-Compatible Hydrogels for Biomedical Applications, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- M. Al-Haydari, Immune Response to Synthesized PNipam-Based Graft Hydrogels Containing Chitosan and Hyaluronic Acid, Open Access Diss. Thesis, McMaster University, 2010, vol. 1, p. 4193.
- J. E. Babensee, J. M. Anderson, L. V. McIntire and A. G. Mikos, Host Response to Tissue Engineered Devices, Adv. Drug Delivery Rev., 1998, 33, 111–139 CrossRef CAS.
- J. Anderson, A. Rodriguez and D. Chang, Foreign Body Reaction to Biomaterials, Semin. Immunol., 2009, 20, 86–100 CrossRef PubMed.
- B. L. Seal, T. C. Otero and A. Panitch, Polymeric Biomaterials for Tissue and Organ Regeneration, Mater. Sci. Eng., R, 2001, 34, 147–230 CrossRef.
- J.-F. Lutz, Thermo-Switchable Materials Prepared Using the OEGMA-Platform, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS PubMed.
- G. Vancoillie, D. Frank and R. Hoogenboom, Thermoresponsive Poly(oligo Ethylene Glycol Acrylates), Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS PubMed.
- S. P. Massia, J. Stark and D. S. Letbetter, Surface-Immobilized Dextran Limits Cell Adhesion and Spreading, Biomaterials, 2000, 21, 2253–2261 CrossRef CAS.
- R. Konradi, C. Acikgoz and M. Textor, Polyoxazolines for Nonfouling Surface Coatings–a Direct Comparison to the Gold Standard PEG, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS PubMed.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Zwitterionic Polymers Exhibiting High Resistance to Nonspecific Protein Adsorption from Human Serum and Plasma, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS PubMed.
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Injectable in Situ Cross-Linking Hydrogels for Local Antifungal Therapy, Biomaterials, 2010, 31, 1444–1452 CrossRef CAS PubMed.
- K. Chawla, T.-B. Yu, S. W. Liao and Z. Guan, Biodegradable and Biocompatible Synthetic Saccharide-Peptide Hydrogels for Three-Dimensional Stem Cell Culture, Biomacromolecules, 2011, 12, 560–567 CrossRef CAS PubMed.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, Free Radical Polymers with Tunable and Selective Bio- and Chemical Degradability, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Fréchet, A Novel Strategy for Encapsulation and Release of Proteins: Hydrogels and Microgels with Acid-Labile Acetal Cross-Linkers, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef CAS PubMed.
- K. Ulbrich, V. Šubr, L. W. Seymour and R. Duncan, Novel Biodegradable Hydrogels Prepared Using the Divinylic Crosslinking Agent N,O-Dimethacryloylhydroxylamine. 1. Synthesis and Characterisation of Rates of Gel Degradation, and Rate of Release of Model Drugs, in Vitro and in Vivo, J. Controlled Release, 1993, 24, 181–190 CrossRef CAS.
- C. Rippe, A. Rippe, O. Torffvit and B. Rippe, Size and Charge Selectivity of the Glomerular Filter in Early Experimental Diabetes in Rats, American Journal of Physiology - Renal Physiology, 2007, 293, F1533–F1538 CrossRef CAS PubMed.
- O. Varghese and W. Sun, In Situ Cross-Linkable High Molecular Weight Hyaluronan− Bisphosphonate Conjugate for Localized Delivery and Cell-Specific Targeting: A Hydrogel Linked Prodrug, J. Am. Chem. Soc., 2009, 4, 8781–8783 CrossRef PubMed.
- M. Deshmukh, Y. Singh, S. Gunaseelan, D. Gao, S. Stein and P. J. Sinko, Biodegradable Poly(ethylene Glycol) Hydrogels Based on a Self-Elimination Degradation Mechanism, Biomaterials, 2010, 31, 6675–6684 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanoparticle PEGylation for Imaging and Therapy, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- B. D. Polizzotti, B. D. Fairbanks and K. S. Anseth, Three-Dimensional Biochemical Patterning of Click-Based Composite Hydrogels via Thiolene Photopolymerization, Biomacromolecules, 2008, 9, 1084–1087 CrossRef CAS PubMed.
- C. A. Deforest, E. A. Sims and K. S. Anseth, Peptide-Functionalized Click Hydrogels with Independently Tunable Mechanics and Chemical Functionality for 3D Cell Culture, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS PubMed.
- Y. Fu and W. J. Kao, In Situ Forming Poly(ethylene Glycol)-Based Hydrogels via Thiol-Maleimide Michael-Type Addition, J. Biomed. Mater. Res., Part A, 2011, 98, 201–211 CrossRef PubMed.
- S. C. Rizzi and J. A. Hubbell, Recombinant Protein-Co-PEG Networks as Cell-Adhesive and Proteolytically Degradable Hydrogel Matrixes. Part I: Development and Physicochemical Characteristics, Biomacromolecules 6, 1226–1238 Search PubMed.
- T. Matsunaga, T. Sakai, Y. Akagi, U. Chung and M. Shibayama, Structure Characterization of Tetra-PEG Gel by Small-Angle Neutron Scattering, Macromolecules, 2009, 10, 1344–1351 CrossRef.
- E. Ruoslahti and M. Pierschbacher, Arg-Gly-Asp: A Versatile Cell Recognition Signal, Cell, 1986, 44, 517–518 CrossRef CAS.
- D. Guarnieri, A. De Capua, M. Ventre, A. Borzacchiello, C. Pedone, D. Marasco, M. Ruvo and P. A. Netti, Covalently Immobilized RGD Gradient on PEG Hydrogel Scaffold Influences Cell Migration Parameters, Acta Biomater., 2010, 6, 2532–2539 CrossRef CAS PubMed.
- C. Zhang, S. Hekmatfer and N. W. Karuri, A Comparative Study of Polyethylene Glycol Hydrogels Derivatized with the RGD Peptide and the Cell-Binding Domain of Fibronectin, J. Biomed. Mater. Res., Part A, 2013, 102, 170–179 CrossRef PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, Bioconjugation by copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- D. A. Ossipov and J. Hilborn, Poly(vinyl Alcohol)-Based Hydrogels Formed by “Click Chemistry”, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. a. DeForest, K. S. Anseth and C. N. Bowman, Spatial and Temporal Control of the Alkyne-Azide Cycloaddition by Photoinitiated Cu(II) Reduction, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- M. Van Dijk, C. F. van Nostrum, W. E. Hennink, D. T. S. Rijkers and R. M. J. Liskamp, Synthesis and Characterization of Enzymatically Biodegradable PEG and Peptide-Based Hydrogels Prepared by Click Chemistry, Biomacromolecules, 2010, 11, 1608–1614 CrossRef CAS PubMed.
- S. Tan, A. Blencowe, K. Ladewig and G. G. Qiao, A Novel One-Pot Approach towards Dynamically Cross-Linked Hydrogels, Soft Matter, 2013, 9, 5239–5250 RSC.
- Y. Okumura and K. Ito, The Polyrotaxane Gel: A Topological Gel by Figure-of-Eight Cross-Links, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, A Strain-Promoted [3 + 2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Sequential Click Reactions for Synthesizing and Patterning Three-Dimensional Cell Microenvironments, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- J. Zheng, L. A. Smith Callahan, J. Hao, K. Guo, C. Wesdemiotis, R. A. Weiss and M. L. Becker, Strain-Promoted Crosslinking of PEG-Based Hydrogels via Copper-Free Cycloaddition, ACS Macro Lett., 2012, 1, 1071–1073 CrossRef CAS PubMed.
- D. C. Dittmer, C. G. Swain, R. E. Benson, M. Friedman, J. F. Cavins and J. S. Wall, Relative Nucleophilic Reactivities of Amino Groups and, J. Am. Chem. Soc., 1965, 87, 3672–3682 CrossRef.
- M. Friedman, Chemistry, Biochemistry, and Safety of Acrylamide. A Review, J. Agric. Food Chem., 2003, 51, 4504–4526 CrossRef CAS PubMed.
- C. F. H. Allen and W. J. Humphlett, The Thermal Reversibility of the Michael Reaction: V. The Effect of the Structure of Certain Michael Adducts on Cleavage, Can. J. Chem., 1966, 44, 2315–2321 CrossRef CAS.
- C. F. H. Allen, J. O. Fournier and W. J. Humphlett, The Thermal Reversibility of the Michael Reaction: IV. Thiol Adducts, Can. J. Chem., 1964, 42, 2616–2620 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Tunable Degradation of Maleimide-Thiol Adducts in Reducing Environments, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Reversible Maleimide-Thiol Adducts Yield Glutathione-Sensitive Poly(ethylene Glycol)-Heparin Hydrogels, Polym. Chem., 2013, 4, 133–143 RSC.
- S. J. Buwalda, P. J. Dijkstra and J. Feijen, In Situ Forming Poly(ethylene Glycol)- Poly(L-Lactide) Hydrogels via Michael Addition: Mechanical Properties, Degradation, and Protein Release, Macromol. Chem. Phys., 2012, 213, 766–775 CrossRef CAS PubMed.
- F. Cellesi, N. Tirelli and J. A. Hubbell, Towards a Fully-Synthetic Substitute of Alginate: Development of a New Process Using Thermal Gelation and Chemical Cross-Linking, Biomaterials, 2004, 25, 5115–5124 CrossRef CAS PubMed.
- J. M. Jukes, L. J. van der Aa, C. Hiemstra, T. van Veen, P. J. Dijkstra, Z. Zhong, J. Feijen, C. A. van Blitterswijk and J. de Boer, A Newly Developed Chemically Crosslinked Dextran-Poly(ethylene Glycol) Hydrogel for Cartilage Tissue Engineering, Tissue Eng., Part A, 2010, 16, 565–573 CrossRef CAS PubMed.
- A. E. Rydholm, N. L. Held, D. S. W. Benoit, C. N. Bowman and K. S. Anseth, Modifying Network Chemistry in Thiol-Acrylate Photopolymers through Postpolymerization Functionalization to Control Cell-Material Interactions, J. Biomed. Mater. Res., Part A, 2008, 86, 23–30 CrossRef PubMed.
- A. Metters and J. Hubbell, Network Formation and Degradation Behavior of Hydrogels Formed by Michael-Type Addition Reactions, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- M. V. Tsurkan, K. Chwalek, S. Prokoph, A. Zieris, K. R. Levental, U. Freudenberg and C. Werner, Defined Polymer-Peptide Conjugates to Form Cell-Instructive starPEG-Heparin Matrices In Situ, Adv. Mater., 2013, 25, 2606–2610 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, A. Krouwels, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Synthesis and Characterization of Hyaluronic Acid-Poly(ethylene Glycol) Hydrogels via Michael Addition: An Injectable Biomaterial for Cartilage Repair, Acta Biomater., 2010, 6, 1968–1977 CrossRef CAS PubMed.
- S. P. Zustiak and J. B. Leach, Characterization of Protein Release from Hydrolytically Degradable Poly(ethylene Glycol) Hydrogels, Biotechnol. Bioeng., 2011, 108, 197–206 CrossRef CAS PubMed.
- C. Hiemstra, Z. Zhong, M. J. van Steenbergen, W. E. Hennink and J. Feijen, Release of Model Proteins and Basic Fibroblast Growth Factor from in Situ Forming Degradable Dextran Hydrogels, J. Controlled Release, 2007, 122, 71–78 CrossRef CAS PubMed.
- C. Hiemstra, L. J. van der Aa, Z. Zhong, P. J. Dijkstra and J. Feijen, Novel in Situ Forming, Degradable Dextran Hydrogels by Michael Addition Chemistry: Synthesis, Rheology, and Degradation, Macromolecules, 2007, 40, 1165–1173 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene Glycol)-Co-Peptide Hydrogels Formed by Michael-Type Addition, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- A. J. García, PEG-Maleimide Hydrogels for Protein and Cell Delivery in Regenerative Medicine, Ann. Biomed. Eng., 2014, 42, 312–322 CrossRef PubMed.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. a. Sulchek, T. H. Barker and A. J. García, Maleimide Cross-Linked Bioactive PEG Hydrogel Exhibits Improved Reaction Kinetics and Cross-Linking for Cell Encapsulation and in Situ Delivery, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed.
- R. D. Levit, N. Landázuri, E. A. Phelps, M. E. Brown, A. J. García, M. E. Davis, G. Joseph, R. Long, S. A. Safley and J. D. Suever, et al., Cellular Encapsulation Enhances Cardiac Repair, J. Am. Heart Assoc., 2013, 2, e000367 Search PubMed.
- J. Yu, X. Xu, F. Yao, Z. Luo, L. Jin, B. Xie, S. Shi, H. Ma, X. Li and H. Chen, In Situ Covalently Cross-Linked PEG Hydrogel for Ocular Drug Delivery Applications, Int. J. Pharm., 2014, 470, 151–157 CrossRef CAS PubMed.
- K. P. Vercruysse, D. M. Marecak, J. F. Marecek and G. D. Prestwich, Synthesis and in Vitro Degradation of New Polyvalent Hydrazide Cross-Linked Hydrogels of Hyaluronic Acid, Bioconjugate Chem., 1997, 8, 686–694 CrossRef CAS PubMed.
- S.-Y. Choh, D. Cross and C. Wang, Facile Synthesis and Characterization of Disulfide-Cross-Linked Hyaluronic Acid Hydrogels for Protein Delivery and Cell Encapsulation, Biomacromolecules, 2011, 12, 1126–1136 CrossRef CAS PubMed.
- H.-L. Wei, Z. Yang, L.-M. Zheng and Y.-M. Shen, Thermosensitive Hydrogels Synthesized by Fast Diels–Alder Reaction in Water, Polymer, 2009, 50, 2836–2840 CrossRef CAS PubMed.
- C. M. Nimmo and M. S. Shoichet, Regenerative Biomaterials That “Click”: Simple, Aqueous-Based Protocols for Hydrogel Synthesis, Surface Immobilization, and 3D Patterning, Bioconjugate Chem., 2011, 22, 2199–2209 CrossRef CAS PubMed.
- H.-L. Wei, K. Yao, Z. Yang, H.-J. Chu, J. Zhu, C.-C. Ma and Z.-X. Zhao, Preparation of Thermosensitive Hydrogels by Means of Tandem Physical and Chemical Crosslinking, Macromol. Res., 2011, 19, 294–299 CrossRef CAS.
- D. C. Rideout and R. Breslow, Hydrophobic Acceleration of Diels-Alder Reactions, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Making Polymeric Nanoparticles Stimuli-Responsive with Dynamic Covalent Bonds, Polym. Chem., 2013, 4, 31–45 RSC.
- Y. Xin and J. Yuan, Schiff's Base as a Stimuli-Responsive Linker in Polymer Chemistry, Polym. Chem., 2012, 3, 3045–3055 RSC.
- A. A. Kale and V. P. Torchilin, Design, Synthesis, and Characterization of pH-Sensitive PEG-PE Conjugates for Stimuli-Sensitive Pharmaceutical Nanocarriers: The Effect of Substitutes at the Hydrazone Linkage on the Ph Stability of PEG-PE Conjugates, Bioconjugate Chem., 2007, 18, 363–370 CrossRef CAS PubMed.
- Y. Luo, J. B. Kobler, J. T. Heaton, X. Jia, S. M. Zeitels and R. Langer, Injectable Hyaluronic Acid-Dextran Hydrogels and Effects of Implantation in Ferret Vocal Fold, J. Biomed. Mater. Res., Part B, 2010, 93, 386–393 CrossRef PubMed.
- M. H. Alves, C. J. Young, K. Bozzetto, L. A. Poole-Warren and P. J. Martens, Degradable, Click Poly(vinyl Alcohol) Hydrogels: Characterization of Degradation and Cellular Compatibility, Biomed. Mater., 2012, 7, 024106 CrossRef CAS PubMed.
- K. Y. Lee, K. H. Bouhadir and D. J. Mooney, Degradation Behavior of Covalently Cross-Linked Poly(aldehyde Guluronate) Hydrogels, Macromolecules, 2000, 33, 97–101 CrossRef CAS.
- R. Zhang, Z. Huang, M. Xue, J. Yang and T. Tan, Detailed Characterization of an Injectable Hyaluronic Acid-Polyaspartylhydrazide Hydrogel for Protein Delivery, Carbohydr. Polym., 2011, 85, 717–725 CrossRef CAS PubMed.
- G. N. Grover, R. L. Braden and K. L. Christman, Oxime Cross-Linked Injectable Hydrogels for Catheter Delivery, Adv. Mater., 2013, 25, 2937–2942 CrossRef CAS PubMed.
- J.-F. Lutz, Polymerization of Oligo(ethylene Glycol) (meth)acrylates: Toward New Generations of Smart Biocompatible Materials, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS PubMed.
- H. Dong and K. Matyjaszewski, Thermally Responsive P(M(EO)2MA-Co-OEOMA) Copolymers via AGET ATRP in Miniemulsion, Macromolecules, 2010, 43, 4623–4628 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, Libraries of Methacrylic Acid and Oligo(ethylene Glycol) Methacrylate Copolymers with LCST Behavior, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS PubMed.
- J.-F. Lutz and A. Hoth, Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene Glycol) Methacrylate, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of poly(NIPAM) Over?, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- S. Sun and P. Wu, On the Thermally Reversible Dynamic Hydration Behavior of Oligo(ethylene Glycol) Methacrylate-Based Polymers in Water, Macromolecules, 2013, 46, 236–246 CrossRef CAS.
- P. Banerjee, D. J. Irvine, A. M. Mayes and L. G. Griffith, Polymer Latexes for Cell-Resistant and Cell-Interactive Surfaces, J. Biomed. Mater. Res., 2000, 50, 331–339 CrossRef CAS.
- J. Hyun, H. Ma, Z. Zhang, T. P. Beebe Jr and A. Chilkoti, Universal Route to Cell Micropatterning Using an Amphiphilic Comb Polymer, Adv. Mater., 2003, 15, 576–579 CrossRef CAS PubMed.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Biocompatible, Thermoresponsive, and Biodegradable: Simple Preparation of “All-in-One” Biorelevant Polymers, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- J. Lei, C. Mayer, V. Freger and M. Ulbricht, Synthesis and Characterization of Poly(ethylene Glycol) Methacrylate Based Hydrogel Networks for Anti-Biofouling Applications, Macromol. Mater. Eng., 2013, 298, 967–980 CrossRef CAS PubMed.
- R. París and I. Quijada-Garrido, Swelling Behaviour of Thermo-Sensitive Hydrogels Based on Oligo(ethylene Glycol) Methacrylates, Eur. Polym. J., 2009, 45, 3418–3425 CrossRef PubMed.
- J. A. Yoon, T. Kowalewski, K. Matyjaszewski, C. Gayathri and R. R. Gil, Comparison of the Thermoresponsive Deswelling Kinetics of Poly(2-(2-Methoxyethoxy)ethyl Methacrylate) Hydrogels Prepared by ATRP and FRP, Macromolecules, 2010, 43, 4791–4797 CrossRef CAS.
- J. A. Yoon, T. Kowalewski and K. Matyjaszewski, Comparison of Thermoresponsive Deswelling Kinetics of Poly (oligo(ethylene Oxide) Methacrylate)-Based Thermoresponsive Hydrogels Prepared by “ Graft-from ” ATRP, Macromolecules, 2011, 44, 2261–2268 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, About the Phase Transitions in Aqueous Solutions of Thermoresponsive Copolymers and Hydrogels Based on 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene Glycol) Methacrylate, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J.-F. Lutz, Thermogelation of PEG-Based Macromolecules of Controlled Architecture, Macromolecules, 2009, 42, 33–36 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed. Engl., 2002, 41, 898–952 CrossRef.
- P. Bulpitt and D. Aeschlimann, New Strategy for Chemical Modification of Hyaluronic Acid: Preparation of Functionalized Derivatives and Their Use in the Formation of Novel Biocompatible Hydrogels, J. Biomed. Mater. Res., 1999, 47, 152–169 CrossRef CAS.
- T. Ito, Y. Yeo, C. B. Highley, E. Bellas and D. S. Kohane, Dextran-Based in Situ Cross-Linked Injectable Hydrogels to Prevent Peritoneal Adhesions, Biomaterials, 2007, 28, 3418–3426 CrossRef CAS PubMed.
- S. Wang, O. P. Oommen, H. Yan and O. P. Varghese, Mild and Efficient Strategy for Site-Selective Aldehyde Modification of Glycosaminoglycans: Tailoring Hydrogels with Tunable Release of Growth Factor, Biomacromolecules, 2013, 14, 2427–2432 CrossRef CAS PubMed.
- N. M. B. Smeets, M. Patenaude, D. Kinio, F. M. Yavitt, E. Bakaic, F.-C. Yang, M. Rheinstädter and T. Hoare, Injectable Hydrogels with in Situ-Forming Hydrophobic Domains: Oligo( D, L -Lactide) Modified Poly(oligoethylene Glycol Methacrylate) Hydrogels, Polym. Chem., 2014, 5, 6811–6823 RSC.
- N. M. B. Smeets, E. Bakaic, F. M. Yavitt, F.-C. Yang, M. C. Rheinstädter and T. Hoare, Probing the Internal Morphology of Injectable Poly(oligoethylene Glycol Methacrylate) Hydrogels by Light and Small-Angle Neutron Scattering, Macromolecules, 2014, 47, 6017–6027 CrossRef CAS.
- M. Patenaude and T. Hoare, Injectable, Mixed Natural-Synthetic Polymer Hydrogels with Modular Properties, Biomacromolecules, 2012, 13, 369–378 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets, H. Dorrington and T. R. Hoare, “Off-the-shelf” thermoresponsive hydrogel design: tuning hydrogel properties by mixing precursor polymers with different lower-critical solution temperatures, RSC Adv., 2015, 5, 33364–33376 RSC.
- Y. Hou, C. A. Schoener, K. R. Regan, D. Munoz-pinto, S. Mariah and M. A. Grunlan, Photo-Crosslinked PDMSstar-PEG Hydrogels: Synthesis, Characterization, and Potential Application for Tissue Engineering Scaffolds, Biomacromolecules, 2010, 11, 648–656 CrossRef CAS PubMed.
- Y. Chung and Y. Chiu, Protein Adsorption and Cell Alignment on Micropatterned Phosphorylcholine Surfaces, J. Med. Biol. Eng., 2009, 29, 320–324 Search PubMed.
- L. D. Unsworth, H. Sheardown and J. L. Brash, Protein Resistance of Surfaces Prepared by Sorption of End-Thiolated Poly(ethylene Glycol) to Gold: Effect of Surface Chain Density, Langmuir, 2005, 21, 1036–1041 CrossRef CAS PubMed.
- E. Tziampazis, J. Kohn and P. V. Moghe, PEG-Variant Biomaterials as Selectively Adhesive Protein Templates: Model Surfaces for Controlled Cell Adhesion and Migration, Biomaterials, 2000, 21, 511–520 CrossRef CAS.
- H. Du, P. Chandaroy and S. W. Hui, Grafted Poly-(ethylene Glycol) on Lipid Surfaces Inhibits Protein Adsorption and Cell Adhesion, Biochim. Biophys. Acta, Biomembr., 1997, 1326, 236–248 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- T. Yamaoka, Y. Tabata and Y. Ikada, Distribution and Tissue Uptake of Poly(ethylene glycol) with Different Molecular Weights after Intravenous Administration to Mice, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |