
Modification of a glassy carbon electrode with a bilayer of multiwalled carbon nanotube/benzene disulfonate-doped polypyrrole: application to sensitive voltammetric determination of olanzapine†
Saeed Shahrokhian*ab,
Mahnaz Azimzadeha and
Pouya Hosseinia
aDepartment of Chemistry, Sharif University of Technology, Tehran 11155-3516, Iran. E-mail: shahrokhian@sharif.edu; Fax: +98-21-66012983; Tel: +98-21-66005718
bInstitute for Nanoscience and Technology, Sharif University of Technology, Tehran, Iran
First published on 19th August 2014
Abstract
A glassy carbon electrode was coated with a thin layer of multi-walled carbon nanotubes and subsequently was electro-polymerized with polypyrrole. The prepared electrode was used for the voltammetric determination of olanzapine (OLZ). The peak current of OLZ increases remarkably on the surface of the modified electrode. This is due to the large increase in the microscopic area of the electrode surface along with the strong adsorption of OLZ on the surface of the electrode. The film modifier was characterized by scanning electron microscopy, atomic force microscopy and cyclic voltammetry techniques. Experimental variables, such as deposited amount of nanotube suspension, pH of the supporting electrolyte, potential and time of the accumulation and the number of electro-polymerization cycles were optimized by monitoring the LSV responses toward OLZ. In the optimal conditions, the electrode showed a linear response to the concentration of OLZ within a 0.02–2.00 μM range with a detection limit of 6.00 nM. The electrode shows advantages such as simple preparation, high stability and uniformity in the composite film and high sensitivity in response to the OLZ. The modified electrode has been applied for accurate determination of trace amounts of OLZ in pharmaceutical and clinical preparations.
1. Introduction
Olanzapine (OLZ, 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno(2,3-b)(1,5) benzodiazepine, Scheme 1) is effective in the treatment of schizophrenia and bipolar disorders due to its affinity to serotonin 5-HT2A receptor, dopamine D1, D2, D4, serotonin 5-HT2c, 5-HT6, 5-HT7, H1, α1-adrenergic and muscarinic receptors, and its blocking of them in the brain and preventing their excessive activity.1–7 In the other words, OLZ controls these receptors to amend behavior, mood and thinking.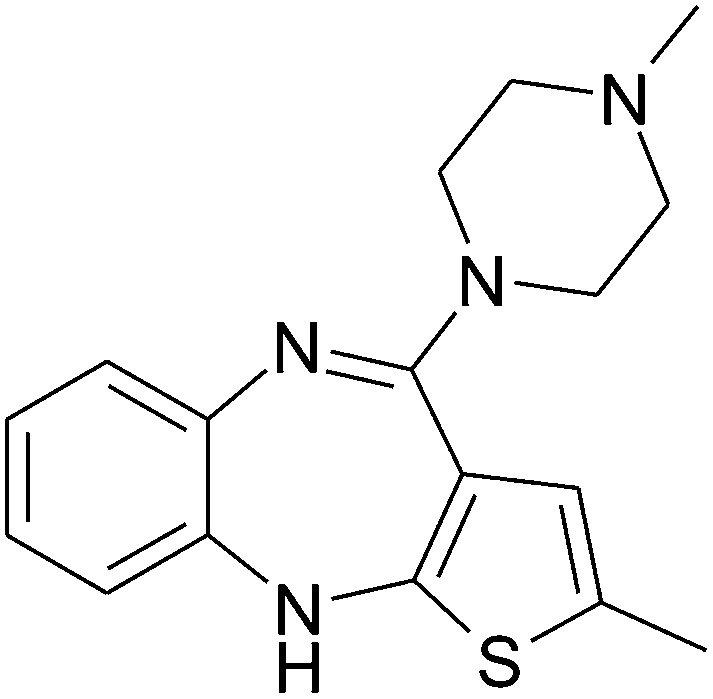 | ||
| Scheme 1 Molecular structure of olanzapine. | ||
Several methods have been used for analysis of OLZ in biological fluids, including spectrophotometric methods,8–10 gas chromatography, high performance liquid chromatography, ultra-performance liquid chromatography and liquid chromatography/tandem mass spectrometry.11–17 However, these methods are generally expensive, time consuming and need sensitive detectors and complicated sample preparation procedures. In contrast, electrochemical methods including cyclic voltammetry, linear sweep voltammetry, differential pulse voltammetry and square wave voltammetry have been shown to be simple, accurate and inexpensive for the analysis of various drug species such as OLZ.18–23
Carbon nanotubes (CNTs) were introduced by Iijima in 1991.24 CNTs due to their excellent mechanical, thermal and chemical stability, high electrical conductivity and high surface area show significant advantages in modification of the surface of electrodes.25 In fact, the elevated electrochemical reactivity enhances the kinetics of the electron transfer reactions by reducing the redox overpotential.26 Also CNTs give a chance for fabrication of multi-functioned electrochemical sensors because of their modifiable edges and sidewalls and ease the determination of biomolecules, pharmacological substances and inorganic ions.27–30 Therefore, the use of CNT modified electrodes as electrochemical sensors and biosensors with higher sensitivities and lower limits of detection is interesting.27,31,32
Conducting polymers (CPs) with unique electronic, chemical and biochemical properties have extensively been used in the electrochemical sensors and biosensors.33–37 CPs are poly-conjugated polymers with features of conventional organic polymers and electronic properties like metals.38,39 Polypyrrole (PPY) is one of the most important CPs as an electrode modifier that have received significant attention because of low cost, high electrical conductivity, reversibility between its conducting and insulating states, high stability, facile synthesis, efficient polymerization, simple formation from aqueous solutions and controllable thickness.40–42 An efficient method for the preparation of PPY film is electrochemical polymerization, using an anodic potential in a solution containing pyrrole and an anionic dopant.43 Presence of anionic dopants is critical for the preparation of adherent and conductive PPY film through the electro-polymerization process. They may establish the electrical neutrality of the polymer film affecting the deposition yield, physical characteristics and morphology of the prepared PPY.44 Electro-deposition of PPY in the presence of aromatic sulfonate dopants will improve the electronic conductivity of polymer films.45,46 Aromatic dopants would make the pyrrole rings to be parallel to the electrode surface and, hence, would lead to a higher conductivity.47
The composites of CNTs and CPs show synergistic influences such as good stability, strong adherence, high number of active sites and homogeneity in electrochemical deposition of CPs together with nanoporosity. Also, high surface area of CNT films leads to a great performance, high charge density, electrical conductivity and electrocatalytic activity in sensory applications.35,48–50 In addition, CNTs operate as a backbone for a homogeneous distribution of conducting polymers in the produced composites.51 The electro-polymerization can be considered as an effective method for preparation of CNT and CP composites as a modifier film, via electro-deposition of PPY on the pre-casted CNT film.33,34 Other methods are based on the resolving of the monomer in a suspension of CNTs and co-deposition of the polymer with the CNTs on the electrode surface.33,52,53
OLZ shows some electrochemical activities, but its electrochemical determination at the bare electrodes is not feasible due to slow electron transfer reaction, electrode fouling problems, and a very low sensitivity in relation of current intensity with the OLZ concentration.54,55 To overcome these problems, the development of the modified electrodes is recommended for electrochemical determination of OLZ.
In the present work, a thin layer of multi-walled carbon nanotubes (MWCNTs) is was casted on the surface of a glassy carbon electrode, and successively was coated with the electrochemically polymerized PPY doped with sodium benzene-1,3-disulfonate. The prepared electrode was applied for the sensitive (can we write “accurate determination”?) determination of OLZ in pharmaceutical formulations and plasma, and also it was used for the determination of OLZ in the presence of clonazepam (CLZ). The surface morphology of the modified electrode was investigated by the scanning electron microscopy (SEM) and atomic force microscopy (AFM). In Comparison to the bare GCE, a significant increase was resulted in the oxidation peak current of OLZ. This improvement can be due to high microscopic surface area of the modified electrode and effective accumulation of OLZ on the porous thin film.
2. Experimental
2.1. Materials and reagents
Olanzapine (OLZ, Scheme 1) and clonazepam (CLZ) were prepared from Bakhtar Bioshimi and Tehran darou companies, respectively. Pyrrole, dimethylformamide (DMF) and sodium benzene-1,3-disulfonate (SBDS) were purchased from Merck. MWCNTs (purity > 95%) were obtained from Nanostructured & Amorphous Materials (USA). All other chemicals were of analytical reagent grade and were purchased from Merck. Pyrrole was purified by distillation, and to avoid any degradation was kept in a dark vial in the refrigerator before use. Stock solutions of OLZ and CLZ (100 μM) were freshly prepared every day in an aqueous solution (containing 0.1 M HCl) and DMF, respectively, and kept in dark at 4 °C between the measurements, to avoid any decomposition. All aqueous solutions were prepared with doubly distilled water. Buffer solutions of different pHs were prepared by the addition of 0.20 M sodium hydroxide to 0.10 M phosphate buffer solution (pHs 2.0, 3.0, 6.0, 7.0, 8.0, 9.0 and 10) and 0.10 M acetic acid (pHs 4.0 and 5.0). These solutions were then used as supporting electrolyte in voltammetric determinations. OLZ tablets (15 mg, Bakhtar Bioshimi Co., Tehran, Iran) were purchased from the local pharmacies. Fresh frozen plasma samples were obtained from the Iranian Blood Transfusion Organization.2.2. Apparatus
All electrochemical measurements were made using a Metrohm Computrace Voltammetric Analyzer (model 797 VA) instrument. A three-electrode system, including a glassy carbon working electrode (unmodified or modified), a KCl saturated Ag/AgCl reference electrode and a platinum wire auxiliary electrode was used. Measurements of the pH and preparation of the buffer solutions were done with a digital pH/mV/Ion meter (Metrohm, pH Lab 827).2.3. Preparation of modified electrode
The bare GCE was first polished to mirror-like surface with 0.05 μm alumina slurry on a polishing cloth, followed by rinsing with doubly distilled water and drying at room temperature. An appropriate amount of pure MWCNTs was functionalized under concentrated nitric acid treatment process for 24 h, in order to obtain more edge sites and better dispersion of nanotubes. The MWCNTs suspension was prepared by dispersing 1 mg of the functionalized MWCNTs (CNT–COOH) in 1 mL of DMF solvent under ultrasonic agitation for 30 minutes, giving a black dispersion that is quite stable for 3–4 months. Then a desired volume of the dispersed CNT was taken with a microsyringe and after casting on the electrode surface, the electrode was placed in an oven at 50 °C for 10 min to evaporate the DMF solvent. This electrode is recognized as CNT/GCE. Subsequently, the electro-deposition of the PPY film on the surface of CNT/GCE was carried out in an aqueous solution containing 5 mM pyrrole and 3 mM SBDS as dopant anion by the potential cycling between 0.00 V and +0.75 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1 for a total of 30 cycles. This electrode is mentioned as PPY/CNT/GCE. Then, this electrode was washed with the distilled water, immersed in phosphate buffer solution (pH 7.0) and cycled 10 times between −0.20 and +1.30 V (scan rate 100 mV s−1) in order to overoxidize the PPY film and obtain a reproducible background current. Over-oxidation of PPY film leads to formation of some carbonyl groups in β-position of the pyrrole rings in the PPY film.56 This electrode, denoted as OPPY/CNT/GCE and was used as the modified electrode in the electrochemical investigations of OLZ.3. Results and discussion
3.1. Characterization of the prepared electrodes
SEM was employed to investigate the surface morphology of the CNT/GCE and OPPY/CNT/GCE as shown in Fig. 1A and B, respectively. As can be seen in Fig. 1A, the GCE surface is coated with a homogeneous layer of MWCNTs. It can be concluded from Fig. 1B that electro-polymerization of PPY occurs on MWCNTs as a nanosized backbone, and most of the MWCNTs have been densely covered with a very thin, nanoporous and homogeneous layer of the polymer. | ||
| Fig. 1 SEM images of (A) CNT coated GCE, and (B) sodium benzene-1,3-disulfonate-doped OPPY film on the surface of GCE pre-coated with 4 μL of MWCNTs and AFM images of (C) PPY/MWCNT/GCE and (D) OPPY/MWCNT/GCE. | ||
The over-oxidation of PPY film improves the porosity of the polymer film and creates some negative sites via carboxylate groups formed in its structure.21 In order to demonstrate the effect of the over-oxidation on the porosity of the PPY film, the AFM images were prepared for MWCNT/GCEs, coated with PPY and overoxidized the PPY. As it is shown in Fig. 1C and D, the over-oxidation has created some grooves and pores in the polymer film, leading to enhancement of the microscopic area of the electrode and facilitating the interlayer diffusion of the analyte species. Therefore, by using this modified electrode higher sensitivities would be expected in the electrochemical measurements.
3.2. Electrochemical deposition of PPY film
During the electro polymerization process the current grows by increasing the number of successive scans, which illustrates formation of an efficient and stable polymer film on the electrode surface. In addition, by formation of PPY on CNT/GCE the increase in current is much greater than that on the bare GCE. This is due to a higher surface area created by the coated MWCNTs (Fig. S1†).On the other hand, increasing the number of scans more than 30 cycles led to a lower sensitivity for the electrode response (Fig. S2†). This is because of the increase in the thickness and resistance of the polymer film. Therefore, 30 cycles were selected as the optimum number of scans for the preparation of the PPY film.
3.3. Effect of different dopants on the response of electrode
As mentioned above, the formation of polymer film is influenced by the anion dopant. We have compared the electrochemical responses of the modified electrode with different anion dopants, containing SBDS, phthalic acid and isophthalic acid. As can be seen in Fig. 2A, PPY growth current increases in the sequence of isophthalic acid < phthalic acid < SBDS, which may be related to the stability of dopant anions in the polymer film. It can be stated that the negative charge has more resonance on SBDS compared with phthalate or isophthalate. On the other hand, the negative charge on the phthalate is stabilized by hydrogen bonding between the negative oxygen and acidic hydrogen of the neighbor carboxyl group. Therefore, anion stability increases with the order of isophthalate < phthalate < SBDS. Higher stability can lead to a more efficient presence of dopant anion in the polymerization process. The voltammetric results for 2.00 μM OLZ are presented in Fig. 2B. It can be observed that the electrochemical response of OLZ on the surface of the electrode containing SBDS is larger (∼3 times) than that of the polymer doped with two other dopants. | ||
| Fig. 2 (A) 30th cyclic voltammograms for the PPY polymerization reaction on CNT/GCE in 5.00 mM pyrrole + 3.00 mM sodium benzene-1,3-disulfonate, phthalic acid and isophthalic acid. (B) Cyclic voltammograms of 2.00 μM OLZ at: OPPY/CNT/GCE doped with sodium benzene-1,3-disulfonate, phthalic acid and isophthalic acid in phosphate buffer solution at pH 3.0. Scan rate was 100 mV s−1 and accumulation time was 80 s. | ||
3.4. Electrochemical behavior of OLZ on the surface of the modified electrode
The oxidation behavior of 2.00 μM OLZ in phosphate buffer solution (pH 3.0) on the bare and modified electrodes was investigated by linear sweep voltammetry (LSV) with a scan rate of 100 mV s−1 after an accumulation time of 80 s with an open circuit (Fig. 3). As can be seen, the bare GCE (inset) shows the oxidation peak current of about 0.053 μA for OLZ, which is very low. However, the peak current of OLZ has increased considerably on the surface of the modified electrodes. In Comparison to the bare GCE, the peak current increased by a factor of 9.89 (Ip,a = 0.524 μA) and 92.64 (Ip,a = 4.91 μA) for the surfaces of CNT/GCE and OPPY/CNT/GCE, respectively. This improvement in the peak current is due to the larger active surface area of the modified electrodes and a better adsorption of OLZ on the nanoporous modifier film. | ||
| Fig. 3 LSVs of 2.00 μM of OLZ on the surface of various electrodes; bare GCE (solid line, inset), CNT/GCE (dotted line) and OPPY/CNT/GCE (dashed-dotted line). Supporting electrolyte was phosphate buffer solution (pH 3.0), potential scan rate was 100 mV s−1 and accumulation time was 80 s. | ||
On the other hand, a subsequent oxidation peak (Ip,a = 0.96 μA) can be observed on the OPPY/CNT/GCE. It can be considered that entrapment of the OLZ oxidation product in the polymer film and then involving in a consecutive electro-oxidation reaction has led to the appearance of this oxidation peak on the OPPY/CNT/GCE (Scheme 2).19
 | ||
| Scheme 2 Proposed mechanism of olanzapine oxidation. | ||
3.5. Effect of pH on the voltammetric responses
The effect of pH of the buffer solution on the electrochemical behavior of OLZ has been investigated in the range of 2.0–10.0 by the LSV method (Fig. 4A). As can be seen in Fig. 4B, a linear negative shift was observed for the variation of the anodic peak potential of OLZ, as pH increases (eqn (1)).| Ep,a (mV) = −58.9(±1.29) pH + 648(±8.44) (R2 = 0.996, n = 9) | (1) |
 | ||
| Fig. 4 (A) LSVs of 2.00 μM OLZ on the surface of OPPY/CNT/GCE in various pHs (from 2.0 to 10.0) of buffer solution; dependence of (B) Ep,a and (C) Ip,a on the pH of solution. The potential scan rate was 100 mV s−1 and accumulation time was 80 s. | ||
The slope value of −58.9 mV per pH unit, which is very close to the theoretical slope of −59 mV, indicates that equal numbers of electrons and protons are involved in the electrode process of OLZ. This confirms the previous work, according which, one proton and one electron are involved in the electrochemical process.19 The anodic peak current of OLZ reaches its maximum value at pH 3.0 (Fig. 4C). Therefore, all voltammetric determinations have been performed in a phosphate buffer solution of pH 3.0, as the supporting electrolyte.
3.6. Optimization of the amount of MWCNTs casted on the electrode surface
In the case of the thin film modified electrodes, the electrochemical response toward the analyte is affected by the thickness of modifier film. It can be due to dependence of the mass transfer mechanism via diffusion through the porous film on the thickness of the modifier film.57,58 In order to investigate this issue, different volumes (from 1 to 5 μL) of a constant concentration of MWCNTs suspension (1 mg mL−1) were casted on the GCE surface. Voltammetric responses of 2.00 μM OLZ on the surface of OPPY/CNT/GCE are shown in Fig. 5. As can be seen, by increasing the volume of the MWCNTs suspension from 1 to 4 μL, the oxidation peak current was increased. However, more casted volumes leads to a decrease of the oxidation peak current. This can be resulted from the slow mass transfer process of OLZ, due to large film thickness of the casted suspension on the GCE surface. Hence, the volume of 4 μL of CNT has been considered as the optimum volume in order to prepare the modified electrode. | ||
| Fig. 5 LSVs of 2.00 μM of OLZ on the surface of GCE coated with various amounts of 1 mg mL−1 of MWCNTs suspension. Supporting electrolyte was phosphate buffer solution of pH 3.0, potential scan rate was 100 mV s−1 and accumulation time was 80 s. | ||
3.7. Effect of time and potential of accumulation
As it was previously mentioned, two consecutive well-separated oxidation peaks for OLZ were observed at OPPY/CNT/GCE. It was realized that, by applying accumulation potential between these two peaks (Ep,a1 = 0.479 V, Ep,a2 = 0.615 V), a gradual decrease in the height of the first peak along with increasing of the second peak is observed. Also by increasing the accumulation time to 80 s, these changes reach to nearly constant values (Fig. 6). In fact, migration of OLZ to the surface of the electrode follows by an adsorption mechanism. Therefore, by giving time to the system, the amount of the adsorbed analyte on the surface of modified electrode increases until reaching the equilibrium adsorption. On the other hand it was observed that a wider linear dynamic range can be obtained through the second peak, hence 0.50 V and 80 s were selected for the accumulation potential and time for determination and analytical applications, respectively. | ||
| Fig. 6 Effect of accumulation time on LSVs of 2.00 μM OLZ on the surface of OPPY/CNT/GCE. Supporting electrolyte was phosphate buffer solution (pH 3.0), potential scan rate was 100 mV s−1 and accumulation potential was 500 mV. | ||
3.8. Voltammetric determinations
The LSVs of solutions containing different concentrations of OLZ under the optimum experimental conditions are shown in Fig. 7A. The oxidation peak currents are proportional to OLZ concentration over a linear range of 0.02–2.00 μM (Fig. 7A inset). The linear regression eqn (2) for this region is:| Ip,a/μA = 3.34(±0.0394)C/μM + 0.0252(±0.0278) (R2 = 0.998) | (2) |
 | ||
| Fig. 7 (A) LSVs for various concentrations of OLZ in the range of (down to up), 0.02–2.00 μM and corresponding linear calibration curve of peak current versus OLZ concentration (inset), (B) LSVs for various concentrations of OLZ in the range of 0.10–2.00 μM in the presence of CLZ (100 μM) and corresponding linear calibration curve of peak current versus OLZ concentration (inset), (C) LSVs for the addition of different concentrations of standard OLZ (from down to up: 0.00, 0.02, 0.04, 0.15, 0.35, 0.45, 0.55, 0.75 μM) in the real sample solution of OLZ and the plot of peak current versus added concentrations of OLZ (inset). Supporting electrolyte was phosphate buffer solution (pH 3.0), scan rate was 100 mV s−1, accumulation time was 80 s and accumulation potential was 500 mV. | ||
In these investigations a detection limit (based on S/N = 3) of 6.00 nM was obtained for the OLZ. A Comparison between the present work with previous electrochemical determinations of OLZ17–20 shows that our prepared modified electrode has a lower detection limit for OLZ determination. The electrochemical investigation of OLZ on the surface of glassy carbon electrode by linear sweep voltammetry17 has shown a linear range within 19.80–160.00 μM with a detection limit of 9.6 μM. However, the voltammetric response properties obtained in the present work (including linear range and detection limit) are better than this report. Also a glassy carbon electrode was used for the determination of OLZ by differential pulse voltammetry.18 This report presents a detection limit of about 10 nM, which is more than the detection limit of the present work. In another work, OLZ has been determined using differential pulse voltammetry on the surface of a gold electrode modified with the oxidized single walled carbon nanotubes.19 The results of this study showed a detection limit of 0.32 μM and linear dynamic range of 0.64–32.00 μM OLZ, which is more restrictive, compared to the present work. Recently, a sensor was fabricated by modification of glassy carbon electrode with amine-functionalized TiO2/multi-walled carbon nanotubes for the detection of OLZ.20 This modification has provided two different linear dynamic ranges (0.12–33.00 and 33.00–124 μM) but resulted in a higher detection limit of 0.09 μM. In addition, the procedure for the preparation of the modified electrode is time consuming, which may be considered as a drawback to this sensor.
On the other hand, chromatographic and mass spectrometric methods exhibit lower detection limits and wider dynamic ranges but these methods are generally expensive, time consuming and need sensitive detectors and complicated sample preparation procedures.10–16 Also, spectrophotometric methods show higher detection limits and relatively lower linear dynamic ranges.8–10
3.9. Reproducibility and repeatability evaluations
In order to study the repeatability of the response of the modified electrode, electrochemical experiment was repeated 5 times with the same OPPY/CNT/GCE in a solution containing 0.20 μM OLZ. The relative standard deviation (RSD) based on these replicates was 5.26%. The reproducibility of the method was evaluated by preparing five modified electrodes at different days with the same fabrication procedure. The RSD value for the peak current determinations using this electrode on a 0.20 μM OLZ solution was calculated to be 5.92%. After storing the modified electrode under ambient conditions for one week, the peak current response of the electrode for a 0.20 μM OLZ solution retained 95.6% of its initial response. This result indicates that the modified electrode has a good repeatability, reproducibility and long term stability.Also after storing the modified electrode under ambient conditions for two weeks, the peak current response of the electrode for a concentration of 2 μM OLZ retained 96.4% of its initial response. This shows the long term stability of the response of the modified electrode.
3.10. Determination of OLZ in the presence of CLZ
In the case of clinical and pharmaceutical determinations, serious interferences may be raised from other components present in the matrix of the samples. Using CLZ together with OLZ can cause a low blood pressure, shallow breathing, weak pulse, muscle weakness, drowsiness, dizziness and slurred speech.59 So, it is a worthy attempt to determine OLZ in the presence of CLZ. Also, the selectivity of the method for the determination of OLZ in the presence of CLZ is very important. Some solutions containing a constant concentration of 100 μM CLZ and various concentrations of OLZ were prepared. The results of voltammetric studies on the surface of OPPY/CNT/GCE, obtained under the optimum experimental conditions, are shown in Fig. 7B. As can be seen, the peak current of OLZ exhibits a gradual increase with its concentration, while the peak current of CLZ has a constant value. Based on the plot of peak currents versus concentrations of OLZ, a linear range of 0.10–2.00 μM in the phosphate buffer solution (pH 3.0) was obtained (Fig. 7B inset). The linear regression equation for this region is:| Ip,a/μA = 3.46(±0.0544)C/μM − 0.270(±0.0489) (R2 = 0.998) | (3) |
According to the obtained results, presence of CLZ has no distinct effect on the determination of OLZ.
3.11. Analytical applications
| Ip,a/μA = 3.30(±0.0680)C/μM + 0.192(±0.0265) (R2 = 0.997) | (4) |
According to this equation, the tablet matrix did not show any interference with the electrochemical analysis of OLZ. The amount of 14.54 mg OLZ with a good accuracy of 97% and a RSD of 4.3% (n = 5) was obtained for the analysis of tablet samples with a labeled value of 15 mg. Moreover, for accuracy studies, recoveries in the lower, middle and higher level concentrations of the spiked OLZ to the pharmaceutical solutions were calculated, which was within the range of 96.8% to 104.9%.
| No. | Spiked (μM) | Found (μM) | Recovery (%) |
|---|---|---|---|
| 1 | 2.00 | 1.96 | 98.0 |
| 2 | 1.00 | 0.97 | 97.0 |
| 3 | 0.80 | 0.75 | 93.7 |
| 4 | 0.40 | 0.37 | 92.5 |
4. Conclusions
In the present work, a sensitive and effective electrochemical sensor was constructed for the electrochemical determination of OLZ. Presence of sodium benzene-1,3-disulfonate as a dopant anion leads to an efficient polymerization of pyrrole on the surface of the glassy carbon electrode pre-casted with MWCNT and formation of a stable and uniform film of the modifier on the electrode surface. The modified electrode with advantages such as, low detection limit, high sensitivity and relatively wide linear dynamic range was successfully applied for the voltammetric determination of sub-micromolar amounts of OLZ in pharmaceutical and clinical preparations.Acknowledgements
The authors gratefully acknowledge the support of this work by the Research Council and the Center of Excellence for Nanostructures of the Sharif University of Technology, Tehran, Iran. They are grateful to Professor Mehdi Jalali-Heravi (Department of Chemistry, Sharif University of Technology) for his valuable suggestions. The authors are gratefully appreciated Bakhtar Bioshimi and Tehran Daru Companies for preparation of OLZ and CLZ samples, respectively.References
- M. Aravagiri, Y. Teper and S. R. Marder, Biopharm. Drug Dispos., 1999, 20(8), 369–377 CrossRef CAS.
- S. Kapur, R. B. Zipursky and G. Remington, Am. J. Psychiatry, 1999, 156, 286–293 CAS.
- S. Kapur, R. B. Zipursky, G. Remington, C. Jones, J. Da Silva, A. A. Wilson and S. Houle, Am. J. Psychiatry, 1998, 155, 921–928 CAS.
- J. Tauscher, B. Küfferle, S. Asenbaum, P. Fischer, L. Pezawas, C. Barnas, S. Tauscher-Wisniewski, T. Brücke and S. Kasper, Psychopharmacology, 1999, 141, 175–181 CrossRef CAS.
- F. P. Bymaster, D. O. Calligaro, J. F. Falcone, R. D. Marsh, N. A. Moore, N. C. Tye, P. Seeman and D. T. Wong, Neuropsychopharmacology, 1996, 14(2), 87–96 CrossRef CAS.
- A. Schotte, P. F. M. Jansssen, W. Gommeren, W. H. M. L. Luyten, P. VanGompel, A. S. Lesage, K. De Loore and J. E. Leysen, Psychopharmacology, 1996, 124(1–2), 57–73 CrossRef CAS.
- B. L. Roth, S. C. Craigo, M. S. Choudhary, A. Uluer, F. J. Monsma, Y. Shen, H. Y. Meltzer and D. R. Sibley, J. Pharmacol. Exp. Ther., 1994, 268(3), 1403–1410 CAS.
- A. Jasinska and E. Nalewajko, Anal. Chim. Acta, 2004, 508(2), 165–170 CrossRef CAS PubMed.
- N. Rajendraprasad, K. Basavaiah, K. Tharpa and K. B. Vinay, Eurasian J. Anal. Chem., 2009, 4(2), 191–203 CAS.
- S. Firdous, T. Aman and A. Nisa, J. Chem. Soc. Pak., 2005, 27(2), 163–167 CAS.
- T. C. Sanchez, M. A. Martinez and E. A. Almarza, Forensic Sci. Int., 2005, 155(2–3), 193–204 CrossRef PubMed.
- C. Krishnaiah, M. V. Murthy, R. Kumar and K. Mukkanti, J. Pharm. Biomed. Anal., 2011, 54(4), 667–673 CrossRef CAS PubMed.
- M. Josefsson, M. Roman, E. Skogh and M. L. Dahl, J. Pharm. Biomed. Anal., 2010, 53(3), 576–582 CrossRef CAS PubMed.
- S. Gopinath, R. S. Kumar, S. Alexander and P. Danabal, Biomed. Chromatogr., 2011, 26(9), 1077–1082 CrossRef PubMed.
- Q. Zheng, F. Wang, H. Li, P. Xu, H. Tang, L. Li and R. Cheng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 905, 127–132 CrossRef CAS PubMed.
- D. S. Patela, N. Sharmab, M. C. Patela, B. N. Patel, P. S. Shrivastavc and M. Sanyald, Acta Pharm. Sin. B, 2012, 2(5), 481–494 CrossRef PubMed.
- M. A. Saracino, A. Koukopoulos, G. Sani, M. Amore and M. A. Raggi, Ther. Drug Monit., 2007, 29(6), 773–780 CrossRef CAS PubMed.
- M. A. Raggi, G. Casamenti, R. Mandrioli, G. Izzo and E. Kenndler, J. Pharm. Biomed. Anal., 2000, 23(6), 973–981 CrossRef CAS.
- M. A. El-Shal, Adv. Pharm. Bull., 2013, 3(2), 339–344 Search PubMed.
- D. Merli, D. Dondi, M. Pesavento and A. Profumo, J. Electroanal. Chem., 2012, 683, 103–111 CrossRef CAS PubMed.
- M. Arvand and B. Palizkar, Mater. Sci. Eng., C, 2013, 33, 4876–4883 CrossRef CAS PubMed.
- M. H. Mashhadizadeh and E. Afshar, Electrochim. Acta, 2013, 87, 816–823 CrossRef CAS PubMed.
- M. H. Mashhadizadeh and E. Afshar, Electroanalysis, 2012, 24(11), 2193–2202 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323(5915), 760–764 CrossRef CAS PubMed.
- F. Xiao, F. Zhao, J. Li, R. Yan, J. Yu and B. Zeng, Anal. Chim. Acta, 2007, 596(1), 79–85 CrossRef CAS PubMed.
- P. Kumari, in Nanomaterials as Promising DNA Biosensors, Biosensors Nanomaterials, ed. S. Li, J. Singh, H. Li and I. A. Banerjee, Wiley-VCH, Germany, 2011, pp. 247–254 Search PubMed.
- S. Fei, J. Chen, S. Yao, G. Deng, D. He and Y. Kuang, Anal. Biochem., 2005, 339(1), 29–35 CrossRef CAS PubMed.
- C. B. Jacobs, M. J. Peairs and B. J. Venton, Anal. Chim. Acta, 2010, 662(2), 105–127 CrossRef CAS PubMed.
- M. Guo, J. Chen, J. Li, B. Tao and S. Yao, Anal. Chim. Acta, 2005, 532(1), 71–77 CrossRef CAS PubMed.
- K. Scida, P. W. Stege, G. Haby, G. A. Messina and C. D. García, Anal. Chim. Acta, 2011, 691(1–2), 6–17 CrossRef CAS PubMed.
- R. T. Kachoosangi, M. M. Musameh, I. Abu-Yousef, J. M. Yousef, S. M. Kanan, L. Xiao, S. G. Davies, A. Russell and R. G. Compton, Anal. Chem., 2009, 81, 435–442 CrossRef CAS PubMed.
- F. Can, S. KorkutOzoner, P. Ergenekon and E. Erhan, Mater. Sci. Eng., C, 2012, 32(1), 18–23 CrossRef CAS PubMed.
- M. D. Shirsat, C. O. Too and G. G. Wallace, Electroanalysis, 2008, 20(2), 150–156 CrossRef CAS PubMed.
- Y. W. Lin and T. M. Wu, Polym. Int., 2011, 60(3), 382–388 CrossRef CAS PubMed.
- S. Shahrokhian, Z. Kamalzadeh and R. S. Saberi, Electrochim. Acta, 2011, 56(27), 10032–10038 CrossRef CAS PubMed.
- S. Shahrokhian, Z. Kamalzadeh and R. S. Saberi, Electroanalysis, 2011, 23(12), 2925–2934 CrossRef CAS PubMed.
- H. Peng, L. Zhang, C. Soeller and J. Travas-Sejdic, Biomaterials, 2009, 30(11), 2132–2138 CrossRef CAS PubMed.
- J. C. Vidal, E. Garcia-Ruiz and J. R. Castillo, Microchim. Acta, 2003, 143(2–3), 93–111 CrossRef CAS.
- J. Li and H. Xie, Mater. Lett., 2012, 78, 106–109 CrossRef CAS PubMed.
- D. P. Dubal, S. V. Patil, W. B. Kim and C. D. Lokhande, Mater. Lett., 2011, 65, 2628–2631 CrossRef CAS PubMed.
- J. Li, W. Wei and S. Luo, Microchim. Acta, 2010, 171(1–2), 109–116 CrossRef CAS.
- J. O. Iroh, Y. Zhu, K. Shah, K. Levine, R. Rajagopalan, T. Uyar, M. Donley, R. Mantz, J. Johnson, N. N. Voevodin, V. N. Balbyshev and A. N. Khramov, Prog. Org. Coat., 2003, 47, 365–375 CrossRef CAS PubMed.
- J. O. Iroh and G. A. Wood, Composites, 1998, 29(2), 181–188 CrossRef.
- D. E. Tallman, C. Vang, G. G. Wallace and G. P. Bierwagen, J. Electrochem. Soc., 2002, 149(3), 173–179 CrossRef PubMed.
- T. Raudsepp, M. Marandi, T. Tamm, V. Sammelselg and J. Tamm, Electrochim. Acta, 2008, 53, 3828–3835 CrossRef CAS PubMed.
- G. R. Mitchell, F. J. Davis and C. H. Legge, Synth. Met., 1998, 26, 247–257 CrossRef.
- C. Meng, C. Liu and S. Fan, Electrochem. Commun., 2009, 11(1), 186–189 CrossRef CAS PubMed.
- Y. Ma, W. Cheung, D. Wei, A. Bogozi, P. L. Chiu, L. Wang, F. Pontoriero, R. Mendelsohn and H. He, ACS Nano, 2008, 2, 1197–1204 CrossRef CAS PubMed.
- G. Chakraborty, K. Gupta, A. K. Meikap, R. Babu and W. J. Blau, Solid State Commun., 2012, 152, 13–18 CrossRef CAS PubMed.
- S. Paul, Y. S. Lee, J. A. Choi, Y. C. Kang and D. W. Kim, Bull. Korean Chem. Soc., 2010, 31(5), 1228–1232 CrossRef CAS.
- S. Shahrokhian and R. S. Saberi, Electrochim. Acta, 2011, 57, 132–138 CrossRef CAS PubMed.
- V. Brânzoi, L. Pilan and F. Brânzoi, Electroanalysis, 2009, 21, 557–562 CrossRef PubMed.
- G. M. Greenway and S. J. L. Dolman, Analyst, 1999, 124, 759–762 RSC.
- N. F. Atta, A. Galal and S. M. Azab, Analyst, 2011, 136, 4682–4691 RSC.
- A. Witkowski and A. Brajter-Toth, Anal. Chem., 1992, 64, 635–641 CrossRef CAS.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15–21 RSC.
- G. P. Keeley and M. E. G. Lyons, Int. J. Electrochem. Sci., 2009, 4, 794–809 CAS.
- http://www.drugs.com/drug-interactions/clonazepam-with-zyprexa-703-0-1744-1113.html.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04584j |
| This journal is © The Royal Society of Chemistry 2014 |