
Effect of pretreatment on ceria-supported cobalt catalyst for ammonia synthesis†
Bingyu Lin*,
Yanchao Qi,
Kemei Wei and
Jianxin Lin
National Engineering Research Center of Chemical Fertilizer Catalyst, Fuzhou University, Fuzhou, Fujian 350002, PR China. E-mail: bylin@fzu.edu.cn; bylinfzu@yahoo.com; Fax: +86 0591-83738808; Tel: +86 0591-83731234
First published on 18th August 2014
Abstract
Cerium in a catalyst may affect the catalytic reaction by providing surface hydroxyl groups. In this work, Co/CeO2 catalysts with different properties of hydroxyl groups were prepared using different pretreatment atmospheres during the heat treatment process of the hydroxide gel with metal and cerium. The heat treatment process of Co/CeO2 precipitate in different pretreatment atmospheres was monitored by mass spectrometry. Pulse chemisorption measurements in combination with XRD, TPR, XPS, TEM were used to reveal the relationship between the properties of the as-obtained samples and their catalytic performances in ammonia synthesis. It was found that the pretreatment atmosphere of Co/CeO2 affected the dehydroxylation pathways. Most hydroxyls in CeO2 can release H2O by reacting with atomic hydrogen during the hydrogen reduction process. On the other hand, oxygen treatment resulted in the conversion of hydroxyls to oxides, and further hydrogen reduction promoted the reduction of Ce4+ to Ce3+ and the formation of the stable surface hydroxyl radicals. The active sites available for nitrogen dissociation and ammonia synthesis may decrease because some redundant hydrogen cannot be removed in time by combination with the stable hydroxyls, and thus the catalyst with oxygen treatment showed lower ammonia synthesis activity.
1. Introduction
Ammonia not only is the foundation for the manufacture of nitrogenous fertilizers, but also is a key chemical for the production of many explosives and polymers.1 Catalytic ammonia synthesis (Haber–Bosch process) is an excellent way to convert nitrogen and hydrogen into ammonia, and this process is the most important invention of the twentieth century.2 Much attention is currently focused on ammonia synthesis in both academia and industry due to the dramatic increase in the demand for ammonia and its production.So far, various transition metal catalysts such as Fe, Ru, Re and Co have been developed for this reaction.3–9 Among them, Fe-based and Ru-based catalysts have been extensively used in ammonia plants all over the world. However, iron catalyst is kinetically sensitive to the changes in ammonia concentration, and the catalytic activity decreased greatly with increasing ammonia concentration of the reactant gas.10,11 On the other hand, it is also difficult for Ru catalyst to give an economic advantage because of the high costs of Ru metal combined with the disadvantages such as the methanation of carbon support at industrially reaction conditions. Consequently, extensive research has been carried out on the search for a new catalytic material with high activity and lower price than Ru.
Hagen et al.12,13 reported that Ba–Co/C catalyst showed higher ammonia synthesis activities and lower ammonia inhibition than the commercial iron catalyst, indicating that this catalyst was a promising candidate for industrial applications. Rarog-Pilecka et al.10,11,14 further confirmed that Co catalyst can be used as a complement because it was less inhibited by ammonia and much cheaper than Ru catalyst. According to the results of Rarog-Pilecka et al.,10,14 cobalt dispersion was a critical parameter to obtain a highly efficient Co catalyst. They also found that ceria can be used a structural promoter for the unsupported cobalt catalysts to hinder the sintering of cobalt oxide.15,16 Recently, Karolewska et al.11 found that the promotion effect of cerium only could be observed for the samples prepared by the co-impregnation of cobalt and cerium precursors. They suggested that the addition of cerium can increase the ammonia synthesis activity because cerium can significantly increase the cobalt dispersion of carbon-supported cobalt catalysts promoted with barium. However, previously, ceria or other lanthanide oxides were also found can release hydrogen poisoning, and thus extensive studies used lanthanide oxides as support materials or promoters for Ru catalyst using in ammonia synthesis.17–22
In recent years, there has been an increasing interest in learning the effect of hydroxyl groups on catalytic reactions or designing highly efficient supported-metal catalyst containing hydroxyl radicals.23–29 Flytzani-Stephanopoulos and workers23,24,28 proposed that Ce ions could stabilize and provide the OH species to the Pt atoms for platinum supported on alumina, silica or multiwalled carbon nanotubes. The surface OH groups can be easily activated by CO, and thus a partially oxidized Pt-alkali-Ox(OH)y species was suggested to be the active site for the low-temperature WGS reaction. Yang et al.26 also claimed that gold species with surrounding extra surface–OH groups were the active sites for the WGS reaction over Au catalyst supported on titania. Chen et al.29 found that OH groups at the Fe3+–OH–Pt interfaces can react with CO to release carbon dioxide, and then the catalyst with hydroxyl groups showed high activity for CO oxidation. According to this knowledge, they successfully prepared an oxide-supported PtFeNi nanocatalyst with excellent performance of CO oxidation. Furthermore, Chen et al.30 reported that atomic hydrogen could adsorb on CeO2 to form surface hydroxyls, and then the hydroxyls undergo different dehydroxylation pathways by releasing H2O or producing hydrogen.
Inspired by the findings in literature mentioned above, one may conclude that cerium would affect the activity of ammonia synthesis by creating surface hydroxyls groups. In the present study, Co/CeO2 catalysts with different properties of hydroxyl groups were prepared using the different pretreatment atmosphere for the heat treatment process of the hydroxide gel with metal and cerium. The aim is to investigate the correlation between the properties and the activity of Co/CeO2 catalyst. The knowledge about this relation will help not only to understand the effect of cerium, but also to obtain the catalyst design concept.
2. Experimental
2.1 Catalyst preparation
Co catalysts were prepared by coprecipitation method. The aqueous solution of Ce(NO3)3 and Co(NO3)3 was stirred at 80 °C for 0.5 h, the weight ratio of Co to CeO2 was ca. 7.9 wt.%. Subsequently, NaOH solution (0.1 mol L−1) was added drop wise to form a suspension with pH of around 10. After aging for 3 h, the suspension was filtered and washed with distilled water until no pH change. After dried at 110 °C overnight, the as-obtained sample was labeled as Co/CeO2, the weight percentages of Co and CeO2 (detected by XRF) were 7.1% and 92.9%, respectively. Some Co/CeO2 was further heated at 600 °C for 4 h in different gases including air or hydrogen, and the catalysts were named as Co/CeO2–O (air) and Co/CeO2–H (hydrogen) depending on the treatment gas. For comparison, CeO2 sample without heat treatment was also prepared by precipitation method.2.2 Catalyst characterization
X-ray powder diffraction (XRD) data were collected on a X'Pert PRO diffractometer (PANalytical) using Co Kα radiation at 40 kV and 40 mA. N2 physisorption measurements were performed using a ASAP 2020 apparatus (Micromeritics) at −196 °C with samples previously outgassed at 180 °C. The specific surface area (SBET) was determined from nitrogen adsorption isotherms. The pore size distributions were obtained from the adsorption branches by the Kruk–Jaroniec–Sayari method.31X-ray phototoelectron spectroscopy (XPS) analyses were carried out on an ESCALAB 250 photoelectron spectroscopy (Thermo Fisher Scientific) with monochromatized Al Kα X-rays (1486.6 eV). The spectra were decomposed by XPSPEAK software (version 4.1, Raymund WM) with Shirley background subtraction and 80% Gaussian/Lorentzian ratio. The peaks were referenced to the C 1s and Ce+4 3d3/2 at 284.6 and 916.7 eV, respectively.
Temperature-programmed reduction (TPR) experiments were conducted in Micromeritics AutoChem 2920 instrument. The catalyst (110 mg, sieve fraction 0.30–0.56 mm) was heated to 600 °C at 10 °C min−1 in a flow of a 10% H2/Ar mixture (30 mL min−1). A temperature-programmed oxidation study (TPO) was carried out in 2% O2/He mixture (30 mL min−1). The outlet gas stream was monitored by a mass spectrometry (Hiden HPR-20).
The H2 temperature programmed desorption (TPD) studies were carried out in the same Autochem 2920 instrument. The catalyst was reduced in hydrogen at 500 °C for 6 h, and then flushed in Ar and cooled down to 430 °C. Subsequently, the catalyst adsorbed hydrogen for 0.5 h and cooled down to room temperature. After purging in an Ar flow for 0.5 h, the sample was heated from room temperature to 600 °C with a rate of 10 °C min−1, and the off-gas was detected by the mass spectrometer.
Pulse chemisorption measurements were performed using an Autochem2920 instrument with mass spectrometry detection. In a typical experiment, samples of 110 mg were first reduced in hydrogen at 500 °C for 6 h, then flushed in Ar and cooled down to 430 °C. 10 vol% H2/Ar (0.3541 mL) was introduced into the sample by pulse injection for 5 times. Then the carrier gas was switched into nitrogen, and 10 vol% H2/Ar was injected again.
2.3 Activity measurements
Ammonia synthesis rates were measured in a stainless steel reactor. Co catalyst (0.30 g, 32–60 meshes) was diluted with quartz powder in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 volumetric ratio. Prior to activity measurements, the sample was reduced in the reacting mixture (H2/N2 = 3.0 v/v) at different temperatures (200, 300, 400, 425, 450 and 500 °C for 6 h, respectively), and then stabilized at 430 °C for 40 h. The rates were measured at a space velocity of 2.4 × 105 cm3 gcat−1 h−1.
:30 volumetric ratio. Prior to activity measurements, the sample was reduced in the reacting mixture (H2/N2 = 3.0 v/v) at different temperatures (200, 300, 400, 425, 450 and 500 °C for 6 h, respectively), and then stabilized at 430 °C for 40 h. The rates were measured at a space velocity of 2.4 × 105 cm3 gcat−1 h−1.
3. Results
3.1 Temperature-programmed oxidation and temperature-programmed reduction
The precipitates of CeO2 or Co/CeO2 had been prepared using Ce(NO3)3 and Co(NO3)3 as precursors. Then various species including of NOx, HxO and NHx would appear when the precipitants were treated at high temperature (Table S1†). Different species with same m/z values would be distinguished by comparing and the shape and the intensity ratio of the mass spectra with different m/z values (Fig. S1 and S2†). O2-TPO and H2-TPR studies were carried out to understand the changes of Co catalysts during the heat treatment process. The possibility that the physisorbed water or other impurities affected the changes of Co catalysts cannot be excluded. Therefore, no pretreatment was conducted before the onset of analysis. Fig. 1 shows the H2-TPR profiles of CeO2. Two broad peaks at about 50–200 °C and 250–500 °C can be observed in the mass spectrum of m/z = 18 during the reduction process of CeO2 which are due to H2O. No strong H2 consumption peak was observed at low temperature, and the ratio of m/z = 17 to 18 was ca. 0.17, which was in accordance with the value of HO/H2O obtained from the dissociative ionization of water (Fig. S2†). These results indicated that the mass signals in this temperature range mainly were water and its dissociated species. The occurrence of H2O peak at 250–500 °C was accompanied by a H2 consumption peak, suggesting that hydrogen was responsible for the generation of water at high temperature. The appearance of water may be due to the reduction of ceria or the interaction of hydroxyls with hydrogen. Three peaks centered at 115, 217 and 313 °C can be found in the mass spectrum with m/z = 16. On the other hand, the signal with m/z = 32 totally disappeared at above 200 °C. These results clearly showed that the appearance of NH2 when CeO2 was reduced in hydrogen. The occurrence of the peaks in the spectra of m/z = 15 and m/z = 30 further confirmed the generation of NHx.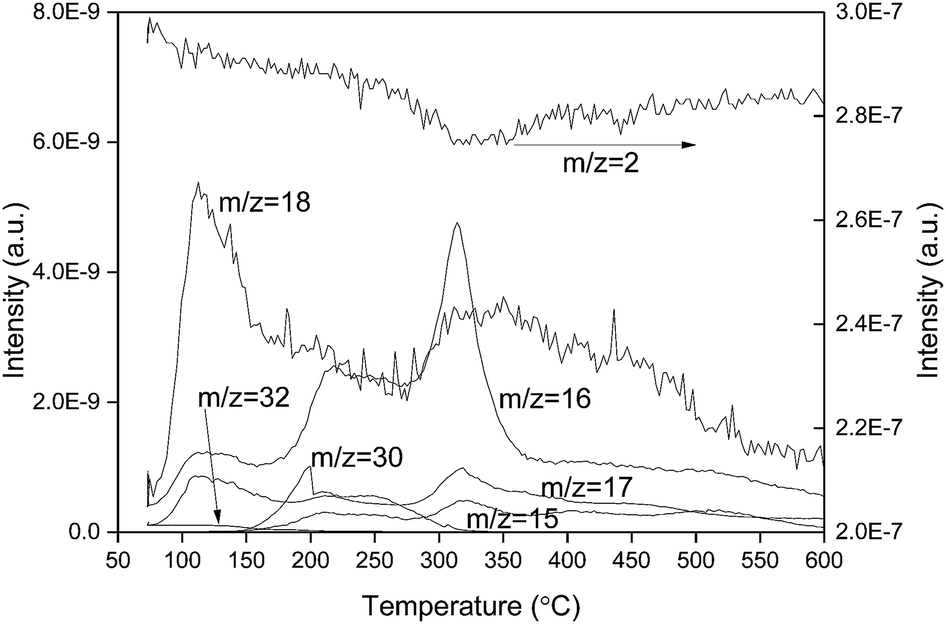 | ||
| Fig. 1 H2-TPR profiles of CeO2. | ||
The presence of cobalt increased the amount of water desorbed at the low temperatures (Fig. 2A). Other two strong water peaks along with H2 consumption peaks can be observed in H2-TPR profiles of Co/CeO2: one peak centered at 202 °C with shoulder at 224 °C, and the other peak show maximum intensity at ca. 331 °C. The former peak may mainly be due to the reduction of cobalt compound. The latter peak has same temperature range as that of the hydrogen reduction of CeO2, indicating that this peak was correlative with the reduction of cerium compound. The strong peaks at above 270 °C in the spectra with m/z = 15 or 16 can be found on Co/CeO2 reduced in hydrogen (Fig. 2A). This result suggested that NHx radicals appeared during the hydrogen reduction of Co/CeO2. The low-temperature H2O peak also can be found during the hydrogen reduction of Co/CeO2–H catalyst with preadsorbed oxygen (Fig. 2B). It can therefore conclude that the peak was relative not only to hydroxyls in hydroxide or residual H2O, but also to the reduction of some easily reduced oxides. The generation of H2O at ca. 248 and 290 °C for oxidized Co/CeO2–H because the presence of multi-step reduction of cobalt oxides (Co3O4 → CoO → Co).32–34 The signal intensity ratios of the m/z = 16 or m/z = 17 to m/z = 18 at 290 °C both were close to the data obtained from the dissociative ionization of water (Fig. S2†), indicating that only water was produced during the reduction of Co/CeO2–H pretreated with oxygen.
 | ||
| Fig. 2 H2-TPR profiles of (A) Co/CeO2 and (B) Co/CeO2–H catalyst pretreated oxygen at 400 °C. | ||
The O2-TPO profiles of Co/CeO2 are shown in Fig. 3A. Before activity measurement, the Co/CeO2–O catalyst should be activated in the reacting mixture (H2/N2 = 3.0 v/v), thus a H2-TPR study was also performed (Fig. 3B). The sample obtained by heat treatment of Co/CeO2 in oxygen showed higher intensity of the low-temperature H2O peak than the catalyst with hydrogen treatment (Fig. 2A). A new strong peak centered at 270 °C can be found in the spectra of m/z = 17 and 18 for Co/CeO2 with oxygen treatment. These results suggested that most hydroxyls desorbed as H2O form when Co/CeO2 was treated in oxygen. The weak peaks can be observed in the spectra with m/z = 15 or 30, indicating that oxygen treatment of Co/CeO2 produced few NH radicals. Furthermore, the increase in the signal corresponding to m/z = 2 was found for the sample treated in oxygen, suggesting that a little amount of hydrogen formed during the heat treatment. Co/CeO2 catalyst with oxygen treatment showed similar H2-TPR profiles as Co/CeO2–H pretreated oxygen, suggesting that most Co existed as Co3O4 form.
 | ||
| Fig. 3 (A) O2-TPO profiles of Co/CeO2 and (B) H2-TPR profiles of Co/CeO2 catalyst with oxygen treatment. | ||
3.2 Physicochemical properties of Co catalysts
Fig. 4 displays the XRD patterns of Co/CeO2, Co/CeO2–H and Co/CeO2–O. Only the distinct peaks corresponding to the fluorite structure of CeO2 (JCPDS 34-0394) can be observed for all samples. No peak of Co-related species on the XRD patterns indicated that Co species were either formed a solid solution in ceria or highly dispersed.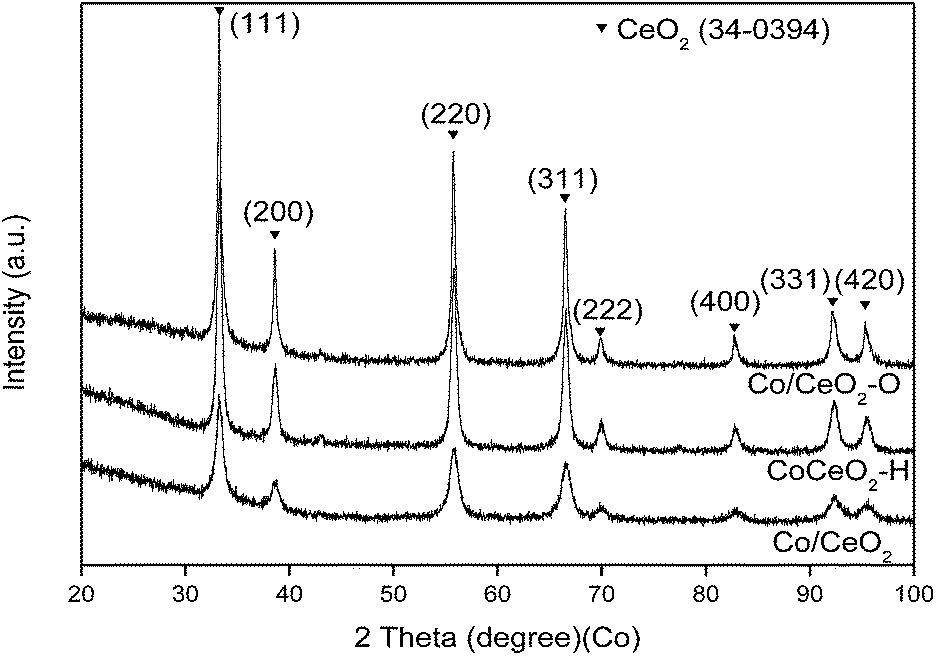 | ||
| Fig. 4 XRD patterns of Co/CeO2, Co/CeO2–H and Co/CeO2–O. | ||
Fig. 5 shows the TEM images of Co catalysts. Most CeO2 crystallites observed in Co/CeO2–H were spheral or polyhedral, with sizes ranging from 5 to 27 nm. The average particle size calculated for this sample was 16 nm. Some rod-like CeO2 crystallites can be found for the samples treated in oxygen, indicating that some CeO2 crystallites sintered together. Therefore, the number of micropores and the surface area both slightly decreased (Table S2 and Fig. S3†).
 | ||
| Fig. 5 TEM images of the catalysts (a) Co/CeO2–H and (b) Co/CeO2–O. | ||
The lattice fringe was calculated to be 0.31 nm for all samples, which corresponded to the (111) plane of CeO2 shown in XRD patterns (Fig. 4). Co particles with various sizes, ranging from 2 to 6 nm can be observed for all Co catalysts (Fig. 5), suggesting the change in treatment gases did not affect the sizes of Co particles.
Fig. 6 shows the XPS spectra of Ce 3d, O 1s and Co 3p of Co catalysts with hydrogen reduction at 450 °C for 2 h. The Ce 3d region of sample is composed of five doublets: u′′′ (916.7 eV), v′′′ (898.3 eV), u′′ (907.6 eV), v′′ (889.0 eV), u (900.8 eV), v (882.3 eV), u0 (898.8 eV), v0 (880.2 eV), u′ (902.8 eV) and v′ (884.4 eV).20,35–37 After deconvolution of the spectra, the degree of ceria reduction was calculated from the ratio between the sum of the peak area of u0, v0, u′ and v′ peaks and the sum of the area of all the peaks (Table 1). It was found that the hydrogen treatment catalyst at 600 °C for 4 h showed the lowest amount of Ce3+ concentration. The Ce3+ concentration for Co/CeO2 or Co/CeO2–O catalysts with hydrogen reduction was ca. 20%, which is in good accordance with the results of Laachir et al.35 and Soykal et al.38 They all reported that a stabilized cerium oxide with about 20% Ce3+ would be obtained when CeO2 was reduced in hydrogen at 400–623 °C in a TPR analysis.
 | ||
| Fig. 6 (A) Ce 3d, (B) O 1s and (C) Co 3p XPS spectra of the catalysts reduced in hydrogen gas. | ||
| Sample | Ce3+ con. | O 1s (eV) | ||
|---|---|---|---|---|
| CeO2 | 20% | 529.1(39) | 531.1(53) | 533.5(8) |
| Co/CeO2–H | 12% | 529.3(60) | 531.3(36) | 533.5(4) |
| Co/CeO2–O | 19% | 529.3(27) | 531.2(59) | 533.5(14) |
The O 1s spectra can be deconvolved into three peaks centered at 529.3, 531.2 and 533.5 eV (Fig. 6B). According to some previous literatures,37,39–41 these three peaks can be assigned to lattice oxygen of CeO2, hydroxide species and hydroxyls bound to Ce3+-related surface defects, respectively. Other studies showed that O 1s component at 533.5 eV should be assigned to only physisorbed H2O (ref. 42 and 43) or both chemisorbed and physisorbed H2O.30,44,45 From Fig. 6B and Table 1, it is clear that the hydrogen treated sample contained a larger amount of lattice oxygen of CeO2 than the catalysts obtained by oxygen treatment, and more hydroxide species were found on Co/CeO2–O. However, it is also difficult to state clearly that the hydroxide species arise as a result of the dissociative adsorption of H2O in air before XPS study or the interaction of hydrogen with CeO2 during the sample reduction from XPS data. Furthermore, the Co 3p binding energy of Co/CeO2–H was similar to that of Co/CeO2–O, indicating that the difference in treatment atmosphere of Co catalyst did not affect the Co 3p binding energy.
3.3 H2-TPD study and pulse chemisorption measurement
Fig. 7 presents the H2-TPD profiles with different m/z values for CeO2 and Co catalysts. The distinct desorption peaks can be observed in the spectra with m/z = 16, 17 and 18. Based on the signal intensity ratios of O/H2O and HO/H2O obtained from H2-TPD with the corresponding values from the dissociative ionization of water (Fig. S2†), one can conclude that some O and OH desorbed from CeO2 and Co/CeO2–O during the heat process. A broad desorption peak at above 335 °C can be observed in the profile of m/z = 17 for the catalyst obtained by oxygen treatment. Furthermore, the signal of m/z = 2 slightly increased with the increase of temperature, but not distinct H2 desorption peak was found for all samples. These results indicated that most hydrogen in the samples was stored as hydroxyl, which agrees with the results of Laachir et al.35 | ||
| Fig. 7 TPD profiles with different m/z values for (A) CeO2, (B) Co/CeO2–H and (C) Co/CeO2–O. | ||
Fig. 8 showed the pulse chemisorption results for CeO2, Co/CeO2–H and Co/CeO2–O. The average variations in the mass signals during the pulse chemisorption process were also calculated in Table 2. The main species detected in the pulse chemisorption were observed at m/z = 2, 15, 16, 17, 18 and 32, which were corresponded to H2, NH, NH (or O), NH3 (or OH), H2O and O2, respectively. As can be seen, when 10% H2/Ar mix gas was introduced into CeO2 with Ar as carrier gas, the increase in the signals at m/z = 2, 17 and 18 can be observed. On the other hand, the signal intensities of m/z = 16 and 32 decreased greatly. It is difficult to determine that a O2 molecule came from the impurity oxygen in gas or the oxygen in ceria. However, these results clearly indicated that the introduction of hydrogen to CeO2 would react with oxygen to form OH or H2O. When the carrier gas was turned into nitrogen, the intensity of the mass signal at m/z = 18 decreased, but other signals (m/z = 15, 16, 17 and 32) all increased significantly. Therefore, it can be concluded that the pulsed hydrogen preferred to exist as NHx or HO rather than H2O for the sample flushed with nitrogen flow. However, significant change in the peak intensities with m/z = 16, 17, 18 and 32 can be found if hydrogen was introduced by pulse injection. Obviously, the presence of nitrogen promoted the generation of HxO by the reaction of hydrogen with oxygen.
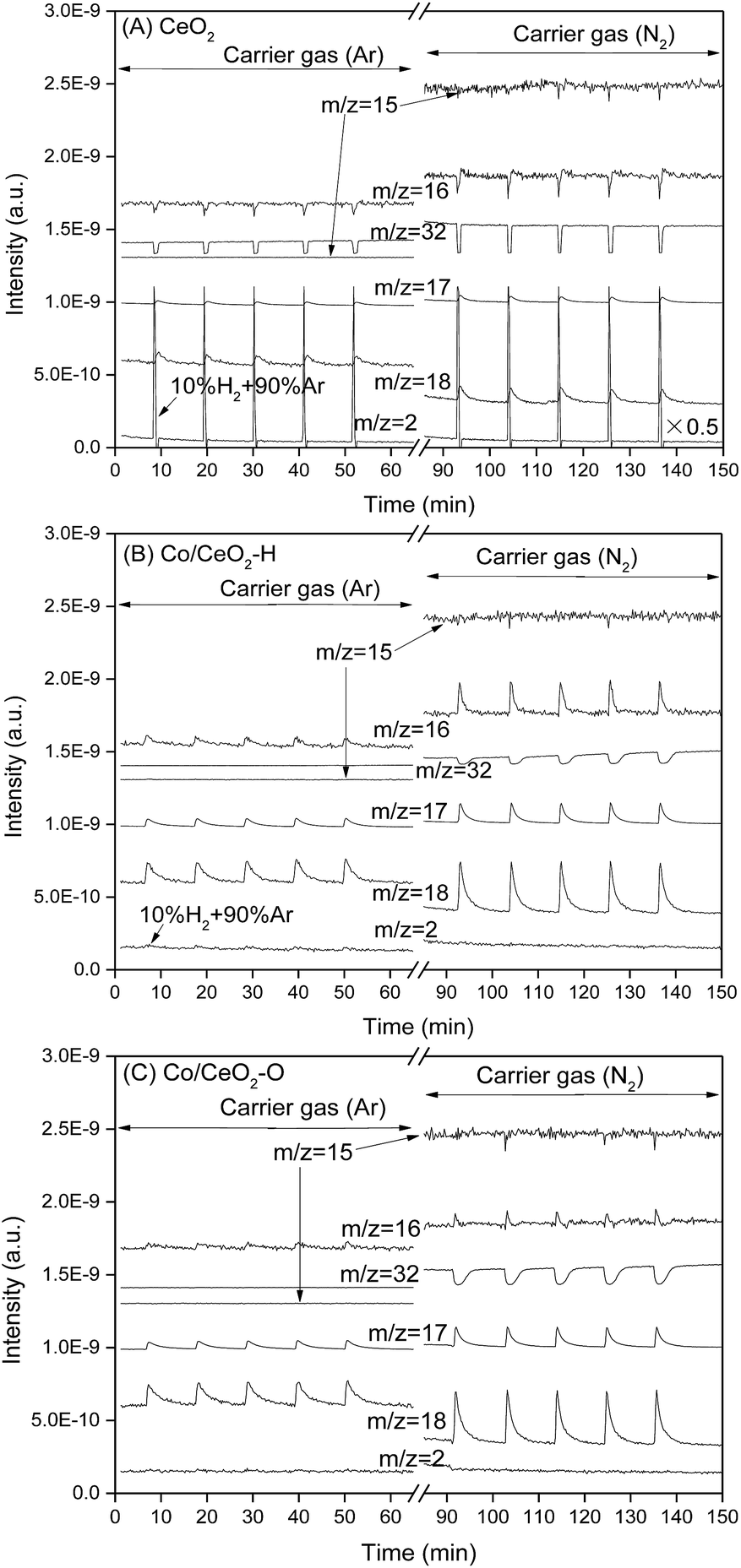 | ||
| Fig. 8 H2/Ar pulse chemisorption results over (A) CeO2, (B) Co/CeO2–H and (C) Co/CeO2–O. | ||
| Sample | Variations of mass signala, a.u. (× 10−10) | |||||
|---|---|---|---|---|---|---|
| Ar carrier gas | N2 carrier gas | |||||
| 16 | 17 | 18 | 16 | 17 | 18 | |
| a The variations of mass signal denotes that the value of curve in one pulse (10% H2–Ar gas) subtracts the original values of mass signal. | ||||||
| CeO2 | −0.8 | 0.22 | 0.70 | −7.5 | 0.37 | 0.98 |
| Co/CeO2–H | 0.5 | 0.54 | 1.5 | 2.2 | 1.4 | 3.4 |
| Co/CeO2–O | 0.33 | 0.53 | 1.5 | 0.76 | 1.4 | 3.6 |
Unlike that of CeO2, regardless of the kind of carrier gas used, no variations in mass signal of m/z = 2 can be observed for Co catalysts. This result suggested that the samples with Co metal adsorbed all hydrogen in the 10 vol% H2/Ar gas during the pulse chemisorption process. However, the addition of hydrogen greatly increased the peak intensities of the signals at m/z = 16, 17, 18 and 32 for all Co catalysts. The signal intensity with m/z = 16 increased if hydrogen was injected into Co catalysts, suggesting that the addition of hydrogen promoted the generation of O or NH2 (carrier gas, nitrogen). The generated atomic oxygen may desorb directly or react with each other to form oxygen. When pure hydrogen was introduced into Co/CeO2–H, more atomic oxygen would be produced and recombine, thus the mass signals corresponding to O2 molecule (m/z = 32) can be observed (Fig. S4†).
The addition of H2 significantly increased the peak intensities with m/z = 16, 17, 18 and 32 for Co catalysts when Ar was replaced by N2 as carrier gas. The ratios among the mass signal variations of m/z = 16, 17 and 18 were −1.1:0.31:1.0, 0.33:0.33:1.0 and 0.22:0.33:1.0 for CeO2, Co/CeO2–H and Co/CeO2–O during the pulse chemisorption process of hydrogen using Ar as carrier gas. These corresponding data were changed to −7.6:0.38:1, 0.64:0.41:1.0 and 0.21:0.38:1.0 with N2 carrier gas. The signals at m/z = 17 may be due to the dissociative ionization of H2O, the produced OH radicals and NH3. The signals at m/z = 16 were correlation with O radicals and NH2 radicals. It is difficult to evaluate the exact amount of different species produced during the pulse chemisorption process. However, one can conclude that Co/CeO2–H produced much more NH2 from N2 carrier gas and the pulsed hydrogen than CeO2 or Co/CeO2–O based on the difference in the ratio of the signal variations. The weak signals at m/z = 30, which correspond to N2H2 can be observed if pure hydrogen was introduced into Co/CeO2–H (Fig. S4†). These results suggested that NH radicals would be produced during the pulse chemisorption process.
3.4 Ammonia synthesis activity
Fig. 9a presents the ammonia synthesis rates for Co catalysts with different treatment atmosphere at 10 MPa and 2.4 × 105 cm3 gcat−1 h−1. The catalytic activities of Co/CeO2–H were unaffected by changing the pellet sizes, the dilution ratio of catalyst and quartz sand or the space velocities (Fig. S6†). This result confirmed the absence of transport artifacts during the activity measurement process. The activities of Co catalysts increased with increase of the reaction temperatures in the range of 400 to 430 °C. At the temperature range studied, Co catalyst obtained from the direct hydrogen reduction showed much higher ammonia synthesis activities than the sample prepared by oxygen treatment. Kinetic studies showed that activation energies for Co/CeO2–H and Co/CeO2–O were 51 and 56 kJ mol−1, respectively. These data both were much lower than the values reported by previous studies. Hagen et al.12,13 and Rarog-Pilecka et al.10,14 all found that the apparent energies of activation were higher than 90 kJ mol−1 for carbon-supported cobalt catalysts. These results can be expected because the activation energies of ammonia synthesis were depended significantly on the type of support materials.46,47 Kitano et al.47 even found the apparent activation energies varied from 40 to 120 kJ mol−1 for Ru catalysts with different support materials, such as C12A7:e−, AC, MgO, CaO or Al2O3.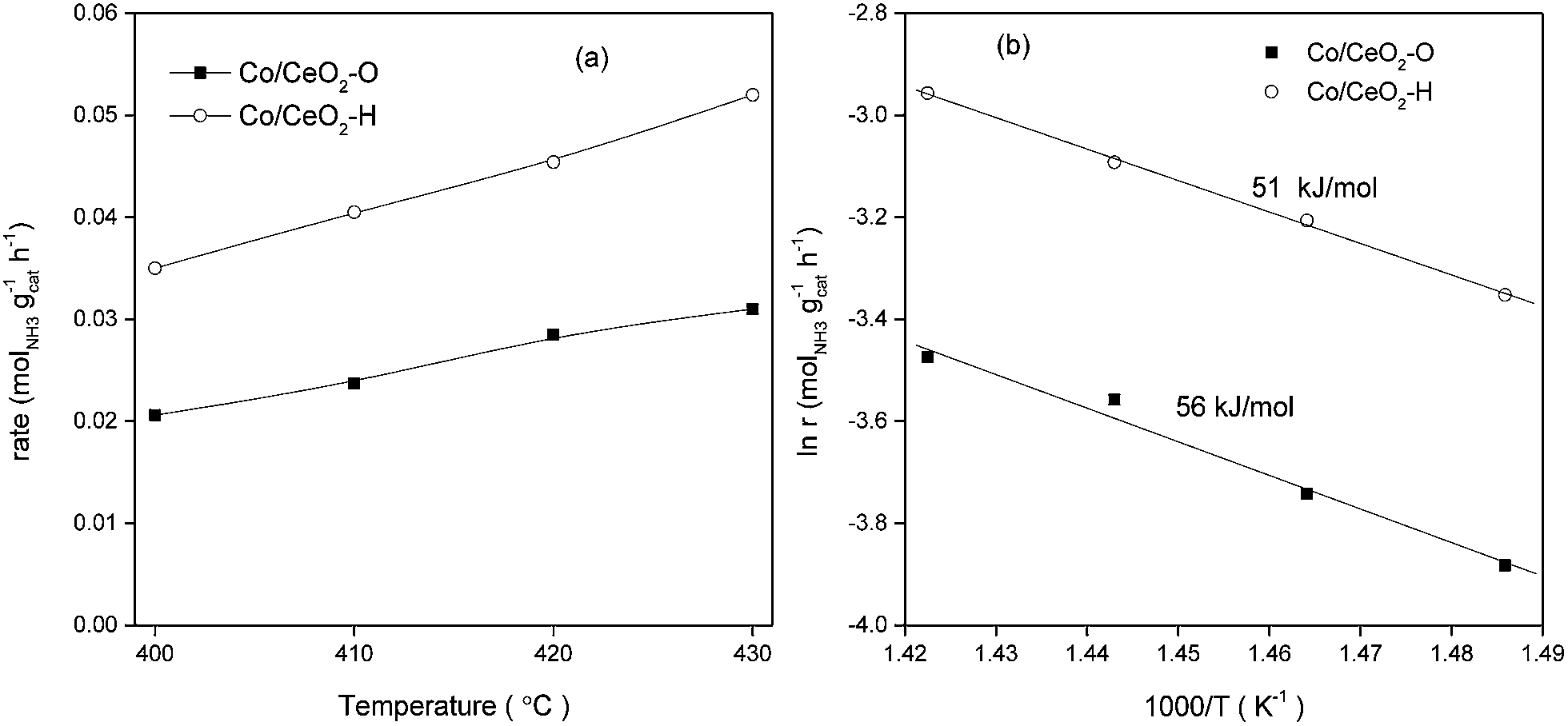 | ||
| Fig. 9 (a) Ammonia synthesis rates and (b) Arrhenius plots of the reaction rate on various Co catalysts. | ||
4. Discussion
4.1 The heat treatment process
The Co/CeO2 precipitate contained a large amount of cerium hydroxide and cobalt hydroxide. Some hydroxides may convert to oxides during the drying process, and some nitrate radicals remain on the sample. The mass data combined with XPS have provided important information about the produced species and the change of the structure of Co catalysts with heat treatment. Chen et al.30 proposed that surface hydroxyls in CeO2 could release H2O by the associative desorption of OH radicals or the generation of hydrogen. Similar reaction pathways may exist during the heat treatment process of Co/CeO2. Furthermore, Lin et al.32 found some CoOx linked to the Co–Ce interaction in cobalt/ceria–zirconia catalysts could be reduced at 50–150 °C. The Co–Ce interaction may exist in our Co/CeO2 catalyst, therefore, a little amount of cobalt compound would convert to Co metal at low temperature (50–200 °C) during hydrogen reduction. Hydrogen molecule can dissociate into atomic hydrogen on Co metal, and atomic hydrogen may react with surface hydroxyls or nitrate radicals to form a chemisorbed H2O or NHx according to reaction (1) or (2), respectively.| OH + H → H2O | (1) |
| NOx + H → NHx + H2O | (2) |
The generated H2O can easily be desorbed from the catalysts. Therefore, The H2O peak at a low temperature can be observed during the hydrogen reduction process of various catalysts, including of the Co/CeO2, Co/CeO2–O or the Co/CeO2–H catalyst with preadsorbed oxygen. Moreover, the required temperatures for the hydrogen reduction of cobalt compound in Co/CeO2 were lower than those in Co/CeO2 or Co/CeO2–H with oxygen treatment. Similar results of adsorption and reaction of atomic H with hydroxyls or oxygen have also been observed for other different catalysts with oxide, such as CeOx/Cu(111) catalyst30,48 and Pt/oxide catalyst.49
On the other hand, some atomic hydrogen can adsorb on surface O atoms to form hydroxyls, which have been confirmed not only by theoretical calculation,50,51 but also by experimental result.30,35 Furthermore, it was reported that some hydroxyls on CeO2 were found to be stable up to ca. 300 °C.30,42,45 Consequently, the H2O peak at above 300 °C can be observed in the H2-TPR profiles of CeO2 or Co catalyst. These results clearly suggested that the presence of atomic hydrogen inhibited the transformation of cerium hydroxide to ceria.
The TPR profiles of Co/CeO2–H preadsorbed oxygen were similar as these of Co/CeO2 with oxygen treatment, which indicated that all hydroxyls had converted to oxides when Co/CeO2 was heated in oxygen. The association of hydroxyls to release H2O (Reaction (3)) had been observed on CeO230 or iron–nickel hydroxide–platinum nanoparticles.29
| 2OH → O + H2O | (3) |
The produced CoO may further react with oxygen to form Co3O4:
| CoO + O2 → Co3O4 | (4) |
The appearance of a little amount of hydrogen indicated that the recombinative desorption of hydroxyls occurred according to reaction (7):30
| 2OH → 2O + H2 | (5) |
The different reaction pathways of cerium hydroxide may be responsible for the differences in the amount of lattice oxygen, hydroxyls and Ce3+ concentration in various Co catalysts. The hydroxyls in Co/CeO2–H mainly desorbed by the interaction with atomic hydrogen to form H2O, which did not change the oxidation state of CeO2.30 On the other hand, most hydroxyls converted into oxides during the oxygen treatment process. When Co/CeO2–O was reduced in hydrogen, some atomic hydrogen will adsorb on ceria to form surface hydroxyls. The formation of surface hydroxyls is accompanied by the reduction of Ce4+ to Ce3+.30,50,51 Consequently, a larger amount of Ce3+ concentration can be found for Co catalysts with oxygen treatment. It is reported by Chen et al.30 that surface hydroxyls on stoichiometric CeO2 can associate with each other to release H2O at the low temperatures, whereas some hydroxyls on partially reduced CeO2 were more stable.30,42,45 Herein all catalysts were reduced in hydrogen at 500 °C for 2 h and then cooled down to room temperature. During this treatment process, more hydroxyls existed in the sample with oxygen treatment, which has been confirmed by XPS results.
4.2 The reaction pathways
After decades' study, a general agreement on the mechanism of the catalytic reaction in the Haber–Bosch process was reached:52| H2 ↔ 2H | (6) |
| N2 ↔ 2N | (7) |
| H + N ↔ NH | (8) |
| NH + H ↔ NH2 | (9) |
| NH2 + H ↔ NH3 | (10) |
Nitrogen dissociation is the rate-determining step for ammonia synthesis.52–54 However, the ammonia synthesis rate was strongly affected by hydrogen adsorption because hydrogen and nitrogen competitively adsorbed on same active sites of Ru catalysts.17–19,22,55–57 A single pulse of known volume (0.3541 mL) of hydrogen and nitrogen was introduced into Co/CeO2–H with Ar as carrier gas. The MS signal response for pulse scheme H2 → N2 → H2 is shown in Fig. S7.† The concurrence of nitrogen and hydrogen during nitrogen pulse and the subsequent H2 pulse experiment suggested hydrogen and nitrogen adsorbed on the same sites for Co/CeO2 catalyst. In such a case, the reaction pathways of ammonia synthesis over Co catalyst can be proposed (Scheme 1): (1) nitrogen or hydrogen molecule adsorbed and dissociated into atomic hydrogen or nitrogen on Co surface (Reaction 6 and 7). No H2 peak was observed in the H2-TPD profiles of Co catalysts (Fig. 7), which suggested that hydrogen was stored as hydroxyl in ceria. Meantime, no adsorbed nitrogen can be detected in the N2-TPD study (Fig. S5†) because Co metal bound N atom weakly.58 (2) The presence of hydroxyls on Co catalyst would react with atomic hydrogen to release H2O (Reaction 1). Hydroxyls can also associate each other to form H2O and oxygen (Reaction 3) or hydrogen (Reaction 5). (3) The dissociation reactions of hydrogen and nitrogen are reversible. Atomic hydrogen undergoes two different reaction pathways: one is to produce NHx by reacting with atomic nitrogen (Reaction 8–10), and the other one is to release H2O via the generation of hydroxyls. The presence of nitrogen increased the number of active sites available for hydrogen dissociation because some hydrogen atoms on active sites can be removed by reacting with atomic nitrogen. Consequently, more HxO was produced using N2 as carrier gas for H2 pulse chemisorption. On the other hand, the presence of atomic hydrogen strongly adsorbed on active sites may inhibit the synthesis of ammonia by decreasing the active site available for nitrogen dissociation.
 | ||
| Scheme 1 Proposed reaction pathways of ammonia synthesis over Co catalyst. | ||
As shown in above results, a larger number of stable hydroxyls existed in the sample with oxygen treatment. Consequently, the redundant amount of atomic hydrogen cannot be removed immediately, and then the number of active sites available for nitrogen dissociation and ammonia synthesis decreased significantly. On the other hand, the sample obtained from the direct hydrogen reduction showed a lower amount of Ce3+ concentration, and some hydrogen atoms can be easily removed by a combination reaction with hydroxyls. Therefore, Co/CeO2–H catalyst showed higher activity for ammonia synthesis than Co/CeO2–O. Our findings have confirmed that the presence of hydroxyls can affect the ammonia synthesis activity of Co catalyst supported on ceria. The recent result of Zugic et al.28 also showed that Na provided the OH species to the metal atoms for platinum supported on carbon nanotubes. These results also remind us that the presence of alkali promoters may not just affect the electronic state of metal surface or the chemisorptive properties of reactant gases,59 but also affect the amount of surface hydroxyl groups for the supported metal catalyst. The potassium promoter effect and the deactivation mechanism of K promoted Ru/carbon catalyst for ammonia synthesis has to be checked by considering the influence of surface hydroxyls. In conclusion, this result deepens the fundamental understanding of the effect of surface hydroxyls on the catalytic activity for ceria-supported metal catalyst, and can also be used to design highly efficient catalysts for ammonia synthesis, hydrogenation or other reactions involving hydrogen.
5. Conclusions
Co/CeO2 precipitate obtained from Ce(NO3)3 and Co(NO3)3 was treated in hydrogen or oxygen. The results showed the pretreatment atmosphere affected the dehydroxylation pathways of hydroxyl. Surface hydroxyls in CeO2 can release H2O not only by the associative desorption of OH radicals or the generation of hydrogen, but also by the recombination with atomic hydrogen. Most hydroxyls were converted to oxides during the oxygen treatment process of Co/CeO2. Further reduction in hydrogen led to the formation of the stable surface hydroxyls with the reduction of Ce4+ to Ce3+. On the other hand, the formation of H2O by reacting hydroxyls with atomic hydrogen did not change the oxidation state of CeO2 for the sample obtained by hydrogen reduction.H2 temperature programmed desorption and pulse chemisorption measurements indicated that no nitrogen adsorbed on Co catalyst, and hydrogen was stored as hydroxyl. Adsorbed hydrogen may be used to produce NHx by reacting with atomic nitrogen or release H2O by generating hydroxyls during the ammonia synthesis process. Hydrogen and nitrogen competitively adsorb on same active sites for Co catalyst. The presence of nitrogen promoted the generation of HxO because the redundant amount of atomic hydrogen can be removed by forming NHx. Co catalyst with oxygen treatment contained a larger number of stable hydroxyls. Some redundant atomic hydrogen cannot be removed immediately, and thus ammonia synthesis was inhibited.
An important finding of the study is that the pretreatment atmosphere of ceria-supported Co catalyst affected the amount of lattice oxygen, hydroxyls and Ce3+ concentration. The presence of atomic hydrogen during the treatment process increased the amount of lattice oxygen and Ce4+ concentration. Consequently, the catalysts treated in pretreatment atmosphere showed different catalytic activities in ammonia synthesis reaction.
Acknowledgements
The authors would like to appreciate the financial support of the National Natural Science Foundation of China (21203028) and School Foundation for Young Scientists of Fuzhou University (XRC-1325).Notes and references
- D. Brown, T. Edmonds, R. Joyner, J. McCarroll and S. Tennison, Catal. Lett., 2014, 144, 545–552 CrossRef CAS PubMed.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS PubMed.
- K. Aika and K. Tamaru, in Ammonia: catalysis and manufacture, ed. A. Nielsen, Springer-Verlag, Berlin, 1995, pp. 103–148 Search PubMed.
- R. Schlögl, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 2501–2575 Search PubMed.
- F. Hayashi, M. Kitano, T. Yokoyama, M. Hara and H. Hosono, ChemCatChem, 2014, 6, 1317–1323 CAS.
- B. Lin and K. Wei, Catal. Commun., 2013, 41, 110–114 CrossRef CAS PubMed.
- B. Lin, K. Wei, J. Lin and J. Ni, Catal. Commun., 2013, 39, 14–19 CrossRef CAS PubMed.
- B. Lin, R. Wang, J. Lin, J. Ni and K. Wei, Catal. Commun., 2011, 12, 1452–1457 CrossRef CAS PubMed.
- X. Yu, B. Lin, B. Gong, J. Lin, R. Wang and K. Wei, Catal. Lett., 2008, 124, 168–173 CrossRef CAS PubMed.
- W. Rarog-Pilecka, E. Miskiewicz, M. Matyszek, Z. Kaszkur, L. Kepinski and Z. Kowalczyk, J. Catal., 2006, 237, 207–210 CrossRef CAS PubMed.
- M. Karolewska, E. Truszkiewicz, M. Wscisel, B. Mierzwa, L. Kepinski and W. Rarog-Pilecka, J. Catal., 2013, 303, 130–134 CrossRef CAS PubMed.
- S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen, K. Stahl and I. Chorkendorff, Chem. Commun., 2002, 1206–1207 RSC.
- S. Hagen, R. Barfod, R. Fehrmann, C. J. H. Jacobsen, H. T. Teunissen and I. Chorkendorff, J. Catal., 2003, 214, 327–335 CrossRef CAS.
- W. Rarog-Pilecka, E. Miskiewicz, L. Kepinski, Z. Kaszkur, K. Kielar and Z. Kowalczyk, J. Catal., 2007, 249, 24–33 CrossRef CAS PubMed.
- W. Raróg-Pilecka, M. Karolewska, E. Truszkiewicz, E. Iwanek and B. Mierzwa, Catal. Lett., 2011, 141, 678–684 CrossRef.
- M. Karolewska, E. Truszkiewicz, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Appl. Catal., A, 2012, 445–446, 280–286 CrossRef CAS PubMed.
- Y. Niwa and K.-i. Aika, J. Catal., 1996, 162, 138–142 CrossRef CAS.
- Y. Izumi, Y. Iwata and K.-i. Aika, J. Phys. Chem., 1996, 100, 9421–9428 CrossRef CAS.
- Y. Kadowaki and K.-i. Aika, J. Catal., 1996, 161, 178–185 CrossRef CAS.
- X. Luo, R. Wang, J. Ni, J. Lin, B. Lin, X. Xu and K. Wei, Catal. Lett., 2009, 133, 382–387 CrossRef CAS.
- X. Wang, J. Ni, B. Lin, R. Wang, J. Lin and K. Wei, Catal. Commun., 2010, 12, 251–254 CrossRef CAS PubMed.
- B. Lin, R. Wang, J. Lin, J. Ni and K. Wei, Catal. Commun., 2011, 12, 553–558 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- C. Zhang, F. Liu, Y. Zhai, H. Ariga, N. Yi, Y. Liu, K. Asakura, M. Flytzani-Stephanopoulos and H. He, Angew. Chem., Int. Ed., 2012, 51, 9628–9632 CrossRef CAS PubMed.
- M. Yang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2013, 135, 3768–3771 CrossRef CAS PubMed.
- B. Zugic, D. C. Bell and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2014, 144, 243–251 CrossRef CAS PubMed.
- B. Zugic, S. Zhang, D. C. Bell, F. Tao and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2014, 136, 3238–3245 CrossRef CAS PubMed.
- G. Chen, Y. Zhao, G. Fu, P. N. Duchesne, L. Gu, Y. Zheng, X. Weng, M. Chen, P. Zhang, C.-W. Pao, J.-F. Lee and N. Zheng, Science, 2014, 344, 495–499 CrossRef CAS PubMed.
- B. Chen, Y. Ma, L. Ding, L. Xu, Z. Wu, Q. Yuan and W. Huang, J. Phys. Chem. C, 2013, 117, 5800–5810 CAS.
- M. Kruk, V. Antochshuk, M. Jaroniec and A. Sayari, J. Phys. Chem. B, 1999, 103, 10670–10678 CrossRef CAS.
- S. S. Y. Lin, D. H. Kim, M. H. Engelhard and S. Y. Ha, J. Catal., 2010, 273, 229–235 CrossRef CAS PubMed.
- A. R. de la Osa, A. De Lucas, A. Romero, J. L. Valverde and P. Sánchez, Catal. Today, 2011, 176, 298–302 CrossRef CAS PubMed.
- X. Chen, S. A. C. Carabineiro, S. S. T. Bastos, P. B. Tavares, J. J. M. Órfão, M. F. R. Pereira and J. L. Figueiredo, Appl. Catal., A, 2014, 472, 101–112 CrossRef CAS PubMed.
- A. Laachir, V. Perrichon, A. Badri, J. Lamotte, E. Catherine, J. C. Lavalley, J. El Fallah, L. Hilaire, F. Le Normand, E. Quemere, G. N. Sauvion and O. Touret, J. Chem. Soc., Faraday Trans., 1991, 87, 1601–1609 RSC.
- M. Romeo, K. Bak, J. El Fallah, F. Le Normand and L. Hilaire, Surf. Interface Anal., 1993, 20, 508–512 CrossRef CAS PubMed.
- A. Pfau and K. D. Schierbaum, Surf. Sci., 1994, 321, 71–80 CrossRef CAS.
- I. I. Soykal, H. Sohn, D. Singh, J. T. Miller and U. S. Ozkan, ACS Catal., 2014, 4, 585–592 CrossRef CAS.
- G. S. Herman, Y. J. Kim, S. A. Chambers and C. H. F. Peden, Langmuir, 1999, 15, 3993–3997 CrossRef CAS.
- A. M. Venezia, G. Pantaleo, A. Longo, G. Di Carlo, M. P. Casaletto, F. L. Liotta and G. Deganello, J. Phys. Chem. B, 2005, 109, 2821–2827 CrossRef CAS PubMed.
- L. Ilieva, G. Pantaleo, I. Ivanov, A. M. Venezia and D. Andreeva, Appl. Catal., B, 2006, 65, 101–109 CrossRef CAS PubMed.
- L. Kundakovic, D. R. Mullins and S. H. Overbury, Surf. Sci., 2000, 457, 51–62 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola, J. Evans, M. Estrella, L. Barrio, M. Pérez, J. Hrbek and J. A. Rodriguez, J. Catal., 2010, 271, 392–400 CrossRef CAS PubMed.
- V. Matolín, I. Matolínová, F. Dvořák, V. Johánek, J. Mysliveček, K. C. Prince, T. Skála, O. Stetsovych, N. Tsud, M. Václavů and B. Šmíd, Catal. Today, 2012, 181, 124–132 CrossRef PubMed.
- D. R. Mullins, P. M. Albrecht, T.-L. Chen, F. C. Calaza, M. D. Biegalski, H. M. Christen and S. H. Overbury, J. Phys. Chem. C, 2012, 116, 19419–19428 CAS.
- B. Lin, K. Wei, J. Ni and J. Lin, ChemCatChem, 2013, 5, 1941–1947 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- J. A. Rodriguez, J. Graciani, J. Evans, J. B. Park, F. Yang, D. Stacchiola, S. D. Senanayake, S. Ma, M. Pérez, P. Liu, J. F. Sanz and J. Hrbek, Angew. Chem., Int. Ed., 2009, 48, 8047–8050 CrossRef CAS PubMed.
- L. Xu, Y. Ma, Y. Zhang, Z. Jiang and W. Huang, J. Am. Chem. Soc., 2009, 131, 16366–16367 CrossRef CAS PubMed.
- G. Vicario, G. Balducci, S. Fabris, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2006, 110, 19380–19385 CrossRef CAS PubMed.
- M. B. Watkins, A. S. Foster and A. L. Shluger, J. Phys. Chem. C, 2007, 111, 15337–15341 CAS.
- G. Ertl, in Encyclopedia of Catalysis, John Wiley & Sons, Inc., 2002, pp. 1–24 Search PubMed.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Tornqvist and J. K. Norskov, Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Norskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- S. E. Siporin and R. J. Davis, J. Catal., 2004, 222, 315–322 CrossRef CAS PubMed.
- S. E. Siporin and R. J. Davis, J. Catal., 2004, 225, 359–368 CrossRef CAS PubMed.
- B. Lin, R. Wang, X. Yu, J. Lin, F. Xie and K. Wei, Catal. Lett., 2008, 124, 178–184 CrossRef CAS.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS.
- B. Lin, K. Wei, X. Ma, J. Lin and J. Ni, Catal. Sci. Technol., 2013, 3, 1367–1374 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The main species detected by mass spectrometry, surface area and pore size of Co catalysts, hydrogen pulse chemisorption results with pure hydrogen for Co/CeO2–H catalyst and N2 temperature programmed desorption profiles of Co/CeO2–H catalyst. See DOI: 10.1039/c4ra06175f |
| This journal is © The Royal Society of Chemistry 2014 |