DOI:
10.1039/D5RA01845E
(Paper)
RSC Adv., 2025,
15, 17241-17247
Mechanistic insights into base-free nickel-catalyzed Suzuki–Miyaura cross-coupling of acid fluoride and the origin of chemoselectivity: a DFT study†
Received
15th March 2025
, Accepted 5th May 2025
First published on 22nd May 2025
Abstract
Palladium-catalyzed Suzuki–Miyaura Coupling (SMC) is a powerful strategy to construct C–C bonds; however, it suffers from the disadvantages of using expensive palladium catalysts and additives. Based on the experimental development of the base-free nickel catalyzed Suzuki–Miyaura coupling of acid fluorides (ArC(O)F) with diboron reagent, we carried out DFT calculations to gain insight into the reaction mechanisms. The coupling reaction proceeds via four stages: (1) oxidative addition of the acid fluoride to the Ni(0) center to break the C–F bond, (2) transmetalation with diboron reagent, (3) carbonyl deinsertion via reverse carbonyl migratory insertion, and (4) reductive elimination to afford the coupling product and regenerate the active catalyst. It was found that the competitive rotation of the Ni–B bond and Ni–C(aryl) bond of the intermediate generated from the oxidative addition of the acid fluoride to Ni(0) center (stage I) determines the chemoselectivity of the catalytic cycle, and carbonyl migratory insertion is the rate-determining step of the coupling. Our study reveals that the PhOMe moiety in TS8 induces greater steric hindrance and reduced electronic stabilization compared to BPin in TS5, resulting in increased geometric distortion, higher distortion energy, and a less favorable transition state with a higher activation barrier. Furthermore, our computational results indicate that transmetalation prefers a concerted mechanism. Detailed analyses reveals that the strong fluorophilicity of boron enables efficient, base-free transmetalation. This study could be helpful for the development of cheap catalysts for Suzuki–Miyaura cross-coupling reactions.
1. Introduction
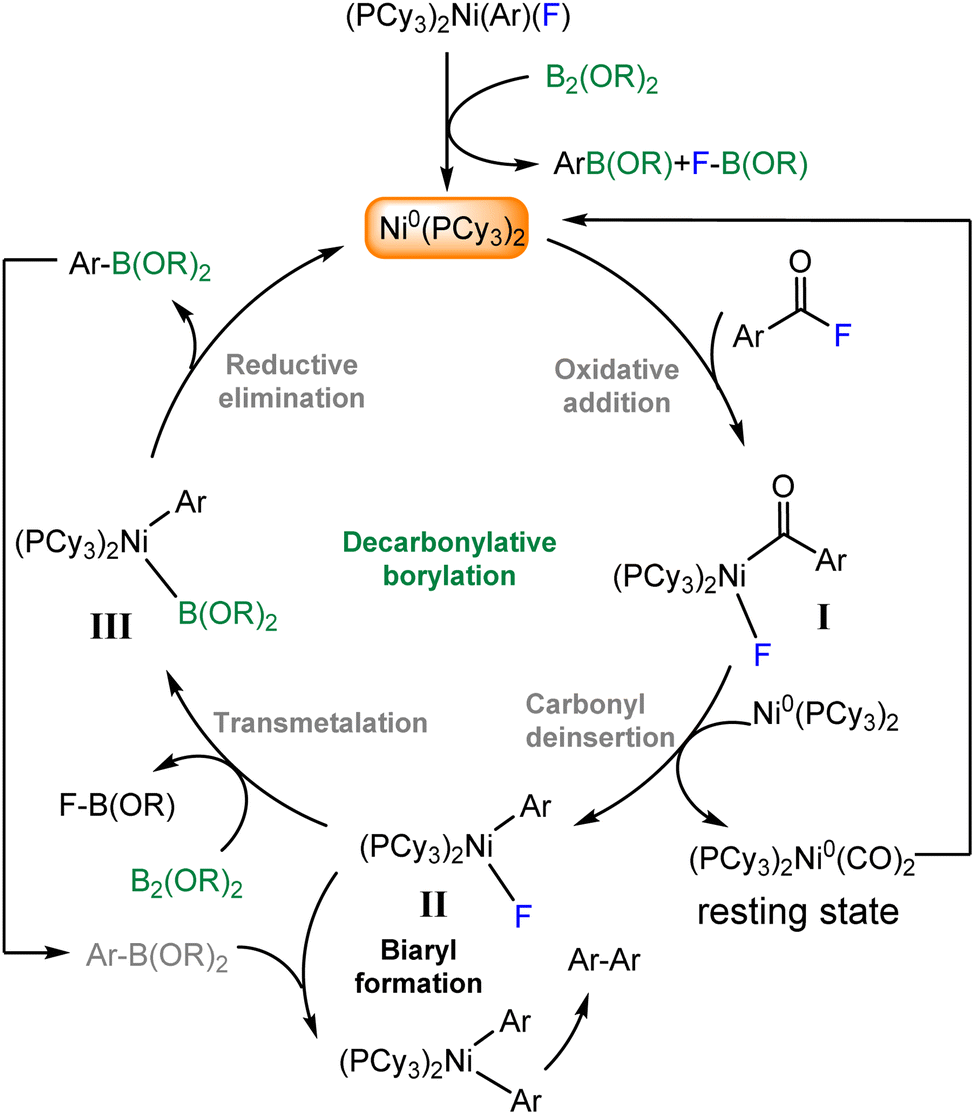
The palladium-catalyzed Suzuki–Miyaura coupling is a powerful strategy to construct C–C bond forming reactions, which consist of the coupling between organoboron nucleophiles and aryl halide (or triflate) electrophiles in the presence of a base,1–5 often used in organic, medicinal and agrichemical industries.6–9 The key challenges associated with this transformation are the use of expensive palladium catalysts, the addition of exogenous base to facilitate transmetalation and requirement of expensive and rare electrophiles.10,11 To further address these challenges significant efforts have been devoted to replacing palladium with nickel for the development of different SMC reactions in the presence of an exogenous base.12–17 Ideally, using cheap and earth–abundant transition metal (e.g., Ni) catalysts with available electrophiles in the absence of an exogenous base is the most preferred way to carry out SMC reactions.18–21 In 2018, Sanford and coworkers made a breakthrough, discovery that a nickel (Ni) complex could serve as a catalyst for the coupling reaction between aryl boronic acids and acid fluorides, in the absence of an exogenous base (Fig. 1 eqn (a)).22 Recently, the same group developed a base free nickel-catalyzed decrbonylative coupling of carboxylic acid fluorides with diboron reagent to selectively afford aryl boronate ester products (Fig. 1 eqn (b)).23 However, a primary challenge associated with this metal-catalyzed borylation reaction is that the competing of organoboron product versus the diboron reagent in the transmetalation step, leading to undesired over cross-coupling to afford biaryl byproduct (Fig. 1 eqn (b)).23
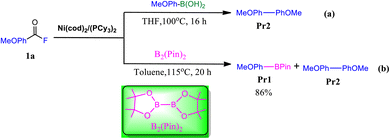 |
| | Fig. 1 Base-free nickel catalysed decarbonylative Suzuki–Miyaura-type reactions.22,23 | |
Recently, we24 reported a DFT study of the base-free Ni-catalyzed Suzuki–Miyaura cross-coupling of acid fluoride by Sanford and co-worker,22 where we found that the transmetalation proceeds through a stepwise mechanism, instead of the conventional concerted one. Additionally, the strong fluorophilicity of boron and the coordination interaction between the oxygen atom of boronic acid with Ni metal plays a vital role in promoting the base free transmetalation. Herein, we theoretically studied the Suzuki–Miyaura cross-coupling mechanism of the base-free nickel-catalyzed decarbonylative borylation of acid fluorides, to reveal the origin of the chemoselectivity to aryl boronate ester over biaryl. Indeed, our calculation showed that the decarbonylation step occurs after transmetalation; which further supports our previous finding.24 Our understanding of the substrate-dependent chemoselectivity should be helpful for development of cheap catalysts for Suzuki–Miyaura cross-coupling reactions between acid fluoride and diboron reagent.
2. Computational methods
All the DFT calculations were carried out using the Gaussian 09 program.25 All structures were optimized in the gas phase at B3LYP26,27/BSI level, where BSI represents a basis set with 6-31G(d,p)28–31 for nonmetal atoms and SDD32,33 for Ni. Harmonic vibrational frequency calculations were subsequently performed to verify the optimized structures to be minimal (no imaginary frequency) or transition states (TSs, having a unique one imaginary frequency). The energies were then improved by M06 (ref. 34–36)/BSII//B3LYP/BSI single-point calculations with solvent effects accounted by SMD37,38 solvent model, using the experimental solvent, toluene. BSII denotes a basis set with SDD for Ni and 6-311++G(d,p) for other atoms. Additional information is relegated to ESI.† Selected optimized structures are illustrated using CYLview.39
3. Results and discussion
Sanford et al.23 have shown their strategy to synthesize aryl boronate ester from acid fluoride and diboron reagent (Fig. 2). At first, the oxidative addition of the acid fluoride substrate to Ni0(PCy3)2 to yield an acyl Ni specious (I), thus carbonyl deinsertion to afford transmetalation active complex (II), then transmetalation with diboron reagent, C–B bond-forming reductive elimination afford the product and regenerate Ni0(PCy3)2. Furthermore, the over cross-coupling of aryl boronate ester leads to biaryl formation. In the following, we report our DFT study results to gain insight into the mechanisms of the reactions and disclose the origin of chemoselectivity.
 |
| | Fig. 2 Experimentally postulated mechanism for organoboron and biaryl synthesis from acid fluoride and diboron reagent by Sanford et al.23 | |
3.1 The mechanism for base-free Ni-catalyzed decarbonylative borylation
Based on our previous theoretical study on nickel-catalyzed Suzuki–Miyaura coupling of acid fluorides (ArC(O)F) with boronic acids (Ar′B(OH),24 Cat is regarded as the real catalyst which can be generated from the Ni(Cod)2 as the catalyst precursor and two equivalents of PCy3 ligand,24 Fig. 3 shows the energy profile for oxidative addition and decarbonylation stages in Fig. 1(b). Similarly, the initial stage of the reaction is an oxidative addition of the acidic fluoride (1a) to Ni(0) center via TS1 with a free energy barrier of 17.6 kcal mol−1 to reversibly form a tetrahedral geometry IM1. Subsequently, IM1 can be easily transformed to a 17.0 kcal mol−1 more stable square planar geometry IM2. To undergo decarbonylation, a PCy3 ligand dissociates from IM2 via TS2 with a barrier of 23.7 kcal mol−1, giving IM3 with a vacant coordination site. Because IM2 is a saturated complex, a ligand should dissociate from Ni-center to allow decarbonylation. Next, a carbonyl migratory insertion occurs via TS3 with a low barrier of 2.1 kcal mol−1 from IM3 to give a carbonyl nickel fluoride complex IM4. Subsequent liberation of CO from IM4 gives IM5 which either associates with a released PCy3 ligand to reach more stable IM6 or proceeds to the next stage of transmetalation with diboron reagent (B2Pin2). However, the possible dissociation intermediate has higher energy barrier of 32.7 kcal mol−1 relative to IM2 and we excluded the possibility without further consideration. The results indicated that direct decarbonylation after oxidative addition is unlikely, encouraging us to find an alternative. Previously, we also ruled out the direct decarbonylation after oxidative addition.24
 |
| | Fig. 3 Free-energy profiles for oxidative addition and decarbonylation stages in Fig. 1(b). | |
Alternatively, we also considered the possibility of IM2 to directly undergo transmetalation. Fig. 4 describes the alternative where the transmetalation takes place first. Note that, in Fig. 3 the decarbonylation occurs after oxidative addition. The optimized structures of selected intermediates and transition state are shown in Fig. 5. Previously, we considered two transmetalation pathways.24 Starting from IM2, the concerted mechanism (black line) occurs via TS4 crosses a barrier of 16.8 kcal mol−1 and is slightly endergonic by 1.5 kcal mol−1 to form IM7. The calculated result agrees with the conventional transmetalation. Previously, we considered a stepwise mechanism.24 Unfortunately, we were not able to locate the transition state after several attempts made. However, the possible coordinative intermediate IM8 (ΔG = 8.4 kcal mol−1) is 2.1 kcal mol−1 higher than the concerted TS4 (ΔG = 6.3 kcal mol−1) and can be excluded. Starting from IM7 the carbonyl migratory insertion requires rotating the Bpin group trans to the carbonyl group, as illustrated by TS5 in Fig. 4 which crosses a barrier of 21.9 kcal mol−1 relative to IM7. Alternatively, we also considered the possibility of rotating the PCy3 to undergo a carbonyl migratory insertion. However, as illustrated in ESI (Fig S.2),† we found that the calculated free energy barrier for the possible CO dissociation intermediate IM11A is 4.6 kcal mol−1 higher than TS5, which revealed that the mechanism following rotating the PCy3 is unfavorable. Then, a carbonyl migratory insertion proceeds via TS6, generates more stable the CO-coordinated Ar[Ni](CO)Bpin intermediate IM10. To confirm both sides of the potential energy surface, we performed the IRC calculation for TS6. The result shows a concerted mechanism involving both carbonyl migratory insertion and reductive elimination takes place simultaneously (as evidenced by the slight elongation of the Ni–CO bond length in the forward IRC pathway, continuing from P3 (1.784 Å) to P4 (1.809 Å) and consequently from P4 (1.809 Å to IM10 (1.813 Å) (see Fig. S.3†). Finally, CO liberation helps to recover the catalyst and the coupling product (Pr1). Overall, the carbonyl migratory insertion has a rate-determining step (RDS) with a barrier of 23.4 kcal mol−1 (TS5 relative to IM2) and is exergonic by 43.9 kcal mol−1.
 |
| | Fig. 4 Free-energy profiles for the mechanism via transmetalation, decarbonylation and catalyst regeneration, leading Pr1. | |
 |
| | Fig. 5 Optimized structures of the key stationary points labeled in Fig. 4. | |
3.2 Mechanism for biaryl formation
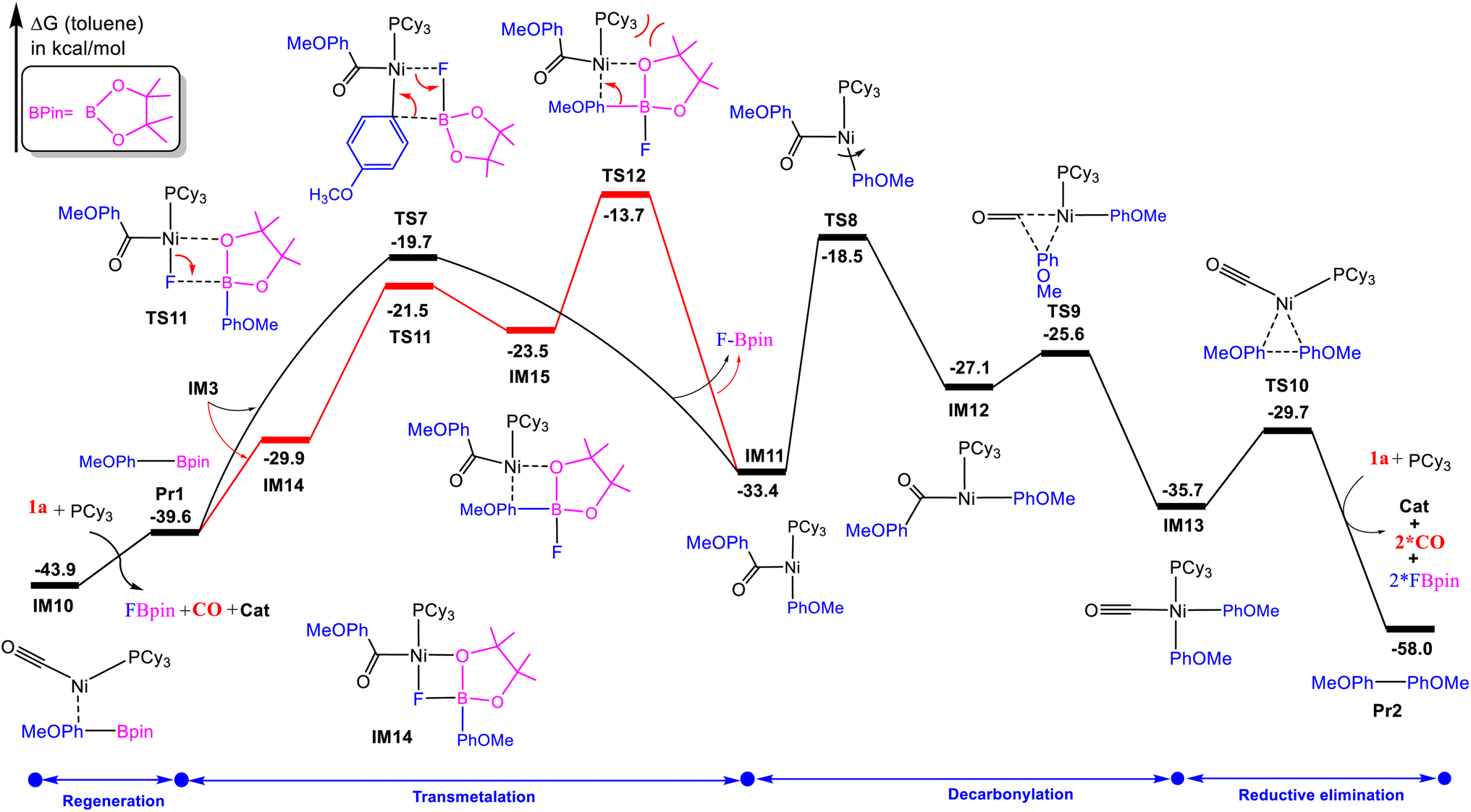
The mechanisms discussed above elucidate the generation of the borylated product. Interestingly, the over cross-coupling of Pr1 leads to biaryl formation. The mechanism of the subsequent biaryl formation was investigated. From CO-coordinated Ar[Ni](CO)Bpin intermediate IM10, transmetalation, decarbonylation, and reductive elimination to deliver the final product and regenerate the active catalyst. Fig. 6 shows the energetics of these four steps; optimized geometries of key stationary points are provided in Fig. 7. The transmetalation process can proceed in a concerted or stepwise fashion, based on our previous studies,24 we first considered a concerted mechanism, as illustrated by the four-membered TS7, the transmetalation proceeds by cleaving the Ni–F and B–C(aryl) bonds concurrently, giving IM11 and FB(pin). Alternatively, for a stepwise mechanism, the electronic deficient boron of the Pr1 moiety extracts the F atom via TS11 to break the Ni–F bond, resulting in IM15. The calculated relative free energy of TS11 is 1.8 kcal mol−1 lower than that of TS7. However, we found that the calculated free energy barrier of the B–C(aryl) bond cleavage and the formation of Ni–C bond via TS12 is 6.0 kcal mol−1 higher than TS7, which revealed that stepwise mechanism is unfavorable. In the stepwise pathway, the steric clash between the bulky Bpin group and the phosphine ligand causes a destabilization, leading to the high energy barrier observed in TS12. This steric hindrance is absent in the concerted mechanism, allowing for a smoother transition. Thus, the concomitant cleavages of the two bonds (i.e. Ni–F and B–C(aryl) bonds) could intrinsically be easier than the sequential cleavages of the two bonds. Although the stepwise transmetalation takes the advantage of the favorable coordination interaction between oxygen in Pr1 and nickel, which is indicated by the short lengths of Ni–O coordination bonds (2.08 Å in TS11), however the formation of IM15), where the Ni–F bond is broken before the B–C bond is fully formed and often less stable and requires additional energy to proceed to the next step. Further, we compared the charge of Ni in TS7 (−0.74) and TS12 (−0.65), suggesting that TS7 may correlate with greater stabilization of the transition state due to stronger coordination of the ligand at the metal center, contributing to a lower activation energy. In TS12 the steric clash between the Bpin moiety and the phosphine ligand causes a relatively high energy barrier than TS7. Similar to the above mechanism, from IM11 the carbonyl migratory insertion requires rotating the aryl group trans to the carbonyl group, a very facial rotation along the Ni–C(aryl) bond of IM11 preferentially takes place to form IM12 via TS8 which crosses a barrier of 14.9 kcal mol−1 relative to IM11. Then, a carbonyl migratory insertion proceeds via TS9, generating more stable the Ar[Ni](CO)Ar′ intermediate IM13. In TS9 the distance of the breaking C–C bond (1.74 Å) and forming C–C bond (2.09 Å) confirm the formation of CO-coordinated intermediate IM13 geometrically. Finally, IM13 undergoes reductive elimination via TS10, affording the coupling product Pr2 and regenerating the catalyst. Overall, based on the calculated free energy changes of the whole catalytic cycle, the resting state of the cycle is the CO-coordinated Ar[Ni](CO)Bpin intermediate IM10, and the carbonyl migratory insertion has a rate-determining step (RDS) with a barrier of 25.4 kcal mol−1 (TS8 relative to IM10) and is exergonic by 58.0 kcal mol−1. Therefore, these results showed that the formation of biaryl product is thermodynamically more favorable than the organoboron product Pr1.
 |
| | Fig. 6 Free-energy profiles for biaryl formation Pr2. | |
 |
| | Fig. 7 Optimized structures of the key stationary points are displayed in Fig. 6. | |
3.3 Origins for different chemoselectivity of part 1(b)
The mechanisms discussed above elucidate the base-free nickel catalyzed Suzuki–Miyaura cross-coupling of acid fluoride and diboron reagent to afford organoboron and biaryl. Analyzing the mechanisms in Fig. 3 and 4, we can find out that, the reaction undergoes sequential oxidative addition, transmetalation, decarbonylation, and reductive elimination. Comparing Fig. 4 and 6, the pathway leading to Pr1 is somewhat similar to Pr2, although the decarbonylation and reductive elimination steps are somewhat different. In Fig. 4 the RDS is the rotation of Ni–B bond via TS5. Similarly, the RDS in Fig. 6 is the rotation of the Ni–C(aryl) bond of IM11 via TS8. Fig. 4 and 6 provide valuable insights into the energetic results that help us understand the origin of chemoselectivity towards aryl boronate esters over biaryl compounds. The RDS barrier (TS5) to give aryl boronate ester (Pr1) is 2.0 kcal mol−1 lower than that (TS8) to give biaryl product (Pr2). We analyzed and compared the relative activation barriers (ΔG‡) associated with the key steps in the reaction pathways of TS5 and TS8. The activation barrier (ΔG‡) for TS8 (∼25 kcal mol−1) relative to IM10 is higher than that of TS5 (∼23 kcal mol−1) relative to IM2 (see Fig. 4 and 6), indicating that the PhOMe-containing intermediate overcomes a larger distortion energy to reach the transition state. To further investigate steric contributions, we performed a distortion interaction analysis on transition states (Fig. S4†). The bond angle at the nickel center is compressed for TS8 (123°) compared to TS5 (127°), and the dihedral angle between one of the PCy ligands, nickel, and the PhOMe moiety varies significantly from planar at 138° to 169° when considering the PCy ligand, nickel, and the BPin moiety. This variation indicates an increase in torsional strain. We also performed NBO (natural bond orbital) analysis to gain insight into the electronic environment of the nickel center. The NBO analysis shows the Ni in the TS5 intermediate is slightly negative (−0.104), while in TS8, it is more positive (+0.18). This suggests greater electron donation from BPin due to its stronger σ-donor character. These findings reveal that TS8 induces greater steric hindrance, resulting in a more distorted geometry and a less energetically favorable transition state compared to the BPin-containing system.Thus, part b of Fig. 1 reactions kinetically prefers to produce aryl boronate ester (Pr1), agreed with the experimental result.23 Note that Pr1 is thermodynamically less favorable than Pr2, thus the selectivity of aryl boronate ester of the reaction is exclusively controlled by kinetics. The chemoselective preference for the borylation product using the B2(Pin)2 substituent was attributed to the following factors: the electron-deficient boron creates an extra stabilization energy and lowers the free energy in TS5. In contrast, using Pr1 as a substituent, the electronic and steric effect of PhOMe moiety in TS8 destabilizes the biaryl formation kinetically. Overall, the chemoselectivity originates from the electronic and steric effects of BPin and PhOMe in TS5 and TS8.
4. Conclusion
In this study, we performed a DFT mechanistic study of a base-free nickel-catalyzed decarbonylative borylation of acid fluorides giving aryl boronate ester or biaryl selectively. The catalysis proceeds in four stages: (1) the oxidative addition of the acid fluoride substrate to Ni(0) center (2) transmetalation with diboron reagent. (3) Carbonyl deinsertion via reverse carbonyl migratory insertion. (4) Reductive elimination to afford the product and regenerate the active catalyst. In our computed mechanism, the transmetalation prefers a concerted mechanism and takes place first before carbonyl deinsertion which is different from the experimentally proposed mechanism. Importantly, the competitive rotation of Ni–B bond via TS5 and Ni–C(aryl) bond of IM1 via TS8, determines the chemoselectivity of the catalytic cycle and carbonyl migratory insertion is the rate-determining step of the coupling. The strong fluorophilicity of diboron assists the base free transmetalation. This study provides valuable insights that could contribute to the development of cost-effective catalysts for Suzuki–Miyaura cross-coupling reactions.
Data availability
The data supporting this article including xyz coordinates have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
References
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963–4972 RSC.
- R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC.
- M. Mondal, T. Begum and U. Bora, Org. Chem. Front., 2017, 4, 1430–1434 Search PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- R. Renzo, B. Fabio, L. Marco, M. Chiara, M. Giulia and A. P. Luca, Curr. Org. Chem., 2015, 19, 1302–1409 Search PubMed.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org Chem., 2013, 2013, 19–30 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- Z. Li and L. Liu, Chin. J. Catal., 2015, 36, 3–14 CrossRef CAS.
- L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed.
- M. Ohashi, H. Saijo, M. Shibata and S. Ogoshi, Eur. J. Org Chem., 2013, 2013, 443–447 CrossRef CAS.
- N. Blanchard and V. Bizet, Angew. Chem., Int. Ed., 2019, 58, 6814–6817 CrossRef CAS PubMed.
- Y. Ogiwara, D. Sakino, Y. Sakurai and N. Sakai, Eur. J. Org Chem., 2017, 2017, 4324–4327 CrossRef CAS.
- Y.-Y. Sun, J. Yi, X. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2014, 50, 11060–11062 RSC.
- C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100–104 CrossRef CAS PubMed.
- C. A. Malapit, J. R. Bour, S. R. Laursen and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 17322–17330 CrossRef CAS PubMed.
- C. Zhang, R. Zhao, W. M. Dagnaw, Z. Liu, Y. Lu and Z.-X. Wang, Theor. Chim. Acta, 2019, 84, 13983–13991 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.
P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 324–333 CrossRef CAS PubMed.
- A. D. Kulkarni and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 2325–2332 CrossRef CAS PubMed.
- P. Śliwa and J. Handzlik, Chem. Phys. Lett., 2010, 493, 273–278 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- U. Domańska, M. Laskowska and A. Pobudkowska, J. Phys. Chem. B, 2009, 113, 6397–6404 Search PubMed.
- C. Y. Legault, CYLView, Version 1.0 B, Université de Sherbrooke, Sherbrooke, Quebec, Canada, 2009 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yohannes Mulugeta Hailu
*a,
Yohannes Mulugeta Hailu