 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The activation of Woollins' reagent. Isolation of pyridine stabilised PhPSe2†
Laura
Ascherl
ab,
Andreas
Nordheider
a,
Kasun S. Athukorala
Arachchige
a,
David B.
Cordes
a,
Konstantin
Karaghiosoff
b,
Michael
Bühl
a,
Alexandra M. Z.
Slawin
a and
J. Derek
Woollins
*a
aSchool of Chemistry, University of St Andrews, St Andrews KY16 9ST, Scotland. E-mail: jdw3@st-andrews.ac.uk
bLudwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 München, Germany
First published on 23rd April 2014
Abstract
Woollins' reagent (WR, (PhPSe2)2) plays an essential role in the selenation of organic compounds. Reaction of WR with pyridine gives the P(V) species PhPSe2 stabilised by pyridine coordination which is the first crystallographically characterised mononuclear RPSe2 system stabilised by an external molecule and has potential as a selenation reagent for reactions under mild conditions.
Species with the molecular formula RPE2 (E = O, S, Se, Te) exhibit a very unusual bonding situation: phosphorus is in the formal oxidation state of +V but only tricoordinated (σ3λ5). The coordination sphere of σ3λ5 phosphoranes is unsaturated, therefore a strong Lewis acidity is expected.1 To prevent these species from dimerisation, two main routes are considered in synthesis: (A) stabilisation of the monomeric form with sterically demanding substituents and (B) provision of an intramolecular or intermolecular species, which is able to fill the electronic gap at the phosphorus atom.2
The monomeric form of Woollins' reagent (WR),3 a phenyldiselenoxophosphorane exhibiting a σ3λ5 phosphorus atom has been postulated.4 In comparison, the sulfur analogue of WR, Lawesson's reagent (LR) (Fig. 1),5 is known to be in equilibrium with its dithiophosphine ylide in solution. This ylide is said to be the reactive intermediate when it comes to thionation processes with LR.6
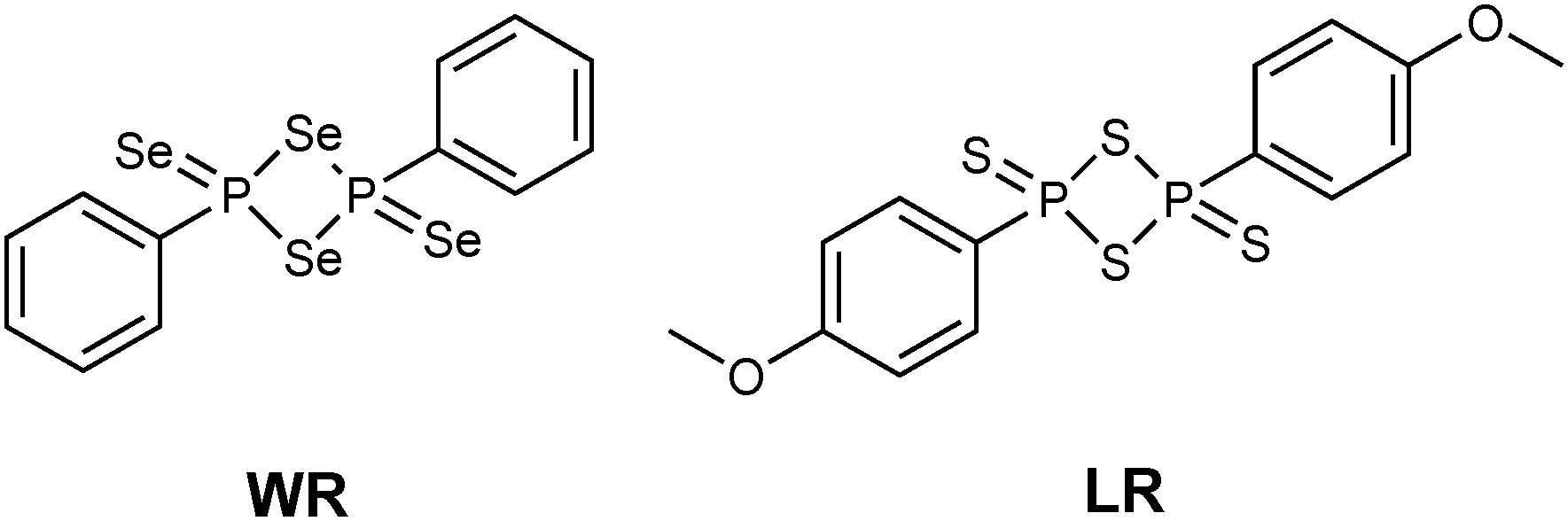 | ||
| Fig. 1 Molecular structures of WR and LR. | ||
This supports the assumption that a similar equilibrium and thus a monomeric form can be found for WR as well.
Stirring WR in pyridine for 30 min indeed is a facile way to stabilise this σ3λ5 bonding situation, thus eliminating the need to use bulky substituents (Scheme 1). The resulting new phenyldiselenoxophosphorane 1a is formed quantitatively.7
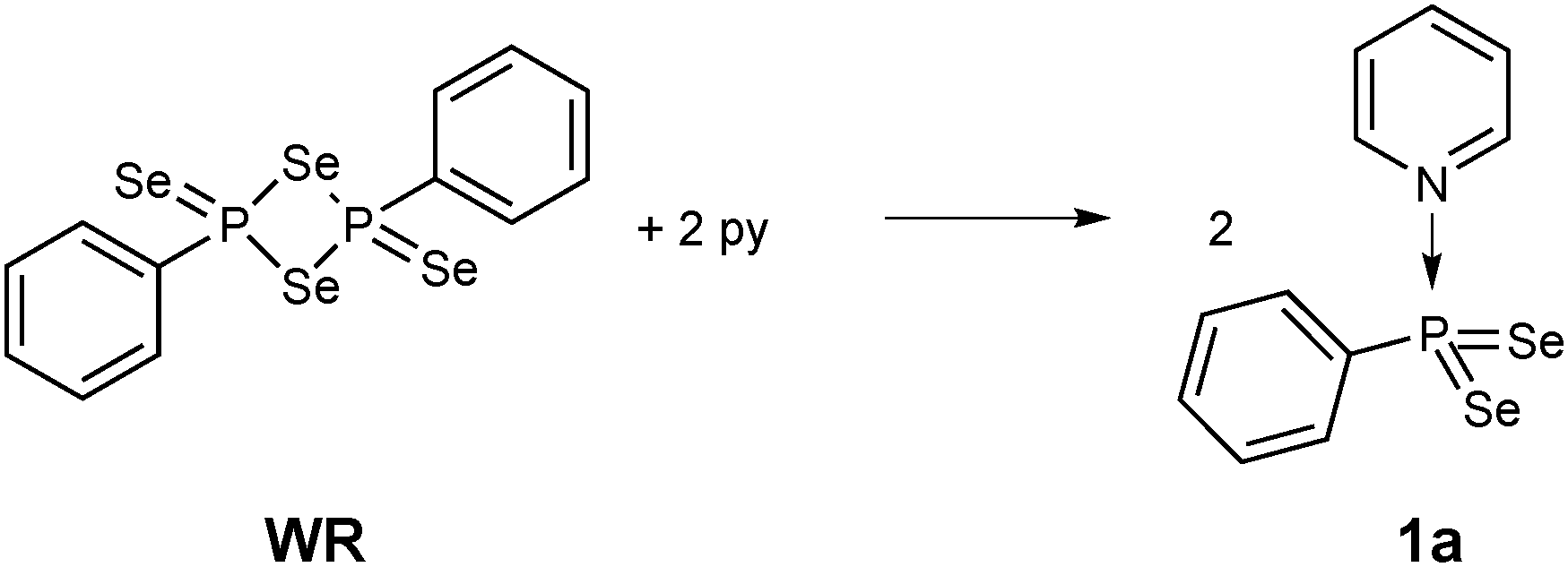 | ||
| Scheme 1 Reaction of Woollins' reagent WR with pyridine to form the pyridine adduct 1a. | ||
The 31P NMR spectrum of 1a consists of a singlet at 101.6 ppm with satellites representing the 1JPSe coupling of −808.4 Hz, adequate for the representation of a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se double bond. Consistently, the 77Se NMR signal appears as a doublet at 91.9 ppm.
Se double bond. Consistently, the 77Se NMR signal appears as a doublet at 91.9 ppm.
Storing the resulting yellow solution at −40 °C for two days yielded yellow, prism shaped crystals of 1a·py (Fig. 2).
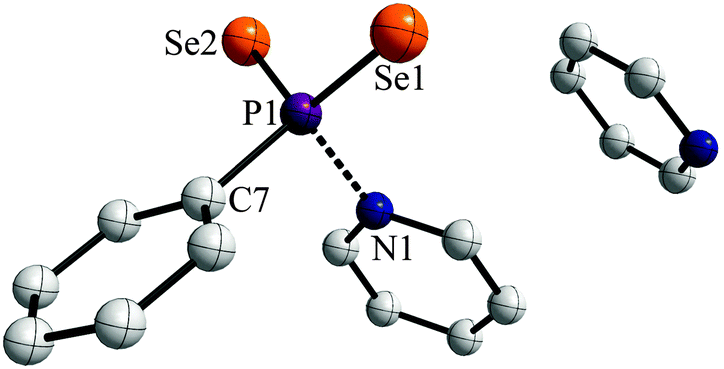 | ||
| Fig. 2 X-ray structure of 1a⋯py in the solid state; hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): P1–Se1 2.108(3), P1–N1 1.886(7), P1–Se2 2.106(3), P1–C7 1.830(7), Se1–P1–Se2 120.30(9), Se1–P1–C7 112.1(3), Se2–P1–C7 111.8(3). | ||
The two PSe double bonds in the range of 2.106(3) to 2.108(3) Å are comparable to those reported for WR (2.102(3) Å).3e Interestingly, the P1–N1 distance (1.886(7) Å) is significantly longer than usual PV–N single bonds reported before (e.g. 1.652(3) Å in PhP(NHCH2Ph)2Se, 1.665(2) Å for [iPrNH2iPr][PhPSe2NHiPr]8a or 1.617(6)–1.688(3) Å in [(tBu(H)N(Se)P)2(μ-NtBu)2]8b or 1.800(4) Å for Na[H3NO3P]8c). This suggests that the pyridine entity is bonded rather weakly to the phenyldiselenoxophosphorane and can be abstracted quite easily in order to release the reactive intermediate. In contrast a N→PV donor–acceptor distance was reported to be 2.039(5) Å for a 2,4-di-tert-butyl-6-(l-piperidino)phenyl stabilized PSe2 system, which prompted us to investigate the bonding situation in 1a in more detail.9
In order to analyse the unusual bonding situation and predict the reactivity of the pyridine adduct 1a compared to WR itself, density functional theory (DFT) and ab initio calculations were performed, calling special attention to the strength of the P–N interaction in 1a. As a result the strength of this interaction depends strongly on the surrounding medium and, to a much lesser extent, on the method that is used.
Compared to the solid state structure, the popular B3LYP functional overestimates the P–N distance significantly by ca. 0.27 Å, when the optimisation is conducted in the gas phase (see Table S1 in the ESI†). The ab initio MP2 method furnishes a shorter P–N bond, but still 0.16 Å longer than in the solid. Adduct 1a is characterised by a large dipole moment, 8.8 D at the B3LYP level. When the molecule is immersed in a polarisable continuum mimicking the solvent pyridine, the P–N bond contracts significantly (by ca. −0.18 Å and −0.12 Å at B3LYP and MP2, respectively, Table S1, ESI†) and the dipole moment increases (to 13.7 D at B3LYP). A further, minor bond-length decrease by ca. 0.01 Å is obtained with increasing polarity of the surrounding medium (as might be expected for a crystal consisting of highly polar molecules). This situation is reminiscent of other donor–acceptor complexes such as BH3NH3 and related species, where even larger gas-to-solid bond contractions can be found.10
The nature of the P–N bond was probed through the Wiberg Bond Index (WBI),11 an indicator for the extent of covalent bonding, which approaches a value close to one for true single bonds. On-going from the gas phase into the continuum modelling pyridine, the P–N WBI increases from 0.31 to 0.44 (B3LYP), suggesting a strong covalent character in addition to the electrostatic interactions and charge-transfer that give rise to the high dipole moment.
According to natural population analysis (NPA),12 the charge transfer from the pyridine to the PhPSe2 moiety amounts to ca. 0.32e (the resulting electrostatic potential, which is free from ambiguities of population analyses, is shown in Fig. 3). In an unconstrained search for the localised natural bond orbitals (NBOs), the key bonding orbital between P and N is labelled as a lone pair on N, but with a rather low occupancy (1.62) and a large donor–acceptor interaction with a low-occupancy NBO on P (according to second-order perturbation analysis). When a P–N NBO is enforced using the CHOOSE option, it is strongly polarised towards N (77% contribution from the latter). Taken together, the P–N interaction shows the characteristics of a highly polar donor–acceptor bond with significant covalent character. In order to assess the strength of the P–N interaction in 1a, we have computed the dissociation energy according to Scheme 2 using a more elaborate computational protocol (including dispersion, thermodynamic and BSSE corrections, see computational details in ESI†).
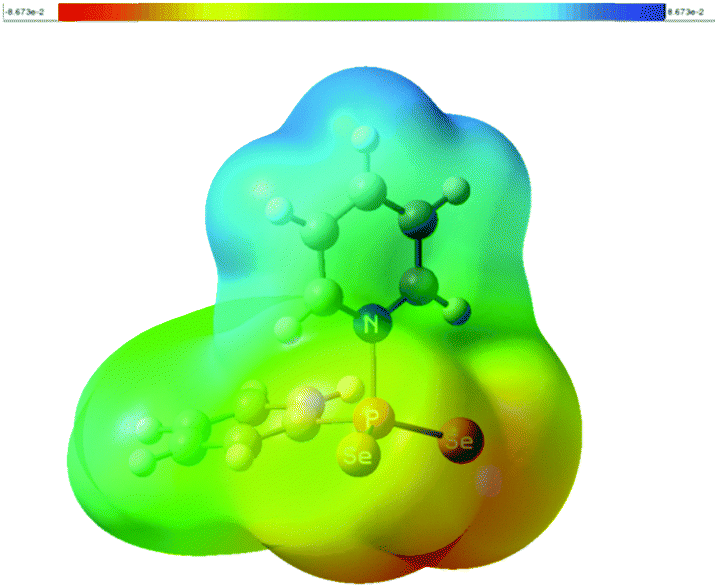 | ||
| Fig. 3 Electrostatic potential of 1a at the B3LYP/CPCM(py) level, plotted on a colour scale from +8.65 × 10−2 a.u. (blue) to −8.65 × 10−2 a.u. (red) and mapped onto an isodensity surface with ρ = 4.10−4 a.u. | ||
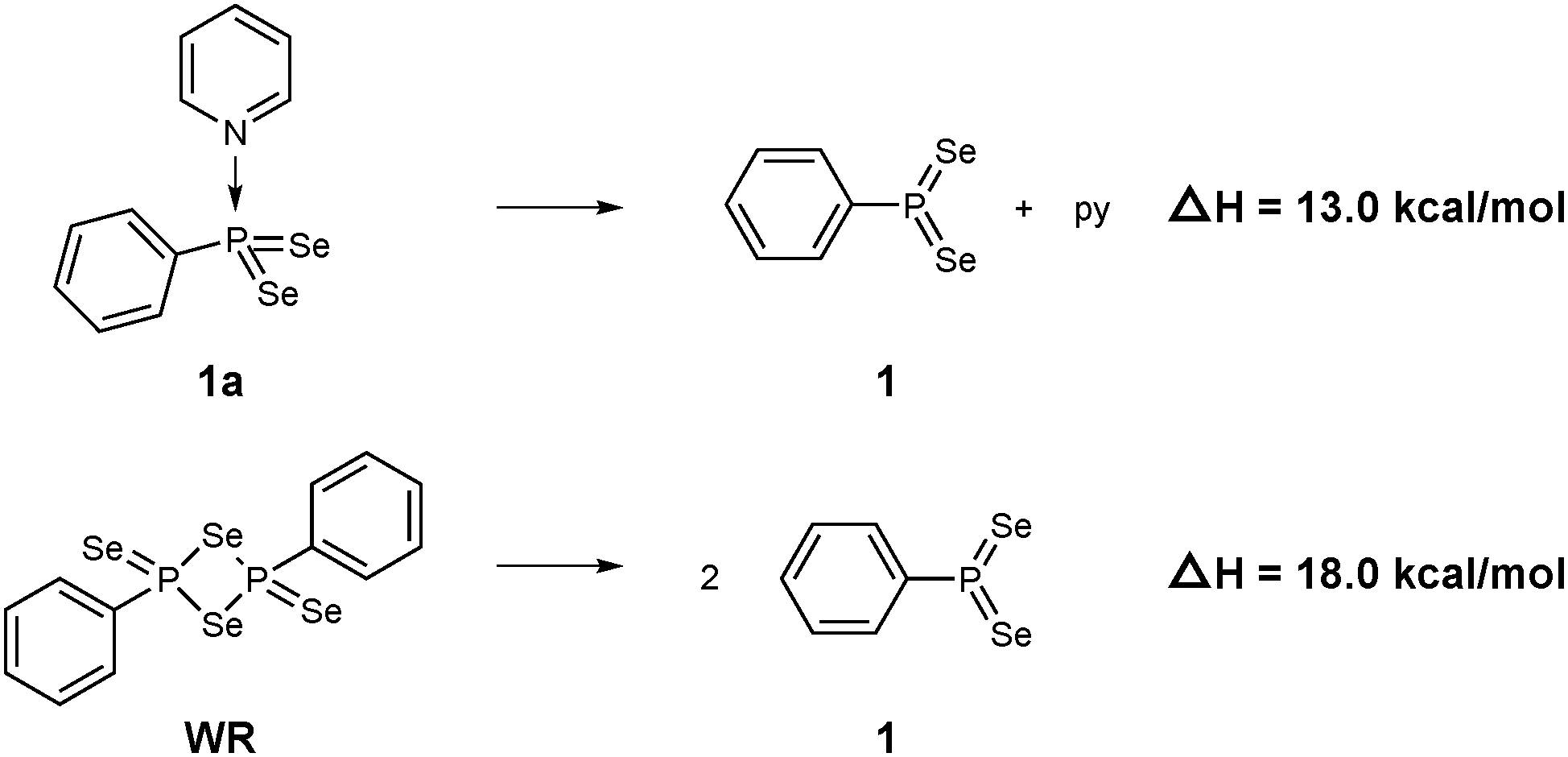 | ||
| Scheme 2 Computed dissociation enthalpies of 1a and WR into the active PhPSe2 moiety 1 (B3LYP-D3-level). | ||
At the B3LYP-D3/CPCM(py) level, the computed dissociation enthalpies and free energies at room temperature are ΔH = 13.0 kcal mol−1 and ΔG = 1.6 kcal mol−1. It should be noted that calculated entropy changes based on the ideal-gas assumption tend to overestimate entropy changes in solution when the number of particles change. This problem notwithstanding, the bond in 1a is predicted to be rather weak, weaker actually than the multicentre bonds that hold WR together: the corresponding dissociation enthalpy and free energy are ΔH = 18.0 kcal mol−1 and ΔG = 6.4 kcal mol−1, i.e. both higher by ca. 5 kcal mol−1. Barring any additional kinetic barriers, 1a should thus liberate the presumed reactive intermediate 1 more easily than WR.
As mentioned above, the coordination sphere of σ3λ5 phosphoranes is unsaturated and a strong Lewis acidity can therefore be expected. This assumption has been proved multiple times by the fact that dithiophosphoranes react willingly with nucleophiles such as, among others, methanol, acetylenes or dienes.1b,13 Furthermore dithiophosphorane species are known to undergo nucleophilic attack from the oxygen of carbonyl groups.6b
In order to get a first insight into the reactivity of 1a towards nucleophiles, the compound has been reacted with selected substrates that had been successfully reacted with WR.
Thus, the reaction of methanol with 1a in pyridine at 50 °C yielded O-methyl Se-hydrogen phenylphosphonodiselenoate as found for the reaction of WR with methanol (Scheme 3).14 Observed were short reaction times, mild conditions and as an advantage the solubility of 1a in pyridine, whereas a suspension has to be used for WR.
 | ||
| Scheme 3 Reaction of WR and 1a yield the same product (O-methyl Se-hydrogen phenylphosphonodiselenoate). | ||
In contrast, the reaction of 1a with diphenylacetylene,13c benzamide15 or DMF16 led to a different product distribution in the 31P and 77Se NMR when compared to WR. This indicates a rather different reactivity of 1a in comparison to WR that provides new possibilities in future investigations.
In terms of air sensitivity 1a seems to be less stable compared to WR. Upon exposure to air for about 15 min 1a decomposed to form the dianionic hexaselenodiphosphonate 2a (Fig. 4).17 The pathway of the formation is most likely a result of a hydrolysis process followed by subsequent oxidation. Although traces of water and oxygen probably lead to the formation of the dianionic species 2a, the compound itself is air sensitive and prone to disproportionation. Thus, after two weeks, the formation of the dipyridiniumphenyltriselenophosphonate 3a (Fig. 4) could be observed by storing a solution of 2a at room temperature.17 This compound is likely to be formed via disproportionation of 2a to 3a and elemental selenium, which precipitates from the solution.
 | ||
| Fig. 4 Molecular structures of 2a and 3a. | ||
However, pyridine is not unique in being able to stabilise the monomeric WR: Using γ-picoline (4-methylpyridine) instead results in similar 31P and 77Se NMR spectra as well as similar decomposition products 2b and 3b.18 This suggests that γ-picoline also forms the stabilised σ3λ5 adduct, analogous with that of pyridine, and opens the possibility to fine tune the reactivity and stability of these new species.
To conclude, the new phenyldiselenoxophosphorane 1a was synthesised and characterised. DFT and ab initio calculations indicate a weakly covalent, but highly polar donor–acceptor bond for the P–N interaction. Furthermore, quantum chemical calculations showed that the reaction of WR with pyridine indeed results in an activation of WR forming a σ3λ5 stabilised species. The release of the reactive intermediate 1a and therefore the corresponding dissociation enthalpy is ca. 5 kcal mol−1 lower for 1a compared to WR.
The authors are thankful to the School of Chemistry and EaStCHEM for support and for access to a research computing facility maintained by Dr H. Fruchtl.
Notes and references
- (a) H. Beckmann, G. Großmann, G. Ohms and J. Sieler, Heteroat. Chem., 1994, 5, 73 CrossRef CAS; (b) J. Navech, J. P. Majoral and R. Kraemer, Tetrahedron Lett., 1983, 24, 5885 CrossRef CAS.
- M. Meisel, J. Pauli and C. Donath, in Phosphorus-31 NMR Spectral Properties in Compound Characterization and Structural Analysis, ed. L. D. Quin and J. G. Verkade, Wiley-VCH, 1994, p. 131 Search PubMed.
- (a) G. Hua and J. D. Woollins, Angew. Chem., 2009, 121, 1394 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1368 ) CrossRef; (b) G. Hua, J. M. Griffin, S. E. Ashbrook, A. M. Z. Slawin and J. D. Woollins, Angew. Chem., 2011, 123, 4209 ( Angew. Chem., Int. Ed. , 2011 , 50 , 4123 ) CrossRef; (c) G. Hua, A. M. Z. Slawin, R. A. M. Randall, D. B. Cordes, L. Crawford, M. Bühl and J. D. Woollins, Chem. Commun., 2013, 2619 RSC; (d) J. D. Woollins, Synlett, 2012, 1154 CrossRef CAS; (e) P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2001, 300 RSC.
- K. Karaghiosoff, Habilitation Dissertation, Munich, 1997 Search PubMed.
- H. Hoffmann and G. Schumacher, Tetrahedron Lett., 1967, 8, 2963 CrossRef.
- (a) H. Z. Lecher, R. A. Greenwood, K. C. Whitehouse and T. H. Chao, J. Am. Chem. Soc., 1956, 78, 5018 CrossRef CAS; (b) T. B. Rauchfuss and G. A. Zank, Tetrahedron Lett., 1986, 27, 3445 CrossRef CAS; (c) M. Jesberger, T. P. Davis and L. Barner, Synthesis, 2003, 1929 CrossRef CAS.
- Woollins' reagent (WR) (532 mg, 1.00 mmol) was stirred in dry pyridine (5 mL) for 30 min. The reaction mixture was filtered (Schlenk sinter, pore 3) to remove traces of the slight excess of elemental selenium within the Woollins' reagent to yield a dark yellow solution of 1a in quantitative yield. Colourless, air sensitive crystals were obtained at −40 °C after two days. 31P NMR (202.5 MHz, C6D6): δ = 101.6, (s, 1JPSe = −808.4 Hz) 77Se NMR (95.4 MHz, C6D6): δ = 91.9 (d, 1JSeP = −809.5 Hz) MS (EI+): m/z = 79.0 [C5H5N]+, 267.8 [C6H5PSe2]+.
- (a) G. Hua, R. A. M. Randall, A. M. Z. Slawin and J. D. Woollins, Z. Anorg. Allg. Chem., 2011, 637, 1800 CrossRef CAS; (b) T. Chivers, M. Krahn and G. Schatte, Inorg. Chem., 2002, 41, 4348 CrossRef CAS PubMed; (c) T. S. Cameron, C. Chan and W. J. Chute, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2391 CrossRef.
- M. Yoshifuji, S. Sangu, K. Kamijo and K. Toyota, Chem. Commun., 1995, 297 RSC.
- (a) M. Bühl, T. Steinke, P. von Ragué Schleyer and R. Boese, Angew. Chem., Int. Ed. Engl., 1991, 30, 1160 CrossRef; (b) R. Bjornsson and M. Bühl, J. Chem. Theory Comput., 2011, 8, 498 CrossRef; (c) S. Aldridge, A. J. Downs, C. Y. Tang, S. Parsons, M. C. Clarke, R. D. L. Johnstone, H. E. Robertson, D. W. H. Rankin and D. A. Wann, J. Am. Chem. Soc., 2009, 131, 2231 CrossRef CAS PubMed; (d) J. A. Phillips, J. A. Halfen, J. P. Wrass, C. C. Knutson and C. J. Cramer, Inorg. Chem., 2005, 45, 722 CrossRef PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- (a) P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J., 2002, 8, 2705 CrossRef CAS PubMed; (b) J. Navech, M. Revel and R. Kraemer, Phosphorus, Sulfur Silicon Relat. Elem., 1984, 21, 105 CrossRef CAS; (c) G. Hua, Y. Li, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2007, 891 CrossRef CAS.
- G. Hua, J. B. Henry, Y. Li, A. R. Mount, A. M. Z. Slawin and J. D. Woollins, Org. Biomol. Chem., 2010, 8, 1655 CAS.
- J. Bethke, K. Karaghiosoff and L. A. Wessjohann, Tetrahedron Lett., 2003, 44, 6911 CrossRef CAS.
- P. Bhattacharyya and J. D. Woollins, Tetrahedron Lett., 2001, 42, 5949 CrossRef CAS.
- Crystals of 2a and 3a suitable for X-ray crystallography were isolated from the reaction mixture. A structural representation and in detail discussion can be found in the ESI†.
- Detailed information on decomposition and behaviour of both solvent systems can be found in the ESI.† NMR data of 1b: 31P NMR (202.5 MHz, C6D6): δ = 97.6 ppm, (s, 1JPSe = −808.8 Hz) 77Se NMR (95.4 MHz): δ = 82.9 ppm (d, 1JSeP = −809.2 Hz). Crystals of 2b and 3b suitable for X-ray crystallography were isolated from the reaction mixture. A structural representation and in detail discussion can be found in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic as well as computational details in pdf form. CCDC 985890–985894. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01073f |
| This journal is © The Royal Society of Chemistry 2014 |