
Rational construction of metal–organic frameworks for heterogeneous catalysis
Sha
Ou
and
Chuan-De
Wu
*
Center for Chemistry of High Performance and Novel Materials, Department of Chemistry, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: cdwu@zju.edu.cn
First published on 16th October 2014
Abstract
Metal–organic frameworks (MOFs) are a class of porous materials that consist of highly ordered organic building blocks and metal ions/clusters. Because the emerging porous materials have the ability to retain and even enhance the individual functionalities of the secondary building units (SBUs), the catalytic functions of porous MOFs can be therefore easily realized by carefully choosing and tuning the functional moieties in building synthons. Accompanying the development on the synthesis strategy and the characterization technology for MOF materials, MOF catalysts have undergone an upsurge and transcended the stage of opportunism. This brief review will give a cross-section of the rational design and synthesis of porous metal–organic framework materials for heterogeneous catalysis.
 Sha Ou | Sha Ou received her B.S. degree in Chemistry from Guizhou University in 2011. Currently, she is a doctoral student in chemistry at Zhejiang University under the direction of Professor Chuan-De Wu. Her research is focused on the development of porous metal–organic frameworks for heterogeneous catalysis. |
 Chuan-De Wu | Chuan-De Wu obtained his Ph.D. degree in Physical Chemistry from Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences in 2003. He worked as a postdoctoral fellow at the University of North Carolina at Chapel Hill during 2003–2005 before joining Zhejiang University in 2005 as a full professor of chemistry. His research interest focuses on the development of porous framework materials and their heterogeneously catalytic application. |
1. Introduction
Metal–organic frameworks (MOFs) are an emerging class of crystalline porous materials built from organic linkers connecting with metal ions/clusters through metal–ligand bonds.1 Because of their fascinating topological structures and remarkable properties, MOF materials have provided multiple entries into technologically useful materials for applications in the fields of gas storage, separation, magnetiism, biomedicine, fluorescence, molecular recognition and heterogeneous catalysis.2–9In principle, crystal engineering of porous MOFs can be regarded as self-assembly and self-organization of connectors of metal ions/clusters and linkers of organic building synthons under mild reaction conditions. The topological structures of MOFs are majorly defined by the coordination geometries of metal nodes and the directionalities of organic ligands. The major objective of current crystal engineering of MOF materials is to vary the metal connectors and the organic linkers for the construction of desired porous functional materials for target applications. Even though many synthesis strategies and characterization technologies for MOF materials have been massively developed, there are however still many difficulties to precisely predict and control the overall topological structures from their chemical compositions, because combination of the metal nodes and organic linkers are affected by many factors, such as solvent, temperature, reaction duration, concentration, counter ion and pH value. Nevertheless, it is feasible to rationally introduce catalytically-active moieties into porous MOFs by carefully modifying the organic/inorganic moieties, and thus realize their heterogeneous catalytic properties.
Considering that many reviews have comprehensively discussed the catalytic applications of MOFs, this short review only gives a cross-section on rational design and synthesis of the functional moiety oriented MOFs for heterogeneous catalysis.9 The selectively grafted functional moieties on the pore surfaces of MOFs let them exhibit interesting catalytic properties with remarkable size- and shape-selectivities, which cannot be matched by the traditional inorganic zeolites.10
2. The principle for design of metal–organic framework catalysts
Generally speaking, most MOFs have a certain degree of catalytic activity for reactions that are dependent on the sites of metal/cluster nodes and/or the organic moieties. Even though the metal sites are fully ligated by strongly coordinated organic ligands, there are still abundant unsaturated metal coordination sites on the surfaces of MOFs because of coordination defects. If the metal coordination sites are partially terminated by volatile solvent molecules, such as water, alcohols, acetonitrile, DMF, etc., these MOF materials will be efficient for some catalytic reactions that are prompted by the metal nodes. This is because these MOFs are easily activated to generate unsaturated metal coordination sites by removal of the volatile solvent molecules under suitable conditions. However, because these metal ions are also the pivotal components of MOFs, the framework structures are easily collapsed during catalysis. For example, the well known porous MOF HKUST-1 is built from paddle-wheel Cu2(COO)4 secondary building units (SBUs) linked by benzene 1,3,5-tricarboxylate.11 The axial coordination sites of CuII ions are occupied by weakly bound water molecules, which can be removed by simply heating the compound in a vacuum. Dehydration makes the copper Lewis acid centers highly catalytically active, and they can coordinate with organic substrates to result in the activation of the catalytic process, such as cyanosilylation of benzaldehyde.12 However, because the CuII ions are easily reduced by aldehyde molecules under the catalytic reaction conditions, HKUST-1 is partially decomposed during catalysis.Obviously, using the above method for the synthesis of MOF catalysts is not a good choice. Therefore, several effective strategies have been well developed and have been gradually maturing. The most straight forward strategy is to use catalytically active moieties as building synthons to synthesize organic linkers. Those catalytically active ligands can well define the catalytic properties of the resulting porous metal–organic materials. To date, numerous catalytically active moieties have been successfully immobilized onto the pore surfaces of porous MOFs, which present remarkable catalytic activities with interesting size- and shape-selectivities.12 The catalytically active moieties include metallosalens, metalloporphyrins, amino acids, carbenes, polyoxometalates, etc. Because there are still many difficulties in the construction of highly porous MOFs in practical procedures, it is impossible to introduce any homogeneous catalytically active moieties into the crystalline porous materials in a “one-pot” synthesis. Therefore, post-synthetic modification has been developed as an alternative strategy for the construction of metal–organic framework catalysts.13 Those porous metal–organic materials, consisting of post-modifiable moieties on the building blocks, can be easily functionalized with suitable functional moieties to generate highly efficient porous MOF catalysts.
3. Coordinatively unsaturated metal nodes as catalytic active sites
There have been a large number of MOFs whose coordination sites of metal nodes are unsaturated or occupied by volatile solvent molecules, such as water, alcohols, acetonitrile, DMF, etc. The former can be directly used for catalysis without any requirement for special activation, whereas the latter ones should be carefully activated to remove the volatile solvent ligands on the metal nodes to generate coordinatively unsaturated metal sites for heterogeneous catalysis.The supramolecular solid, [Cu2L12(DMF)2(H2O)2] (1), built from 1,8-naphthalenecarboxylic anhydride-4,5-dicarboxylicate (L1) linking binuclear copper(II) synthons, is very stable in common organic solvents because the binuclear copper(II) units are strongly connected via multiple supramolecular contacts (Fig. 1).14 The copper(II) ion in the supramolecular solid is in a distorted square-planar coordination geometry, which can directly provide vacant axial positions to coordinate with substrate molecules during heterogeneous catalysis. Prompted by the supramolecular solid 1, arylboronic acid and imidazole substrates were smoothly transformed to the desired cross-coupling products (up to 96.8% yield) under mild conditions. Moreover, the solid catalyst 1 can be simply recovered by filtration, which was reused for six consecutive cycles without significantly diminishing the catalytic efficiency.
 | ||
| Fig. 1 Supramolecular solid 1 built from [Cu2L12(DMF)2(H2O)2] units (Cu, cyan). | ||
The 1D coordination polymer [Cu(L2)2] (2) is built from an amino acid derivative L2 (HL2 = N-(4-pyridyl)-D,L-valine) bridging CuII ions.15 In the crystal structure of 2, the copper ion coordinates to two carboxyl oxygen atoms and two pyridyl groups of four L2 ligands in a square-planar coordination geometry. Two L2 ligands trans-link two CuII centers to form an 18-membered macrocycle ring, which further propagates into a 1D polymeric chain (Fig. 2). Since the square-plane coordinated CuII ion has two vacant axial positions, compound 2 efficiently prompted the catalytic cross-coupling reaction of arylboronic acids with imidazole at room temperature, which generated the anticipated products of N-phenylimidazoles in good yields (up to 97%). The catalytic activity of 2 is highly dependent on the solvent media. When different solvents, such as DMF, THF, CH2Cl2 and H2O, were used instead of MeOH, almost no product was produced except for trace product from the solvent of DMF, which suggests that there was a competing coordination to Cu ions between the solvent and substrate molecules.
 | ||
| Fig. 2 The 1D polymeric chain network of compound 2 (Cu, cyan). | ||
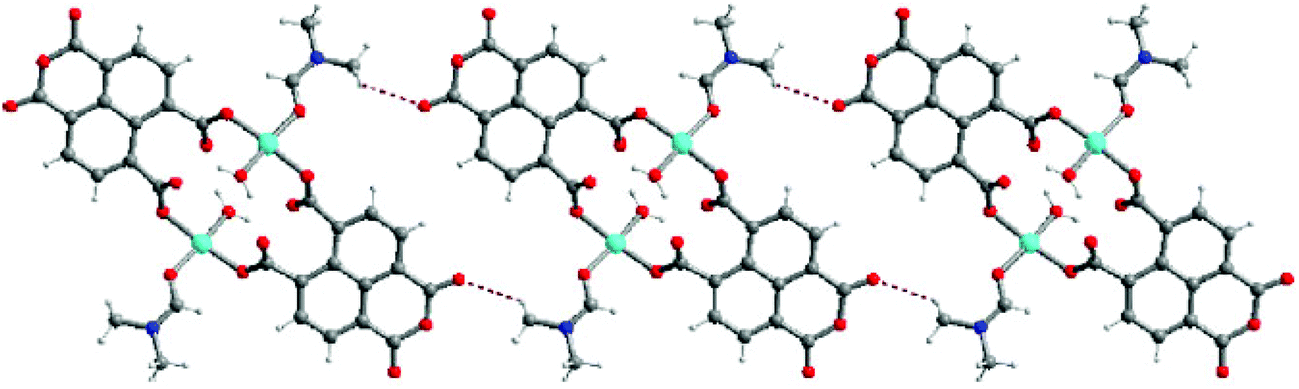
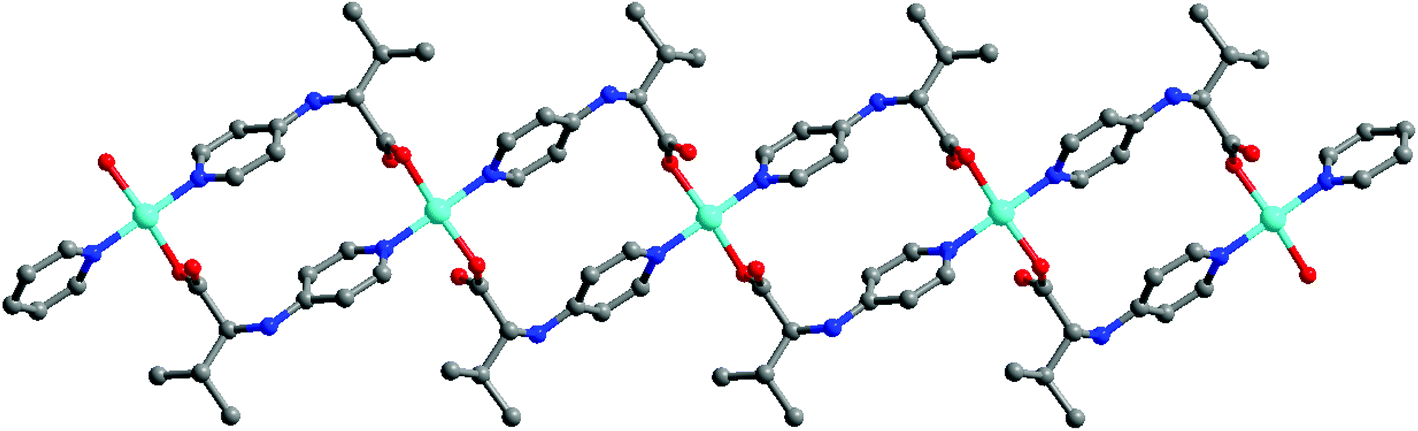
With the help of mixed ligands in multiple coordination modes, different coordination geometries of the metal nodes can be simultaneously generated in a porous MOF, which often consists of the active metal sites that are either coordinatively vacant or coordinated to volatile solvent molecules. In the structure of [Cu3(pdtc)L32(H2O)3]·guest (3; H4pdtc = pyridine-2,3,5,6-tetracarboxylic acid; HL3 = (E)-4-(2-(pyridin-4-yl)vinyl)benzoic acid), two kinds of pyridine carboxylate ligands connect with copper nodes to form an interesting 3D porous framework.16 As shown in Fig. 3, one third copper ions, which are exposed on the interior surface of the 1D channel walls (13.76 × 23.62 Å2), coordinate to volatile water molecules that are readily substituted by suitable molecules. Single crystal structural analysis demonstrates that the water ligands can be replaced by pyridine and EtOH molecules, whereas the overall framework is almost intact. The immobilized catalytically active copper(II) centers in the channel walls can trigger the Henry reaction of benzaldehydes with nitroalkanes, in which up to 85% product yield has been achieved. The catalytic properties of 3 are much superior to those of simple copper salts because the homogeneous metal coordination sites are easily ligated by the counter ions.
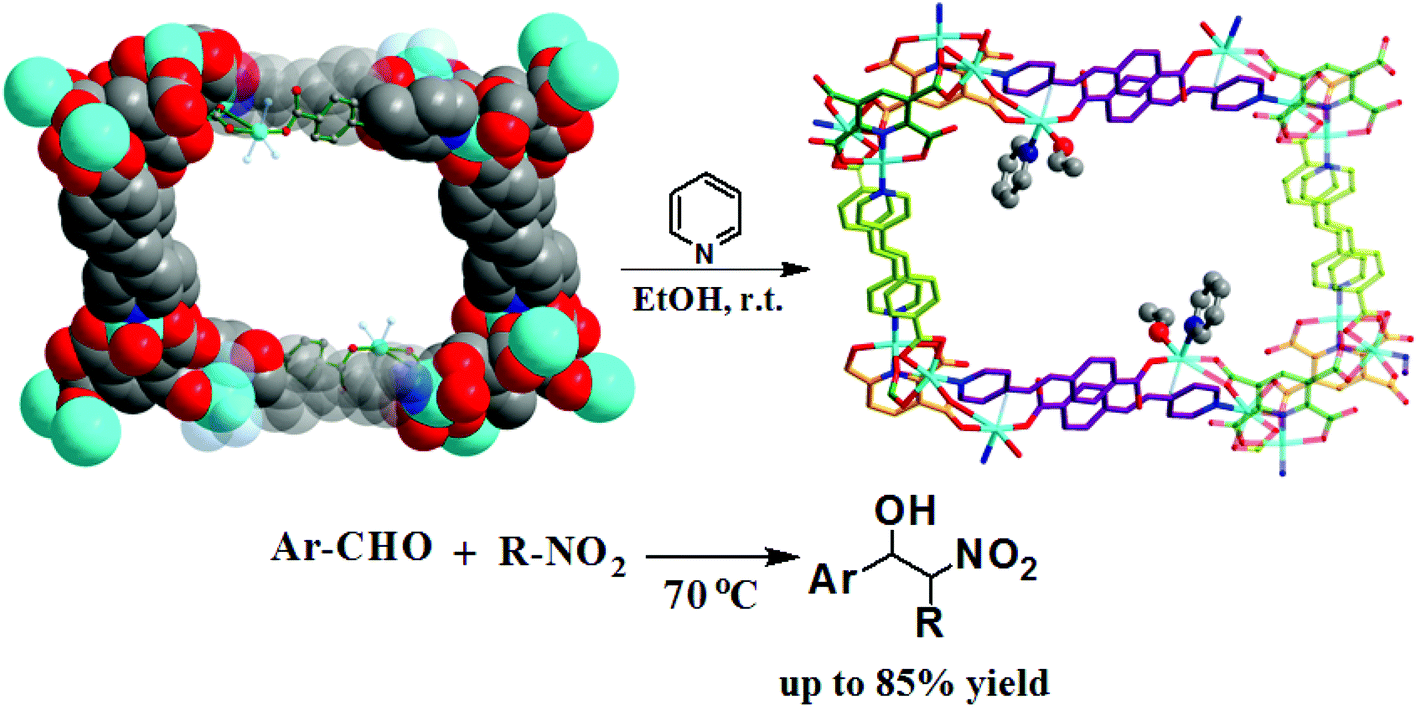 | ||
| Fig. 3 The structural transformation of 3 in pyridine/EtOH solution, and the catalytic Henry reaction of benzaldehydes with nitroalkanes. | ||
The catalytic properties of MOFs are very sensitive to the pore structures, even though their pore structures are slightly tuned, because the substrate and product molecules are transported through the open channels. The neutral 2D MOF [Mn2L42(H2O)2]·3H2O (4; H2L4 = E-5-(2-(pyridin-4-yl)vinyl)isophthalic acid) is built from octahedrally coordinated MnII ions linked by L4 ligands with cavity dimensions of 7.80 × 8.94 Å2 (Fig. 4).17 In the crystal structure of 4, the MnII center is oriented towards the 1D channels, and coordinates to one labile water molecule. Therefore, the Mn centers are active in the oxidation of phenylmethanol, which produced the oxidation product benzaldehyde in 64% yield when the reaction was performed at 60 °C for 18 h. It is interesting that two neighboring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C centers in two layers are at a reasonable distance, which lets compound 4 undergo topochemical [2 + 2] cycloaddition in a single-crystal-to-single-crystal (SCSC) manner from a 2D to a 3D structure transformation under UV light radiation. Compared with the structure of 4, two CC centers from two original neighboring layers form a cyclobutane ring in the porous network of [Mn2L5(H2O)2]·3H2O (5; H4L5 = 5,5′-(3,4-diphenylcyclobutane-1,2-diyl)diisophthalic acid). Even though the pore volume is slightly increased (from 24.3% to 27.0%), the catalytic activity of 5 is significantly improved, which generates the oxidation product benzaldehyde in 97% yield under otherwise identical conditions. The above results demonstrate that the pore structures of MOF catalysts play a very important role in the catalytic performance of active sites inside the pores.
C centers in two layers are at a reasonable distance, which lets compound 4 undergo topochemical [2 + 2] cycloaddition in a single-crystal-to-single-crystal (SCSC) manner from a 2D to a 3D structure transformation under UV light radiation. Compared with the structure of 4, two CC centers from two original neighboring layers form a cyclobutane ring in the porous network of [Mn2L5(H2O)2]·3H2O (5; H4L5 = 5,5′-(3,4-diphenylcyclobutane-1,2-diyl)diisophthalic acid). Even though the pore volume is slightly increased (from 24.3% to 27.0%), the catalytic activity of 5 is significantly improved, which generates the oxidation product benzaldehyde in 97% yield under otherwise identical conditions. The above results demonstrate that the pore structures of MOF catalysts play a very important role in the catalytic performance of active sites inside the pores.
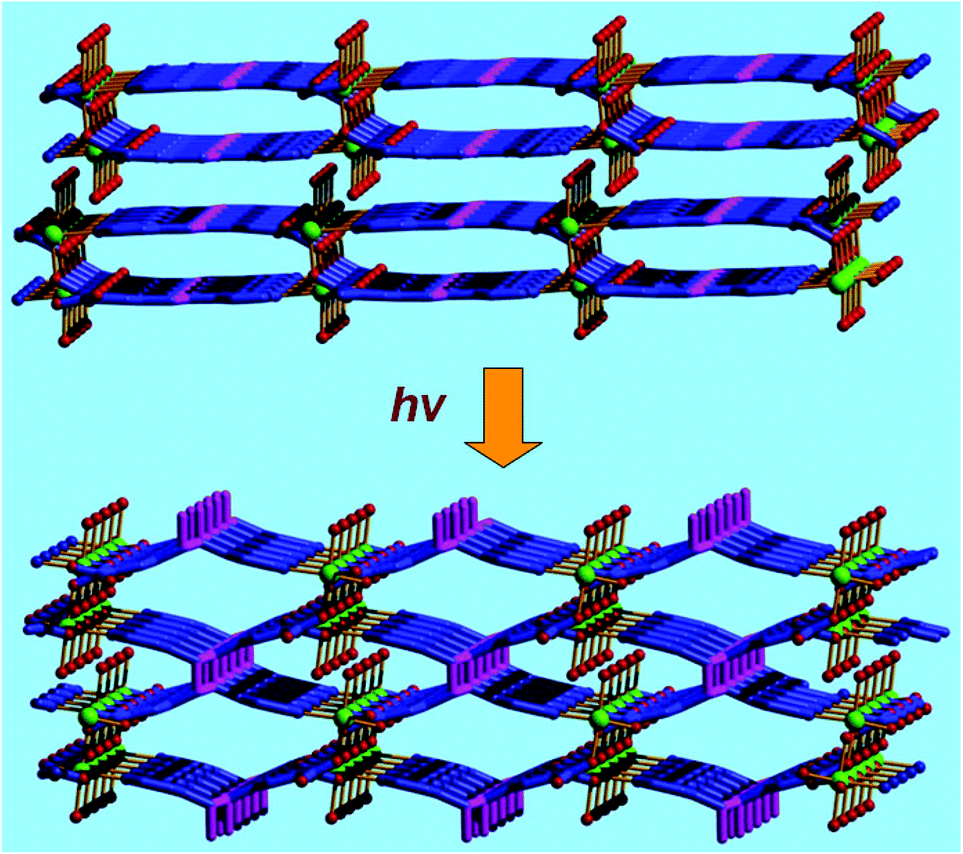 | ||
| Fig. 4 The UV irradiated SCSC structural transformation from 2D layered network of 4 (above) to 3D porous framework of 5 (below). | ||
4. Metal–organic framework catalysts constructed from metallosalens
Salens, the condensation products of aryl aldehydes and diamines, are a class of universal and excellent ligands to chelate various metal ions for the synthesis of different metallosalen catalysts.18 Metallosalens have realized different catalytic properties with remarkable enantio-selectivities in a variety of reactions. Therefore, metallosalens are the excellent synthons for the synthesis of porous MOF catalysts, which have been realized for heterogeneous catalytic applications on alkene epoxidation, cyclopropanation and hydrolytic kinetic resolution of epoxides with interesting size-, shape- and enantio-selectivities.A chiral bipyridyl Mn-salen ((R,R)-(2)-1,2-cyclohexanediamino-N,N′-bis(3-tert-butyl-5-(4-pyridyl)salicylidene)MnIIICl = L6) together with a biphenyldicarboxylate (bpdc) was used to connect paddle-wheel Zn2(COO)4 SBUs for the construction of a homochiral porous MOF [Zn2(bpdc)2L6]·guest (6), which is highly efficient for asymmetric epoxidation of olefins (Fig. 5).19 The catalytically active MnIII species, incorporated in the Mn-salen units, are accessible to substrate molecules transported through the opening channels (6.2 × 15.7 Å2 in dimensions). The heterogeneous catalytic oxidation of 2,2-dimethyl-2H-chorome by 2-(tertbutylsulfonyl)iodosylbenzene gives the epoxide product with an ee value of 82%. To prove that the catalysis occurs inside the pores of 6, a mixture of large porphyrin substrate (larger than the channel sizes of 6) and a small ethyl 4-vinyl benzoate substrate (smaller than the channel sizes of 6) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 olefinic unit ratio was reacted with 2-(tertbutylsulfonyl) iodosylbenzene in the presence of heterogeneous 6 or homogeneous Mn-salen. The different catalytic properties of 6 and homogeneous Mn-salen on oxidation of the mixed substrates have proved that the heterogeneous catalysis chiefly occurs inside the pores of solid catalyst 6.
:1 olefinic unit ratio was reacted with 2-(tertbutylsulfonyl) iodosylbenzene in the presence of heterogeneous 6 or homogeneous Mn-salen. The different catalytic properties of 6 and homogeneous Mn-salen on oxidation of the mixed substrates have proved that the heterogeneous catalysis chiefly occurs inside the pores of solid catalyst 6.
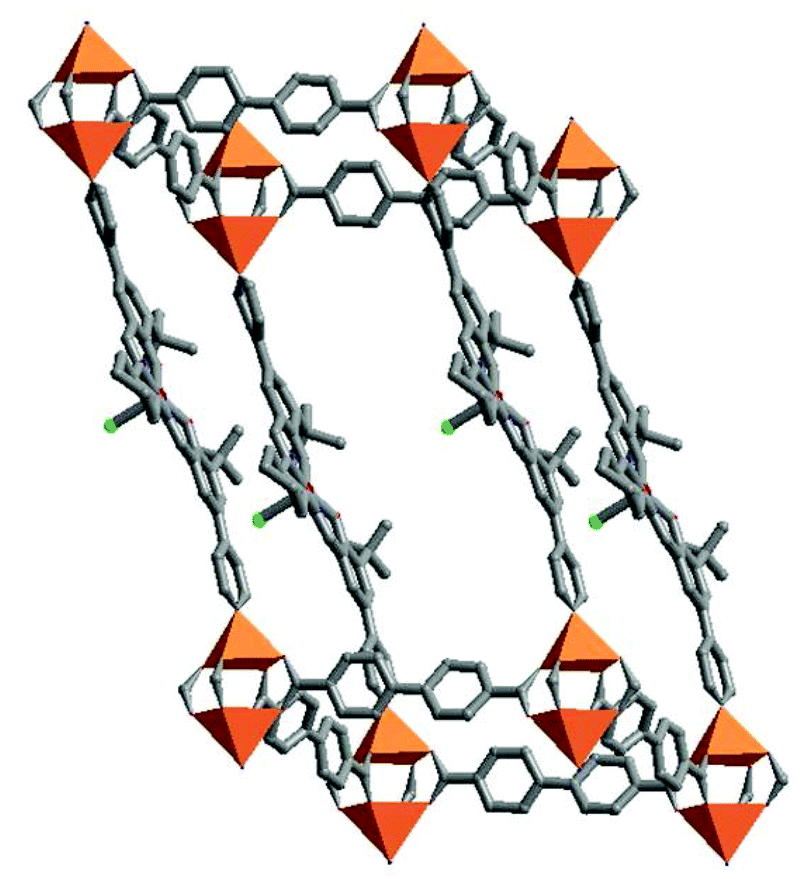 | ||
| Fig. 5 The 3D porous structure of 6 constructed from Mn-salen and paddle-wheel Zn2(COO)4 SBUs. | ||
As shown in Fig. 6, the Lin group has synthesized a family of isoreticular chiral metal–organic frameworks (CMOFs) [Zn4(μ4-O)(Mn-salen)3]·guest (CMOFs 1–5), which are built from [Zn4(μ4-O)(O2CR)6] SBUs and chiral Mn-Salen catalytic centers ((R,R)-1,2-cyclohexanediamino-N,N′-bis(3-tert-butyl-salicylidene)MnIIICl) with systematically elongated dicarboxylate bridges (–COOH, –CHCHCOOH, and –C6H4COOH groups terminated on the 4,4′-positions of Mn-Salen).20 The pore structures and the pore sizes of these CMOFs are systematically dependent on the spacers of the tunable dicarboxylate bridges and the steric sizes of the solvents with the open channel dimensions varied from 8 × 6 Å2 to 25 × 23 Å2. CMOFs 1–5 are highly efficient at catalyzing asymmetric epoxidation of a number of olefins, whereas the epoxidation rates were shown to be strongly dependent on the open channel sizes of CMOFs, in which up to 92% ee has been achieved for the asymmetric epoxidation of chromene to generate (1R,2S)-indene oxide. These results demonstrate that larger open channels can facilitate diffusion of the reactant and product molecules to increase the catalytic reaction rates.
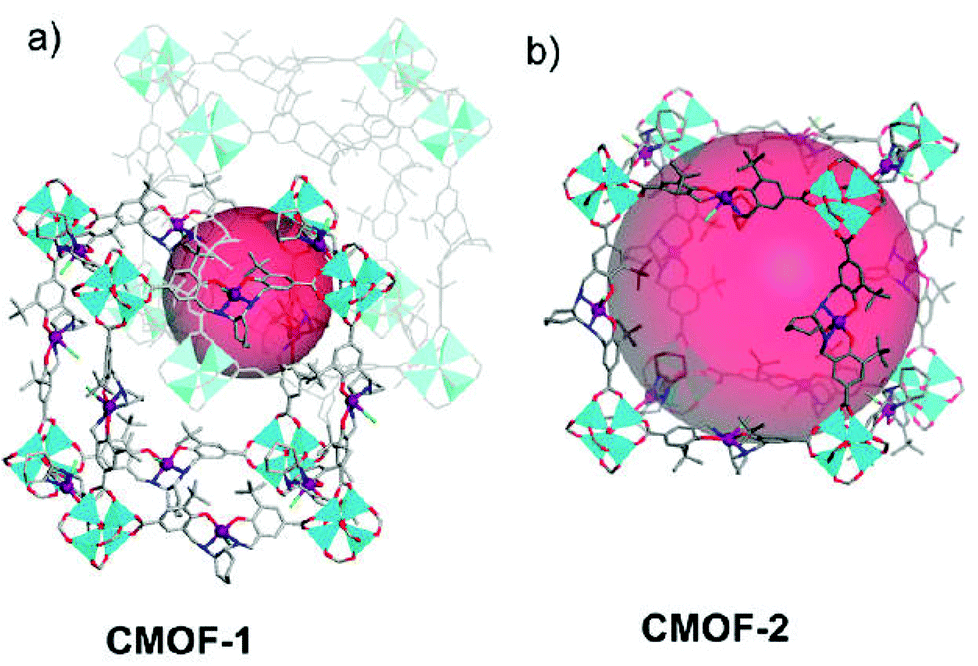 | ||
| Fig. 6 The structures of the interpenetrated network of CMOF-1 and the non-interpenetrated network of CMOF-2. | ||
They also studied the catalytic properties of a pair of CMOFs {Zn4(μ4-O)-[(RuIII-salen)(py)2Cl]3}·guest (CMOFs 7 and 8) based on Ru-salen-dicarboxylate.21 The largest cavities in the interpenetrated network of CMOF-7 are 8 Å in diameter with corresponding pore window dimensions of 4 × 3 Å2, whereas those of the non-interpenetrated framework of CMOF-8 are 17 Å and 14 × 10 Å2, respectively. The RuIII centers in the as-synthesized CMOFs 7 and 8 can be reduced to RuII centers by strong reducing agents such as LiBEt3H or NaB(OMe)3H in a reversible SCSC manner. The resulting non-interpenetrated RuII-CMOF was highly active for the asymmetric cyclopropanation of substituted alkenes with very high diastereo- and enantio-selectivities (dr = 7:1 (trans/cis); ee = 91% (trans) and 84% (cis)). In contrast, the interpenetrated RuII-CMOF was nearly inactive (less than 1% yield), probably because the small channels cannot transport the substrate molecules, thus preventing the diffusion of the reagents to access the RuII-salen active sites in the interior pore surfaces.
The Cui group has reported a homochiral MOF, [Cd4(CoIII-salen)4(DMF)(OAc)4]·4H2O (7), based on a Co-salen-dicarboxylate (Fig. 7).22 The 3D chiral porous framework of 7 is built from square-planar tetrameric [Cd4(O2C)8] SBUs connected with Co-salen linkers with open channel dimensions of 12 × 8 Å2 along the a axis. The catalytically active Co-salen sites are accessible to substrate molecules via the open channels, which can efficiently prompt the heterogeneous asymmetric hydrolytic kinetic resolution of a range of benzyloxy epoxide derivatives with ee value up to 99.5%. Since the heterogeneous catalysis mainly occurs inside the pores of the Co-salen MOF, it has size-selective property. The conversion of the small substrate phenoxy epoxide is of 56%, whereas the conversion for the sterically bulky substrate triphenyl glycidyl ether is less than 5% under the same reaction conditions because the bulky substrate has difficultly accessing the catalytic sites inside the pores.
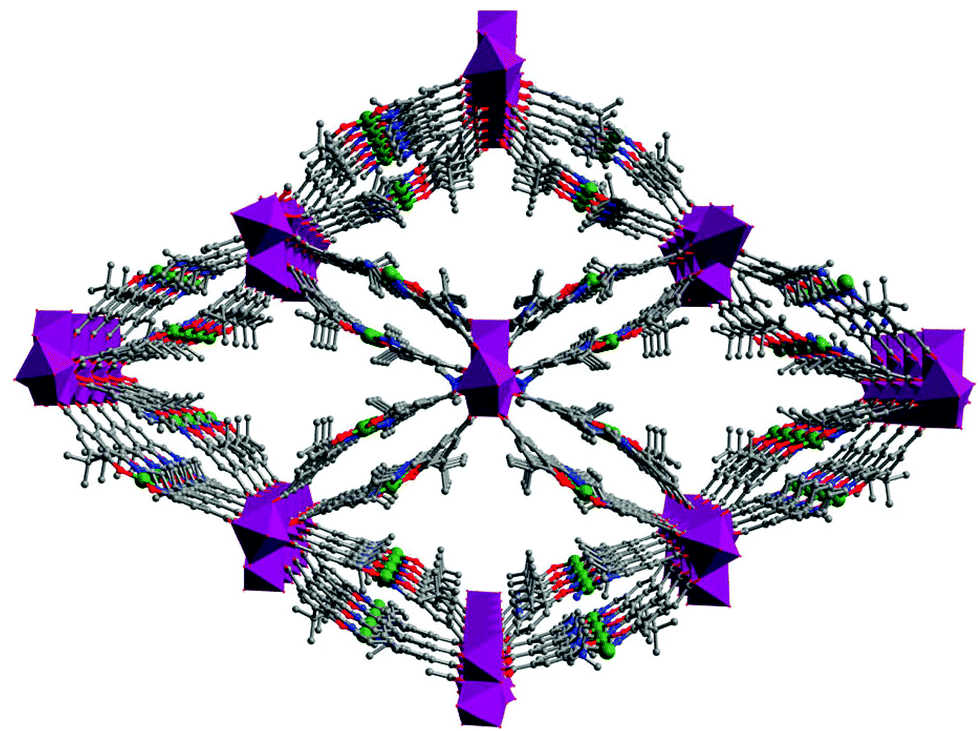 | ||
| Fig. 7 The 3D porous structure of 7 (Cd, purple polyhedra). | ||
5. Metal–organic framework catalysts constructed from metalloporphyrins
Metalloporphyrins are the active sites in monooxygenases which can efficiently oxidize a variety of organic molecules under mild conditions.23 The rigid macrocycle rings make metalloporphyrin moieties an ideal class of building blocks for the construction of highly porous MOFs. Incorporation of metalloporphyrin moieties into the emerging porous MOF materials not only can retain and even enhance the functionalities of the building blocks, but can also improve the stability of metalloporphyrins in oxidation catalysis.24 Furthermore, the different catalytic functionalities of the porous porphyrinic framework materials can be easily targeted by modulating the porphyrin metal sites and tailoring the peripheral environments of metalloporphyrins. Among the reported porphyrinic MOFs, the bridging moieties and the configurations of metalloporphyrins have played crucial roles on tuning and stabilizing the pore structures of porphyrinic MOFs.The porphyrinic MOF [Zn2(HCOO)(FeIII(H2O)-TCPP)] (8) is a 3D porous structure that is built from formate pillars to connect with layered networks of FeIII-TCPP moieties linking paddle-wheel Zn2(COO)4 SBUs (Fig. 8).25 In the crystal structure of 8, the square-pyramidally coordinated iron(III) ions are accessible to the included solvent molecules in the pores. The solid catalyst is highly active for the aldol reaction of aldehydes and ketones. The yields of aldol products are excellent (up to 99%), which are superior to those of its homogeneous constituents, such as FeCl-Me4TCPP, Zn(NO3)2 and FeCl3.
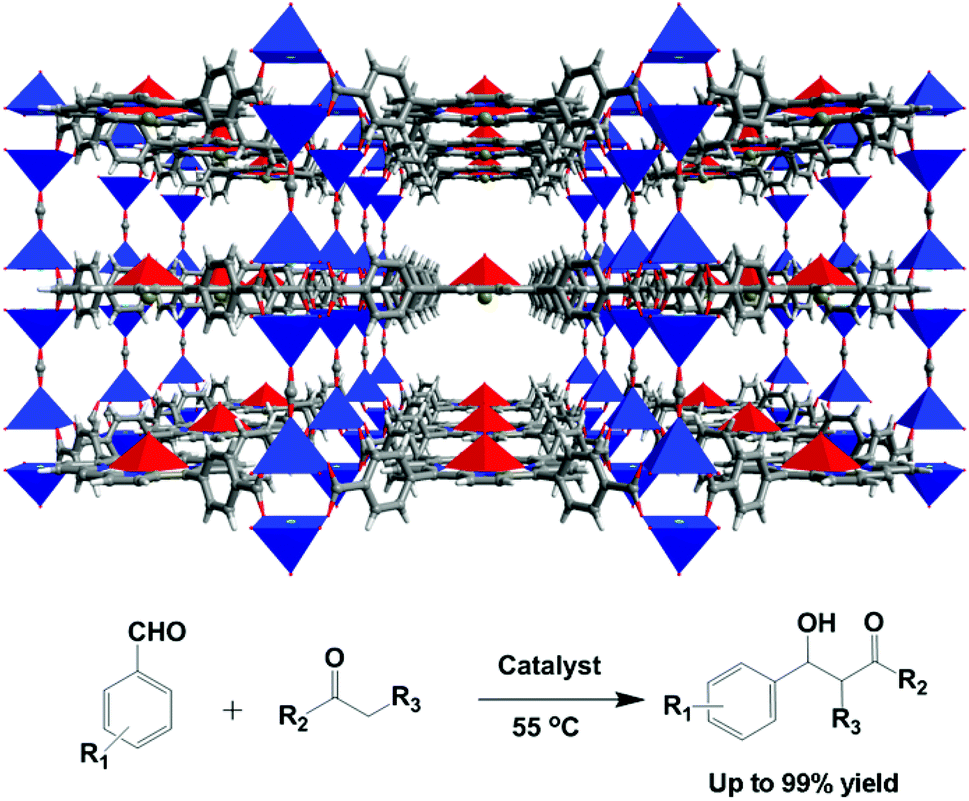 | ||
| Fig. 8 Crystal structure of 8, and the catalytic aldol reaction of aldehydes with ketones. | ||
In some cases, the axial coordination sites of the porphyrin metal ions are ligated by coordination donors in porphyrinic MOFs, which will obstruct their catalytic efficiency. As shown in Fig. 9, the active MnIII sites in two isomorphous porphyrinic frameworks [(CH3)2NH2][Zn2(HCOO)2(MnIII-TCPP)]·guest (9) and [(CH3)2NH2][Cd2(HCOO)2(MnIII-TCPP)]·guest (10) are blocked by formate ligands, which make the active sites inaccessible to substrate molecules through the opening channels.25 Even though they are highly active for the epoxidization of a number of olefins with different sizes and dimensions, because the catalytic active sites are only available on the exterior solid surfaces with coordinative defect, the catalysis thus occurs on the solid surfaces of the porphyrinic MOFs, which present significant particle size effect on the oxidation rates.
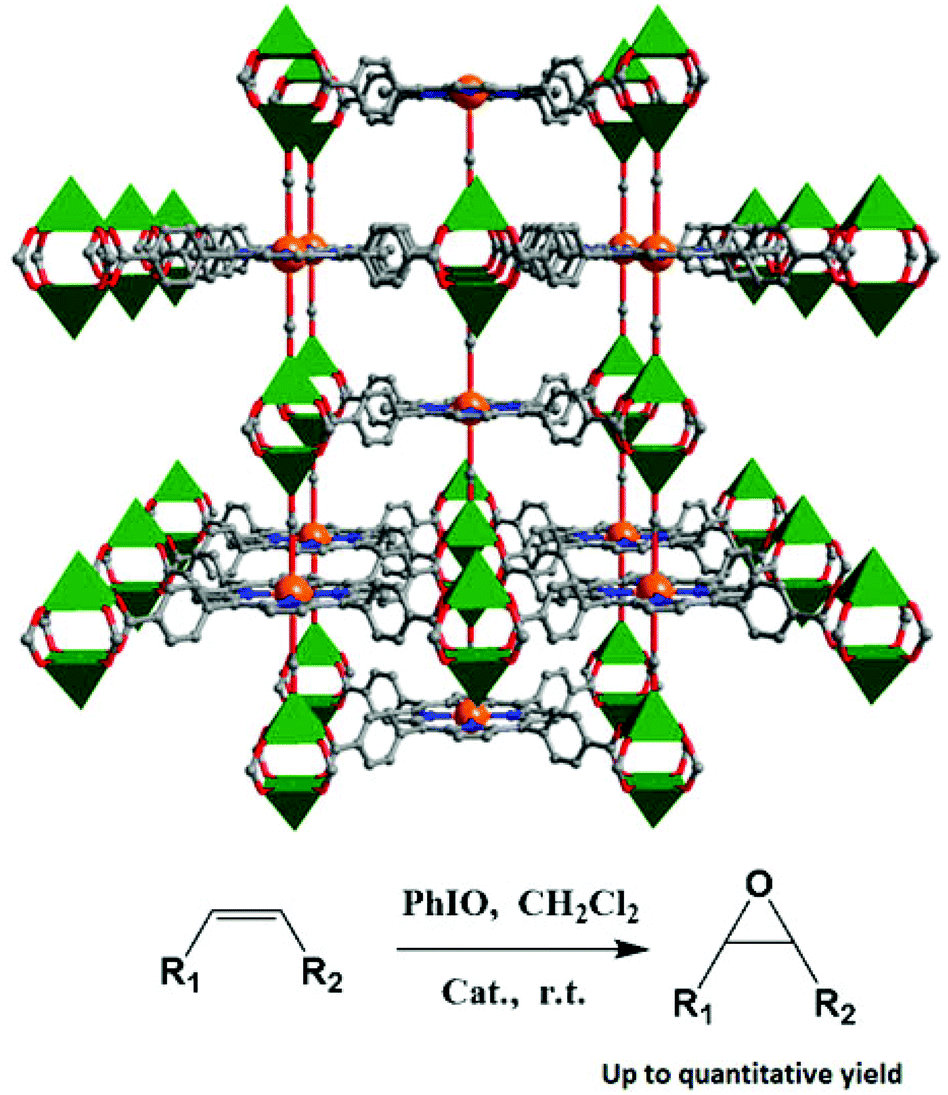 | ||
| Fig. 9 A perspective view of the 3D porphyrinic framework 9 (Mn, orange), and epoxidation of olefins catalyzed by solid 9. | ||
Because the preferred coordination geometry of palladium(II) ions is square-planar, the axial coordination sites of the porphyrin palladium(II) ions are therefore not ligated by coordination donors, as demonstrated in the porous coordination network [Cd1.25(Pd-H1.5TCPP)(H2O)]·guest (11), in which the square-coordinated palladium(II) sites are exposed on the channel walls along with the tailored zeolitic periphery (Fig. 10).26 The styrene substrate can be fully oxidized into a mixture of 91% acetophenone and 9% benzaldehyde with H2O2 oxidant under mild conditions. The catalytic property of 11 is superior to that of its components Pd-H4TCPP, PdCl2, Pd(OAc)2 and Pd/C in the cases of substrate conversions and product selectivities. The single crystal structure and 1H NMR spectroscopy clearly indicate that the styrene molecules are readily incorporated into the pores of 11, which thus demonstrates that the catalytic reaction chiefly occurs inside the pores.
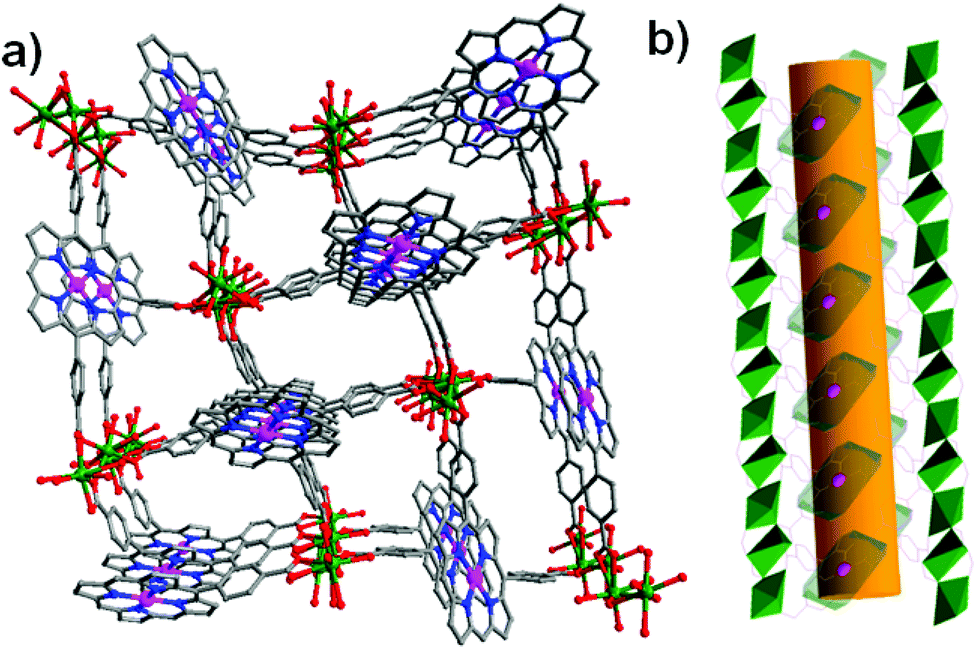 | ||
| Fig. 10 (a) The 3D structure of 11 as viewed down the a axis, and (b) the 1D opening channel with accessible PdII sites in 11. | ||
The three isostructural porous porphyrinic MOFs, [Mn5Cl2(MnCl-OCPP)(DMF)4(H2O)4]·guest (ZJU-18), [Mn5Cl2(Ni-OCPP)(H2O)8]·guest (ZJU-19) and [Cd5Cl2(MnCl-OCPP)(H2O)6]·guest (ZJU-20) demonstrate that metal-5,10,15,20-tetrakis(3,5-biscarboxylphenyl)porphyrin (M-H8OCPP), containing four m-benzenedicarboxylate moieties, is powerful for construction and stabilization of highly porous coordination frameworks.27 In the crystal structure of ZJU-18, MnIIICl-OCPP is linked by two kinds of binuclear MnII2(COO)4Cl2 and trinuclear MnII3(COO)4(μ-H2O)2(H2O)6 metal carboxylate SBUs to form a 3D porous structure of tbo net, which consists of large pore cages of about 21.3 Å and the pore windows are about 11.5 Å in diameter (Fig. 11). ZJU-18 is highly efficient for selective oxidation of alkylbenzenes (>99% yield of acetophenone). The high oxidation efficiency of ZJU-18 should be attributed to the cooperative effect between the catalytically active metal nodes and the porphyrin metal ions. This conclusion has been proved by the catalytic properties of ZJU-19 and ZJU-20, because the conversion of ethylbenzene is 9% when oxidized by ZJU-19 with active metal nodes only, and 69% when oxidized by ZJU-20 with active MnIII-porphyrins only. The catalytic activity and stability of ZJU-18 are also much superior to those of the homogeneous MnIIICl-Me8OCPP counterpart (>99% conversion for ZJU-18 vs. 16% for MnIIICl-Me8OCPP, and sustained 15 cycles vs. deactivated at the third cycle).
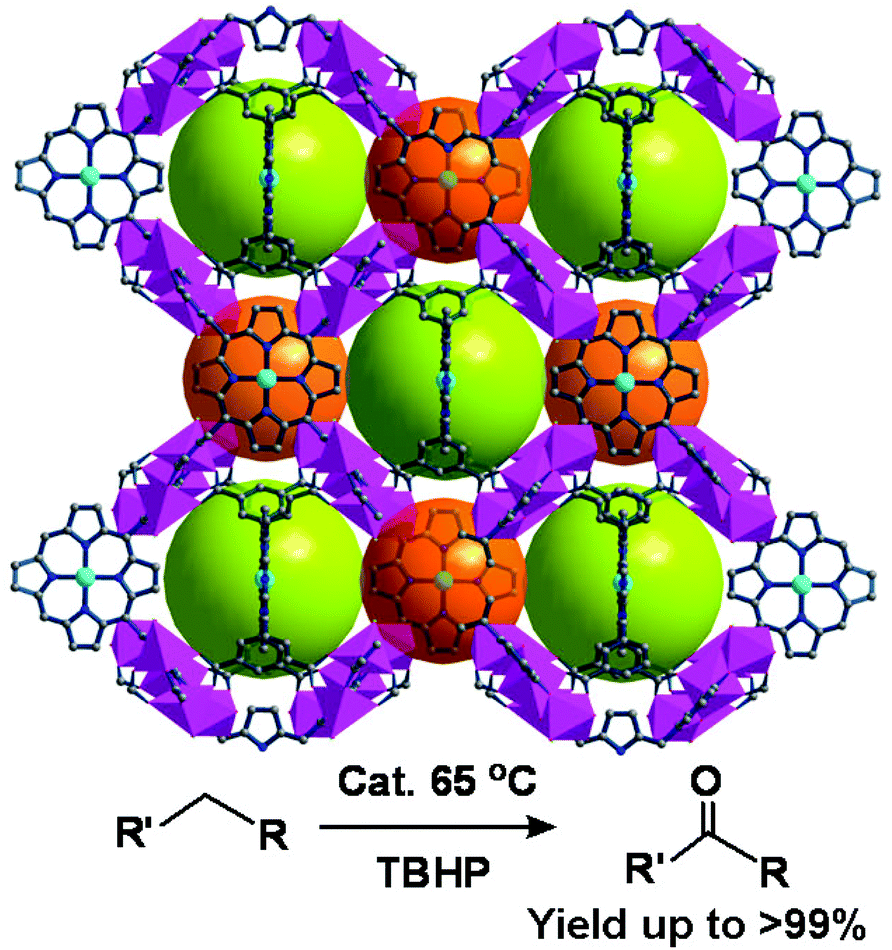 | ||
| Fig. 11 Crystal structure of ZJU-18 built from MnIIICl-OCPP linking up binuclear and trinuclear Mn-carboxylate SBUs, and catalytic oxidation of alkylbenzenes by ZJU-18. | ||
The two robust porphyrinic frameworks [Cu4(Ni-OCPP)(H2O)4]·guest (ZJU-21) and [Cu16(MnIIIOCPP)3(OH)11(H2O)17]·guest (ZJU-22) are very stable in various different solvents, even in acidified aqueous solution.28 As shown in Fig. 12, ZJU-21 is built from paddle-wheel Cu2(COO)4 SBUs and octatopic metalloligand Ni-OCPP, forming nanopore cages (2.1 nm in diameter). The structure of ZJU-22, containing 1D nanotubular channels of 1.5 nm in diameter, is a unique 3D framework constructed from three independent nets which are interconnected and stabilized by Mn–OH–Cu bridges. ZJU-21 and ZJU-22 exhibit pore structure dependent catalytic activities in the cross-dehydrogenative coupling (CDC) reaction of tertiary amines and nitroalkanes under solvent free conditions (up to 97% yield) or in water (up to 93% yield) at room temperature.
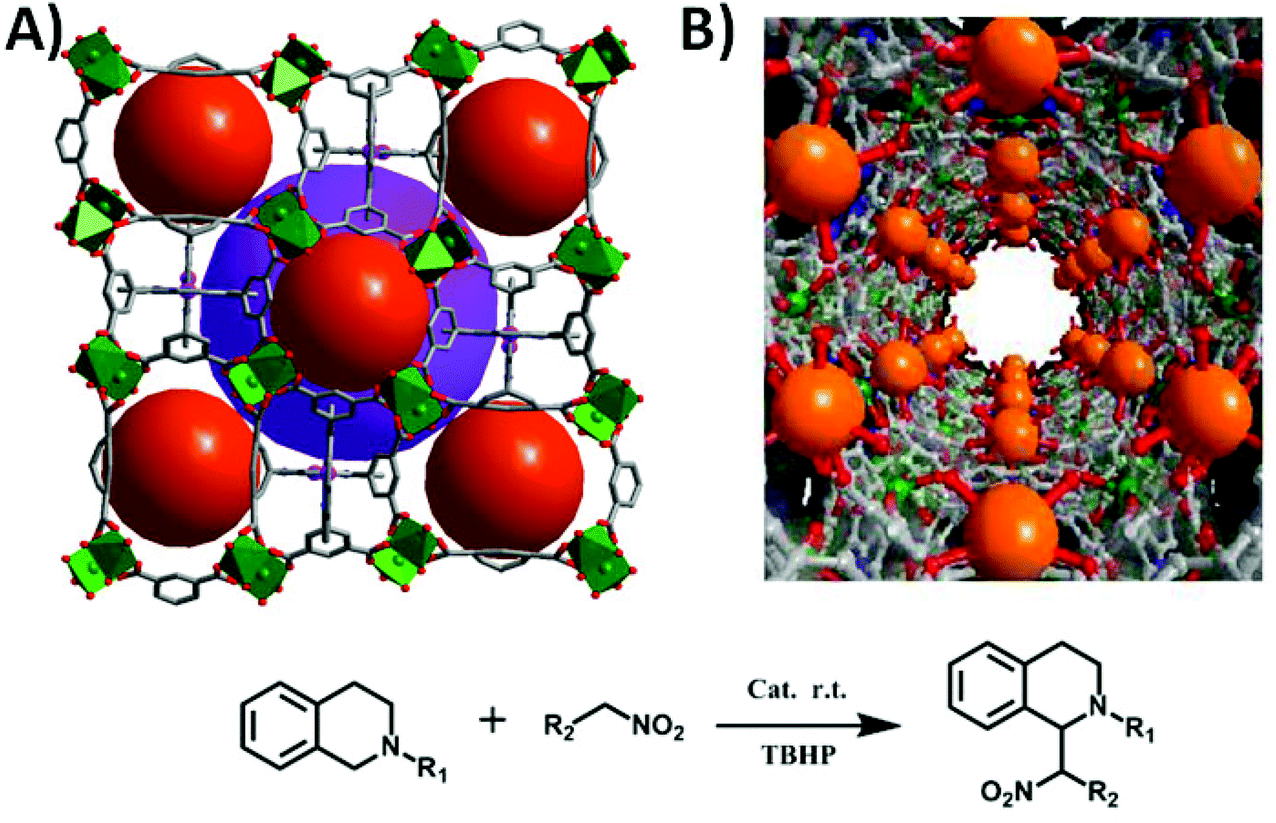 | ||
| Fig. 12 Crystal structures of the 3D porous ZJU-21 (A) and ZJU-22 (B), and the catalytic CDC reaction of 1,2,3,4-tetrahydroisoquinoline derivatives and nitroalkanes. | ||
To make use of MnIIICl-OCPP connecting with paddle-wheel Zn2(COO)3 SBUs, a porous porphyrinic framework material CZJ-4, whose structure consists of two kinds of cages and open windows of about 1.0 nm in diameter, is successfully constructed (Fig. 13).29 With immobilization of MnIII-porphyrins onto the pore surfaces of MOF, CZJ-4 exhibits high catalytic efficiency, selectivity and stability on epoxidation of olefins, in which high conversions (up to 100%) and selectivities (up to 94% epoxide product) have been achieved in the oxidation of small olefins. The catalytic property of heterogeneous CZJ-4 is superior to that of the homogeneous MnIIICl-Me8OCPP in terms of substrate conversion and product selectivity. Because the large substrates have difficulty in accessing the interior pores, CZJ-4 demonstrates excellent substrate size-selectivity in the heterogeneous epoxidation catalysis.
 | ||
| Fig. 13 Crystal structure of CZJ-4, containing two kinds of cages and substrate accessible MnIII sites inside the cages. | ||
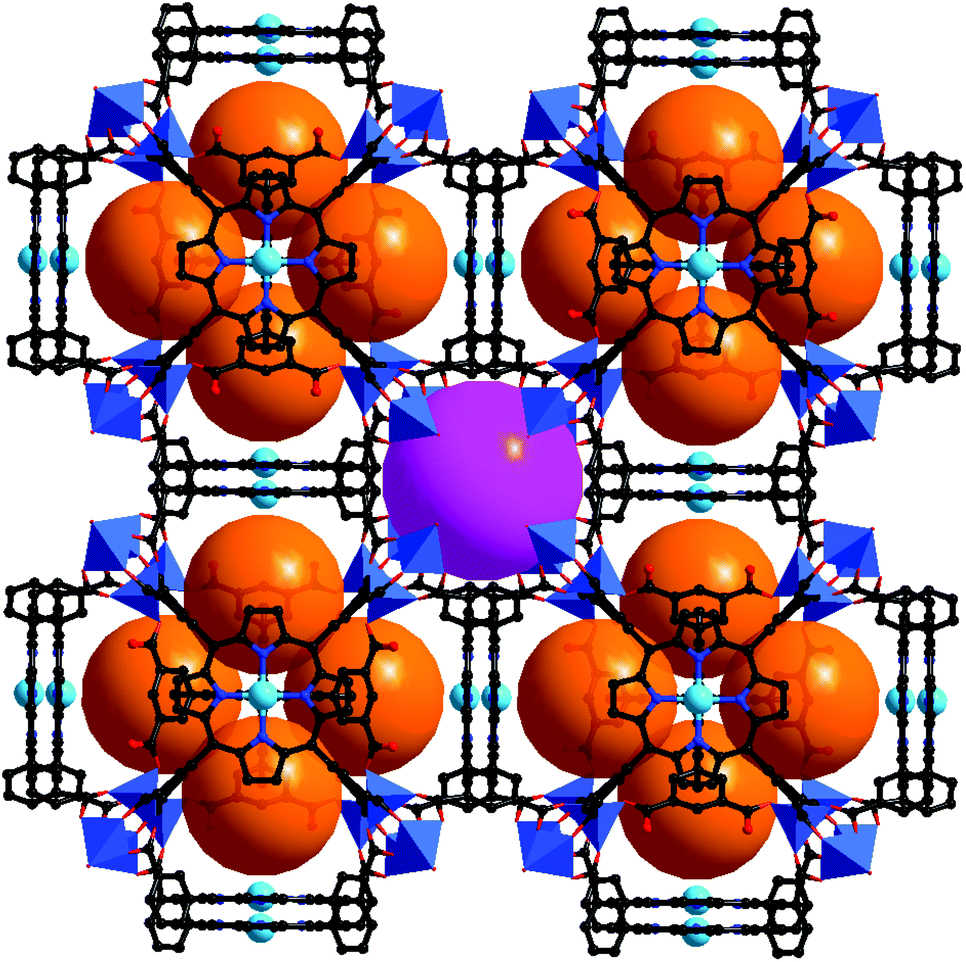
The porphyrinic framework [Zn2(MnIIIOH-TCPP)(DPNI)]·guest (CZJ-1; DPNI = N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide) is a doubly interpenetrated structure (Fig. 14).30 In the structure of CZJ-1, the paddle-wheel Zn2(COO)4 SBUs are bridged by MnIIIOH-TCPP to form 2D sheets which are further connected by DPNI to form the 3D porous structure with substrate accessible pore channels of about 6.1 × 7.8 Å2. CZJ-1 exhibits high catalytic activity for the epoxidation of styrene at room temperature, in which styrene can be almost fully oxidized into the epoxide product (97% selectivity), and the turnover number is up to 14068. Remarkably, the most inert cyclohexane substrate can be almost quantitatively oxidized by CZJ-1 at room temperature (94% conversion). The high efficiency of CZJ-1 for the activation of the inert substrate should be attributed to the collaborative work effects of multiple species, including the secondary catalytically active DPNI moieties, in the pores. The fundamental basis, to prove that the catalysis mainly takes place inside the pores and the inside active sites are accessible to the included substrate molecules, is evident from the single crystal structures of CZJ-1⊃2styrene and CZJ-1-Ti that are transformed from CZJ-1 in SCSC transformations.
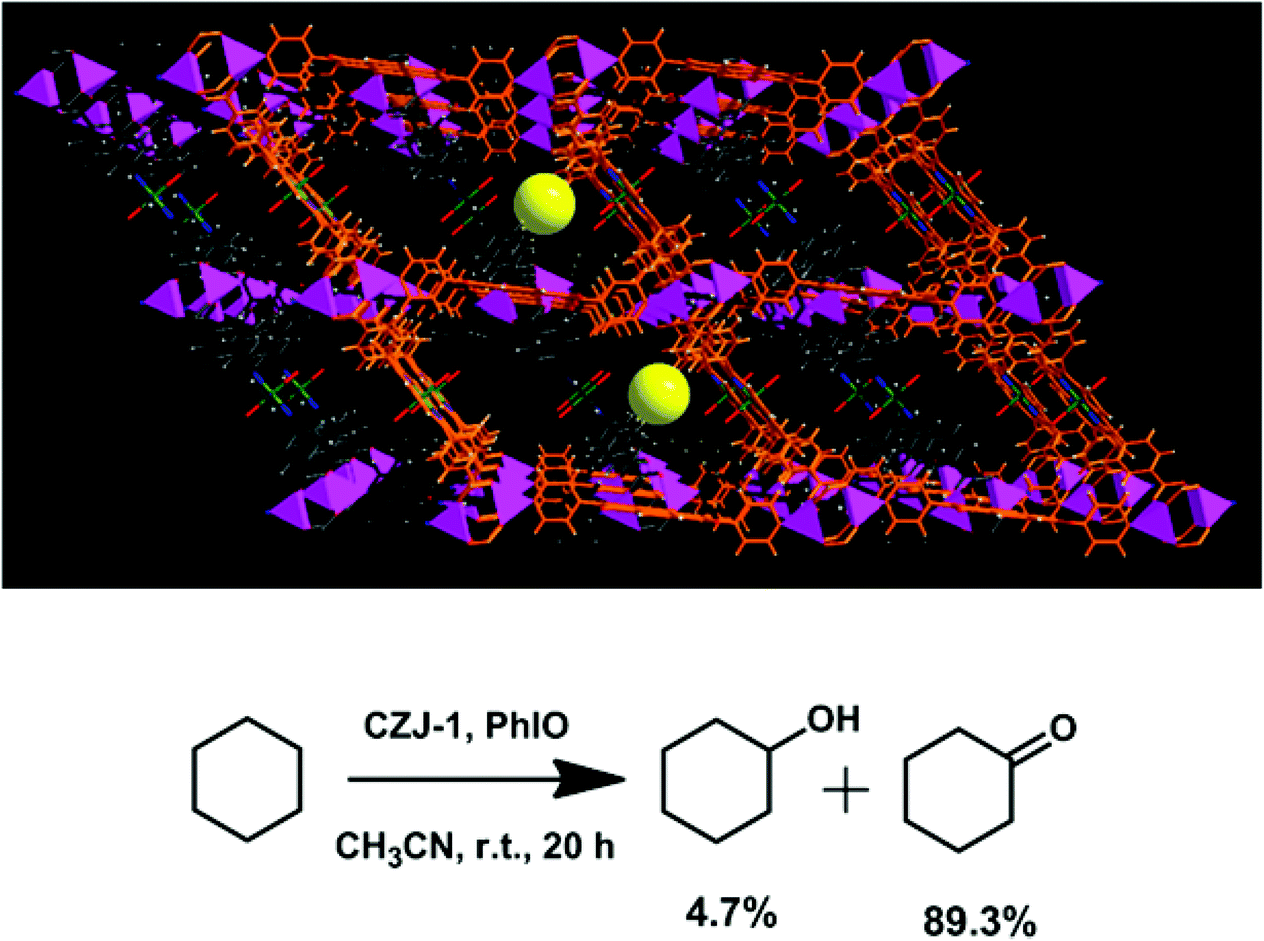 | ||
| Fig. 14 Crystal structure of CZJ-1 with uniformly immobilized MnIII sites on the pore surfaces, and highly efficient oxidation of cyclohexane catalyzed by CZJ-1. | ||
The porphyrin-POM hybrid framework, {[Cd(MnTPyP)](PW12O40)}, is built from Cd-Mn-TPyP layers and interlayer inserted [PW12O40]3− anions (Fig. 15). The hybrid material can efficiently oxidize alkylbenzene with tertbutylhydroperoxide (TBHP) as an oxidant.31 {[Cd(MnTPyP)](PW12O40)} oxidizes ethylbenzene to form one product of acetophenone (92.7% yield), which is superior to those of its precursors, such as MnIIICl-TPyP (73.6% yield) and [PW12O40]3− (inactive). Because oxidation mainly occurs inside the pores, {[Cd(MnTPyP)](PW12O40)} presents excellent size selectivity in the oxidation of a series of alkylbenzenes.
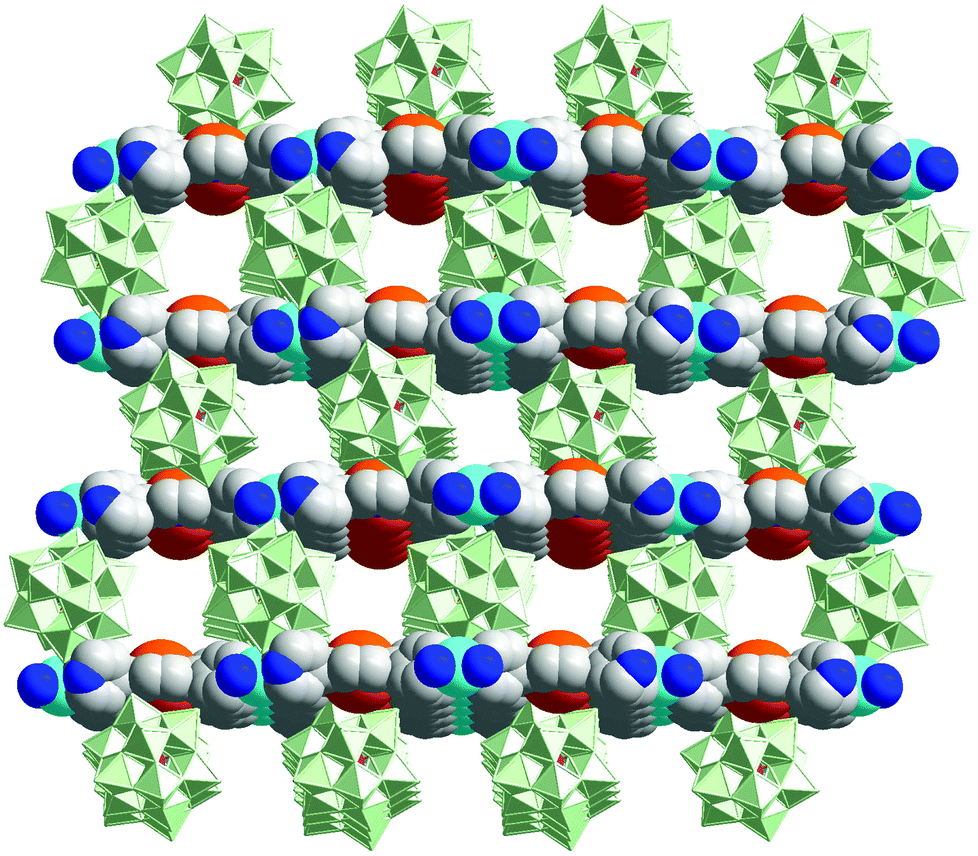 | ||
| Fig. 15 The porous structure of {[Cd(MnTPyP)](PW12O40)}, consisting of alternate layers of anionic POMs and cationic MnIII-porphyrin nets (MnIII, orange). | ||
Metalloporphyrins are excellent light sensitizers in photosynthesis. Immobilization of metalloporphyrins onto the pore surfaces of MOFs can achieve outstanding stability and photoactivity in heterogeneous phases. The 3D porous porphyrinic MOF, [Zn2(H2O)4SnIV(TPyP)(HCOO)2]·guest (12), is constructed from lamellar networks of photoactive tin(IV)-porphyrin (SnIVTPyP) building blocks linking ZnII ions, which are further connected by formates to coordinate with the SnIV centers (Fig. 16).32 MOF 12 exhibits remarkable photocatalytic activities on photo-oxygenation of phenol and sulfides under visible light irradiation, resulting in almost quantitative conversions with remarkable selectivity (>99%) in heterogeneous phases. The catalytic property of the heterogeneous catalyst 12 is much superior to that of the homogeneous counterpart SnIV(OH)2TPyP under identical conditions in terms of the catalytic activity and stability.
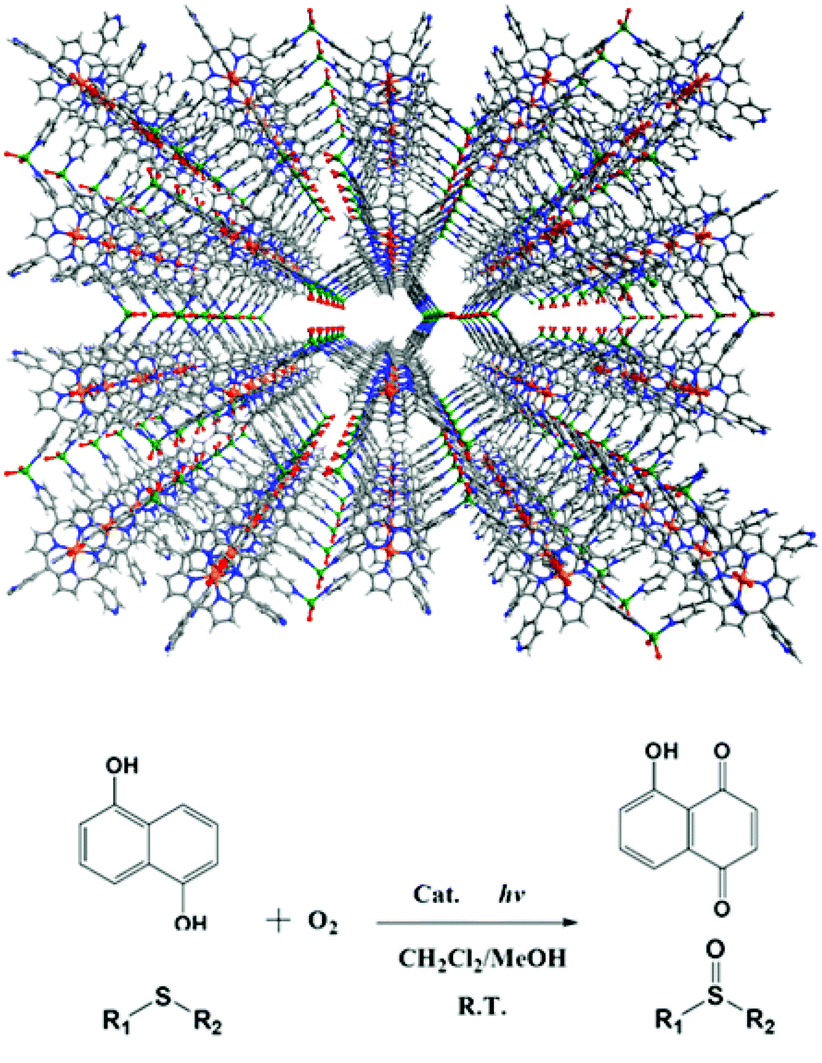 | ||
| Fig. 16 The structure of 12 and its application in photo-oxidation of phenol and sulfides. | ||
Encapsulating porphyrins and metalloporphyrins as guests or templates in the pores of MOFs in “one-pot” synthesis is an alternative strategy to stabilize the active metalloporphyrin sites in a “ship-in-a-bottle” configuration, which can prohibit self-dimerization and oxidative degradation of the metalloporphyrins. Eddaoudi et al. have encapsulated 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin (H2TMPyP) into an indium imidazoledicarboxylate-based rho-ZMOF host to generate a unique and tunable catalyst platform of H2RTMPyP (Fig. 17).33 The catalytic property of the platform can be systematically tuned by metallation of the metal-free porphyrins with different metal ions of Mn, Cu, Zn and Co ions via post-synthesis modification. They proved that Mn-RTMPyP is active for oxidation of cyclohexane to cyclohexanol/cyclohexanone in 91.5% yield with turn over number of 23.5 (based on the consumed oxidant).
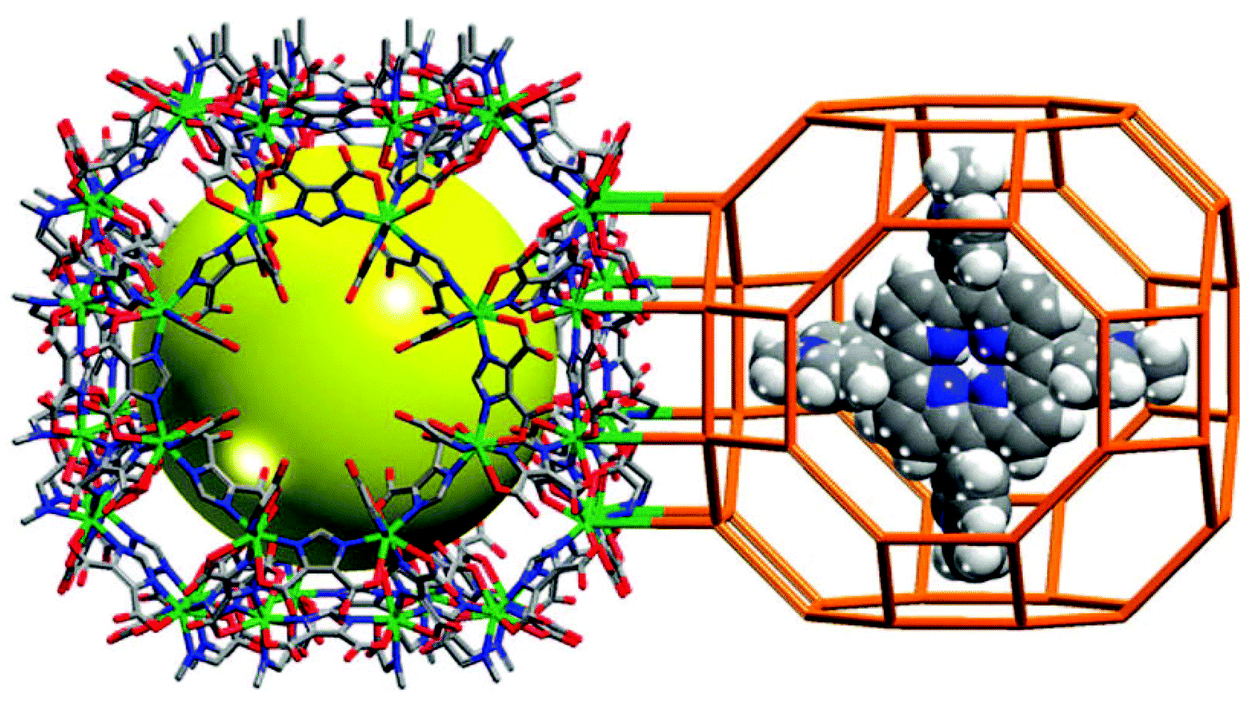 | ||
| Fig. 17 Crystal structure of rho-ZMOF (left) and schematic presentation of [H2TMPyP]4+ porphyrin enclosed in rho-ZMOF cage (right). | ||
6. Metal–organic framework catalysts constructed from amino acid moieties
Natural amino acids are the basic building units and the functional moieties in biological organisms, which have special biological activities and selective substrate-binding functions. Limited by their few available coordination donors and flexible backbones, most amino acids do not have the ability to create and stabilize highly porous MOFs. Therefore, several effective strategies have been developed to immobilize amino acid synthons onto porous MOFs.34We have reacted amino acids with aldehydes and further reduced to synthesize a series of amino acid derivatives.35 The pyridyl group on the Py-Ser ligand links copper-amino carboxylate SBUs into a 2D bilayer framework [Cu2(Py-Ser)2Cl2]·H2O (13), containing multiple chiral centers (Fig. 18).36 Compound 13 is highly active for catalyzing the Biginelli reaction of benzaldehyde, urea and ethyl acetoacetate with 90% yield of dihydropyrimidinone; however, it does not exhibit any enantioselectivity. It is interesting that 13 efficiently catalyzes the reaction of asymmetric 1,2-addition of a Grignard reagent to α,β-unsaturated ketone/aldehyde: up to 93% conversion and 99% ee value have been achieved, which are superior to those of Py-Ser ligand with 84% conversion and 51% ee value.
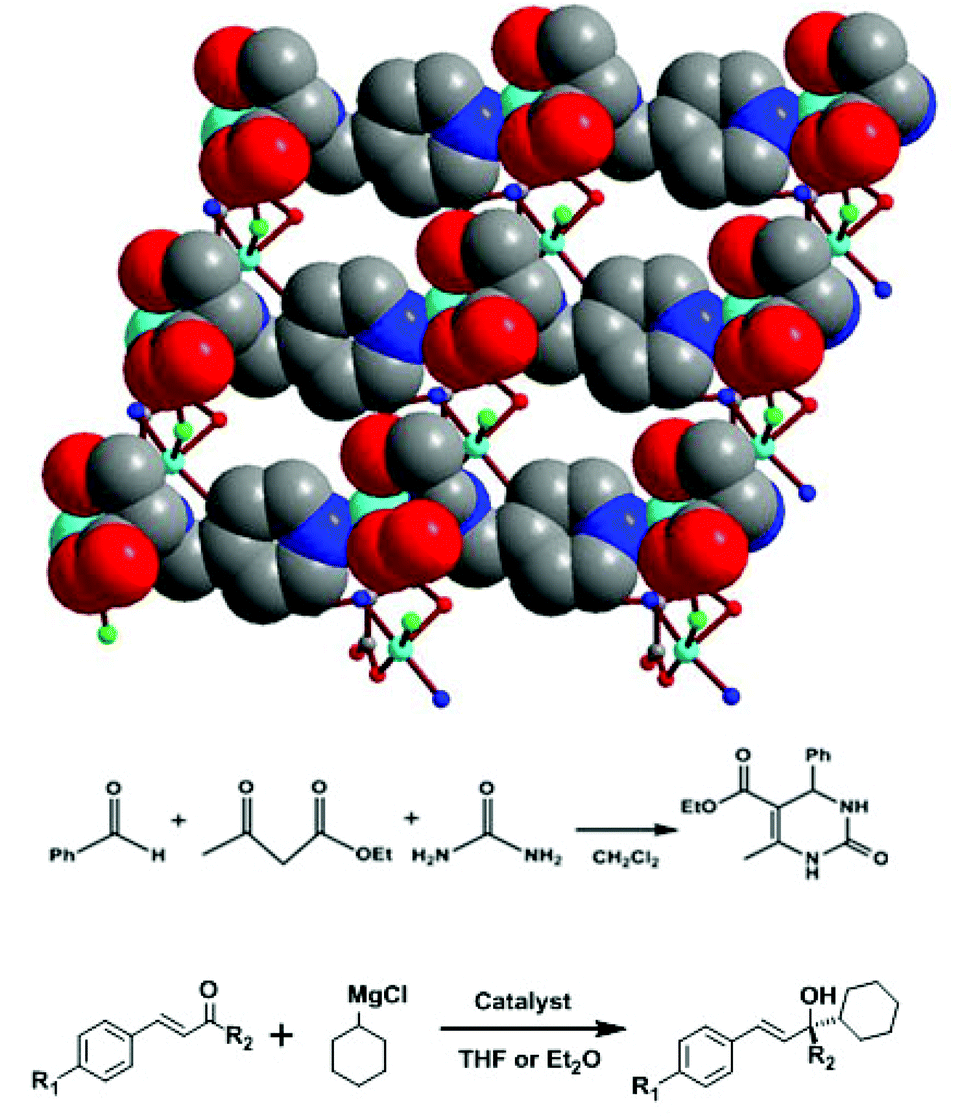 | ||
| Fig. 18 Crystal structure of 13, and Biginelli reaction of benzaldehyde, urea and ethyl acetoacetate, and 1,2-addition of α,β-unsaturated ketones catalyzed by compound 13. | ||
The Duan group has reported a homochiral 2D honeycomb coordination network Cd-BTB, which is built up from 1,3,5-tris(4-carboxyphenyl)benzene (H3BTB) linking cadmium(II) ions with rhombus channels of 4.0 × 6.5 Å2 (Fig. 19).37 The chiral L-PYI (L-pyrrolidin-2-ylimidazole) moiety, derived from spontaneous deprotection of the Boc (tert-butoxycarbonyl) group on L-N-tertbutoxycarbonyl-2-(imidazole)-1-pyrrolidine (L-BCIP), terminally coordinates to cadmium ions. It is interesting that L-PYI moieties are located above and below the 2D layer in Cd-BTB, which lets Cd-BTB be highly active for the aldol reaction, in which 61% ee value has been achieved in the reaction between 3-nitrophenyl and cyclohexanone. Because the small pore cannot accommodate substrate molecules, the catalytic reaction only occurs on the exterior solid surfaces of Cd-BTB.
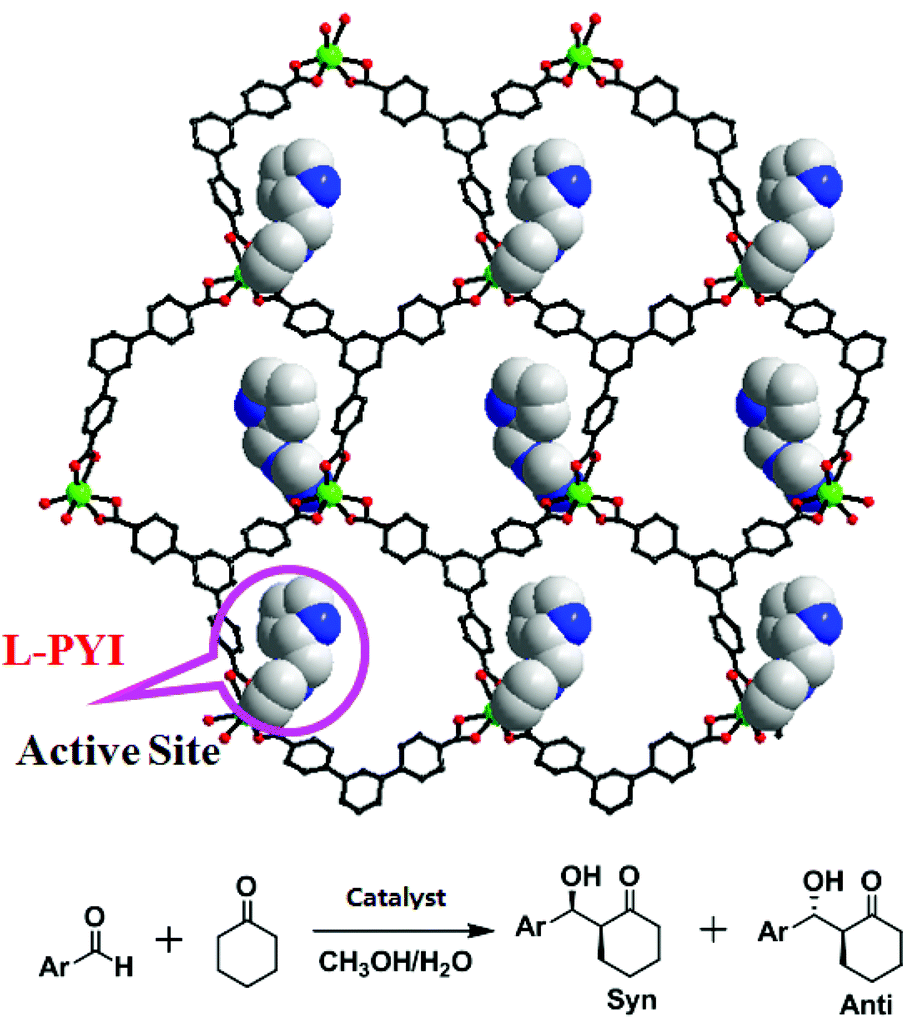 | ||
| Fig. 19 The 2D honeycomb network of Cd-BTB with active L-PYI sites (space-filling model) in the pores, and the catalytic aldol reaction of aldehydes with cyclohexanone. | ||
The photoactive MOF Zn-BCIP1 is a 2D brick wall lamellar network, built from 3-connected binuclear zinc nodes and 4,4′,4′′-nitrilotribenzoate (H3TCA) bridges (Fig. 20).38 The dimensions of the porous channels are 12 × 16 Å2, which contain chiral L-BCIP moieties that coordinate with zinc ions. The homochiral MOF can be simply activated to deprotect the proline unit by heating. The resulting new MOF Zn-PYI1 is efficient for photocatalytic alkylation of aldehydes coupled with diethyl 2-bromomalonate. Zn-PYI1 catalyzes the reaction of phenylpropyl aldehyde with 2-bromomalonate to generate the corresponding product in 74% yield with excellent enantioselectivity of 92%. Similar results have been achieved when the enantiomorph Zn-PYI2 was used to prompt the reaction, besides producing the product of opposite chirality.
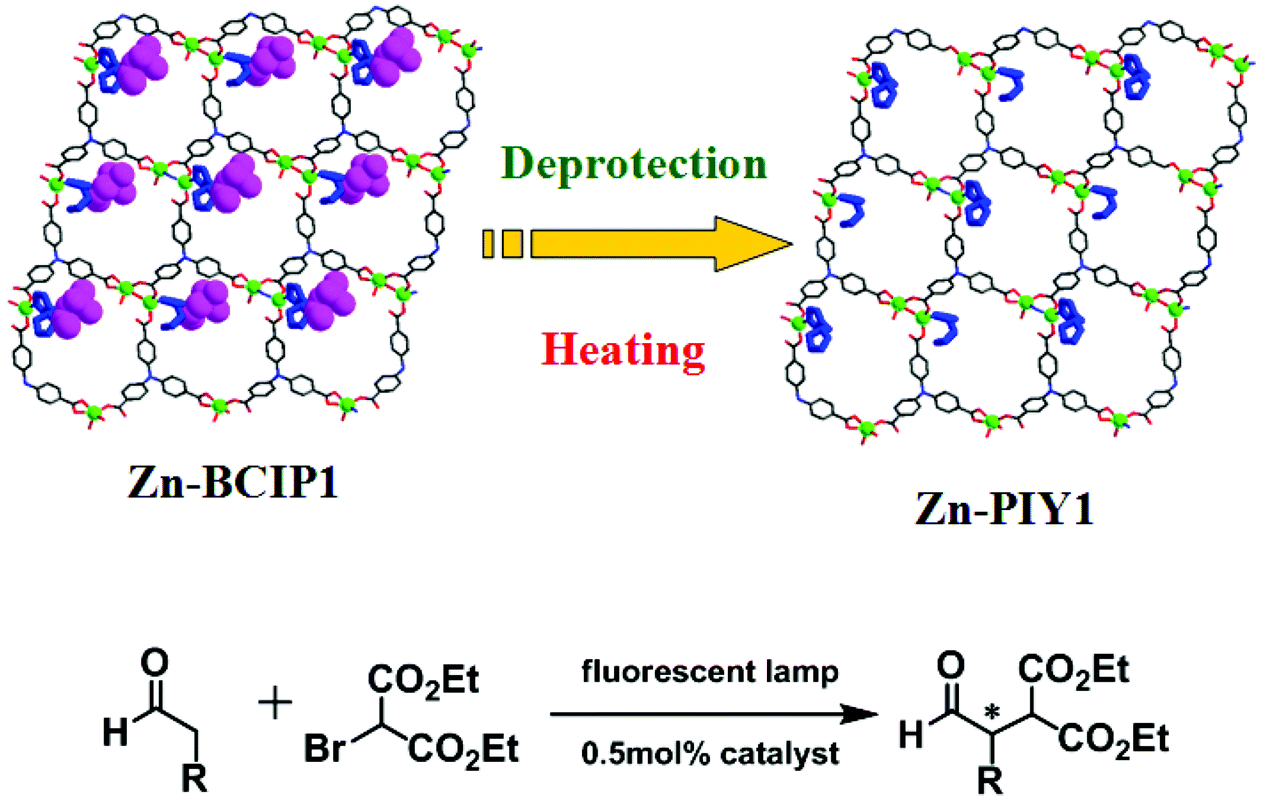 | ||
| Fig. 20 Illustration of the structural transformation from Zn-BCIP1 to Zn-PYI1 by removal of the protective groups on the pyrrolidine moieties, and photocatalytic α-alkylation of aliphatic aldehydes. | ||
7. Post-modification of metal–organic frameworks for heterogeneous catalysis
Limited by current technologies on growing high quality single crystals and single crystal X-ray diffraction structural analysis, it is not a reality to immobilize any desired functional groups onto porous MOFs by in situ synthesis. Fortunately, we can graft some reactive moieties on organic linkers during the first step. These moieties, exposed on the pore surfaces of MOFs, can be further reacted with some specific moieties to generate metal–organic framework catalysts by post-synthesis.The Lin group has designed a chiral ligand based on the BINOL moiety, ((R)-6,6′-dichloro-2,2′-dihydroxy-1,1′-binaphthyl-4,4′-bipyridine, L14), which was used to construct a homochiral porous MOF [Cd3Cl6(L14)3]·guest (14).39 In the crystal structure of 14, the polymeric 1D zigzag [CdCl2]n chain SBUs, formed from chloride doubly bridging Cd(II) ions, are linked by L14 to form a non-interpenetrated 3D network with large 1D chiral open channels of ∼1.6 × 1.8 nm2 (Fig. 21). Homochiral MOF 14 was readily post-modified by taking advantage of the chiral dihydroxy groups that are accessible to Ti(OiPr)4 transferred through the large open channels. The resulting solid (designated as 15) is highly active for the heterogeneous asymmetric catalysis of addition of diethylzinc to aromatic aldehydes with high conversions and ee values, which rival to those of the homogeneous analog under identical conditions. Control experiments with a series of dendritic aldehydes of different sizes (from 0.8 to 2.0 nm) demonstrate that catalyst 15 has an interesting size-selective property, because the catalysis chiefly occurs inside the open channels.
 | ||
| Fig. 21 (Left) Crystal structure of 14 in space-filling model, showing the large 1D chiral open channels and (right) a schematic representation of the active (BINOLate)Ti(OiPr)2 catalytic sites in the open channels of 15. | ||
N-heterocyclic carbenes (NHCs) and their metalated complexes are highly efficient catalysts in numerous reactions.40 Incorporation of metal–NHCs onto the pore surfaces of porous MOFs should generate highly efficient heterogeneous catalysts. The two MOFs [Cu2L162(MeOH)2]·guest (16; H2L16 = 1,1′-methylenebis(3-(4-carboxyphenyl)-1H-imidazol-3-ium)) and [CuL16Cl2]·guest (17) are built from the same bifunctional aromatic azolium derivative L16 and CuII nodes (Fig. 22).41 In the crystal structure of 16, two copper ions are coupled by four carboxylate groups to form a paddle-wheel SBU, and further connected with L16 to form a wave-like lamellar network, containing 80-membered macrocycles (with the cross dimensions of 18.05 × 34.4 Å2). In the crystal structure of 17, the CuII centers are linked by L16 to extend into a 1D zigzag chain. The NHC precursors in 16 are able to pocket palladium sites by unmasking the imidazolium moieties under mild conditions. The post-modified solid (designated as 18) from 16 presents excellent heterogeneous catalytic activity in the Suzuki–Miyaura coupling reaction of phenylhalides with phenylboronic acids. The catalytic activity of 18 is much superior to Me2L16 and H2L16 supported Pd catalysts, Pd(OAc)2 and Pd/C (8–37% yields). However, the post-metalated solid (designated as 19) from 17 catalyzes the coupling reaction less efficiently under identical conditions (43% yield). The low catalytic efficiency of 19 could be a consequence of the imidazolium groups of L16 that are held very close to the {CuO2Cl2} units linked by strong hydrogen bonds to obstruct the post-metallation and subsequent catalysis.
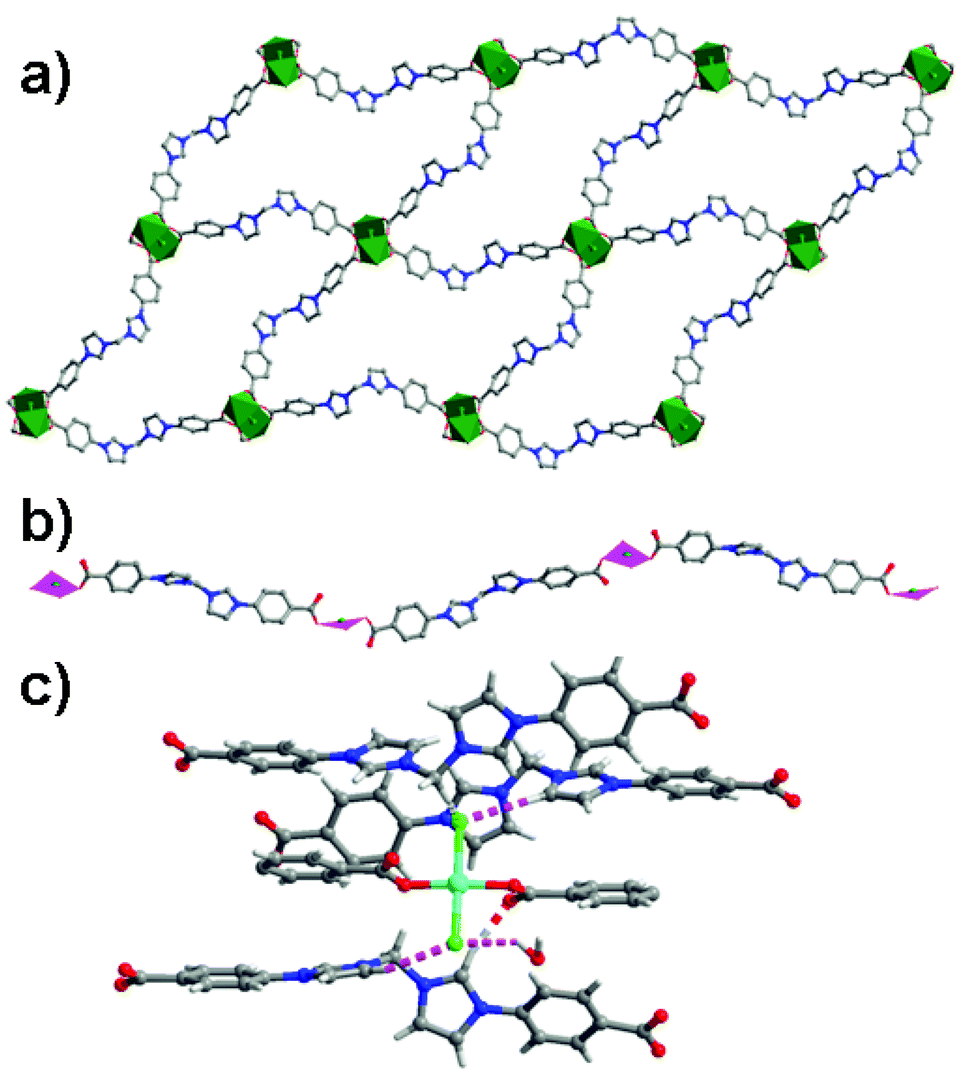 | ||
| Fig. 22 (a) A view of the lamellar network of 16 consisting of 80-membered macrocycles with post-modifiable imidazolium moieties. (b) The linear network of 17. (c) A close view of the steric congestion environments around the azolium moieties in 17. | ||
The metal–organic nanotube (MONT) [ZnL20Cl2]·guest (20; H2L20 = 1,1′-methylenebis(3-(4-carboxy-2-methylphenyl)-1H-imidazol-3-ium)) is built from L20 linking tetra-coordinated zinc ions (Fig. 23).42 MONT 20 has large 1D nanotubular channels with an exterior wall diameter of 4.9 nm and interior channel diameter of 3.3 nm. Interlocking of the nanotubes gives rise to a 3D chiral framework containing 1D helical cylindered channels with diameter of 2.0 nm. The palladium metalated solid (designated as 21) from 20 was a highly active heterogeneous catalyst for a number of reactions. Solid 21 catalyzes the coupling reaction of a range of phenylhalides with phenylboronic acids in toluene at 70 °C for 5 h, leading to almost quantitative product yields (Scheme 1). The catalytic property of 21 is superior to that of the Me2L20 supported Pd catalyst, Pd(OAc)2 and Pd/C (1.1–42% yields) for the Suzuki–Miyaura coupling reaction under identical conditions. Moreover, solid 21 is also highly active on the Heck coupling reaction of various aryl halides and olefins for 1 h with almost quantitative product yields (Scheme 2). The high efficiency and versatility of catalyst 21 were further demonstrated for the catalytic hydrogenation of olefins. Reactions proceeded smoothly at room temperature with 0.5 mol% of 21 under 1 atm H2 atmosphere, in which quantitative yields were achieved for a wide range of substrates after 1 h. These results cannot be reached by its constituents, such as Me2L20 supported Pd catalyst, Pd(OAc)2 and Pd/C (<1–5.3% yields). Moreover, solid 21 can effectively promote the reduction of large scale of (E)-ethyl cinnamate (17.6 g, 100 mmol) with 0.005 mol% catalyst loading, and the turnover number is up to 13746 after 12 h. Furthermore, with 0.5 mol% of 21, nitrobenzene was fully reduced by H2 under atmospheric pressure for 1 h at room temperature, leading to a quantitative yield of aniline.
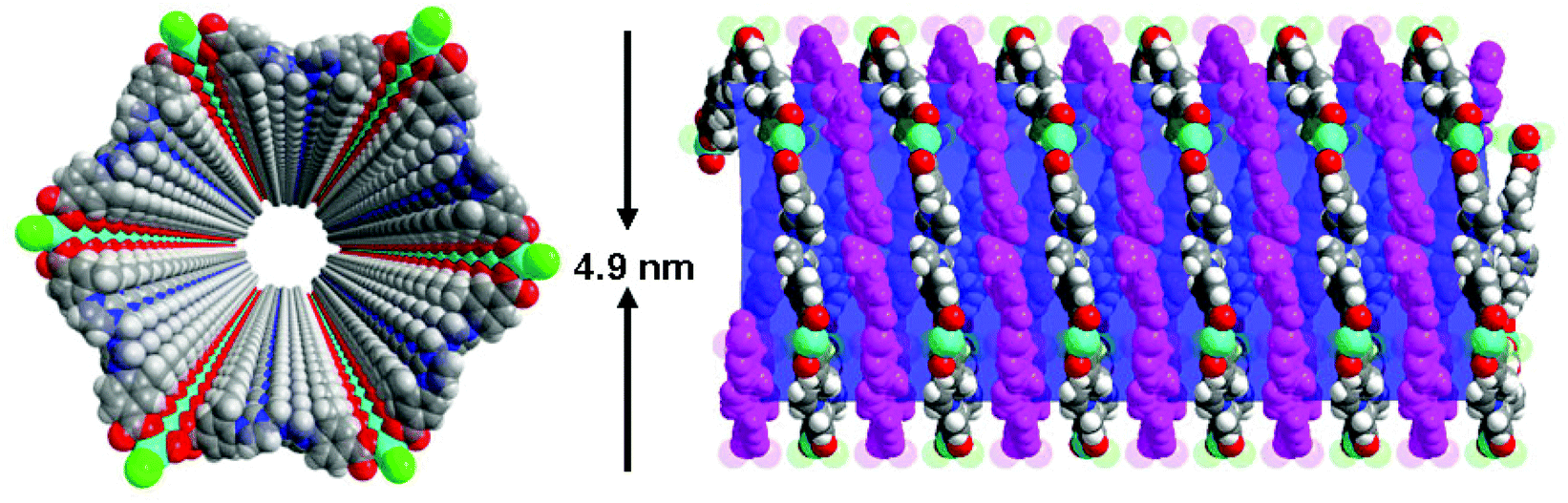 | ||
| Fig. 23 Perspective (left) and side (right) views of the open-ended hollow chiral nanotube 20. | ||
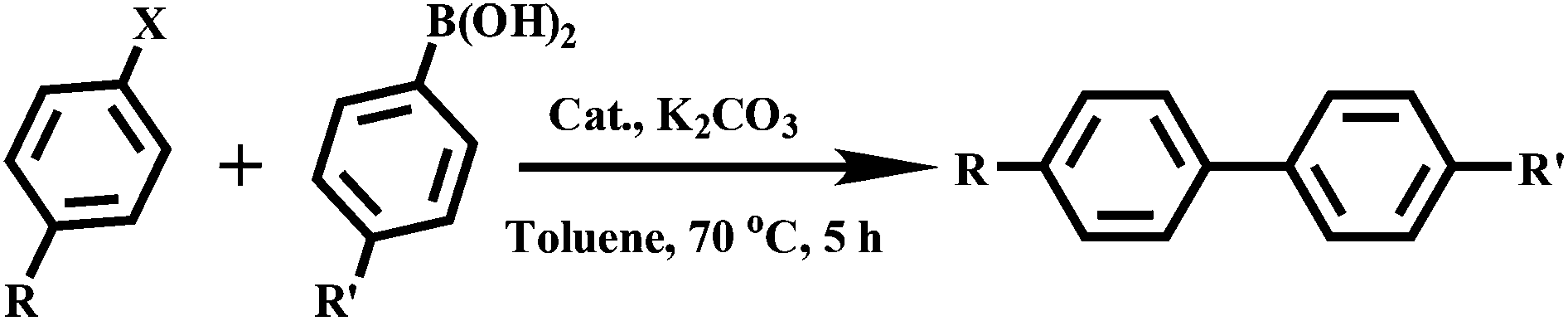 | ||
| Scheme 1 Suzuki–Miyaura coupling reaction of phenylhalides and phenylboronic acids with 0.5 mol% catalyst 21. | ||
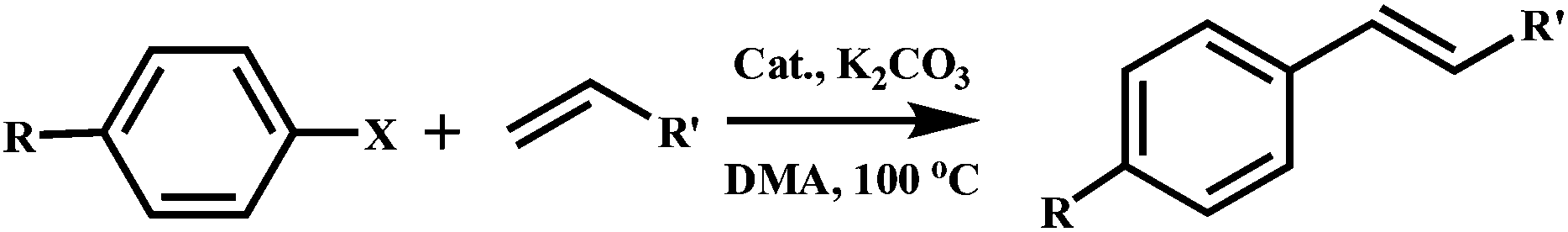 | ||
| Scheme 2 Heck coupling reaction of aryl halides and olefins with 0.5 mol% catalyst 21. | ||
8. Conclusions and outlook
This short review highlights the approaches of rational assembly of metal–organic frameworks for heterogeneous catalysis. Even though the traditional synthesis methods have the opportunity to construct some metal–organic frameworks that contain metal sites coordinated to the volatile solvent molecules, because the catalytically active metal sites are also the pivotal components of MOFs, the dissociation of the coordinating solvent molecules inevitably destroys the porous structures in some cases. Moreover, most structural arrangements of the building synthons in MOFs are not desirable for application in heterogeneous catalysis because connecting the metal sites to the organic linkers often blocks metal active sites. Therefore, the use of molecular building blocks for the synthesis of metal–organic framework catalysts has been developed by fixing the catalytic active sites on organic ligands. The facile tunability of the molecular modules allows us to precisely engineer a multitude of MOF catalysts. It is evident from the examples illustrated in this short review that the catalytic functionalities of MOFs can be designed and modulated rationally. Because their topologies are controlled by the connection between the metal coordination sites and the orientation and number of coordinating donors on the multifunctional linkers, precise selection of the structural SBUs and control of their connection ways allow us systematic modification of the pore structures. These porous materials make the active sites freely accessible to reagent molecules transferred through the opening channels. Considering that numerous homogeneous catalytically active moieties have been readily available for the synthesis of functional linkers, we foresee that there may be a burgeoning field of porous metal–organic framework catalysts, which should have a brilliant future and are undergoing explosive growth.Acknowledgements
We thank the NSF of China (Grant no. 21373180) and the research fund of Key Laboratory for Advanced Technology in Environmental Protection of Jiangsu Province (AE201312) for financial support.References
- (a) J. R. Long and O. M. Yaghi, ed., Chem. Soc. Rev., 2009, 38, 1213 RSC; (b) H.-C. Zhou, J. R. Long and O. M. Yaghi, ed., Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (c) H.-C. Zhou and S. Kitagawa, ed., Chem. Soc. Rev, 2014, 43, 5415 RSC.
- (a) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (b) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- (a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (b) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed; (c) H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed.
- E. Coronado and G. Minguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- (a) Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed; (b) M.-J. Dong, M. Zhao, S. Ou, C. Zou and C.-D. Wu, Angew. Chem., Int. Ed., 2014, 53, 1575 CrossRef CAS PubMed; (c) Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468 RSC.
- (a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed; (b) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS PubMed; (c) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- (a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC; (b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (c) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; (d) Y. Liu, W. M. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS PubMed; (e) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; (f) H. R. Moon, D.-W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807 RSC; (g) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC; (h) T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- D. W. Breck, Zeolite molecular sieves, structure, chemistry, and use, John Wiley & Sons, New York, 1974 Search PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2011, 112, 970 CrossRef PubMed.
- G.-Q. Kong and C.-D. Wu, Inorg. Chem. Commun., 2009, 12, 731 CrossRef CAS PubMed.
- C.-D. Wu, L. Li and L.-X. Shi, Dalton Trans., 2009, 6790 RSC.
- L.-X. Shi and C.-D. Wu, Chem. Commun., 2011, 47, 2928 RSC.
- M.-H. Xie, X.-L. Yang and C.-D. Wu, Chem. – Eur. J., 2011, 17, 11424 CrossRef CAS PubMed.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC.
- S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563 RSC.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef CAS PubMed.
- J. M. Falkowski, C. Wang, S. Liu and W. Lin, Angew. Chem., Int. Ed., 2011, 50, 8674 CrossRef CAS PubMed.
- C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058 CrossRef CAS PubMed.
- J. Jiang, K. Kasuga and D. P. Arnold, in Supramolecular Photosensitive and Electroactive Materials, ed. H. S. Nalwa, Academic Press, San Diego, 2001, p. 113 Search PubMed.
- (a) C. Zou and C.-D. Wu, Dalton Trans., 2012, 41, 3879 RSC; (b) M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199 CrossRef CAS PubMed; (c) W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841 RSC.
- C. Zou, T. Zhang, M.-H. Xie, L. Yan, G.-Q. Kong, X.-L. Yang, A. Ma and C.-D. Wu, Inorg. Chem., 2013, 52, 3620 CrossRef CAS PubMed.
- M.-H. Xie, X.-L. Yang and C.-D. Wu, Chem. Commun., 2011, 47, 5521 RSC.
- X.-L. Yang, M.-H. Xie, C. Zou, Y. He, B. Chen, M. O'Keeffe and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 10638 CrossRef CAS PubMed.
- X.-L. Yang, C. Zou, Y. He, M. Zhao, B. Chen, S. Xiang, M. O'Keeffe and C.-D. Wu, Chem. – Eur. J., 2014, 20, 1447 CrossRef CAS PubMed.
- X.-L. Yang and C.-D. Wu, Inorg. Chem., 2014, 53, 4797 CrossRef CAS PubMed.
- M.-H. Xie, X.-L. Yang, Y. He, J. Zhang, B. Chen and C.-D. Wu, Chem. – Eur. J., 2013, 19, 14316 CrossRef CAS PubMed.
- C. Zou, Z. Zhang, X. Xu, Q. Gong, J. Li and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 87 CrossRef CAS PubMed.
- M.-H. Xie, X.-L. Yang, C. Zou and C.-D. Wu, Inorg. Chem., 2011, 50, 5318 CrossRef CAS PubMed.
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639 CrossRef CAS PubMed.
- X.-L. Yang and C.-D. Wu, CrystEngComm, 2014, 16, 4907 RSC.
- (a) X.-L. Yang, M.-H. Xie, C. Zou, F.-F. Sun and C.-D. Wu, CrystEngComm, 2011, 13, 1570 RSC; (b) X.-L. Yang, M.-H. Xie, C. Zou and C.-D. Wu, CrystEngComm, 2011, 13, 6422 RSC.
- M. Wang, M.-H. Xie, C.-D. Wu and Y.-G. Wang, Chem. Commun., 2009, 2396 RSC.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321 CrossRef CAS PubMed.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991 CrossRef CAS PubMed.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed.
- (a) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- G.-Q. Kong, X. Xu, C. Zou and C.-D. Wu, Chem. Commun., 2011, 47, 11005 RSC.
- G.-Q. Kong, S. Ou, C. Zou and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 19851 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |