
Sequential single-enzyme oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid by an engineered lanthanide-dependent alcohol dehydrogenase†
Ke
Liu
 a,
Ling
Jiang
ab,
Lun
Wang
ab,
Qunfeng
Zhang
a,
Lirong
Yang
ab,
Jianping
Wu
ab and
Haoran
Yu
*ab
a,
Ling
Jiang
ab,
Lun
Wang
ab,
Qunfeng
Zhang
a,
Lirong
Yang
ab,
Jianping
Wu
ab and
Haoran
Yu
*ab
aInstitute of Bioengineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, China. E-mail: yuhaoran@zju.edu.cn
bZJU-Hangzhou Global Scientific and Technological Innovation Centre, Hangzhou, Zhejiang 311200, China
First published on 25th March 2025
Abstract
2,5-Furandicarboxylic acid (FDCA) is a potential platform chemical available from renewable feedstocks to make various polymers and high-value compounds. Bioconversion of 5-hydroxymethylfurfural (HMF) to FDCA with biocatalysts has the advantages of high selectivity, cost-effectiveness, and eco-friendliness. However, the full oxidation of HMF often requires a combination of two or three enzymes, as the production of FDCA involves three consecutive oxidation steps. In this study, we identified a pyrroloquinoline quinone (PQQ) and lanthanide-dependent alcohol dehydrogenase, PedH, capable of directly converting HMF into FDCA, representing a rare and intriguing discovery. We then engineered the enzyme for improved thermostability and activity against HMF to improve the FDCA yield. Using a computational design method, the thermostability was first significantly improved, with the 4M variant obtained showing a Tm value increased by 9.6 °C, and a half-life improved by 10.3-fold compared to the wild type. With 4M as the template, potential mutation sites for engineering enzyme activity were identified based on calculations of the binding free energy and a deep learning model, MutCompute. An automatic high-throughput platform for site-saturation mutagenesis library construction and screening was developed and applied to the mutation targets identified. After five rounds of evolution, a variant was obtained that produced FDCA in a yield 474-fold higher than that of 4M. Under optimized reaction conditions, the optimal variant achieved a FDCA yield of 96.4% with 40 mM HMF as the substrate. Molecular dynamics simulations revealed that the mutations expanded the substrate binding pocket and shortened the reaction distances between substrate and cofactors. This study provides a highly efficient evolution approach for PedH, and several variants that can potentially be used for the high-yield production of FDCA in industrial applications.
Green foundation1. This work identified a pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenase, PedH, with the unique capability to continuously oxidize HMF to FDCA. The use of PedH for FDCA production eliminates the need for metal catalysts, avoids exogenous hydrogen peroxide, reduces by-products, and contributes to the advancement of green chemistry.2. By using various protein engineering strategies, the Tm value of PedH was increased by 9.6 °C, its half-life was extended by 10.3 times and the FDCA yield was improved by 474-fold. Under optimized reaction conditions, the yield of 40 mM FDCA reached 96.4%. 3. Overexpressing the optimal mutant in Pseudomonas putida KT2440 rather than in Escherichia coli may result in a more economically feasible whole-cell biocatalyst. This approach would eliminate the need for external PQQ and artificial electron acceptors, while also enhancing tolerance to higher concentrations of furfural. |
Introduction
The widespread depletion of nonrenewable fossil resources and the emission of greenhouse gases have triggered significant global changes. As a result, there is increasing interest in developing renewable and sustainable fuels and platform chemicals.1 Lignocellulosic biomass is a crucial renewable carbon resource, and its carbohydrate derivatives can be transformed into various biobased platform compounds for producing high-value products. For example, the sugar components of biomass can be chemically dehydrated to give furfural and 5-hydroxymethylfurfural (HMF), of which the latter contains a furan ring, a hydroxymethyl group, and an aldehyde group, making it an attractive starting material for catalytic upgrades.2 HMF can be selectively oxidized to versatile building blocks, including 5-(hydroxymethyl)-furoic acid (HMFA), 5-formylfurfural (FFF), 5-formylfuroic acid (FFA), and 2,5-furandicarboxylic acid (FDCA) (Fig. 1A). Notably, FDCA produced through the full oxidation of the hydroxymethyl and aldehyde groups in HMF has significant market potential as it serves as a ‘green’ substitute for terephthalic acid in producing polyethylene terephthalate (PET).3–5 Additionally, FDCA can be condensed with other monomers to make poly-imines, -amides, -esters, and -urethanes as plastics, resins and porous organic frameworks. Both HMF and FDCA have been identified by the U.S. Department of Energy as being among the top 12 potential platform chemicals available from renewable feedstocks.6 | ||
| Fig. 1 Activity of PedH in transforming HMF into FDCA. (A) Scheme of the oxidation pathways from HMF to FDCA. HMF is oxidized either at the aldehyde group to yield HMFA or at the alcohol group to yield FFF. Both routes proceed via FFA to FDCA upon further oxidation. (B) Specific activity of PedH against various substrates. (C) Biocatalysis of a low concentration of HMF. Conversion was performed in triplicate in 100 mM Tris-HCl buffer (pH 8.0), HMF 0.25 mM, PedH 1 μM, 30 °C. (D) Biocatalysis of a high concentration of HMF. Conversion was performed in triplicate in 100 mM Tris-HCl buffer (pH 8.0), HMF 10 mM, PedH 1 μM, 30 °C. (E) Biocatalysis of various substrates including HMF, FFF, HMFA and FFA at a concentration of 0.25 mM. Conversions were performed in triplicate in 100 mM Tris-HCl buffer (pH 8.0), PedH 1 μM, 0.25 mM of various substrates, 30 °C, for 24 h. | ||
In recent years, extensive research efforts have focused on synthesizing FDCA by oxidizing HMF through chemical pathways.7,8 Traditional thermocatalytic methods for FDCA synthesis often involve harsh reaction conditions, including high temperatures and pressures, along with the presence of metal salts, organic oxidants, and inorganic bases, making the process costly and environmentally harmful.7 In contrast, recent studies have led to the development of highly stable non-precious metal catalysts or heterogeneous catalysts capable of efficiently and selectively catalyzing the oxidation of HMF to FDCA under base-free conditions. For example, Wojcieszak et al. developed an Au/MgF2-MgO-based catalyst that achieved a 99% conversion rate of HMF to FDCA without the need for any added base.9 The non-precious metal catalyst CoOx-MC, under conditions of NaHCO3 additive, O2, and a temperature of 80 °C, achieved a conversion rate of 98.3% for HMF, with a selectivity of 95.3% for FDCA.10 Several studies have also indicated that non-precious single-atom catalysts (SACs) with tailored basicity or acidity have potential for the efficient base-free oxidation of HMF to FDCA.11,12 Recently, a Fe–N–C/γ-Al2O3 catalyst catalyzed the oxidation of 0.8 mM HMF to FDCA under mild, base-free conditions, achieving 99.8% yield of FDCA.12 This provides a compelling demonstration of green chemistry. Nevertheless, the yield of FDCA in base-free catalytic systems still lags behind that of base-dependent catalytic systems.
Electrocatalytic oxidation of HMF for the production of FDCA does not require high-pressure oxidative atmospheres or other environmentally harmful chemical oxidants. Powered by an external potential, electrocatalysis can utilize inexpensive transition metals as highly active catalysts, addressing the thermodynamic and/or kinetic challenges associated with thermocatalysis.7 Chen and co-workers achieved a FDCA yield of 99% through the electrocatalytic oxidation of HMF using a Ni-VN/NF catalyst.13 However, due to the poor conductivity of aqueous solutions of HMF, conductive electrolytes are often necessary for electrocatalysis, which can lead to energy consumption during product separation. In contrast, photocatalytic and photoelectrocatalytic oxidation leverage photons instead of thermal or electrical energy, providing a natural advantage in energy input. Li et al. enhanced the oxidative performance of HMF using BiVO4/CoPi in a TEMPO-mediated reaction, achieving 99.8% yield of FDCA from 1 mM HMF.14 Nevertheless, designing and screening efficient, highly selective photocatalysts with suitable structures for the conversion of HMF into the desired products remains a significant challenge.7
Biocatalysis is emerging as a valuable tool to address these issues, as it typically operates under mild conditions, requires fewer and less toxic reagents and solvents, and offers excellent selectivity.15,16 Currently, producing FDCA through biocatalysis primarily involves microbial and enzymatic conversion approaches. Due to the low FDCA concentration produced by wild-type strains, gene modification or metabolic engineering become essential for the microbial conversion process. Fermentative processes for the microbial bioconversion of HMF to FDCA have been only established using the recombinant strain Raoultella ornithinolytica BF60.17,18 However, these processes generally rely on the activities of oxidases in the cells, and the oxidases require equimolar amounts of molecular oxygen as a co-substrate, which is a limiting factor in high-density cultures. Oxidizing HMF to produce FDCA with isolated enzymes or whole-cell biocatalysts is another promising approach. The full oxidation of HMF often requires a combination of two or three enzymes, as the production of FDCA involves three consecutive oxidation steps. For instance, combinations like aryl-alcohol oxidase (AAO) with an unspecific peroxygenase, galactose oxidase with lipase, and galactose oxidase with alcohol dehydrogenases and horseradish peroxidase have been recently reported.19–21 Additionally, the combination of laccase with chemical catalysts is often employed for the oxidation of HMF. Such oxidation methods, consisting of a laccase-mediator system (LMS), allows for an oxidative step via the oxidized form of the mediator, which can be regenerated using an appropriate laccase.22 For instance, Yang et al. developed a one-pot oxidation system using Comamonas testosteroni SC1588 cells and a laccase-TEMPO system to synthesize FDCA, achieving a yield of 87% over 36 h, equivalent to approximately 0.4 g L−1 h−1.23 Recently, Do Nascimento et al. applied laccase in combination with chemical catalysts in a cascading manner to obtain FDCA under mild reaction conditions via a photocatalytic reactor.24 Production of FDCA from HMF using a single enzyme is quite challenging due to the complexity of the oxidation process. Most enzymes are limited to either alcohol or aldehyde oxidations, while the full oxidation of HMF to FDCA requires an enzyme that can accept both groups. To date, only two enzymes, a 5-hydroxymethylfurfural oxidase (HMFO) and an AAO, have been reported to perform the three-step oxidations to transform HMF into FDCA.25–27 However, both enzymes are flavin adenine dinucleotide (FAD) dependent and can only convert low concentrations of substrate, even with their evolved variants. A thermostable variant of HMFO could convert over 90% of HMF into FDCA within 24 h at a substrate concentration of 5 mM, whereases an AAO variant with improved activity only converted 3% of HMF with a concentration of 2 mM to FDCA within 48 h.26,28
Pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenases (PQQ-ADHs) bear the redox cofactor PQQ and a Lewis acidic metal ion (Ln3+ or Ca2+) in the active site that can catalyze the oxidation of alcohols.29–31 PQQ-ADHs exhibited several advantages in oxidation reaction compared to other oxidases; thus, they have potential for industrial applications. PQQ-ADHs rely on cations as well as PQQ as cofactors to oxidize the substrate, which can even operate in the absence of oxygen and do not generate hydrogen peroxide, which is toxic to cells or enzymes.32,33 Additionally, PQQ-ADHs catalyze the oxidation in an irreversible fashion, leading to the unhindered accumulation of the products. Alternatively, for whole-cell biotransformation, PQQ-ADHs are located in the periplasmic space, which avoids rate-limiting transport across the cytoplasmic membrane for substrates or products. Recently, a PQQ-ADH from Pseudomonas putida KT2440 (PedH) was engineered for the oxidation of HMF, FFF and HMFA for the generation of FDCA.34 Although the activity of PedH was improved against these three substrates, the variants obtained were unable to accept FFA and complete the consecutive oxidation of HMF to FDCA.
In this study, we found that PedH could covert HMF directly into FDCA, although the concentration of substrate and product yield were low; this represents a rare and intriguing finding. We then engineered the enzyme for improved thermostability and activity against HMF. Using a computational design method, the thermostability was first significantly improved. With the thermostable variant 4M as the parent, potential mutation sites for engineering enzyme activity were identified based on calculations of the binding free energy and a deep learning model, MutCompute. After this, an automatic high-throughput site-saturation mutagenesis library building and screening platform was developed and applied to those mutation targets identified. The variant 8M significantly improved the FDCA yield compared to 4M. We also carried out molecular dynamics (MD) simulations to illustrate the mechanisms by which the variants improved activity.
Results and discussion
Sequential oxidation of HMF to FDCA by PedH
We first measured the specific activity of purified PedH against HMF, FFF, HMFA, and FFA, with methanol and ethanol as controls (Fig. 1B). Consistent with previous studies, PedH showed a higher activity toward ethanol than other substrates, and no activity against HMFA. Specifically, the specific activities were 0.031 U mg−1 for HMF, 0.053 U mg−1 for FFF, 0.014 U mg−1 for FFA, and 0.034 U mg−1 for methanol. However, the specific activity values detected were lower than those measured in previous studies; this was attributed to the different method used for measuring the enzyme activity.34 The enzyme activity was measured based on monitoring the phenazine methosulfate (PMS)-mediated reduction of 2,6-dichlorophenol-indophenol (DCPIP). Addition of imidazole to the reaction system led to a higher activity being detected as reported previously, but we did not add imidazole to the experiment as it clearly showed a background activity.35 Despite our adjustments to substrate concentrations and temperature, our attempts to determine the kinetic parameters of PedH were unsuccessful, as accurately measuring the enzyme activity of PedH at lower substrate concentrations proved to be challenging (Fig. S1†).To further confirm the enzyme activity detected, we conducted biocatalysis of HMF over 24 h using the purified enzyme in the presence of the artificial electron mediator/terminal acceptor pair PMS/DCPIP. Surprisingly, in the reaction system with an initial HMF concentration of 0.25 mM, we detected not only the intermediate products FFF (37.3 μM), HMFA (9.4 μM), and FFA (14.1 μM), but also a small amount of FDCA (1.1 μM) (Fig. 1C). This experiment was repeated multiple times to ensure the reliability of the results (Fig. S2†). Most enzymes only perform a single oxidation of either alcohol or aldehyde. To the best of our knowledge, two FAD-dependent enzymes, HMFO and AAO, were previously reported to continuously oxidize HMF to FDCA. Here, PedH is found to exhibit similar properties; this is rare and intriguing. The continuous oxidation capability of PedH may be partially attributed to the unique properties of lanthanides, which acts as strong Lewis acids in the reaction. Actually, Ln-dependent methanol dehydrogenase can convert methanol directly into formate through a formally four-electron process.36 Replacing lanthanide ions with alkaline Earth metal ions or removal of the lanthanide ions resulted in the loss of activity in PedH, confirming the critical role of lanthanides for catalysis (Fig. S3†). We also tested the biocatalysis of HMF and FFA by using PedE, a homologous Ca2+ dependent alcohol dehydrogenase of PedH, and no FDCA production was detected, indicating that the unique properties of lanthanides played an important role in continuously oxidizing HMF to FDCA (Fig. S4†).35 Nonetheless, the continuous oxidation ability of wild-type PedH is very weak. When the concentration of HMF was increased from 0.25 mM to 10 mM, only FFF was apparently produced, with a yield of 0.24 mM after 24 h. Very little HMFA and FFA were produced, and no FDCA was observed (Fig. 1D), and this might be due to the toxicity of the high concentration of aldehyde substrates to the enzyme. This could also be resulted in by competitive binding with active sites between the substrates through which the active center is mainly occupied by the high concentration of HMF rather than other substrates.
To clearly understand the activity of PedH against different substrates, we conducted bioconversion reactions with 0.25 mM of HMF, FFF, HMFA, and FFA as substrates (Fig. 1E). When FFF was used as the substrate, 70.7 μM FFA and 2.2 μM FDCA were produced within 24 h, whereas when HMFA was the substrate, no products were detected, consistent with the specific activity detected (Fig. 1B). This indicated that the oxidation of HMF to FDCA by PedH followed the first route of HMF–FFF–FFA–FDCA. Interestingly, when FFA was the substrate, only 3.4 μM FDCA was detected after 24 h of reaction, suggesting a low conversion rate. The mechanism by which lanthanide-dependent alcohol dehydrogenases oxidize alcohols to aldehydes via an “addition–elimination–protonation” has been elucidated, but this mechanism is evidently not applicable to the oxidation of aldehydes to carboxyl acids.36 We speculated that PedH, similar to several reported oxidases capable of aldehyde oxidation, oxidized aldehydes via their hydrated geminal diol form to yield the final carboxyl product. Studies have indicated that the hydration level of FFA is relatively low, which would also affect its conversion to FDCA.25 These findings suggest that the conversion of FFA to FDCA is the rate-limiting step in the continuous oxidation of HMF to FDCA by PedH.
Computational design to enhance the thermal stability of PedH
Although the continuous oxidation capability of PedH is promising, the specific activity of wild-type PedH toward the aforementioned furan substrates is very low, and the yield of the end product FDCA is barely detectable; this needs to be improved by enzyme engineering. Studies have shown that thermostable proteins tolerate more mutations than mesophilic ones, which makes them a better starting point in protein engineering.37,38 However, PedH showed a low thermal stability and partial insolubility during protein expression, suggesting that it may not be an ideal enzyme scaffold (Fig. S5†). Also, increased enzyme thermostability is often accompanied by improved stability in the presence of organic cosolvents, a high concentration of substrates (or products), and/or other denaturing agents and reaction conditions.39 Therefore, we first attempted to improve PedH's thermal stability through computational design to obtain a stable starting scaffold.Two computational tools, PROSS and FireProt, were applied to predict thermostable single mutants and combinatory mutants for PedH, both of which made predictions based on evolutionary information from multiple sequence alignment (MSA) and free energy changes calculated by Rosetta or FoldX.40,41 PROSS and FireProt predicted 61 and 44 thermostabilizing single-point mutations, respectively, with 13 mutations overlapping between them (Fig. S6†). We constructed all the single-point mutations predicted by FireProt and also synthesized genes of four multiple-point variants including PedH5 containing 24 mutations, PedH6 containing 32 mutations, PedH7 containing 41 mutations, and PedH8 containing 50 mutations predicted by PROSS for experimental characterization (Table S1†).
The variants were purified and their melting temperature, Tm, values were measured. All the variants could be purified well except PedH8, which was not expressed in solution (Fig. S7†). However, only four mutants, namely, S181T, S242T, A401G, and G445P, showed higher Tm values compared to wild-type PedH, with an increase of 3.0 °C, 0.7 °C, 4.7 °C, and 1.8 °C, respectively (Fig. 2A). Structure analysis revealed that the distances between these four sites and rare Earth ions in the active center were greater than 5 Å (Fig. 2B). We then combined these four improved single variants, resulting in a total of 11 multiple-point mutants, whose Tm values and activities against HMF were measured (Fig. 2C). All the combined mutants showed increased Tm values compared to the wild type, but several variants, such as S242T/A401G and A401G/G445P, did not improve Tm relative to their parents, indicating a certain degree of negative epistasis. The quadruple mutant S181T/S242T/A401G/G445P (4M) had the highest Tm value of 72.1 °C, a significant increase of 9.6 °C compared to 62.5 °C of the wild type. This was followed by S181T/S242T/G445P and S181T/G445P, with increases of 8.9 °C and 7.1 °C, respectively. Additionally, the mutants S181T, S181T/G445P, S242T/G445P, S181T/S242T/A401G, and S181T/S242T/A401G/G445P showed a certain increase in activity against HMF compared to the wild type, while variants such as A401G, S242T/A401G/G445P, and S181T/S242T/G445P exhibited weakly decreased specific activities. The half-life times of several improved variants at 55 °C against ethanol as a substrate have also been measured. The quadruple mutant possessed a half-life of 323.1 min, 10.3-fold higher than 31.5 min of the wild type. The half-life times of S242T/G445P, S181T/S242T/A401G, S181T/G445P and S181T were 219.4 min, 202.7 min, 183.9 min and 112.2 min, respectively, all of which were higher than the wild type (Fig. 2D). The 50 ns molecular dynamics (MD) simulations were then performed to understand the structural difference between the wild type and the 4M variant, with the root mean square fluctuation (RMSF) of the backbone atoms calculated over the last 10 ns of the equilibrium trajectory. It was found that the variant exhibited lower RMSF values in the local regions around S242T, A401G and G445P, implying a more stable local conformation (Fig. S8†). It was also found that the mutations S181T, S242T, and G445P caused a higher hydrogen bond occupancy in the 4M variant compared to the wild type, which would contribute to the improved stability (Table S2†).
 | ||
| Fig. 2 Computational design of thermostable PedH variants and their performance in the bioconversion of HMF. (A) Melting temperatures of single variants predicted by PROSS and FireProt. (B) PedH structure showing the positions of four stable single mutants. (C) Activity and thermostability of combinatory variants. The activity was measured against HMF and relative activity with respect to the wild type was reported. (D) Half-life values of thermostable variants at 55 °C against ethanol as a substrate. (E) Biocatalysis of FFA using thermostable variants. Conversions were performed in triplicate in 100 mM Tris-HCl buffer (pH 8.0), FFA 10 mM, PedH 1 μM, 30 °C. | ||
The five mutants showing improved thermostability and high activity were then used for converting HFM at a concentration of 10 mM. After 24 h of reaction, analysis revealed that the main product in all experimental groups was FFF, with trace amounts of HMFA and FFA produced (<0.1 mM). It was also clear that product yields of the thermostable mutants were higher than that of the wild-type (3.0%), with 4.2%, 4.1% and 4.0% for S181T, S181T/S242T/A401G and S181T/S242T/A401G/G445P, respectively (Fig. S9†). As the conversion of FFA to FDCA was the rate-limiting step, we also tested these variants for converting 10 mM FFA within 72 h. Both the wild type and the variants showed a clear increase in the yield of FDCA as the reaction proceeded. The quadruple mutant 4M produced the highest amount of FDCA, reaching 98.2 μM after 24 h and 119.7 μM after 72 h, 13.6% and 19.7% higher than the wild type, respectively. S181T and S181T/G445P also showed weakly improved yields of FDCA compared to the wild type (Fig. 2E). It was speculated that the improved thermal stability enabled the variants to be stable in the presence of substrate and perform an extended period of catalytic reaction, and hence led to a more significant improvement in FDCA yield after 72 h than 24 h. Considering both improved thermostability and activity, we selected the quadruple mutant 4M (S181T/S242T/A401G/G445P) for subsequent enzyme engineering for improved conversion of HMF into FDCA.
Development of an automatic mutation library construction and screening platform for evolving PedH
An efficient and reliable mutation library construction and screening method is critical for the success of the directed evolution of an enzyme. Here, we developed a highly streamlined, robust and general pipeline for automated construction of a site-directed saturation mutation library with NNK randomization, site-directed mutagenesis using the QuikChange method, gene transformation, protein expression and enzyme activity measurements (Fig. 3A). We utilized iBioFoundry for this task, an integrated platform that consisted of high-throughput core instruments like liquid handlers, thermocyclers, fragment analyzers, along with peripherals such as plate sealers, shakers, and incubators, all coordinated by robotic arms and scheduling software. In the initial step, PCR preparation was carried out using the automated workstation (Evo), and as many as 96-PCR tubes were subsequently sealed by a plate sealer (ALPS) for the PCR reaction carried out by an automatic thermocycler. Following this, the tubes were unsealed by automated plate seal removal and the acoustic liquid handler (Echo) was used for DpnI digestion. BL21(DE3) competent cells used for enzyme expression were mixed with the PCR products by using an automated workstation (Evo) and cultured for 1 h. Transformant plating was conducted using another automated workstation (Fluent) and the plates were tagged by a microplate labeler (Agilent Labeler), then incubated in the automated incubator (Cytomat 10C) for 14 h. On the second day, the clones were picked into 800 μL of LB culture medium using an automated workstation colony picker (Fluent), and cultured for 8 h in the automated incubator with orbital shaking (Cytomat 2C Tos), and used for Sanger sequencing to verify the correct mutants and for glycerol stock preparation. We assessed the quality of the mutation library by sequencing a total of 94 transformants selected from the F412 site saturation mutagenesis library. All 20 possible natural amino acid variants were obtained, indicating the reliability of the automatic method (Fig. S10†). Additionally, to evaluate the accuracy of the automated site-directed mutagenesis process, 94 single point variants were constructed and sent for Sanger sequencing. Of these, 88 displayed the correct mutations, yielding an accuracy rate of 93.6% (Fig. S11†). | ||
| Fig. 3 Fully automated screening of PedH variants using iBioFoundry. (A) Process flow diagram for automatic construction and screening of mutation libraries. (B) Schematic diagram showing a colorimetric assay of different variants. The color was detected after the reaction with WB reagent. The high-activity mutants are marked with circles, which display a lighter color, while the low-activity mutants have a darker color. (C) The correlation between the specific activities of purified WT and 10 selected mutants of PedH against HMF with colorimetric data collected during the automation process. | ||
The mutation library was screened based on micro-plate cell culture and a colorimetric assay. The enzyme activity of PQQ-ADH is primarily assayed with alkylated phenazines such as phenazine ethosulfate (PES) or phenazine methosulfate (PMS) as primary electron acceptors, further coupled to dyes for electron transfer. However, previous studies reported issues with background reactions and the reproducibility of these assays due to the instability of electron acceptors and dyes. Wurster's Blue (WB) has been reported to serve as both an electron acceptor and a dye, offering greater stability. Using WB, we developed an automatic high-throughput whole-cell catalytic screening method for PedH (Fig. S12†). The clones on agar plates were picked into 800 μL of LB culture medium using an automated workstation colony picker (Fluent 780) and cultured for 8 h in the automated incubator with orbital shaking (Cytomat 2C Tos). Then, 50 μL of the bacterial culture was transferred using a reagent dispenser (EVO 200) into 750 μL of LB medium containing 10 g L−1 lactose, with the rest stored in glycerol stock. The cultures were incubated in a Cytomat 2C Tos instrument for 12 h. After incubation, the cultures were centrifuged to remove the supernatant. Fresh reaction solution (500 μL) was added to each well, and the cells were resuspended and incubated in the dark for 24 h. On the third day, the cultures were centrifuged, and 200 μL of the supernatant was transferred to a clear-bottom 96-well plate for OD610 measurement using a plate reader (CLARIOstar Plus) (Fig. 3B). The data were analyzed by using Momentum DataMiner.
To verify the reliability of this automated directed evolution method, we analyzed the correlation between the specific activities of purified WT and 10 mutants at position 412 against HMF with colorimetric data being collected during the automation process. The Pearson correlation coefficient (r) was found to be 0.86, indicating a good correlation (Fig. 3C). This suggests that the automated platform can be effectively used for preliminary screening of variants with improved variants, although some false positives might still be recorded. In our biofoundry system, twenty 96-deep well microplates could be cultivated simultaneously, which allowed 20 site-directed saturation mutagenesis libraries to be built and screened in a batch. The procedure for building and screening 20 such mutation libraries takes around 1 d for primer synthesis, 1 h for PCR, 0.5 h for DpnI digestion, 15 h for transformation and colony picking, 20 h for protein expression, 24 h for screening variants with improved activities and 12 h for sequencing verification, and around 96 h overall to go from the requested mutation library to the physical enzyme samples to the improved variants with corresponding enzyme activity data. Using a biofoundry to construct and characterize protein variants is much more efficient than manual methods. The facility aims to support the advancement of artificial intelligence in protein engineering, as the timely testing of algorithm-designed mutants will significantly contribute to refining and optimizing the algorithm.
Directed evolution of PedH aided by computational design, a deep learning model and automatic facilities
With the reliable and efficient mutation library construction and screening method available, we analyzed the PedH structure and attempted to identify the potential mutation target sites based on calculations of the binding free energy and a deep learning model constructed based on protein structure information (Fig. 4A). The substrate HMF was first docked into the PedH structure, which was then analyzed by using Discovery Studio software to calculate the binding free energy change for all single variants of 62 amino acids within 8 Å of the substrate HMF (Fig. 4B). The conserved amino acids critical for catalysis, including E199, N281, D323, D325, C131 and C132, were not included for calculation. Analysis of the binding free energy of the variants revealed that the amino acids could significantly influence substrate binding. For example, mutations at W285, F412, and G556 generally decreased the binding energy, with the mutants W285T, F412A, and G556K showing mutation energies of −2.48 kcal mol−1, −2.26 kcal mol−1, and −2.33 kcal mol−1, respectively. Conversely, mutations at A261 and A557 generally increased the binding energy, with mutants A261Y and A557I showing energies of 2.96 kcal mol−1 and 2.25 kcal mol−1, respectively (Fig. 4B). These amino acids could be the hot spots that influenced the activity of the PedH toward HMF. Consequently, 20 residues with significant mutation energy changes with absolute values larger than 1.0 were identified. In addition to the five amino acids mentioned above, these include G197, G198, G259, W263, G280, R350, L413, G414, N417, W418, W437, L455, I461, W493, and W561 (Fig. 4A & B). Additionally, a bulky amino acid, F459, positioned above the active site pocket, was also selected as a mutation target, as it was close to the previously validated effective site F412 (Fig. 4A).34 | ||
| Fig. 4 Identification of hot-spots for engineering PedH to improve its activity against HMF. (A) PedH structure showing the positions of hot-spot residues influencing PedH activity. Blue sphere, hot spots identified by binding free energy calculations; purple sphere, hot spots identified by MutCompute. (B) Heatmap of the binding free energy changes of all possible single mutations at amino acids within 8 Å around the substrate HMF. Mutations with higher binding energy are shown in red, while those with lower binding energy are shown in blue. The mutation energy was calculated using Discovery Studio 4.0. (C) The relative activities of the top 20 mutants predicted by MutCompute. | ||
The amino acids identified based on binding free energy changes or empirical analysis are typically in proximity to the catalytic center. We also attempted to identify some residues that were distant from the catalytic sites, but still influenced the enzyme activity, which actually remained challenging. MutCompute is a structure-based 3D convolutional neural network model that was trained to associate local protein microenvironments with their central amino acids.42 MutCompute can predict mutations for optimizing the protein structure, and has the potential to identify distant variants that are critical for enzyme stability and activity. Actually, it has been used for the engineering of PET-degrading enzymes, resulting in significant improvements in PET-degrading enzyme activity and thermal stability.43 The PedH structure (6ZCW.pdb) was submitted to the MutCompute sever, and the model prediction mutants were sorted based on the probability change between the predicted amino acid and the wild-type amino acid (Table S3 & Fig. S13†). The top 20 single point mutants were then selected for experimental characterization (Table S4†). We constructed and purified these 20 variants, and evaluated them for specific activities toward two substrates, HMF and ethanol (Fig. 4C). The variants I135V and W554F displayed a 2.0-fold and 1.3-fold increase in ethanol activity, respectively, compared to the WT, and their activity toward HMF was also enhanced by 1.7-fold and 1.6-fold, respectively. The L254M variant increased enzyme activity against ethanol by 1.8 times but showed a slight decrease toward HMF, compared to the WT. Conversely, the A75S variant increased activity against HMF by 1.4 times but reduced activity toward ethanol to 67.4% of the WT. Other variants that showed enhanced activity toward at least one substrate included S30T, S316T, Q528D, and Q586E. Additionally, we observed that N136H and N144D variants dramatically reduced activity against both substrates, and R300Q completely lost enzyme activity. Consequently, these 11 residues, which significantly impacted enzyme activity, were selected for subsequent saturation mutagenesis (Fig. 4A & C).
Using the automated mutation library construction and screening method, we performed site-directed saturation mutagenesis and high-throughput screening on the selected 32 amino acids, namely, 20 sites predicted by binding free energy calculations, 11 sites predicted by MutCompute, and F459. Starting with 4M as the template, saturation mutagenesis libraries were constructed using degenerate primers with “NNK” at the corresponding positions, and then screened for improved activity against the substrates HMF and FFA, respectively (Fig. 5A & B). The mutants showing OD610 values lower than 4M were selected for sequencing verification. Although most of the libraries contained variants with OD610 values lower than 4M, sequencing revealed that some of these strains still retained the 4M genotype, indicating a certain level of false positives in the screening method. We finally identified a total of 47 unique single-point mutants that showed improved enzyme activity compared to 4M, including five mutations at the A557 position and four mutations at the F412 position.
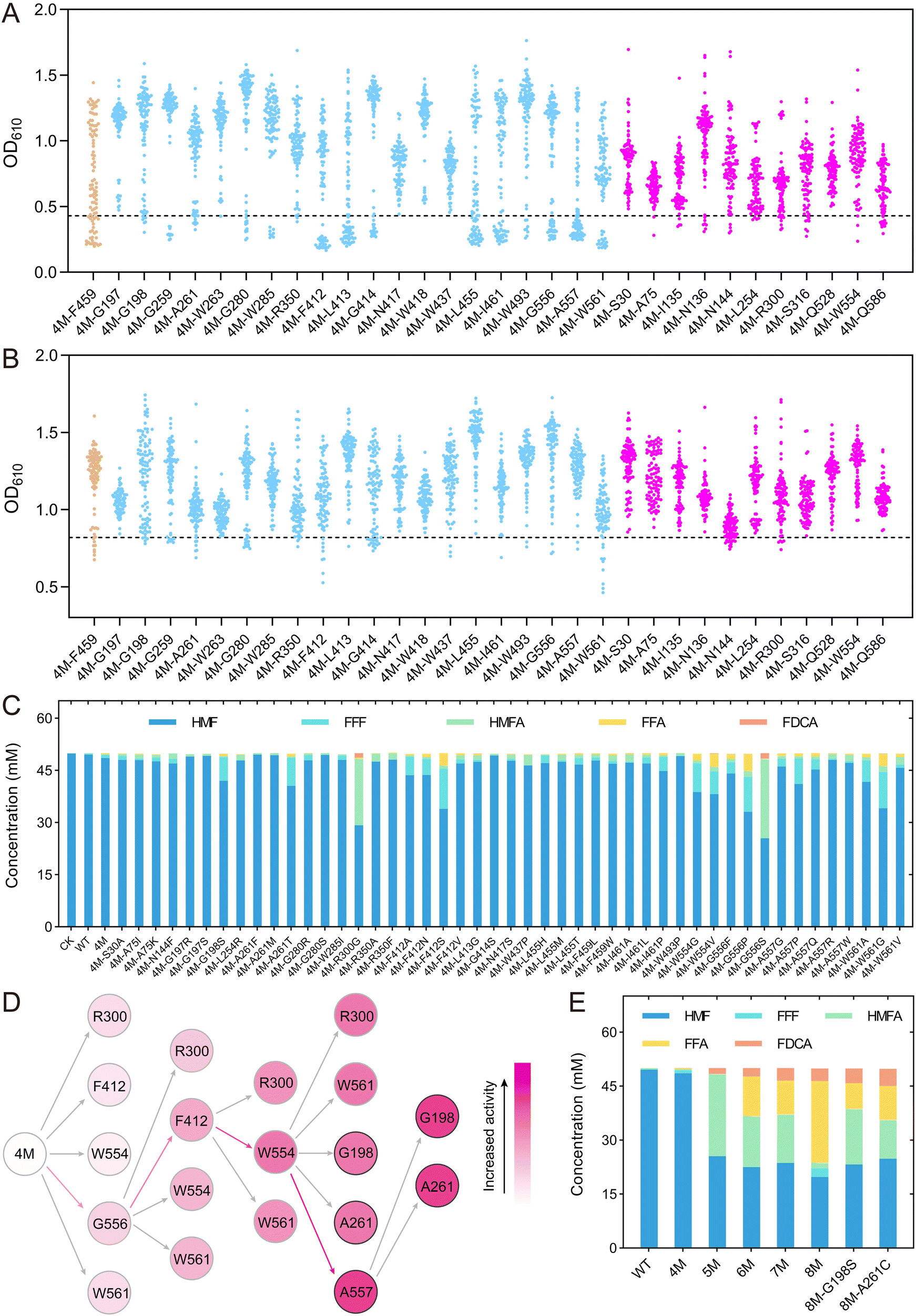 | ||
| Fig. 5 The activities of PedH variants. (A) Screening of saturation mutation libraries against HMF. (B) Screening of saturation mutation libraries against FFA. (C) Biocatalysis of HMF using E. coli cells expressing improved single variants after 24 h. Conversions were performed in 100 mM Tris-HCl buffer (pH 8.0), HMF 50 mM, cells OD600 = 10, 30 °C. (D) Iterative saturation mutagenesis performed at eight sites. The color indicated the yield of FDCA, with deeper color showing higher yield. (E) Biocatalysis of HMF using E. coli cells expressing improved combinational variants after 24 h. Conversions were performed in 100 mM Tris-HCl buffer (pH 8.0), HMF 50 mM, cells’ OD600 = 10, 30 °C. | ||
The single-point mutants obtained from the initial screening were then subjected to whole-cell catalysis to convert 50 mM HMF (Fig. 5C). After reaction for 24 h, we detected the conversion of HMF and the production of various intermediate products for all the variants. Most of the variants showed an improved conversion rate of HMF compared to 4M. The extraordinary variants 4M-R300G and 4M-G556S exhibited 37.6% and 41.5% conversion of HMF, respectively, 10.7-fold and 11.9-fold higher than that of 4M. These two variants produced 1.32 mM and 1.59 mM of FDCA, respectively, representing 132-fold and 159-fold increase compared to 4M, while no FDCA was detected in the WT. Other variants, namely, 4M-F412S, 4M-W561G, 4M-W554 V, 4M-A261T and 4M-G198S, exhibited HMF conversion rates of 32.2%, 31.7%, 23.7%, 18.9% and 16.0%, respectively, significantly higher than the wild type (0.9%) and 4M (3.5%), although the FDCA end product was not produced in high yield.
To further improve the conversion of HMF into FDCA, the amino acids R300, F412, W554, G556, and W561, where the mutations showed a high FDCA yield, were selected for further iterative saturation mutagenesis (Fig. 5D). Since we had established an automatic platform for constructing single-point mutations, an iterative saturation mutagenesis (ISM) was applied to construct combinatory mutations, in which all mutations at one amino acid site were constructed by site-directed mutagenesis instead of saturation mutagenesis using NNK.44,45 Theoretically, for ISM of five mutation sites, a total of 285 (19 × (5 + 4 + 3 + 2 + 1)) mutations would be constructed. In the first round of ISM, all possible single point variants on the five sites (19 × 5) were constructed and the mutant with the highest activity was identified as 4M-G556S (Fig. S14†). With 5M (4M-G556S) as the template, a second round of mutagenesis was performed on the other four amino acids and several mutants showing higher activity than 5M were indeed obtained based on a colorimetric assay (Fig. S15†). Twenty-one mutants with OD610 values lower than or comparable to 5M were selected for whole-cell conversion reactions, leading to the identification of the optimal mutant, 6M (4M-G556S–F412S) (Fig. S16†). From the third round of ISM onward, each mutant underwent whole-cell conversion assays to ensure the reliability of the screening results. Mutant 7M (4M-G556S–F412S–W554S) was identified in the third round, which showed 1.5-fold and 348-fold increase in FDCA compared to 6M and 4M, respectively, although it showed a slight decrease in HMF conversion compared to 6M (Fig. S17†). In the fourth round of ISM, saturation mutagenesis was performed on the R300 and W561 sites starting from 7M, but no further increase in FDCA production was observed (Fig. S18†). Consequently, for the fourth round of evolution, three new sites (G198, A261, and A557) identified in the initial screening were included in the iterative saturation process, ultimately leading to the mutant 8M (4M-G556S–F412S–W554S–A557N) (Fig. 5D & Fig. S19†). The FDCA yield of the 8M mutant is similar to that of the 7M mutant, yet the FFA yield is over 2.4 times greater than that of the 7M mutant. This suggests that 8M has a higher potential for FDCA production. In the fifth round, mutants 8M-G198S and 8M-A261C were obtained, which showed 17.5% and 36.1% increase in FDCA production compared to 8M, 409-fold and 474-fold improvement compared to 4M (Fig. S20† &Fig. 5E).
The whole-cell catalysis reaction for converting a low concentration of HMF was carried out for the improved variants. At a concentration of 10 mM HMF, 8M produced 3.15 mM FDCA within 24 h, which was lower than the yields of 4.14 mM and 4.31 mM produced by 8M-G198S and 8M-A261C, respectively. The HMFA intermediate product produced by 8M-G198S and 8M-A261C was more than 2.5 times higher than that of 8M. After 48 h, the FDCA yield of all three mutants increased to over 6 mM, but the HMFA concentration remained nearly unchanged (Fig. S21†). Based on these results, we hypothesized that these mutants had low activity toward HMFA. Several mutants, including the best mutant from each round, were selected for whole-cell catalysis experiments using HMFA as the substrate. The results confirmed that all of these mutants had low activity toward HMFA (Fig. S22†), indicating that these mutants primarily catalyzed the conversion of HMF into FDCA via the HMF–FFF–FFA–FDCA pathway. Consequently, the high concentration of HMFA produced by the fifth-round mutants would limit the production of FDCA, leading to the halting of the evolutionary process (Fig. 5E & S22†).
Modification of reaction conditions for improving the synthesis of FDCA
Since 8M revealed a high yield of FDCA and low production of HMFA, we modified the reaction conditions to improve the production of FDCA, including WB concentration, type of rare Earth element, pH, concentrations of PQQ and rare Earth elements, temperature, cell concentration, and substrate concentration (Table 1 & Fig. 6). Initially, we determined the optimal WB concentration by examining a range from 0.5 to 8 mM. The highest HMF conversion rate and FDCA yield of 64.4% and 5.2%, respectively, were achieved at a WB concentration of 1 mM (Fig. 6A & Table 1 T1). Since it has been reported that the activity of rare Earth-dependent alcohol dehydrogenases is influenced by the type of rare Earth element, we also assessed the effect of adding various lanthanides—namely, La3+, Ce3+, Nd3+, Sm3+, Eu3+, Gd3+, and Tb3+—in addition to Pr3+ (Fig. 6B). We found that while the addition of Tb3+ reduced the FDCA yield, other lanthanides had no significant effect on FDCA production. By contrast, the addition of lanthanides did influence the conversion of HMF, and under the addition of Nd3+, the highest HMF conversion rate of 66.6% was achieved. Consequently, Nd3+ was selected for subsequent conversion experiments (Table 1 T2).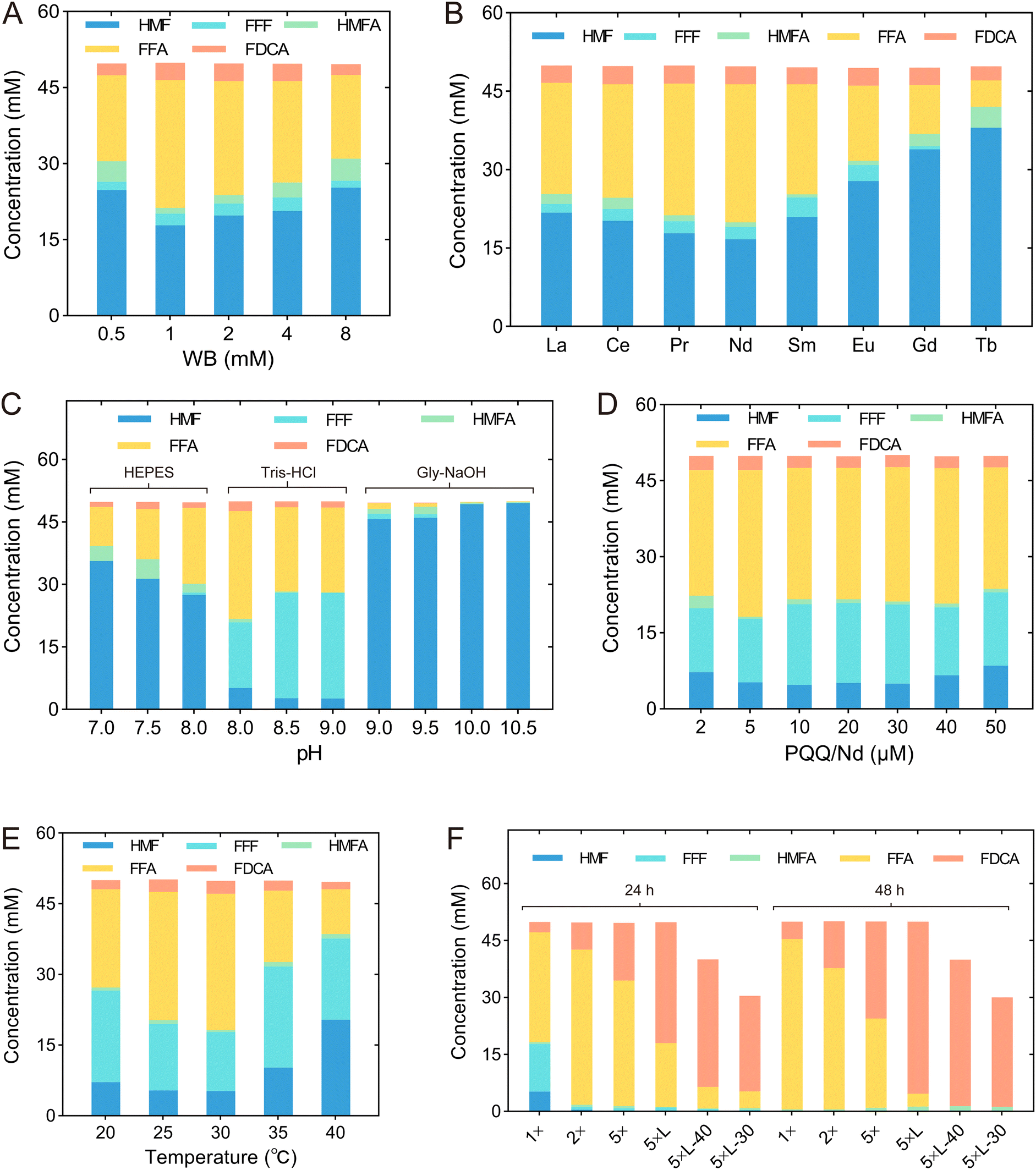 | ||
| Fig. 6 Optimization of reaction conditions, including WB concentration (A), type of rare Earth element (B), pH (C), concentrations of PQQ and rare Earth elements (D), temperature (E), cell concentration and form of biocatalysts (F). 1×, whole cells with OD600 of 10 used in the reaction; 2×, whole cells with OD600 of 20 used in the reaction; 5×, whole cells with OD600 of 50 used in the reaction; 5 × L, cell lysates with OD600 of 50 used in the reaction; 5 × L-40, cell lysates with OD600 of 50 used in the reaction with the 40 mM HMF; 5 × L-30, cell lysates with OD600 of 50 used in the reaction with the 30 mM HMF. | ||
| Tests | WB concentration (mM) | REE ions | pH Buffer | PQQ/Nd (μM) | Temperature (°C) | Cell amount | |
|---|---|---|---|---|---|---|---|
| Whole cell | Cell lysates | ||||||
| Initial conditions | 2 | Pr3+ | 8.0 | 20 | 30 | OD600 = 10 | — |
| 100 mM Tris-HCl | |||||||
| T1 | 1 | Pr3+ | 8.0 | 20 | 30 | OD600 = 10 | — |
| 100 mM Tris-HCl | |||||||
| T2 | 1 | Nd 3+ | 8.0 | 20 | 30 | OD600 = 10 | — |
| 100 mM Tris-HCl | |||||||
| T3 | 1 | Nd3+ | 8.0 | 20 | 30 | OD600 = 10 | — |
| 400 mM Tris-HCl | |||||||
| T4 | 1 | Nd3+ | 8.0 | 5 | 30 | OD600 = 10 | — |
| 400 mM Tris-HCl | |||||||
| T5 | 1 | Nd3+ | 8.0 | 5 | 30 | OD600 = 10 | — |
| 400 mM Tris-HCl | |||||||
| T6 | 5 | Nd3+ | 8.0 | 25 | 30 | OD600 = 50 | — |
| 400 mM Tris-HCl | |||||||
| T7 | 5 | Nd3+ | 8.0 | 25 | 30 | — | OD600 = 50 |
| 400 mM Tris-HCl | |||||||
We also optimized the pH within a range of 7.0 to 10.5. Since FDCA is an acid, the pH kept dropping as the reaction proceeded, and 100 mM Tris-HCl buffer was insufficient to maintain stable pH during the reaction. Therefore, the buffer concentration was increased to 400 mM to provide sufficient buffering capacity. As shown in Fig. 6C, HMF conversion was influenced not only by pH but also by the type of buffer. At pH 8.0, Tris-HCl buffer outperformed HEPES buffer, and a similar trend was observed at pH 9.0, where Tris-HCl was more effective than Gly–NaOH buffer. Under the conditions of 400 mM Tris-HCl at pH 8.0, the HMF conversion rate reached 89.7%, which was slightly lower than the conversion rates observed at pH 8.5 and 9.5. However, the highest FDCA and FFA yields were obtained at pH 8.0, suggesting that pH differentially affected each step of the sequential oxidation process (Fig. 6C & Table 1 T3). Furthermore, the effects of PQQ and Nd3+ concentrations were investigated. The highest yields of FFA and FDCA were observed when both PQQ and Nd3+ were at a concentration of 5 μM (Fig. 6D & Table 1 T4). Subsequently, we examined the effect of temperature, ranging from 20 °C to 40 °C, and found that the optimal temperature was 30 °C (Fig. 6E & Table 1 T5).
Finally, we adjusted cell concentrations and the biocatalyst form to further improve FDCA yields. The ratio of WB, PQQ and Nd3+ to the cells remains constant. When using whole cells for catalysis, after 24 h of reaction, increasing the cell OD600 value from 10 to 50 led to an increase in FDCA yield from 5.4% to 30.3% (Table 1 T6). By contrast, when the cell lysate was used for catalysis at an OD600 of 50, the FDCA yield reached 63.7% after 24 h and further increased to 90.4% after 48 h (Table 1 T7). This increase was likely due to the removal of the process for transporting substrate and product across the cells, which accelerated the reaction rate. When the concentration of HMF was reduced to 40 mM and 30 mM, the FDCA yield after 48 h reached 96.4% and 96.1%, respectively, and the only by-product detected in the reaction system was a small amount of HMFA (Fig. 6F).
Molecular dynamics simulations to reveal the improved mutants’ mechanisms
To explore the mechanism by which the mutants 8M, 8M-G198S, and 8M-A261C significantly enhanced activity toward HMF and other substrates, we measured the enzyme specific activities and kinetic parameters for HMF, FFF, HMFA, and FFA by using purified enzymes (Table 2). Due to the low activity of WT and 4M toward these substrates, their kinetic parameters could not be determined. Three variants showed similar Km values against HMF, with values of 0.46 ± 0.04 mM, 0.48 ± 0.02 mM, and 0.48 ± 0.03 mM, for 8M, 8M-G198S, and 8M-A261C, respectively, suggesting their similar binding affinity for HMF. 8M exhibited the highest specific activity toward HMF and FFF, with increases of 26.6-fold and 23.1-fold compared to the WT, respectively. Additionally, 8M showed weak activity toward HMFA, while the WT and 8M-G198S and 8M-A261C mutants displayed no activity against this substrate (Table 2). The 8M-A261C mutant had the highest activity toward FFA, 37.5 times higher than that of WT, which was 50% and 6.5% higher than 8M and 8M-G198S, respectively, consistent with its highest observed catalytic efficiency kcat/Km of 1.15 mM−1 s−1. This explained why a higher FDCA yield was observed for 8M-A261C than the other two variants (Fig. 5E).| Substrates | Enzymes | |||||
|---|---|---|---|---|---|---|
| WT | 4M | 8M | 8M-G198S | 8M-A261C | ||
| a Activities below the detection limit are indicated as n.d. | ||||||
| K m (mM) | HMF | n.d.a | n.d. | 0.46 ± 0.04 | 0.48 ± 0.02 | 0.48 ± 0.03 |
| FFF | n.d. | n.d. | 1.19 ± 0.22 | 0.98 ± 0.14 | 1.01 ± 0.21 | |
| HMFA | n.d. | n.d. | 4.76 ± 0.77 | n.d. | n.d. | |
| FFA | n.d. | n.d. | 0.45 ± 0.07 | 0.42 ± 0.01 | 0.39 ± 0.04 | |
| k cat (s−1) | HMF | n.d. | n.d. | 0.71 ± 0.02 | 0.55 ± 0.01 | 0.49 ± 0.01 |
| FFF | n.d. | n.d. | 1.10 ± 0.07 | 0.86 ± 0.04 | 0.91 ± 0.06 | |
| HMFA | n.d. | n.d. | 0.08 ± 0.01 | n.d. | n.d. | |
| FFA | n.d. | n.d. | 0.3 ± 0.02 | 0.41 ± 0.01 | 0.45 ± 0.02 | |
| k cat/Km (mM−1 s−1) | HMF | n.d. | n.d. | 1.54 ± 0.09 | 1.15 ± 0.03 | 1.02 ± 0.04 |
| FFF | n.d. | n.d. | 0.92 ± 0.11 | 0.88 ± 0.08 | 0.90 ± 0.13 | |
| HMFA | n.d. | n.d. | 0.02 ± 0.00 | n.d. | n.d. | |
| FFA | n.d. | n.d. | 0.67 ± 0.03 | 0.98 ± 0.00 | 1.15 ± 0.07 | |
| Specific activity (U mg−1) | HMF | 0.031 ± 0.007 | 0.028 ± 0.005 | 0.826 ± 0.07 | 0.588 ± 0.09 | 0.565 ± 0.1 |
| FFF | 0.053 ± 0.011 | 0.053 ± 0.012 | 1.226 ± 0.1 | 0.968 ± 0.12 | 1.022 ± 0.12 | |
| HMFA | n.d. | n.d. | 0.069 ± 0.02 | n.d. | n.d. | |
| FFA | 0.014 ± 0.004 | 0.017 ± 0.004 | 0.35 ± 0.07 | 0.493 ± 0.08 | 0.525 ± 0.07 | |
To further understand the molecular mechanisms by which mutations affect the interaction of PedH with substrates, we conducted 50 ns MD simulations for variants 4M and 8M complexed with the substrates HMF or FFA, namely, 4M complexed with HMF (4M/HMF), 8M complexed with HMF (8M/HMF), 4M complexed with FFA (4M/FFA) and 8M complexed with FFA (8M/FFA) (Fig. S23 & 24†). According to the reaction mechanism revealed for PQQ-ADHs, PedH might not directly oxidize aldehyde groups.36 Instead, PedH could oxidize aldehyde groups by oxidizing glycols similar to several oxidases reported previously.25 Thus, for the rate-limiting substrate FFA, we used its gem-diol form for molecular docking and MD simulations (Fig. S25†). It was observed that the mutations W554S, G556S, A557N, and W561 in 8M were located on the same ‘lid loop’, which lay above the substrate binding pocket, restricting the entry of bulky substrates (Fig. 7A–D & S26†). These mutations reshaped the ‘lid loop’, which enlarged the binding pocket, thereby facilitating the entry and exit of the substrate HMF. Using the CavityPlus tool to calculate the binding pocket volume, we determined that the pocket volume of the 4M/HMF system was 282 Å3, while that of the 8M/HMF system was 376 Å3. Similar results were also observed for the 4M/FFA and 8M/FFA systems (Fig. 7E–H).46 A previous study indicated that mutating from F412 to a smaller amino acid could expand the binding pocket and hence enhance the enzyme activity against large substrates, consistent with our findings.34 The extension of the binding pocket might facilitate the hydroxyl oxygen of bulky substrates approaching Pr3+, thereby enhancing the likelihood of the reaction occurring.
 | ||
| Fig. 7 MD simulations to reveal the mechanisms of the improved variants. Representative catalytic center structures of 4M/HMF (A), 8M/HMF (B), 4M/FFA (C), and 8M/FFA (D) in MD simulations. The structure was obtained from last frame of the MD simulations. The volumes of the substrate binding pockets of 4M/HMF (E), 8M/HMF (F), 4M/FFA (G), and 8M/FFA (H). Statistical analysis of reaction distances between reacting atoms for the 4M/HMF (I), 8M/HMF (J), 4M/FFA (K) and 8M/FFA (L) systems. Distances include the distance, d(O–Pr), between the substrate hydroxyl oxygen and Pr3+, and the distance, d(O-C5), between the substrate hydroxyl oxygen and the C5 atom of PQQ. | ||
We also found a hydrogen bond newly formed between the N557 residue and the aldehyde oxygen of HMF in 8M, which was not observed between HMF and A557 in 4M (Fig. 7A, B & Table S5†). This hydrogen bond would stabilize the substrate conformation and contribute to the occurrence of the reaction. However, in the 8M/FFA system, the N557 residue did not form a hydrogen bond with the carboxyl oxygen of FFA (Fig. 7C and D). This might partially explain why the activity of the mutant against carboxyl-containing substrates like HMFA and FFA was significantly lower than that toward HMF and FFF.
Based on the catalytic mechanism of lanthanide-dependent alcohol dehydrogenase, the distances between the hydroxyl oxygen of the substrate and Pr3+ metal ion or the C5 atom of PQQ should be less than 4 Å for a catalytic reaction to occur. We separately assessed the distance, d(O–Pr), between the substrate hydroxyl oxygen and Pr3+ as well as distance d(O-C5) between the substrate hydroxyl oxygen and the C5 atom of PQQ in the 4M/HMF and 8M/HMF systems. The results showed that the probability of a conformation conducive to catalysis in the 4M/HMF system was nearly zero, whereas, in the 8M/HMF system, this probability was 9.2% (Fig. 7I and J). This was consistent with improved activity against HMF observed for 8M compared to 4M. Similarly, the probability of conformations conducive to catalysis in the 8M/FFA system was 20.9%, significantly higher than that of 4M/FFA, which was consistent with the activity observed (Fig. 7K and L). The MM/GBSA method was then used to calculate the substrate binding free energy. The binding free energies of HMF to 4M and 8M were −14.77 ± 0.32 kcal mol−1 and −16.26 ± 0.25 kcal mol−1, respectively, while those of the gem-diol form of FFA to 4M and 8M were −14.46 ± 0.39 kcal mol−1 and −16.59 ± 0.35 kcal mol−1, respectively (Table S6†). These results indicate that, compared to 4M, the 8M mutant has enhanced affinity for the substrates, consistent with the activities observed.
Conclusions
In this study, we identified a PQQ and lanthanide-dependent ADH, PedH, which possessed the rare ability to continuously oxidize HMF to produce FDCA. This rare property may be related to the strong Lewis acidity of the lanthanides. To engineer PedH for improved yield of FDCA, we developed a fully automated workflow for constructing and screening the PedH mutant library, significantly enhancing the efficiency compared to manual efforts, while keeping overall costs manageable. It took less than 4 days to construct and screen 20 site-directed saturation mutagenesis libraries, which significantly improved the efficiency of protein engineering. The PedH 8M mutant obtained in this study showed significantly enhanced specific activities toward HMF, FFF, HMFA, and FFA. E. coli overexpressing the 8M mutant was an efficient whole-cell biocatalyst for converting HMF into FDCA. Compared to other types of ADHs, PedH can oxidize substrates under anaerobic conditions without producing hydrogen peroxide, which is toxic to cells or enzymes, thus facilitating high-density cultures. Additionally, PedH could be expressed in the periplasmic space, avoiding rate-limiting transport across the cytoplasmic membrane for substrates or products and eliminating the need for co-expression of transporters in whole-cell biotransformation. In summary, this study would be useful for developing whole-cell catalysts for high-efficiency production of FDCA.Experimental
Microorganisms and materials
The gene encoding PedH without the signal peptide (PedH28-595) was codon-optimized for expression in Escherichia coli (E. coli) and synthesized by GENEWIZ (Suzhou, China). The expression vector used in this study was pET28a. HMF was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. FFF, HMFA, FFA, FDCA and PQQ were obtained from TCI (Shanghai) Development Co., Ltd. WB was purchased from Sigma-Aldrich. DCPIP and PES were provided by Shanghai Macklin Biochemical Co., Ltd. DNA polymerase (PrimeStar Max) was obtained from Takara Biomedical Technology (Beijing) Co., Ltd. Other reagents and solvents were purchased from Sinopharm Chemical Reagent Co., Ltd.Expression and purification of PedH and variants
The nucleic acid sequence of PedH was placed in plasmid pET28a, with a 6 × His tag linker fused to its C-terminus. The variants were generated by the polymerase chain reaction (PCR) with a PedH plasmid as a template. E. coli BL21 (DE3) was used for protein expression. Cells were added to Luria–Bertani (LB) medium (5 mL) containing kanamycin (50 μg mL−1) and cultured at 37 °C, 220 rpm for 12 h. A seed culture (2%, v/v) was then inoculated in LB media, supplemented with 1% lactose and kanamycin (50 mg L−1). After 12 h of cultivation at 30 °C, cells were harvested by centrifugation (3500g, 15 min, 4 °C), resuspended in 15 mL of buffer A (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 50 mM imidazole), and then lysed by sonication. The lysate was clarified by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 30 min at 4 °C, and the supernatant was applied to a 1 mL Ni-NTA affinity chromatography column. After washing with 20 column volumes (CVs) of buffer A, proteins were eluted with three CVs of buffer B (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 250 mM imidazole). The eluate was desalted using Amicon Ultra-10K centrifugal filters with buffer (100 mM Tris-HCl, pH 8.0) to a final volume of 0.3 mL. Protein concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific NanoDrop One, Thermo Fisher Scientific Inc., Wilmington, USA).
000×g for 30 min at 4 °C, and the supernatant was applied to a 1 mL Ni-NTA affinity chromatography column. After washing with 20 column volumes (CVs) of buffer A, proteins were eluted with three CVs of buffer B (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 250 mM imidazole). The eluate was desalted using Amicon Ultra-10K centrifugal filters with buffer (100 mM Tris-HCl, pH 8.0) to a final volume of 0.3 mL. Protein concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific NanoDrop One, Thermo Fisher Scientific Inc., Wilmington, USA).
T m measurements
T m measurements were conducted using a Prometheus™ NT. Plex instrument (NanoTemper Technology, Munich).47,48 A mixture containing 10 μM PedH, 30 μM metals, and 30 μM PQQ was prepared in 100 mM Tris-HCl buffer (pH 8.0) and equilibrated at room temperature for 1 h. The samples were then transferred to a 384-well plate with a conical bottom (Greiner, 82051-320) and loaded into Prometheus™ NT.Plex nanoDSF Grade Standard Capillary Chips (PR-AC002). Laser power was adjusted during the discovery scan. Intrinsic fluorescence was recorded at 330 nm and 350 nm while heating the samples from 20 °C to 95 °C at a rate of 2 °C per minute. The data were subsequently analyzed using PR. ThermControl software.Measurements of enzyme kinetics
The activities of purified PedH and its variants were measured using a dye-linked colorimetric assay in 96-well microtiter plates, as described in previous studies.49,50 Briefly, 200 μL of assay solution containing 100 mM Tris-HCl (pH 8), 200 μM 2,6-dichlorophenol indophenol (DCPIP), 1 mM phenazine ethosulfate (PES), 1.5 μM PrCl3, 1.5 μM PQQ, and 0.5 μM of enzyme was prepared. The enzyme and assay solutions were incubated at 30 °C in the microplate reader until the background reaction subsided before adding the substrate. Enzyme activity was determined by measuring the change in OD600 within the first minute after substrate addition. Results were expressed as the mean ± standard deviation (n = 3). Kinetic parameters of PedH and its mutants were determined by fitting enzyme activities at various substrate concentrations to the Michaelis–Menten equation. The kinetic values are presented as mean values with corresponding standard deviation.Biocatalysis and compound analysis
Bioconversions were conducted in duplicate using 500 μL reaction solution in sealed 1.5 mL reaction vials at 30 °C and 220 rpm. The reaction solution consisted of 100 mM Tris-HCl (pH 8), 30 μM PQQ, 30 μM PrCl3, 1 mM Wurster's blue (WB), and 10 mM substrate (HMF, FFF, HMFA or FFA), along with purified PedH or its mutants at a final concentration of 10 μM. Control reactions were carried out without protein. In this setup, electrons are transferred from the enzyme's active site to the electron mediator WB, which is slowly re-oxidized by oxygen. Samples were incubated in the dark to minimize light-induced decomposition of WB. Samples were collected at various time points, filtered by using a 0.22 μm pore size filter membrane, and stored at −80 °C for subsequent product analysis.Whole-cell biocatalysis were performed with 10 mL reaction solution in 50 mL centrifuge tubes at 30 °C and 220 rpm shaking. The reaction solution contained 100 mM Tris-HCl (pH 8), 20 μM PQQ, 20 μM PrCl3, 1 mM WB, and 50 mM HMF as well as whole-cell biocatalyst (OD600 = 10). Reactions were stopped by centrifugation at 12000g for 10 min and the supernatant was diluted to an appropriate ratio and filtered to obtain the sample to be measured.
The reaction conditions were modified to improve the synthesis of FDCA, including WB concentration, type of rare Earth elements, pH, concentrations of PQQ and rare Earth elements, temperature, cell concentration, and substrate concentration. The impact of WB concentration was examined over the range of 0.5–8 mM. For the optimization of rare Earth element types, various ions, including La3+, Ce3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, and Pr3+, were evaluated. To optimize the pH, reactions were conducted in HEPES buffer (pH 7.0–8.0), Tris-HCl buffer (pH 8.0–9.0), or Gly–NaOH buffer (pH 9.0–10.5). The influence of PQQ/Nd3+ was investigated over a concentration range of 2–50 μM. The effect of temperature was studied between 20 °C and 40 °C. The OD600 value of the cells was increased from 10 to 50 to assess the impact of cell concentration. Whole cells were sonicated to create a lysate to examine the influence of catalyst form. The effect of substrate concentration on FDCA yield was investigated within the range of 30–50 mM.
Compound quantification was performed by high-performance liquid chromatography (HPLC) on an Agilent 1260 series instrument (Santa Clara, CA) with an Aminex HPX-87H column (300 × 7.8 mm, 9 μm). Sulfuric acid (5 mM) was used as the mobile phase at a flow rate of 0.8 mL min−1. The column temperature was maintained at 60 °C. Compounds were quantified from 5 μL sample injections using an ultraviolet detector at a fixed wavelength of 264 nm. The retention times were 21.02 min, 26.83 min, 15.83 min, 16.83 min and 11.13 min for HMF, FFF, HMFA, FFA and FDCA, respectively.
Construction of mutation libraries and high-throughput screening by iBioFoundry
Using the Fluent 780 pipetting workstation, the necessary PCR components were added to a 96-well PCR plate, and PCR was carried out by ATC PCR. The PCR product was then digested with DpnI at 37 °C for 30 min to eliminate templates. Then, 5 μL of the digested product was added to a 96-well PCR plate containing 20 μL of E. coli BL21 (DE3) competent cells, mixed, cooled at 0 °C for 30 min, and heat-shocked at 42 °C for 90 s. Using the Fluent 780 pipetting workstation, 150 μL of LB medium was added to each well, and the plate was incubated at 37 °C for 1 h in a shaker (Cytomat 2C Tos). After incubation, the cells were centrifuged at 3000g for 2 min, the supernatant was removed, and 100 μL was retained for resuspension. Then, 80 μL of the resuspension was plated onto LB agar plates containing kanamycin using a colony picker and incubated at 37 °C in a Cytomat 10C instrument until colonies appeared. Single colonies were picked with a colony picker and used to inoculate a 96-deep-well microtiter plate (MTP) containing 800 μL LB medium with 50 μg mL−1 kanamycin per well, and cultured overnight at 37 °C. Subsequently, 50 μL of the overnight culture was transferred to a new 96-deep-well MTP containing 750 μL LB medium, 10 g L−1 lactose, and 50 μg mL−1 kanamycin per well. The plates were cultured in a Cytomat 2C Tos instrument at 30 °C and 600 rpm for 12 h. After incubation, the plates were centrifuged at 3000g for 5 min, and the supernatant was removed. Fresh reaction solution (500 μL) was added to each well, and the cells were resuspended and incubated at 30 °C and 220 rpm in the dark for 24 h. Following the reaction, the plates were centrifuged at 3000g for 5 min, and 200 μL supernatant was transferred to a clear bottom 96-well plate to measure OD610 by using a plate reader (CLARIOstar Plus). The reaction solution was developed in this study. It consists of 100 mM Tris-HCl, 1 mM WB, 10 mM HMF, 10 μM PQQ, and 10 μM PrCl3.Molecular docking and computer-assisted virtual screening
The 3D structural data of HMF were obtained from the PubChem database. Molecular docking of PedH (PDB ID: 6ZCW) with the substrate HMF was carried out by the Autodock vina, and the operation methods refer to the tutorial on the official website (Tutorial–AutoDock Vina (scripps.edu)).34 The protein was pretreated and then placed in the CHARMM force field. Site-saturation mutagenesis was performed on amino acids within 8 Å around HMF using the Calculate Mutation Energy/Binding module of Discovery Studio 4.0 according to the manufacturer's protocol with default settings.Molecular dynamics simulations
Molecular dynamics (MD) simulations refer to our previous research.49 Briefly, MD simulations were performed using the CHARMM36 protein force fields within the GROMACS 2023 package.51,52 The force field parameters for PQQ were generated by using the CGenFF web server with the CHARMM general force field.53 The simulation process included solvation, ion addition, energy minimization, heating, equilibration, and production MD. Energy minimization was conducted using the steepest descent algorithm. During the heating protocol, the temperature was gradually increased from 0 K to 300 K with time steps of 2 fs. Equilibration involved NVT and NPT ensembles, each performed for 250 ps with time steps of 2 fs. In the production phase, simulations were run for 50 ns with time steps of 2 fs, and the output structures were saved at 2 ps intervals. Root-mean-square deviation (RMSD) of the backbone atoms of the protein relative to the initial structure was analyzed by using GROMACS 2023. The system was considered equilibrated when the RMSD values converged. The root-mean-square fluctuations (RMSFs) of the backbone atoms were analyzed using the last 10 ns of the trajectories. The occurrence of hydrogen bonds was analyzed for the last 10 ns of the MD simulation trajectories, with a hydrogen bond being defined by a donor–acceptor distance cutoff of 3.5 Å. The MM/GBSA (molecular mechanics/generalized Born surface area) method was employed for calculations of the binding free energy.Author contributions
H. Y. and K. L. conceived and designed the project. K. L. and L. W. designed and conducted experiments. K. L., H. Y. and L. J. analyzed the results and drafted the manuscript. L. W., L. Y. and J. W. provided ideas for writing and revised the manuscript. K. L. and L. J. performed the molecular dynamics simulations. Q. Z. provided experimental assistance and helped collect the data. All authors have given their approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the “Pioneer” and “Leading Goose” R&D Programs of Zhejiang (Grant No. 2025C01097 & 2024C03013), the Key Research and Development Program of China (Grant No. 2022YFA0913000) and the National Natural Science Foundation of China (Grant No. 22378351). We would like to thank the iBioFoundry and Core Facility at the Institute for Intelligent Bio/Chem Manufacturing, ZJU-Hangzhou Global Scientific and Technological Innovation Centre. The authors would like to thank AI + High Performance Computing Center of ZJU-ICI.References
- D. Troiano, V. Orsat and M. J. Dumont, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS PubMed.
- M. Sajid, X. B. Zhao and D. H. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- H. B. Yuan, H. L. Liu, J. K. Du, K. Q. Liu, T. F. Wang and L. Liu, Appl. Microbiol. Biotechnol., 2020, 104, 527–543 CrossRef CAS PubMed.
- E. de Jong, H. A. Visser, A. S. Dias, C. Harvey and G. J. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- C. L. Chen, L. C. Wang, B. Zhu, Z. Q. Zhou, S. I. El-Hout, J. Yang and J. Zhang, J. Energy Chem., 2021, 54, 528–554 Search PubMed.
- T. Oyegoke, F. Dumeignil, B. E. Y. Jibril, C. Michel and R. Wojcieszak, Catal. Sci. Technol., 2024, 14, 6761–6774 Search PubMed.
- C. P. Ferraz, M. Zielinski, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- X. Y. Liu, M. Zhang and Z. H. Li, ACS Sustainable Chem. Eng., 2020, 8, 4801–4808 CrossRef CAS.
- A. Modak, Chem. – Asian J., 2023, 18, e202300671 CrossRef CAS.
- S. Hameed, W. G. Liu, Z. N. Yu, J. F. Pang, W. H. Luo and A. Q. Wang, Green Chem., 2024, 26, 7806–7817 RSC.
- W. Q. Jia, B. W. Liu, R. Gong, X. X. Bian, S. C. Du, S. Y. Ma, Z. C. Song, Z. Y. Ren and Z. M. Chen, Small, 2023, 19, e2302025 Search PubMed.
- D. J. Chadderdon, L. P. Wu, Z. A. McGraw, M. Panthani and W. Z. Li, Chemelectrochem, 2019, 6, 3387–3392 CrossRef CAS.
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Green Chem., 2021, 23, 3154–3171 Search PubMed.
- X. Fei, J. G. Wang, X. Q. Zhang, Z. Jia, Y. H. Jiang and X. Q. Liu, Polymers, 2022, 14, 625 CrossRef CAS.
- G. S. Hossain, H. Yuan, J. Li, H. D. Shin, M. Wang, G. Du, J. Chen and L. Liu, Appl. Environ. Microbiol., 2017, 83, e02312–e02316 CrossRef CAS.
- F. Koopman, N. Wierckx, J. H. de Winde and H. J. Ruijssenaars, Bioresour. Technol., 2010, 101, 6291–6296 CrossRef CAS.
- J. Carro, P. Ferreira, L. Rodriguez, A. Prieto, A. Serrano, B. Balcells, A. Arda, J. Jimenez-Barbero, A. Gutierrez, R. Ullrich, M. Hofrichter and A. T. Martinez, FEBS J., 2015, 282, 3218–3229 CrossRef CAS PubMed.
- Y. Z. Qin, Y. M. Li, M. H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- H. Y. Jia, M. H. Zong, G. W. Zheng and N. Li, ChemSusChem, 2019, 12, 4764–4768 CrossRef CAS.
- N. Cascelli, V. Lettera, G. Sannia, V. Gotor-Fernandez and I. Lavandera, ChemSusChem, 2023, 16, e202300226 CrossRef CAS PubMed.
- Z. Y. Yang, M. Wen, M. H. Zong and N. Li, Catal. Commun., 2020, 139, 105979 CrossRef CAS.
- M. A. Do Nascimento, B. Haber, M. R. B. P. Gomez, R. A. C. Leao, M. Pietrowski, M. Zielinski, R. O. M. A. de Souza, R. Wojcieszak and I. J. r. Itabaiana, Green Chem., 2024, 26, 8211–8219 RSC.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef CAS PubMed.
- J. Viña-Gonzalez, A. T. Martinez, V. Guallar and M. Alcalde, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140293 CrossRef PubMed.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS.
- C. Martin, A. O. Maqueo, H. J. Wijma and M. W. Fraaije, Biotechnol. Biofuels, 2018, 11, 56 CrossRef PubMed.
- P. M. Goodwin and C. Anthony, Adv. Microb. Physiol., 1998, 40, 1–80 CrossRef CAS PubMed.
- Y. Zheng, J. Huang, F. Zhao and L. Chistoserdova, mBio, 2018, 9, e02430–e02417 CrossRef CAS PubMed.
- W. Versantvoort, A. Pol, L. J. Daumann, J. A. Larrabee, A. H. Strayer, M. S. M. Jetten, L. van Niftrik, J. Reimann and H. J. M. Op den Camp, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 595–603 CrossRef CAS.
- J. A. Garciahorsman, B. Barquera, J. Rumbley, J. X. Ma and R. B. Gennis, J. Bacteriol., 1994, 176, 5587–5600 CrossRef CAS PubMed.
- K. Takeda, H. Matsumura, T. Ishida, M. Samejima, K. Igarashi, N. Nakamura and H. Ohno, Bioelectrochemistry, 2013, 94, 75–78 CrossRef CAS.
- M. Wehrmann, E. M. Elsayed, S. Köbbing, L. Bendz, A. Lepak, J. Schwabe, N. Wierckx, G. Bange and J. Klebensberger, ACS Catal., 2020, 10, 7836–7842 CrossRef CAS.
- M. Wehrmann, P. Billard, A. Martin-Meriadec, A. Zegeye and J. Klebensberger, mBio, 2017, 8, e00570–e00517 Search PubMed.
- M. Prejano, N. Russo and T. Marino, Chem. – Eur. J., 2020, 26, 11334–11339 CrossRef CAS PubMed.
- H. J. Wijma, R. J. Floor and D. B. Janssen, Curr. Opin. Struct. Biol., 2013, 23, 588–594 CrossRef CAS PubMed.
- N. Tokuriki and D. S. Tawfik, Curr. Opin. Struct. Biol., 2009, 19, 596–604 CrossRef CAS PubMed.
- J. A. Iannuzzelli, J. P. Bacik, E. J. Moore, Z. Shen, E. M. Irving, D. A. Vargas, S. D. Khare, N. Ando and R. Fasan, Biochemistry, 2022, 61, 1041–1054 CrossRef CAS.
- A. Goldenzweig, M. Goldsmith, S. E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, R. L. Lieberman, A. Aharoni, I. Silman, J. L. Sussman, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2016, 63, 337–346 CrossRef CAS PubMed.
- M. Musil, J. Stourac, J. Bendl, J. Brezovsky, Z. Prokop, J. Zendulka, T. Martinek, D. Bednar and J. Damborsky, Nucleic Acids Res., 2017, 45, W393–W399 CrossRef CAS.
- R. Shroff, A. W. Cole, D. J. Diaz, B. R. Morrow, I. Donnell, A. Annapareddy, J. Gollihar, A. D. Ellington and R. Thyer, ACS Synth. Biol., 2020, 9, 2927–2935 CrossRef CAS PubMed.
- H. Y. Lu, D. J. Diaz, N. J. Czarnecki, C. Z. Zhu, W. T. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- G. Qu, A. Li, C. G. Acevedo-Rocha, Z. Sun and M. T. Reetz, Angew. Chem., Int. Ed., 2020, 59, 13204–13231 CrossRef CAS PubMed.
- M. T. Reetz, G. Qu and Z. T. Sun, Nat. Synth., 2024, 3, 19–32 Search PubMed.
- S. Wang, J. Xie, J. Pei and L. Lai, J. Mol. Biol., 2023, 435, 168141 CrossRef CAS PubMed.
- L. Wang, A. Hibino, S. Suganuma, A. Ebihara, S. Iwamoto, R. Mitsui, A. Tani, M. Shimada, T. Hayakawa and T. Nakagawa, Enzyme Microb. Technol., 2020, 136, 109518 CrossRef CAS.
- W. Strutz, Biophys. J., 2016, 110, 393a CrossRef.
- K. Liu, L. Jiang, S. Ma, Z. D. Song, L. Wang, Q. F. Zhang, R. H. Xu, L. R. Yang, J. P. Wu and H. R. Yu, Bioresour. Bioprocess., 2023, 10, 92 Search PubMed.
- B. Jahn, N. S. W. Jonasson, H. Hu, H. Singer, A. Pol, N. M. Good, H. J. M. Op den Camp, N. C. Martinez-Gomez and L. J. Daumann, J. Biol. Inorg. Chem., 2020, 25, 199–212 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr., J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Yu, X. He, K. Vanommeslaeghe and A. D. MacKerell Jr., J. Comput. Chem., 2012, 33, 2451–2468 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00157a |
| This journal is © The Royal Society of Chemistry 2025 |