 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tripodal molecules for the promotion of phosphoester hydrolysis†
Jennifer R.
Hiscock
a,
Mark R.
Sambrook
b,
Philippa B.
Cranwell
a,
Pat
Watts
b,
Jack C.
Vincent
b,
David J.
Xuereb
a,
Neil J.
Wells
a,
Robert
Raja
a and
Philip A.
Gale
*a
aChemistry, University of Southampton, Southampton, UK SO17 1BJ. E-mail: philip.gale@soton.ac.uk; Fax: +44 (0)23 8059 6805; Tel: +44 (0)23 8059 3322
bDetection Department, Dstl Porton Down, Salisbury, SP$ 0JQ, UK. E-mail: m.sambrook@dstl.gov.uk; Tel: +44 (0)1980613306
First published on 30th April 2014
Abstract
A series of low molecular weight tripodal amide/histidine-containing compounds (1–2) have been synthesised and shown to increase the rate of bis-(p-nitrophenyl) phosphate (BNPP) and soman (GD) breakdown in buffered aqueous solution.
Organophosphorus (OP) nerve agents (NAs) are a class of chemical warfare agents that include the G-series such as sarin (GB) and soman (GD), and the V-series including VX. These compounds are highly toxic to biological systems as they inhibit the function of the enzyme acetylcholinesterase.1 Our interest in anion (and in particular phosphate) complexation led us to study the application of hydrogen bond donor systems in the recognition of OP NAs such as GD.2,3 Over the last thirty years much effort has been devoted to the synthesis of catalysts capable of mimicking the rate enhancing properties of naturally occurring enzymes.4–11 More recently there has been a concerted effort to utilise synthetic, peptide based molecules as organocatalysts, eliminating the need for metal ions in these systems.12–14 Histidine contains an imidazole group that when incorporated into a molecular/peptide structure can result in the catalysis of hydrolysis reactions due to its acid/base properties.15–17 Breslow and co-workers pioneered the use of bis-imidazoles for phosphate ester hydrolysis.18,19 Later work by Schmuck and co-workers showed that the presence of a histidine residue in the structure of a catalyst is able to promote phosphoester hydrolysis.20
In this article we report the synthesis of two new organic tripodal molecules (1 and 2) appended with N-acetyl-protected histidine residues and a model compound 3. There are a number of examples of tripodal hydrogen bond donating receptors for tetrahedral guest species including phosphates.21,22 The design of the molecules reported here was inspired by these previous reports in that the compounds may bind the nerve agent allowing the appended N-acetyl-protected histidine residues to interact with the substrate. Compound 1 was shown to significantly enhance the hydrolysis rate of GD at pD 6.5 in buffered solution (where pD = pH + 0.4). We designed the tripodal systems to interact with NAs via multiple hydrogen bonding interactions that would both bind the substrate and potentially activate it towards hydrolysis. We have shown that these compounds enhance the breakdown rate of GD and BNPP (an activated DNA model23) in aqueous buffered solutions.
Compound 1 was synthesised by the reaction of tris(2-aminoethyl)amine (tren) with N-acetyl-L-histidine monohydrate using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) as the peptide coupling agent in a solution of N,N-dimethylformamide and N,N-diisopropylethylamine affording the product in 61% yield. Compound 2 was synthesised by the reaction of mono-BOC protected tris(2-aminoethyl)amine with two equivalents of methoxyacetic acid in chloroform, using 1,1′-carbonyldiimidazole (CDI) as the amide coupling agent affording the intermediate in 83% yield. Following a deprotection the resultant amine was reacted with N-acetyl-L-histidine monohydrate under the same conditions resulting in a yield of 56%. Compound 3 was synthesised by the reaction of tris(2-aminoethyl)amine with methoxyacetic acid in chloroform using CDI as the amide coupling agent affording the product in 22% yield.
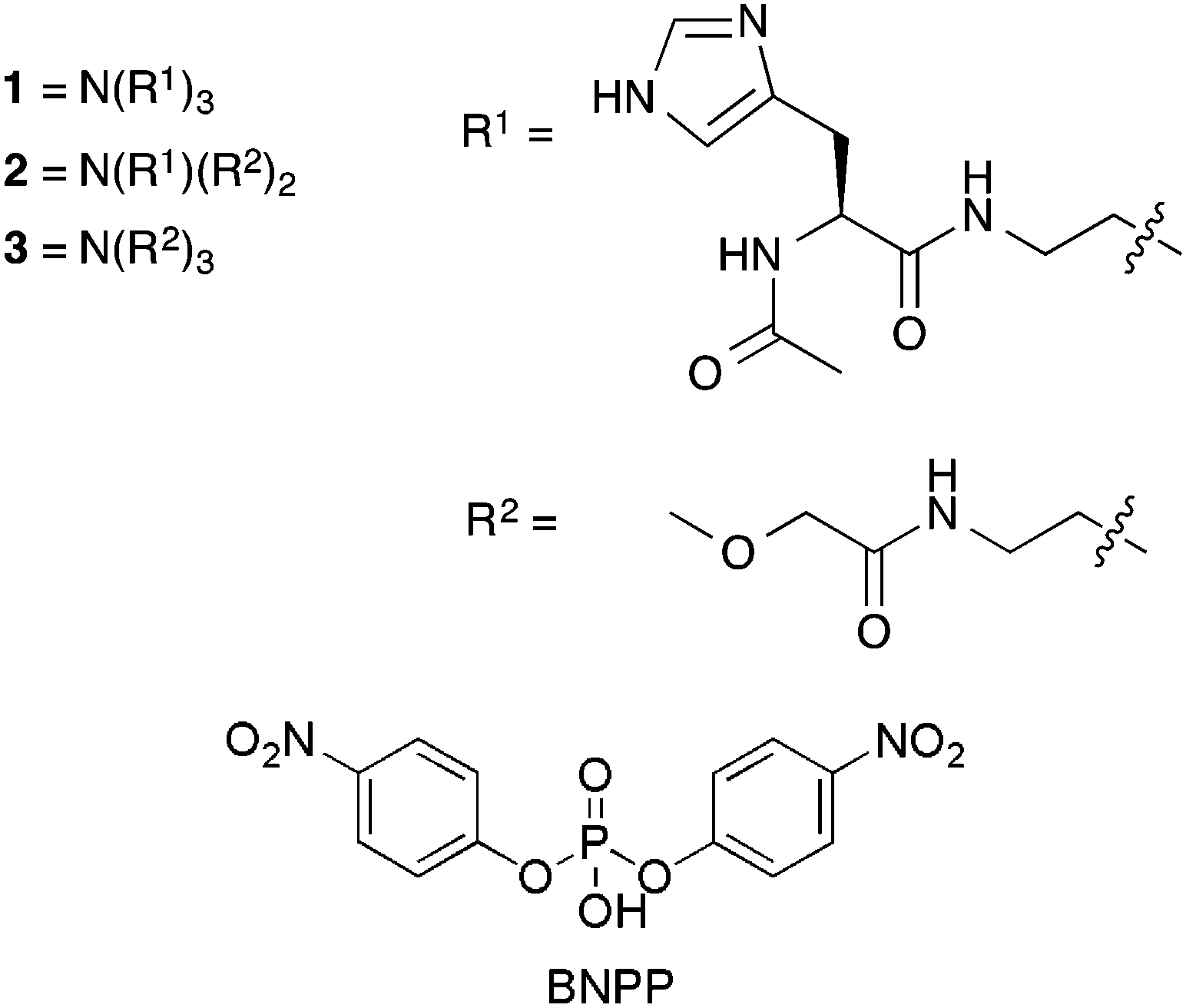
Studies of the breakdown of BNPP (5 mM) were conducted in aqueous 2,2-bis(hydroxymethyl)-2,2′,2′′-nitrilotriethanol (bis–tris) (20 mM) buffered stock solutions at pH 6.1 or 7.1 and 20–23 °C. The compounds were added to portions of these stock solutions in 20, 50 and 100 mol% with respect to BNPP. Imidazole was added in 60, 150 and 300 mol% as compound 1 contains three histidine groups per molecule therefore the mol% of imidazole is matched to the maximum amount of histidine residues in solution in any one experiment. The breakdown of BNPP was monitored by UV-Vis spectroscopy, following the evolution of an absorbance band centred at 400 nm that corresponds to the formation of the breakdown product p-nitrophenolate.
A mechanism for the catalytic hydrolysis of BNPP by two histidine residues has been reported by Dixon and co-workers.24 In this example one of the histidine imidazole residues acts as a nucleophile while the second histidine imidazole residue acts as an acid, protonating the p-nitrophenolate leaving group.
Fig. 1 shows the formation of p-nitrophenolate as BNPP is hydrolysed over time at pH 6.1 and 7.1 in either the absence or presence of 100 mol% of compound 1 and 300 mol% imidazole. The greatest enhancement to the BNPP breakdown by compound 1 is observed at pH 6.1. This may be due to the increased proportion of protonated histidine residues available to supply a proton to the p-nitrophenolate leaving group. The same trend was observed upon addition of imidazole to the BNPP solution. BNPP breakdown was found to be enhanced to a much greater extent with the addition of compound 1 over imidazole. We postulate that this may be due to the structure of compound 1 which holds the three histidine residues in close proximity to each other and also to the substrate through the formation of hydrogen bonds via the amide donor groups, thus increasing the latter's effective concentration.
 | ||
| Fig. 1 Increasing concentration of p-nitrophenolate formed as BNPP is broken down under different pH conditions upon the addition of 100 mol%/300 mol% compound 1 and imidazole respectively with respect to BNPP. The background rate of BNPP breakdown under these pH conditions was also monitored. Residual R2 values for the individual fits ≤1%. The background is taken from an average of two experiments. Individual fits and calculations can be found in the ESI.† | ||
Fig. 2 shows the results of the same experiment conducted with compounds 2 and 3. Compound 2 contains a single histidine group and shows contrasting behaviour to compound 1. At pH 6.1 a lower degree of activity is observed when compared to compound 1. However at pH 7.1 after 750 h a higher proportion of the BNPP has been broken down than observed with compound 1. This may be because compound 2 is increasing the rate of BNPP breakdown through the utilisation of basic properties rather than a combination of acidic and basic properties as expected in the case of compound 3. Compound 3 contains no histidine groups however increased rates of BNPP breakdown are still observed at both pH 7.1 and 6.1 with the greatest enhancement occurring at pH 6.1.
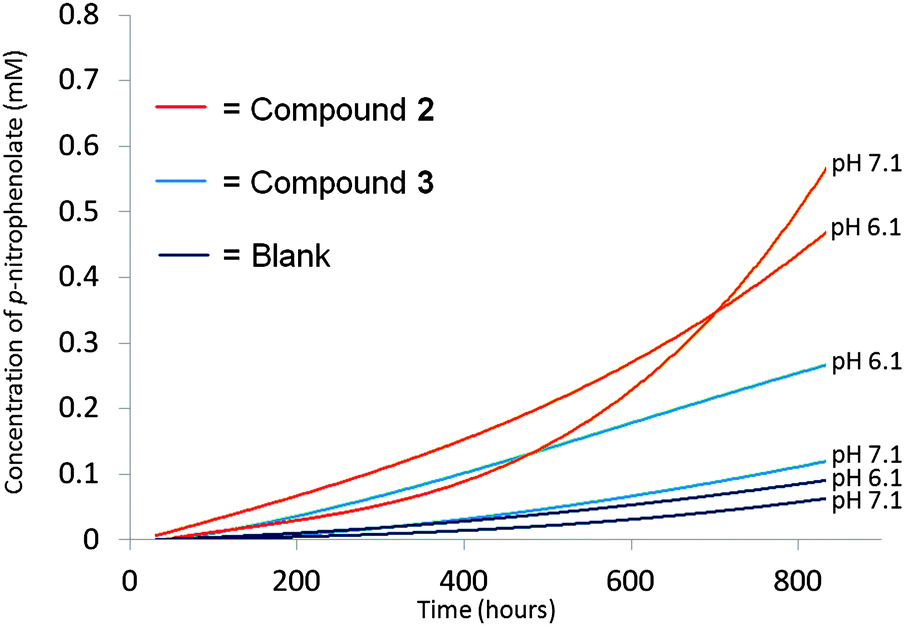 | ||
| Fig. 2 Increasing concentration of p-nitrophenolate formed as BNPP is broken down under different pH conditions upon the addition of 100 mol% compound 2 and 3 with respect to BNPP. The background rate of BNPP breakdown under these conditions was also monitored. Residual R2 values for the individual fits ≤1%. The background is taken from an average of two experiments. Individual fits and calculations can be found in the ESI.† | ||
As compound 1 was found to be the most active of those compounds tested towards the breakdown of BNPP, it was also investigated for the potential to accelerate the hydrolysis of soman (GD). Compound 3 was used as a control in these experiments. Studies for the hydrolytic breakdown of GD (6 mM) were conducted in deuterated bis–tris (20 mM) buffered stock solutions at pD 6.5 (where pD = pH + 0.4) and 20 °C. The conditions chosen were identical to those found to enhance the breakdown of the model compound BNPP to the greatest extent. Compounds 1 and 3 were added to portions of these stock solutions in 100 mol% to GD as appropriate and the breakdown of GD monitored using both 1H and 31P NMR.
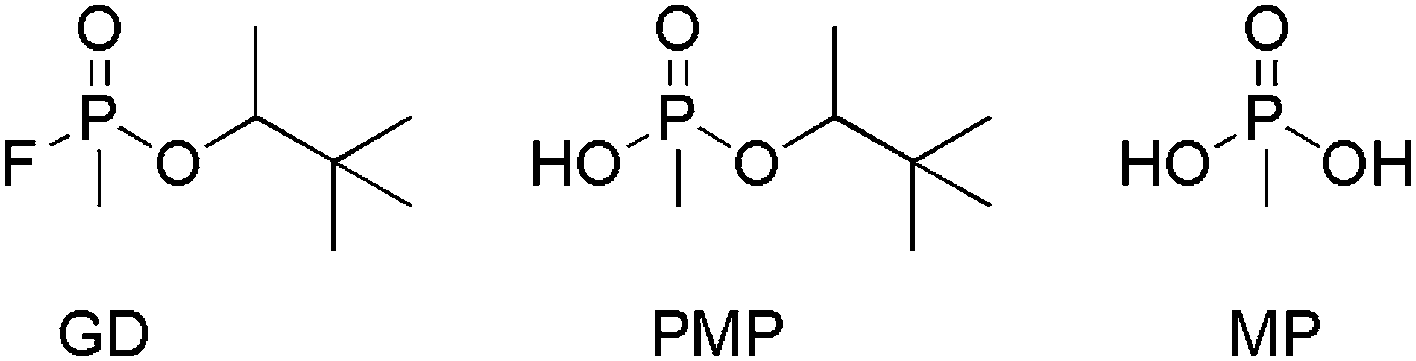
Under the experimental conditions two hydrolysis products were identified. The main product was found to be pinacolyl methylphosphonate (PMP) formed via hydrolysis of the P–F bond. Methylphosphonic acid (MP) was also detected in small quantities due to further hydrolysis of PMP.
Experiments with GD were run for 16–18 hour periods. A stack plot of 1H NMR spectra from one of the hydrolysis experiments is shown in Fig. 3, with the resonances for GD and PMP labelled (see ESI† for an analogous 31P NMR plot). The concentrations of the different species present were calculated by integrating the 1H and 31P NMR spectra and relating these to the starting concentration of GD.
 | ||
| Fig. 3 1H NMR stack plot of the breakdown of GD in bis–tris (20 mM) buffered solutions at pD 6.5 in the presence of 100 mol% of compounds 1. Time = (a) 0 h (b) 3 h (c) 7 h (d) 12 h and (e) 18 h. ©Crown Copyright, Dstl. | ||
The percentage breakdown of GD far exceeds the rate of BNPP breakdown under similar conditions. This is in part due to the lower stability of GD vs. BNPP. The experimental data from the breakdown of GD in the presence of 100 and 50 mol% compound 1 is shown in Fig. 4. After 16 h there is approximately half the starting concentration of intact GD present in the samples that contain 100 mol% compound 1, and with compound 3 only approximately 5% of the GD present has been hydrolysed. Although the results are less clear with the addition of 50 mol% compound 1 it does appear that the rate of GD breakdown is reduced by decreasing the mol% of compound 1 present. This data has been fitted to linear equations which can be found along with the relevant R2 values in the ESI.†
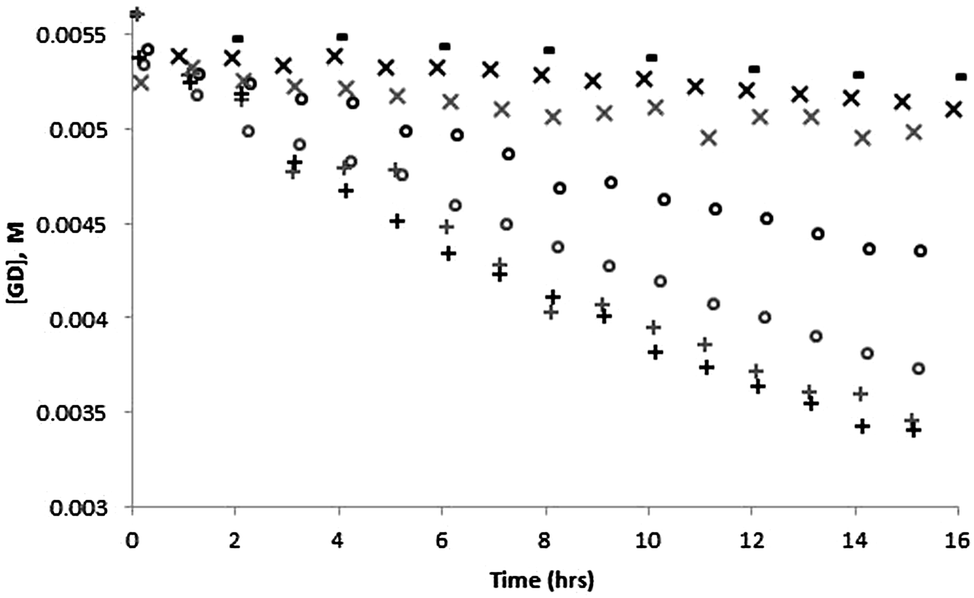 | ||
| Fig. 4 Concentration of GD present as a function of time in bis–tris buffered solutions (20 mM) at pD 6.5 in the absence (−) and presence of 100 (+) and 50 (○) mol% compound 1 (+) and 100 mol% compound 3 (×) as determined by 1H NMR analysis. ©Crown Copyright, Dstl. Two runs were performed for each compound at each concentration with the runs distinguished in the figure by the black and grey symbols. | ||
The association of dimethyl methylphosphonate (DMMP) and PMP with compound 3 was explored with the use of 1H NMR titrations in MeCN-d3. DMMP and PMP have similar structures to GD. Due to solubility and protonation issues host–guest combinations incorporating compounds 1, 2 and BNPP could not be explored. The results of these studies are shown in Fig. 5. Binding constants were calculated using WINEQN MR225 fitting the data to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm. An association between compound 3 and PMP was observed and fitting of the titration data to a 1:1 binding model gave a stability constant of 14 M−1, through the formation of hydrogen bonds from the amide NH groups to PMP. No interaction was observed between compound 3 and DMMP. The higher affinity of compound 3 to PMP is presumably due to the higher polarity of PMP compared to DMMP and the presence of an extra hydrogen bond accepting/donating group in PMP.
:1 binding isotherm. An association between compound 3 and PMP was observed and fitting of the titration data to a 1:1 binding model gave a stability constant of 14 M−1, through the formation of hydrogen bonds from the amide NH groups to PMP. No interaction was observed between compound 3 and DMMP. The higher affinity of compound 3 to PMP is presumably due to the higher polarity of PMP compared to DMMP and the presence of an extra hydrogen bond accepting/donating group in PMP.
 | ||
| Fig. 5 Change in chemical shift of amide NH from compound 3 with the addition of PMP/DMMP (◆/■). | ||
These observations support the theory that compounds 1–3 are capable of forming host:guest complexes via hydrogen bonding interactions that may both bind the target organophosphate ester and activate it towards hydrolysis.
Single point 1H NMR studies were also carried out with compound 3 and GD in CDCl3 in order to observe any potential host:guest association. Upon addition of two molar equivalents of GD to a solution of compound 3 (0.01 M) a downfield shift of 0.014 ppm was observed for the amide NH resonance, evidence supporting the formation of compound 3:GD complex, and pre-concentration of GD.
We have shown that compound 1 is capable of enhancing the rate of GD hydrolysis. These results lead us to suggest that simple tripodal compounds containing amino acids such as histidine could be used for NA remediation.
We would like to thank the Defence Science and Technology Laboratory for funding through the Centre for Defence Enterprise scheme (JRH). We would also like to thank the ESPRC for a PhD studentship (DJX). MRS would like to thank Dstl for Research Scholarship funding. PAG thanks the Royal Society and the Wolfson Foundation for a Royal Society Wolfson Research Merit Award. The authors thank Dr James Jones (Dstl) for assistance with NMR.
Notes and references
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- M. R. Sambrook, J. R. Hiscock, A. Cook, A. C. Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC.
- J. R. Hiscock, F. Piana, M. R. Sambrook, N. J. Wells, A. J. Clark, J. C. Vincent, N. Busschaert, R. C. D. Brown and P. A. Gale, Chem. Commun., 2013, 49, 9119–9121 RSC.
- A. Blasko and T. C. Bruice, Acc. Chem. Res., 1999, 32, 475–484 CrossRef CAS.
- P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2011, 50, 396–400 CrossRef CAS PubMed.
- F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406–2447 RSC.
- R. Salvio, R. Cacciapaglia and L. Mandolini, J. Org. Chem., 2011, 76, 5438–5443 CrossRef CAS PubMed.
- B. M. Smith, Chem. Soc. Rev., 2008, 37, 470–478 RSC.
- A. Sreedhara and J. A. Cowan, J. Biol. Inorg. Chem., 2001, 6, 337–347 CrossRef CAS.
- J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; N. H. Williams, B. Takasaki, M. Wall and J. Chin, Acc. Chem. Res., 1999, 32, 485–493 CrossRef CAS.
- F. Brandhuber, M. Zengerle, L. Porwol, A. Bierwisch, M. Koller, G. Reiter, F. Worek and S. Kubik, Chem. Commun., 2013, 49, 3425–3427 RSC.
- X. H. Cao, G. Wang, R. C. Zhang, Y. Y. Wei, W. Wang, H. C. Sun and L. G. Chen, Org. Biomol. Chem., 2011, 9, 6487–6490 CAS.
- G. D. Joly and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 4102–4103 CrossRef CAS PubMed.
- M. Wiesner, J. D. Revell and H. Wennemers, Angew. Chem., Int. Ed., 2008, 47, 1871–1874 CrossRef CAS PubMed.
- Y. Bai, Y. B. Ling, W. G. Shi, L. F. Cai, Q. Y. Jia, S. B. Jiang and K. L. Liu, ChemBioChem, 2011, 12, 2647–2658 CrossRef CAS PubMed.
- Y. Ma, X. Chen, M. Sun, R. Wan, C. Zhu, Y. Li and Y. Zhao, Amino Acids, 2008, 35, 251–256 CrossRef CAS PubMed.
- C. Schmuck, U. Michels and J. Dudaczek, Org. Biomol. Chem., 2009, 7, 4362–4368 CAS.
- R. Breslow, P. Bovy and C. L. Hersh, J. Am. Chem. Soc., 1980, 102, 2115–2117 CrossRef CAS.
- R. Breslow, J. B. Doherty, G. Guillot and C. Lipsey, J. Am. Chem. Soc., 1978, 100, 3227–3229 CrossRef CAS.
- M. Merschky and C. Schmuck, Org. Biomol. Chem., 2009, 7, 4895–4903 CAS.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2011, 41, 480–520 RSC.
- R. Kramer, Coord. Chem. Rev., 1999, 182, 243–261 CrossRef.
- A. E. Rudolph, J. A. Stuckey, Y. Zhao, H. R. Matthews, W. A. Patton, J. Moss and J. E. Dixon, J. Biol. Chem., 1999, 274, 11824–11831 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, characterisation data for compound 1. See DOI: 10.1039/c4cc00333k |
| This journal is © The Royal Society of Chemistry 2014 |