
A “lignin-first” biorefinery towards efficient aromatic monomer conversion from coconut shells using mild TMAH-based alkaline deep eutectic solvents†
Chenjun
He
a,
Fengqi
Luo
a,
Yongzhi
Zhu
a,
Ao
Zhan
a,
Jiajun
Fan
 c,
James H.
Clark
c,
Jie
Lv
*b and
Qiang
Yu
*a
c,
James H.
Clark
c,
Jie
Lv
*b and
Qiang
Yu
*a
aSouth China Agricultural University, Institute of Biomass Engineering, Key Laboratory of Energy Plants Resource and Utilization, Ministry of Agriculture and Rural Affairs, Guangdong Engineering Technology Research Center of Agricultural and Forestry Biomass, Guangzhou 510642, China. E-mail: yuqiang@scau.edu.cn
bGuangdong Polytechnic Normal University, School of Mechatronic Engineering, Guangzhou 510450, China. E-mail: lvjie1225@163.com
cGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK
First published on 11th January 2025
Abstract
To achieve “lignin-first” dissolution and efficient aromatic monomer conversion from coconut shells, tailored alkaline deep eutectic solvents (DESs) were screened and designed by conductor-like screening model for real solvents (COSMO-RS) calculations. The results indicated that tetramethylammonium hydroxide (TMAH)-based DESs (TMAH-urea, TMAH-ethanolamine, TMAH-lysine and TMAH-imidazole) provide high solubility for lignocellulose and exhibit an excellent fractionation effect for real lignocellulose under mild conditions. This may be attributed to their higher excess enthalpy and strong hydrogen bonding forces. A high delignification rate of 63.33%–67.37% with a good hemicellulose retention of more than 90% could be achieved in 30% TMAH-based DESs at 50 °C for 3 h. An in-depth insight into the evolution of the lignin structure demonstrated that the cleavage of the lignin–carbohydrate ester bond was the main route for lignin extraction, rather than the β-O-4 ether bonds. Therefore, the extracted lignin fragments retained more than 84% of β-O-4 ether bonds, which was conducive to subsequent depolymerization to produce aromatic monomers. The strong oxidation effect of TMAH-imidazole contributed to its superior catalytic oxidation performance and resulted in a high aromatic monomer yield of 74.54%. Overall, this study designed and screened a solvent with strong hydrogen bonding and oxidation abilities, which enabled a feasible “lignin-first” strategy for efficient aromatic monomer production under milder conditions. A “win–win” situation of low energy consumption and high yield was achieved, highlighting a sustainable energy future through the advanced valorization of lignin.
Green foundation1. This study employed COSMO-RS calculation to achieve rapid screening of target deep eutectic solvents (DESs), which is more time-saving and green. Reusable TMAH-based DESs were screened to achieve “lignin-first” dissolution and efficient aromatic monomer conversion from coconut shells under mild conditions with lower solvent consumption, which has broken through the problems of hemicellulose degradation and lignin condensation in the DES process simultaneously. A “win–win” situation of low energy consumption and high yield was achieved.2. A high delignification rate of 67.37% with a good hemicellulose retention (95.14%) was achieved at 50 °C. The extracted lignin with a high β-O-4 bond retention (88.78%), high purity (88.39%) and strong oxidation of TMAH-imidazoles resulted in a high aromatic monomer yield of 74.54%. 3. Future research will be dedicated to coupling DESs with solid catalysts to achieve a more efficient and green process. |
1. Introduction
With the increasing imbalance between the supply and demand of fossil resources and persistent and increasing environmental challenges, it is imperative to exploit alternative and sustainable feedstocks for materials, energy and chemicals.1,2 Lignocellulosic biomass, as a renewable and widely available raw material, holds significant potential in the sustainable energy industry.3,4 Lignin, alongside cellulose and hemicellulose, is one of the primary components of lignocellulose. As the most abundant natural aromatic polymer, lignin has received extensive attention in recent years and is no longer regarded as a by-product.5 A rich array of aromatic units and oxygen-containing functional groups of lignin give it the potential for valorization through depolymerization into high-value aromatic monomers.4,6,7 As a by-product of the pulp and paper industry, industrial lignin has a high yield and is widely used. However, it is difficult to obtain satisfactory results from industrial lignin due to its condensed structure.8,9 Therefore, extracting native lignin from lignocellulose to realize efficient depolymerization would be a better choice. Considering the inherent recalcitrance of lignocellulose due to its complex cross-linking structure, it is essential to develop effective fractionation technologies for effective extraction and conversion.10,11Deep eutectic solvents (DESs), emerging solvents composed of a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA), are usually prepared by physical mixing of two or more components without chemical reactions (proton exchange and acid–base reaction).12 A drop in the melting point in particular combinations would occur due to the hydrogen bonding, resulting in the melting point of the mixture being lower than the melting point of its components, and in general, at least one of HBD or HBA is solid.13–15 As green, low-cost and easily synthesisable solvents, DESs have been widely used in the fractionation of lignocellulose due to their ability to selectively dissolve lignin while only swelling cellulose.11 The type and lignin extraction ability of DESs are determined by their modular composition of HBD and HBA properties. To date, a host of DESs have been prepared to be employed in lignocellulose-based biorefining. Unfortunately, lignin condensation often occurs during the DES process, particularly with acidic DESs, which hinders subsequent depolymerization,16,17 because the breaking of chemical bonds is accompanied by the lignin extraction process. Lignin fragments generated by the cleavage of the β-O-4 linkages are easily condensed to form new C–C bonds by the dehydration reaction to form benzylic carbenium ions under acidic conditions. Unlike acidic DESs, alkaline DESs usually break the lignin–carbohydrate complex (LCC) bond, preserving a more intact lignin structure and yielding less condensed lignin.18 While most research focuses on achieving a high lignin removal rate, hemicellulose recovery is often overlooked. The degradation of hemicellulose in DESs would increase the viscosity of DESs and lead to carbon loss. Achieving effective lignin removal while simultaneously recovering hemicellulose is challenging due to the conflicting characteristics of the two polymers. Tian et al. employed the technique of hydrothermal coupling with DES pretreatment to retain hemicellulose before lignin extraction.16 However, the generation of pseudo lignin would be inevitable during hydrothermal treatment.2,10 Alkaline DESs have been shown to retain hemicellulose more effectively than acidic DESs due to the sensitivity of hemicellulose to acidic conditions.18 In our previous study, the method of “lignin-first” dissolution based on alkaline DESs was proposed to achieve efficient lignin dissolution without compromising the recovery of cellulose and hemicellulose during the DES process, which demonstrated the huge potential of a one-pot alkaline DES biorefinery for lignocellulose fractionation and lignin upgrading.8
Although alkaline DESs are less reported compared with acidic DESs, efficient screening of DESs is still a challenge. At present, the selection of DESs with targeted functions is typically achieved through trial and error, which is time-consuming and increases experimental costs due to the vast array of HBAs and HBDs available. Thus, a fast and accurate computational tool screening method is urgently needed to screen target solvents. Conductor-like screening model for real solvents (COSMO-RS) is a model based on the continuum solvation theory for a specific problem in the molecular structures. It combines quantum chemistry and statistical thermodynamics, which has proven to be a powerful and efficient tool for predicting solvent properties and screening solvents.19,20 It can quickly predict the solubility by calculating the logarithmic activity coefficient (ln(γ)) of the solute in the solvent.21 Casas et al. employed COSMO-RS to predict the solubility of lignin and cellulose in ionic liquids (ILs) and verified it by experiments.22 The results show that hydrogen bonding plays an important role in the dissolution of cellulose and lignin. In the study by Mattern et al., lignocellulose models were established using COSMO-RS and a DES with higher lignin solubility was screened by calculating the ln(γ).23 Yu et al. achieved solvent screening by predicting the solubility of the lignin models established by COSM-RS in different solvents, and the selection of the optimal lignin model was realized by experiment.24 Although many research studies employed COSMO-RS to screen ionic liquids for lignin, it still exhibits good potential for screening DESs for efficient lignin extraction and lignocellulose fractionation due to the designability of DESs.
Furthermore, although DESs have shown excellent potential in biomass refining, the high viscosity of traditional DESs poses a major obstacle to its further development.25 Studies have shown that adding moderate amounts of water to DESs can enhance solvent hydrogen bonding, reduce viscosity and improve mass transfer, facilitating lignin extraction.26–28 The overall supramolecular structure of DESs is still maintained even when 40%–50% water is added.29 In addition to improving the lignin extraction rate, incorporating water in DESs can also reduce the solvent cost, making aqueous DESs an attractive option for biomass treatment. In this study, COSMO-RS was employed to predict the solubility of the lignocellulosic model in multiple DESs composed of different HBDs and HBAs. The predicted results of these established lignocellulosic models were verified by conducting the solubility test. The mechanism between solubility and solvent properties was analyzed by calculating the σ-profile, excess enthalpy and Kamlet–Taft solvatochromic parameters. The “lignin-first” dissolution potential of the target solvent selected by COSMO-RS was investigated by pretreating coconut shells. The fractionation effect of these DESs was regulated by altering the solvent concentration, reaction temperature and reaction time. The structural evolution of the obtained synthesized cellulose and lignin was explored to reveal the fractionation mechanism. Furthermore, the depolymerization performances of the DESs and the derived lignin were evaluated using a catalytic oxidation reaction.
2. Materials and methods
2.1 Solvent screening
 | (1) |
| π* = 0.314 × (27.52 − vNEt2) | (2) |
 | (3) |
 | (4) |
 | (5) |
2.2 Preparation of DESs and “lignin first” dissolution
Coconut shells were employed as the biomass materials in this study. After being from a coconut factory, they were crushed to <500 μm for the subsequent fractionation process. The target DES was selected according to the simulation results. Tetramethylammonium hydroxide (TMAH) was used as the HBA, and urea, lysine, ethanolamine and imidazole were used as HBDs. The HBA and HBD were mixed according to the preset molar ratio, and some distilled water was added to adjust the solvent concentration. As water was used, the TMAH-based DESs could be prepared at lower temperatures (<80 °C) quickly. The coconut shells and DESs were mixed according to the mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 and added to the flask, and continuously stirred at the designated temperature and time. After the reaction, the mixture was separated via vacuum filtration to obtain the liquid fraction rich in lignin and the solid part rich in cellulose and hemicellulose. The solid part was washed with ethanol aqueous solution and water until neutral, and collected for analyzing the fractionation effect of the DES. Part of the liquid fraction was acidified with 1 mol L−1 HCl to precipitate the lignin. Subsequently, the precipitated lignin was washed to neutral and vacuum-dried for structure characterization. The milled wood lignin (MWL) extracted from coconut shells by ball milling and using the 1,4-dioxane/water (96/4, v/v) solvent system was used as a control (ESI†). Another part of the liquid fraction containing lignin and DES was directly used for subsequent depolymerization. The lignin content in the liquid fraction can be calculated according to the delignification ratio. The waste DES liquid was recovered via rotary evaporation and reused for the next cycle of processing by adding alkali to the initial pH and filtering out the precipitated salt. The preparation route is shown in Fig. S1.†
:15 and added to the flask, and continuously stirred at the designated temperature and time. After the reaction, the mixture was separated via vacuum filtration to obtain the liquid fraction rich in lignin and the solid part rich in cellulose and hemicellulose. The solid part was washed with ethanol aqueous solution and water until neutral, and collected for analyzing the fractionation effect of the DES. Part of the liquid fraction was acidified with 1 mol L−1 HCl to precipitate the lignin. Subsequently, the precipitated lignin was washed to neutral and vacuum-dried for structure characterization. The milled wood lignin (MWL) extracted from coconut shells by ball milling and using the 1,4-dioxane/water (96/4, v/v) solvent system was used as a control (ESI†). Another part of the liquid fraction containing lignin and DES was directly used for subsequent depolymerization. The lignin content in the liquid fraction can be calculated according to the delignification ratio. The waste DES liquid was recovered via rotary evaporation and reused for the next cycle of processing by adding alkali to the initial pH and filtering out the precipitated salt. The preparation route is shown in Fig. S1.†
2.3 Characterization of (hemi)-cellulose-rich substrates and DES extracted lignin
A method of two-step acid hydrolysis process according to the National Renewable Energy Laboratory was employed to analyze the chemical composition of the raw material and DES-processed lignocellulose.34 A high-performance liquid chromatography system (LC-2030C LT Plus, Shimadzu, Japan) equipped with a sugar column (SH1011, Shodex, USA) and a refractive index detector was used for determining the content of glucose and xylose. The contents of cellulose and hemicellulose are represented by the contents of glucan and xylan, which were calculated according to the determined concentrations of glucose and xylose, respectively. The content of insoluble lignin was measured by the mass method. The cellulose and hemicellulose recovery rate and delignification rate can be calculated according to the mass of each component before and after DES treatment (ESI†). An X-ray diffractometer (D8 Advance, Bruker, Germany) was employed to analyze the crystal characteristics of the solid part.The molecular weight based on the average weight (Mw) and the average number (Mn) of DES-extracted lignin was determined after acetylation by gel permeation chromatography (ACQUITY APC, Waters, USA). Fourier transform infrared spectroscopy (FTIR) (Nicolet 560 spectrophotometer, USA) was used to determine the functional groups on the surface of the lignin. The measurement range was 400–4000 cm−1 with a resolution of 4 cm−1. More detailed structural changes in the aromatic regions and connecting bonds between the lignin units were captured by the nuclear magnetic resonance (NMR) spectrometer (AV II 600 MHz spectrometer, Bruker, Germany). The quantitative 31P NMR, and 2D-HSQC NMR spectra were collected and calculated according to reported methods.18 The sugar content of lignin was analyzed by the two-step acid hydrolysis process.
2.4 Catalytic oxidation depolymerization
To avoid the effect of acidification on the lignin structure during lignin reprecipitation, the catalytic oxidation performances of the solvent and extracted lignin were investigated by using the pretreated liquid. As reported, metal oxides show excellent properties in oxidative catalytic depolymerization, and different metal oxides were employed as catalysts in this study.9,35,36 Certain amounts of catalyst and 25 mL of liquid containing lignin and the DES were put in a 100 mL autoclave reactor (AHHT-YKJ100-C, HTLAB, China) with a Teflon insert inside. The reactor was charged with O2 and continuously mechanically stirred at the designated times and temperatures. After the reaction, the catalyst was separated by vacuum filtration. Subsequently, the liquid was acidified and the unreacted lignin was precipitated with 1 mol L−1 HCl. The products were identified by gas chromatography mass spectrometry (GCMS, GCMS-QP2010 ULTRAS, SHIMADZU, Japan) and gas chromatography (GC2030, SHIMADZU, Japan). The aromatic monomer yield was calculated according to the following equation:37 | (6) |
The oxidation–reduction properties of different DESs were determined by cyclic voltammetry (CV) analysis. CV analyses were performed on an electrochemical performance test workstation (Zahner, Zennium, Germany) using a three-electrode system at the scan rate of 20 mV s−1, and different DESs were used as the electrolyte solution.
2.5 Model compound verification
The lignin extraction mechanism of TMAH-based DESs was further investigated by using model compounds. Three model compounds, 2-phenoxy-1-phenylethanol (1a), phenyl β-D-glucopyranoside (1b), and benzyl benzoate (1c), were employed as the lignin aryl ether bond, the lignin–carbohydrate ether bond, and the lignin–carbohydrate ester bond, respectively.38 Three types of model compounds (50 mg) were added to the prepared DES (10 g) respectively and reacted at 50 °C for 3 h. The samples before and after treatment were characterized by GCMS. Similarly, 2-phenoxy-1-phenylethanol (1a) was employed to verify the cleavage of the β-O-4 bond during the catalytic oxidation depolymerization.3. Results and discussion
3.1 Solvent screening and validation
Firstly, the hemicellulose, cellulose, and lignin models for calculating the solubility were built. For hemicellulose, six models containing different xylose units were established. As shown in Fig. S2a,† the surface charge of the repeating part was similar in different models. Subsequently, σ-potential, which is used in COSMO-RS to represent the affinity of the system to a surface with a polarity of σ, was calculated. The σ-potential can be divided into three regions: the H-bond donor region (σ < −0.0082 e Å−2), the non-polar region (−0.0082 e Å−2 < σ < +0.0082 e Å−2), and the H-bond acceptor region (σ > +0.0082 e Å−2).30 According to Fig. S2b,† compared with the xylose (Model-1), the σ-potential curves of model 2–6 were close or even overlapped, which indicated that they will exert similar effects under the solvent system based on COSMO-RS calculation. Moreover, the σ-potential of model-1 possessed a more negative value in the H-bond acceptor region, indicating that it has a stronger affinity for the hydrogen bond receptor surface. The cellulose model containing different chain lengths was established by using the same method (Fig. S3†). Unsurprisingly, as the cellulose chain length increases, the σ-potential curves of the cellulose model tend to overlap, which demonstrated that the model containing multiple repeating units can better reflect the solubility of real substances. These results were similar to the previous study.19,30 On the other hand, lignin is made up of three different units that are connected in different linkages. Therefore, the three typical units (S, G, H) of lignin and the dimer-, trimer- and tetramer-models linked by β-O-4, β-5 and β–β linkages were established (Fig. S4†).Subsequently, ln(γ) representing the solubility of the lignocellulosic model in 195 binary alkaline deep eutectic solvents composed of 15 types of HBAs and 13 types of HBDs was predicted by COSMO-RS, where a smaller ln(γ) value indicates higher solubility.39 As shown in Fig. S5,† a similar solubility trend of different models belonging to the same component was obtained in the solvent. For hemicellulose and cellulose models, the different solubilities obtained in the models may be related to their size, molecular weights, and chain lengths, while it could be affected by the type and number of units and chemical bonds in lignin.8 However, the similar solubility trend demonstrated that solubility trends of different solvents were hardly changed by adjusting the unit or connecting bond of these models. Then, the solubility of hemicellulose, cellulose and lignin tested by experiment was linearly fitted with ln(γ) obtained by COSMO-RS simulation (Fig. 1a–e). According to Fig. 1a and b, the actual solubility of xylan exhibited good agreement with the simulation results of ln(γ) of hemicellulose models 2–6 (R2 > 0.7), indicating that these models can be used to represent hemicellulose. Similarly, the cellulose model containing multiple glucose units obtained a higher degree of fit than that with a single glucose unit (Fig. 1c), which demonstrated that models containing multiple sugar units can better simulate the dissolution of real cellulose in solvents. Therefore, the hemicellulose model-3 and cellulose model-3 can be employed to represent hemicellulose and cellulose, which was consistent with previous studies.19,32 As for the lignin model, S-unit (R2 = 0.66) and model-4 (R2 = 0.78) obtained a superior degree of fit (Fig. 1d and e). However, considering the variety of units and connecting bonds of lignin, model-4 will be more suitable to represent the real lignin. The high fit results also showed that the solubility predicted by COSMO-RS is reliable.
 | ||
| Fig. 1 The linear fitting results between lignocellulose solubility and ln(γ) of different model: (a) hemicellulose model-1 to model-3, (b) hemicellulose model-4 to model-6, (c) cellulose model, (d) lignin unit and (e) lignin model-1 to model-4; COSMO-RS σ surface of the hemicellulose model (mid-dimer of xylotetrose), cellulose model (mid-dimer of glucotetrose), and the lignin model. ln(γ) of (f) hemicellulose model, (g) cellulose model and (h) lignin model in different solvent systems predicted by COSMO-RS. | ||
Based on the above analysis, hemicellulose model-3 (mid-dimer of xylotetrose), cellulose model-3 (mid-dimer of glucotetrose) and lignin model-4 were chosen to analyze the influence of HBD and HBA on solubility (Fig. 1f). In general, the type of HBD plays an important role in the fractionation effect of the solvent. As shown in Fig. 1g–i, lignocellulose obtained a lower ln(γ) in solvents using alcoholamines as HBD compared with the other three types of HBDs, which may be related to the number of hydroxyl and amino groups and/or their alkalinity. Previous studies have shown that amino groups can achieve similar delignification effects with carboxyl functional groups, which may be due to amine/amide groups exhibiting alkaline properties, resulting in the deprotonation of the phenolic hydroxyl group of lignin.40 However, excess hydroxyl/amino groups would form more hydrogen bonds with HBA, which would reduce the number of free and active groups that can interact with lignocellulose and weaken the lignocellulose fractionation efficiency.41 Therefore, an inferior solubility was obtained in the diethanolamine-based solvent than in ethanolamine-based solvent, which was consistent with the previous report.40 For the same HBD, choline hydroxide (ChOH) and quaternary ammonium hydroxides (R4NOH, including tetramethylammonium hydroxide (TMAH), tetraethylammonium hydroxide (TEAH), tetrapropylammonium hydroxide (TPAH), and tetrabutylammonium hydroxide (TBAH)) employed as the HBA achieved superior solubility of the three components of lignocellulose, while a higher ln(γ) was noted in the choline chloride (ChCl)- and betaine-based solvents. This may be related to the strong alkalinity of ChOH and R4NOH. As for R4NOH-HBD, the solubility of lignocellulose possessed a decreasing trend with the increase of the alkyl chain of quaternary ammonium base. The HBAs containing shorter alkyl chains possess superior solubility, possibly due to their smaller steric hindrance.41 A imilar result was obtained in quaternary ammonium salts-based DESs. Moreover, the type of halogen ion that forms quaternary ammonium salts also affects the solubility of lignocellulose.40 However, when a quaternary ammonium salt was employed as the HBA, an inferior solubility was obtained in urea-based solvents, while ChOH-urea and TMAH-urea could obtain lower ln(γ) compared with ChOH-HBD/R4NOH-HBD under the same conditions. The possible reason is that urea will decompose and generate NH3 under strong alkaline conditions at lower temperature, which enhances the hydrogen bond interaction of the DES.42 However, NH3 is generated at temperatures higher than 150 °C in the ChCl–urea system38 because the decomposition temperature of urea could be significantly decreased under alkaline conditions.
Subsequently, the σ-profiles of HBAs, HBDs and lignocellulosic models were calculated to analyze the affinity between the lignocellulosic model and polar σ surface to elucidate the influence of HBD and HBA types on the solubility.30,31 The σ-profiles can be divided into three regions: the HBD region (σ < –0.0082 e Å−2), the non-polar region (−0.0082 e Å−2 < σ < 0.0082 e Å−2) and the HBA region (σ > 0.0082 e Å−2). Generally, superior solubility can be obtained in a solvent if it possesses a similar curve between the lignocellulosic components and the HBD and HBA of the solvent system.31 As shown in Fig. 2, compared with cellulose and lignin, the σ-profiles of hemicellulose possessed smaller peaks, almost lower than those of HBAs and HBDs, which could explain why hemicellulose possesses a greater solubility in solvent systems and a smaller range of solubility variation. For HBDs, urea and thiourea exhibit a weaker peak strength in the HBA and HBD region, which corresponds to their poor lignocellulose solubility. As shown in Fig. 2d–f, similar peaks were obtained in Cl− and Br−; thus there was no significant difference in lignocellulose solubility between [TEMA]Cl and [TEMA]Br. Therefore, the different solubility that appeared in [TEMA]Cl/[TEMA]Br and TMAH may be due to the peak variation in Cl−/Br− and OH−.
 | ||
| Fig. 2 σ-Profiles of HBDs and (a) hemicellulose, (b) cellulose and (c) lignin model; σ-profiles of HBAs and (d) hemicellulose, (e) cellulose and (f) lignin model. | ||
However, σ-profiles pose a difficulty for analyzing the polar σ surface affinity between the mixed solvent system and the lignocellulosic model; therefore, the excess enthalpy and Kamlet–Taft parameters of these solvent systems were further analyzed. According to Fig. 3a and b, these solvents present a negative excess enthalpy, demonstrating that their synthesis process is exothermic.8 Their different H-bond values indicate that the hydrogen bonding strength formed by solvents was significantly affected by the type of HBA and HBD. Superior hydrogen bonding was obtained in the solvents prepared with types of amino acids and straight chain amines as HBAs, while the nitrogen heterocyclic-based solvents possessed weaker hydrogen bonding. Moreover, compared with the quaternary ammonium salt–urea, ChOH–urea and TMAH–urea represented stronger hydrogen bonding (−8.26 kJ mol−1 and −4.68 kJ mol−1) and excess enthalpy (−5.57 kJ mol−1 and −6.53 kJ mol−1), which possibly contributed to their superior solubility for lignocellulose.22 As shown in Fig. 3c–h, the excess enthalpy between these solvents and three compositions of lignocellulose exhibited similar trends. In contrast to the quaternary ammonium salts and betaine when TMAH is employed as the HBA, the excess enthalpy between the solute and solvent was mainly contributed by hydrogen bonding, which demonstrates that hydrogen bonding plays a key role in achieving greater solubility of lignocellulose in TMAH-based solvents.8 Furthermore, when the excess enthalpy and hydrogen bonding was lower than 0, a strong force would exist between solvents and lignocellulose.24 Therefore, these solvents exhibited greater solubility for lignin than hemicellulose and cellulose, which also showed that hydrogen bonding has an important role in driving lignin dissolution. Meanwhile, these results create the hope to screen out the solvent for realizing the “lignin-first” dissolution. The properties of these solvents can also be determined using the Kamlet–Taft parameters (α, β, and π*). α, β, and π* represent the hydrogen bond supply capacity (hydrogen bond acidity), hydrogen bond acceptability (hydrogen bond basicity), and dipolarity/polarizability of a solvent, respectively.33,43 As shown in Fig. S6,† the α value of the solvent was positively correlated with the solubility of lignin and cellulose, and the β value was positively correlated with the solubility of the three components, demonstrating that the hydrogen bond supply capacity and acceptability play an important role in the dissolution of lignocellulose.
 | ||
| Fig. 3 The excess enthalpy of (a and b) different solvent systems and the excess enthalpy of (c and d) hemicellulose, (e and f) cellulose, and (g and h) lignin in these solvents. Arg, Lys, His and Thiu represent arginine, lysine, histidine and thiourea, and Imi, Pyra, Pyri and Pyrr represent imidazole, pyrazole, pyridine and pyrrole. | ||
In summary, the differences in properties of the HBAs and HBDs contributed to the differences in hydrogen bond force of multiple DESs. The superior solubility of DESs for lignocellulose could possibly be attributed to their strong hydrogen bond force. The reliability and feasibility of COSMO-RS screening DESs were proved according to the fitting of the solubility experiment and simulation results. Based on the predicted results of COSMO-RS, TMAH-HBD exhibited greater lignocellulose solubility, and it can be regulated by setting the concentration of TMAH.44 Therefore, TMAH-HBDs are expected to achieve “lignin-first” dissolution from real lignocellulose biomass.
3.2 “Lignin-first” dissolution by TMAH based alkaline DESs
As mentioned earlier, TMAH-based solvents exhibited excellent solubility for lignin, which are further employed for biomass fractionation. Therefore, urea (amides), MEA (alcohol amine), lysine (amino acid) and imidazole (N-heterocyclic) were employed as HBDs to prepare solvents with TMAH for “lignin-first” dissolution. According to previous reports, as a strong base, TMAH can realize the separation of lignocellulose composition at low temperatures.44–46 Moreover, TMAH would be an excellent HBA for forming alkaline DESs due to strong ionization ability and hydrogen bonding acceptability.47 As mentioned earlier, DESs are prepared by physical mixing without accompanying chemical reactions; therefore, a TMAH-based DES is usually prepared by using an alkaline HBD. The alkaline DES such as TMAH-urea and TMAH-MEA was developed for natural polymer dissolution and processing in previous studies. The 1H NMR results also proved that no new chemical substances were produced in TMAH-based solvents, indicating that no chemical reaction occurred during the solvent preparation (Fig. S7†), which demonstrates that TMAH-based alkaline DESs were successfully prepared. However, the high concentrations of TMAH and serious reaction conditions may cause carbohydrate loss.48 Li et al. employed a TMAH·5H2O-urea DES to dissolve cellulose at room temperature.49 The results showed that lower concentrations of TMAH-based DES may be conducive to obtaining higher carbohydrate retention. Moreover, an appropriate amount of water can decrease the viscosity of DES and improve the lignin extraction effect.50 The low values of viscosity are beneficial for enhancing the conductivity of DES and accelerating the fractionation of lignocellulose.29 Thus, to achieve effective lignin removal without compromising on the efficient recovery of hemicellulose and cellulose, the concentrations of TMAH-urea were regulated at a lower temperature. Moreover, according to Fig. 4a, TMAH-urea can achieve efficient lignocellulose fractionation at a lower concentration. With the increase in TMAH-urea concentration, the delignification ratio increased from 53.33% to 72.92%. However, the cellulose recovery decreased from 95.13% to 80.2% and hemicellulose recovery decreased from almost 100% to 72.33%, demonstrating that high alkaline concentration could achieve effective lignin removal but compromise on the high recovery of hemicellulose and cellulose. It appeared that the lignocellulose fractionation ability can be enhanced at higher TMAH-urea concentrations, which could possibly be attributed to the increased hydrogen bond strength and active sites. Based on the results of lignin removal and carbohydrate retention, 30% was selected as the concentration for subsequent biomass fractionation. As shown in Fig. 4b, more than 60% lignin removal was obtained in four types of TMAH-based solvents at low temperature and low solvent concentration, which was consistent with the COSMO-RS simulation result. The enhanced crystallinity of the pretreated coconut shells also demonstrated the efficient removal of lignin in the amorphous state (Fig. S8a†). Meanwhile, higher hemicellulose and cellulose recovery can also be achieved, demonstrating their desired “lignin-first” dissolution potential. | ||
| Fig. 4 Carbohydrate recovery and delignification ratio of (a) substrates treated with TMAH-urea with different concentrations and (b) substrates treated with different solvents (50 °C, 3 h). | ||
Subsequently, the effects of reaction temperature and time on the lignocellulose fractionation of solvents were investigated. As shown in Fig. 5a, the delignification ratio of TMAH-urea increased from 53.80%–73.92% as the reaction temperature rose from 30 °C to 90 °C. Nevertheless, cellulose and hemicellulose dissolution also increased with enhanced temperature, especially hemicellulose, which leads to a loss in value of carbohydrates. When the pretreatment temperature reached 90 °C, the hemicellulose recovery rate was almost less than 50%, indicating that higher reaction temperature is not conducive to the retention of hemicellulose.48 Moreover, the deconstruction ability of lignocellulose could also be promoted by extending the reaction time, which suggests that the reaction temperature and time modulate the efficiency of lignocellulose fractionation. Similarly, the delignification, cellulose and hemicellulose loss of TMAH-MEA/lysine/imidazole also increased with the reaction temperature and enhanced time (Fig. 5b–d). Therefore, the crystallinity of pretreated substrates (Fig. S8b†) increased with the increase in reaction temperature and time. Under the treatment conditions of near room temperature (30 °C), the lignin removal of the four TMAH-based solvents can reach nearly 60% with almost no loss of cellulose and hemicellulose. As the temperature rises, the lignin removal could reach more than 70% at the treatment temperature of 90 °C. Unlike the serious reduction in cellulose and hemicellulose recovery from TMAH-urea at higher treatment temperatures, ∼80% cellulose recovery and ∼70% hemicellulose recovery could be achieved in TMAH-MEA/lysine/imidazole. Additionally, the more serious loss of hemicellulose may be caused by the exothermic reaction that occurred between TMAH-urea and the cellulose/hemicellulose (Fig. 3b). Another possible reason is that NH3 and cyanate generated could interact with the DES to form hydrogen bonds and ionic bonds facilitating the dissolution of hemicelluloses. It is known that the decomposition temperature of urea could be significantly decreased under alkaline conditions, which was also conducive to the permeation of DES into the biomass internal structure.42
 | ||
| Fig. 5 Effect of the reaction temperature and time on delignification, cellulose and hemicellulose recovery: (a) TMAH-urea; (b) TMAH-MEA; (c) TMAH-lysine and (d) TMAH-imidazole. | ||
Hence, different from the compromise in the cellulose and hemicellulose recovery observed with TMAH-urea, the other three TMAH-based DESs present superior potential for “lignin-first” dissolution. Moreover, compared with the results of reported alkaline DESs (Table S2†), TMAH-imidazole still exhibited multiple advantages, such as lower reaction temperature, lower solvent concentration and satisfactory delignification rate along with superior cellulose and hemicellulose recovery.8,11,51–53
3.3 Insight into the structural evolution of the DES-extracted lignin
The evolution in the chemical structure of DES-extracted lignin was investigated in depth to reveal the effect of the DES and provide valuable information for enhancing the downstream lignin products. As shown in Fig. 6a, the basic functional group information of DES-extracted lignin was captured by FTIR. The absorption bands of –OH vibration (3411 cm−1), –CH vibration (2923 and 2856 cm−1) and the absorption peaks of the typical aromatic ring skeleton of phenylpropane structures (1615, 1524 and 1446 cm−1) were observed in these lignin samples. The absorption bands at 1356, 1290 and 835 cm−1 represent the stretching of the S–H–G unit.18 Different from MWL, the enhanced absorption bands at the range of 1560–1640 cm−1, 1030–1230 cm−1 and 785 cm−1 were observed in DES-extracted lignin, which were attributed to the N–H/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/CO, the C–N vibration and the N–H bond.51,54,55 The enhancement of carboxyl functional groups may be because hydroxyl groups were oxidized by the effect of the solvent,56 while the nitrogen-containing functional groups may be introduced by the solvent.57 Notably, the lignin extracted by TMAH-imidazole exhibited stronger signal peaks of nitrogen and oxygen-containing functional groups, which may be related to its stronger nucleophilic effect.58,59
N/CO, the C–N vibration and the N–H bond.51,54,55 The enhancement of carboxyl functional groups may be because hydroxyl groups were oxidized by the effect of the solvent,56 while the nitrogen-containing functional groups may be introduced by the solvent.57 Notably, the lignin extracted by TMAH-imidazole exhibited stronger signal peaks of nitrogen and oxygen-containing functional groups, which may be related to its stronger nucleophilic effect.58,59
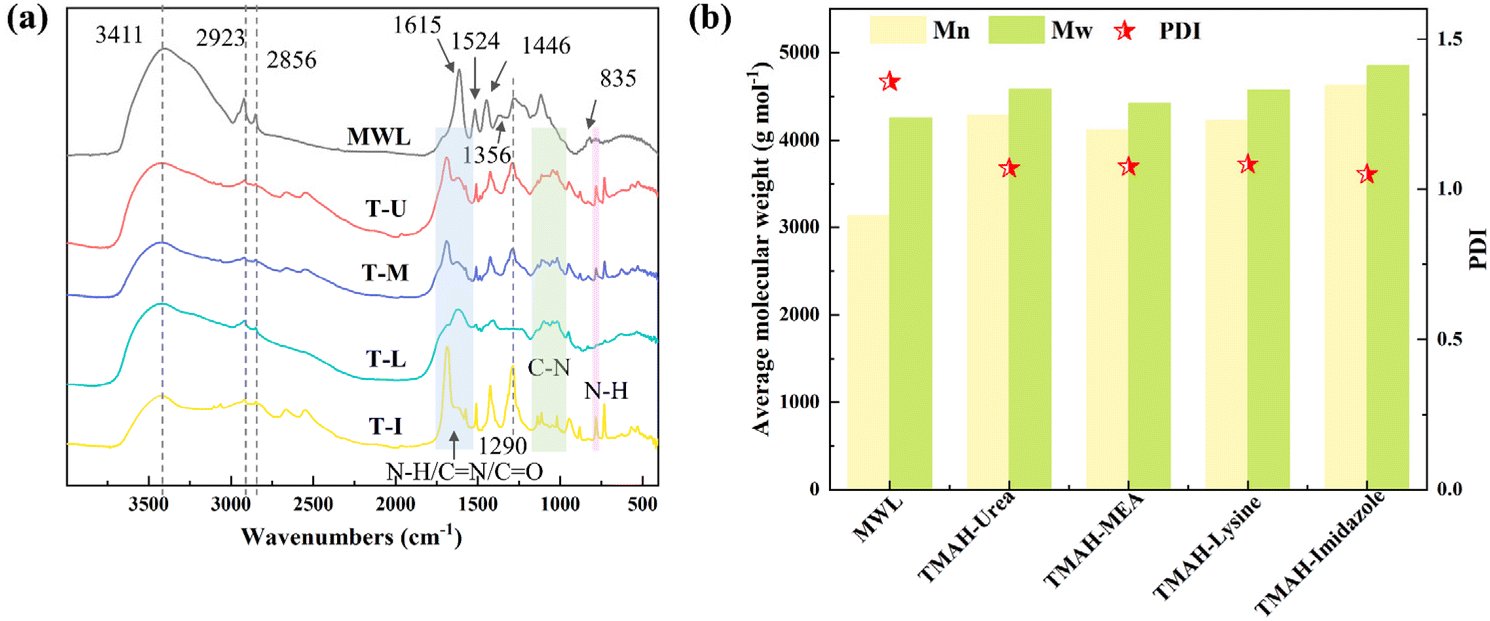 | ||
| Fig. 6 (a) FTIR spectra and (b) the average molecular weights and the polydispersity index (PDI) of MWL and DES-extracted lignin. T represents tetramethylammonium hydroxide, U, M, L and I represent urea, ethanolamine, lysine and imidazole, respectively. | ||
The average molecular weight was measured to investigate the lignin fragmentation or condensation during the DES extraction process. Compared with the average molecular weight of MWL (Mn: 3133 g mol−1, Mw: 4255 g mol−1), the average molecular weight of DES-extracted lignin was increased slightly rather than decreased significantly (Fig. 6b), which corroborated that the lignin extracted by TMAH-based DES attained a relatively complete structure.18 The increased average molecular weight may be attributed to polysaccharide structure linkages associated with lignin introduced through LCC bonds.60 This result demonstrated that TMAH-based DES extracted lignin molecules mainly by cleaving the lignin–carbohydrate linkages rather than lignin fragmentation.53 The PDI of DES-extracted lignin was slightly decreased to 1.05–1.07 compared to that of the MWL (1.36), which concluded that no significant depolymerization occurred in the lignin extraction process.
Subsequently, 2D-HSQC NMR and 31P NMR were employed to understand the structural variations in DES-extracted lignin. The 2D-HSQC NMR spectra could be divided into the side-chain (δC/δH 50–90/2.5–6.0) and aromatic (δC/δH 100–135/5.5–9.0) regions (Fig. 7a), and the corresponding signal peaks are listed in Table S3.†18 In the side-chain region, apparent signals of methoxy can be observed at δC/δH 55.2/3.74 in these lignin samples, showing that abundant G and S units existed in coconut shell lignin, which is consistent with the FTIR results.8 The obvious signals of ether bonds (β-O-4, A), resinol (β–β, B), phenylcoumaran (β-5, C) and the cinnamyl alcohol terminal group (Cγ-OH, I) were captured in MWL. A similar structure was obtained in the spectra of DES-extracted lignin, and the signal of Aα still could be observed clearly, demonstrating that β-O-4 linkage was retained during the DES treatment. According to the quantitative results, the main chemical linkages of lignin are listed in Table 1. Unlike other previous works that extracted lignin by lignin fragmentation, the mild TMAH-based DES achieved lignin-first dissolution target with high content of β-O-4 linkage (50.67–57.80/100 Ar). The slight decrease of β-O-4 linkage in this DES-extracted lignin indicated that the cleavage of the β-O-4 linkage is not the main way for lignin removal, and the lignin was extracted possibly by monolithic stripping rather than fragmenting. Meanwhile, the contents of β–β and β-5 bonds in DES-extracted lignin were decreased compared with that of MWL, demonstrating that no new C–C bonds were generated during the reaction and a less condensed structure existed in DES-extracted lignin.
 | ||
| Fig. 7 (a) 2D-HSQC NMR spectra of the MWL- and DES-extracted lignin and (b) quantification of the content of hydroxyl groups by 31P NMR (mmol g−1). | ||
| Sample | β-O-4 | β–β | β-5 | S/G | Sugar content |
|---|---|---|---|---|---|
| MWL | 60.42 | 12.67 | 10.88 | 0.55 | 3.75 |
| TMAH-urea | 50.67 | <0.1 | <0.1 | 0.41 | 10.17 |
| TMAH-MEA | 52.80 | <0.1 | <0.1 | 0.88 | 12.31 |
| TMAH-lysine | 55.92 | <0.1 | <0.1 | 1.02 | 17.05 |
| TMAH-imidazole | 53.64 | <0.1 | <0.1 | 1.61 | 11.61 |
More blue-gray signal peaks due to residual polysaccharides linked to lignin by LCC bonds were observed in DES-extracted lignin.53,61 As reported, LCC bonds mainly include lignin–carbohydrate ether bonds (benzyl ether bonds), lignin–carbohydrate ester bonds (γ-ester bonds), and phenyl glycoside bonds.62 The benzyl ether bond was stable in an alkaline environment, while they were easily broken in acidic systems.63 Therefore, more sugar content can be detected in DES-extracted lignin compared with that in MWL (Table 1). In contrast, the lignin carbohydrate ester bond was more sensitive to alkaline environments, which can help account for the TMAH-based DESs’ unique ability for precise lignin extraction.
In the aromatic regions, obvious signal peaks of S2,6 (δC/δH 103.4/6.68), G2 (δC/δH 110.5/6.92), G5 (δC/δH 114.7/6.71), G6 (δC/δH 118.8/6.75) and H2,6 (δC/δH 127.8/7.16) representing the basic unit of lignin can be captured in MWL. In addition, the signal peaks of FA2, FA6, PCE2,6 and PB2,6 were also observed. After DES treatment, the signal peaks of H and G units were weakened to different degrees, indicating that lignin may be oxidized during the DES extraction process. Compared with TMAH-urea, TMAH-MEA/lysine/imidazole exhibited a stronger oxidation effect, especially TMAH-imidazole, which corresponds to its higher S/G value (Table 1). Furthermore, no signal peak of Cond-S units can be observed in these DES-extracted lignin samples, which demonstrates that mild TMAH-based DESs can extract native lignin by cleaving the target chemical bonds.18
The functional hydroxyl groups in these lignin samples were quantitatively analyzed by 31P NMR. Aliphatic-OH, phenolic-OH and carboxyl groups can be observed in these lignin samples. According to Fig. 7b, a significant increase in the content of the carboxyl group occurred in DES-extracted lignin, corresponding to the oxidation process of DES as shown in the FTIR and 2D-HSQC results.18 Imidazole molecules contain nitrogen atoms and nitrogen atoms have high electronegativity. At the same time, the aromatic ring structure of the imidazole molecule also gives it a certain stability. In the oxidation reaction, imidazole can release electrons to oxidize other substances, while a reduction reaction occurs within itself, which shows superior oxidation. In contrast, a relatively weak oxidation was shown to occur in urea, MEA and lysine. The absence of an increase in phenolic-OH content in the DES-extracted lignin indicates that the fragile β-O-4 linkages were protected during the DES treatment.18 The increased aliphatic OH may be derived from the polysaccharide that was connected with lignin through the uncleaved LCC bonds.
Based on the above analysis, TMAH-based alkaline DES extracted lignin may be formed by cleavage of the LCC bonds rather than the β-O-4 linkage. To verify this hypothesis, related model compounds containing different linkages were employed in the DES process. According to Fig. 8a, only the signal peak of 1a can be detected in the pretreated sample, demonstrating the favorable stability of lignin β-O-4 linkages in these solvent systems. Although phenyl β-D-glucopyranoside was not detected by GCMS, no monomeric products were captured in the pretreated sample (Fig. 8b), which suggests that no breaking of the lignin–carbohydrate ether bond occurred during the DES process. However, when benzyl benzoate was pretreated, the new products benzyl alcohol (2c) and benzoic acid (3c) along with the obviously reduced 1c can be detected in the pretreated samples (Fig. 8c). These results demonstrate that the effective breaking of the LCC bond, mainly the lignin–carbohydrate ester bond, occurred during the DES extraction process. Meanwhile, the stretching vibration peaks of the CO (1738 cm−1) and C–O (1273 cm−1), both characteristic of the ester group, were significantly weakened or even almost absent in the pretreated samples (Fig. 8d). These results corroborated that mild TMAH-based DESs can extract lignin effectively by breaking the lignin–carbohydrate ester bond and retaining abundant β-O-4 ether linkages.
 | ||
| Fig. 8 GCMS results of (a) 2-phenoxy-1-phenylethanol (1a), (b) phenyl β-D-glucopyranoside (1b), and (c) benzyl benzoate (1c) before and after the reaction in TMAH-based alkaline DESs; (d) FTIR spectra of the coconut shells before and after pretreatment; (e) the lignin extraction mechanism of TMAH-based DESs. | ||
The lignin extraction mechanism of the TMAH-based alkaline DESs is proposed according to these multiple pieces of evidence (Fig. 8e). Lignin consists of different units connected by ether bonds and C–C bonds, and the resulting lignin is connected to hemicellulose by LCC bonds. TMAH-based alkaline DESs with strong hydrogen bonding and alkalinity, extracted lignin by selectively attacking lignin–carbohydrate ester bonds rather than the β-O-4 ether linkages, resulting in the harvested lignin fragments showing high β-O-4 linkage retention. Moreover, due to the retention of the phenylglucoside and benzyl ether bonds, a small amount of xylan may remain attached to the extracted lignin.
3.4 Oxygen depolymerization performance of DES-extracted lignin in the TMAH-based alkaline solvents
Based on the results obtained, lignin extracted using TMAH-based alkaline DESs demonstrated a higher retention rate of β-O-4 linkages, which is favorable for lignin depolymerization and subsequent aromatic monomer production.8 The depolymerization performance of the extracted lignin using TMAH-based DESs was verified by the catalytic oxidation process. According to Fig. 9a, the similar types of aromatic monomers obtained in these four DES systems include phenol, guaiacol, benzyl alcohol, benzaldehyde, 4-hydroxybenzaldehyde, vanillin, syringaldehyde, vanillin acetone, benzonitrile, etc. However, the content of aromatic monomers showed varied results across the four systems. The content of aromatic monomers was 35.34%, 20.11% and 30.24% in TMAH-urea, TMAH-MEA and TMAH-lysine, respectively, while it could reach 56.23% in TMAH-imidazole. Moreover, phenol and aromatic aldehydes were the main products when employing TMAH-MEA, TMAH-lysine and TMAH-imidazole as the reaction solvents, while the main aromatic monomer derived from TMAH-urea was benzonitrile. Due to the difference in the depolymerization product content, the solvent properties could be speculated as the governing factor for the lignin depolymerization process. As reported, HCN could be generated from urea under high temperature and strong oxidation conditions, and then could react with benzaldehyde to form benzonitrile,64,65 which would support the higher benzonitrile content in TMAH-urea. The higher aromatic monomer content in the TMAH-imidazole system may be attributed to the superior oxidation effect of imidazole.59 According to the CV analysis (Fig. S9†), only one slight hump was seen in the CV curve of TMAH-urea and no peak appeared in TMAH-MEA and TMAH-lysine. However, two obvious oxidation peaks appeared between 0.6 and 1.2 V in the CV curve of TMAH-imidazole, which proved the excellent oxidation effect of TMAH-imidazole.37 | ||
| Fig. 9 (a) Types and contents of in different TMAH-based DES systems (reaction conditions: 100 °C, 2 h, 1 MPa O2, catalyst/lignin (C/L) mass ratio: 1:1); effect of reaction conditions on aromatic monomers yield of the main products in TMAH-imidazole: (b) type of catalyst, (c) temperature, (d) time, (e) C/L ratio and (f) oxygen loading; the 2D-HSQC NMR of lignin after the catalytic oxidation reaction: (g) side chain regions and (h) aromatic regions; (i) the GCMS results of 2-phenoxy-1-phenylethanol before and after the catalytic oxidation reaction. | ||
Based on the better performance of the TMAH-imidazole system, the catalytic oxidation performance of lignin with different catalysts in this solvent system was investigated. As shown in Fig. 9b, the yield of aromatic monomer was 30.84% when no catalyst was added at 100 °C for 2 h under an O2 atmosphere at 1 MPa, while it increased to 31.36–74.96% after adding different metal oxide catalysts. The highest monomer yield of 74.96% was obtained when MnO2 was employed as a catalyst. MnO2 has also shown relatively excellent lignin catalytic oxidation performance in previous studies.35,36,66 Additionally, MnO2 is widely available in nature and has no harmful effects on the environment.67 Subsequently, the effects of different reaction conditions on the catalytic oxidation performance of lignin using MnO2 as the catalyst were investigated. According to Fig. 9c, with the reaction temperature increasing from 60 °C to 100 °C, the aromatic monomer yield was significantly increased from 40.28% to 74.96%. However, lower monomer yield was obtained when the temperature increased further. This may be due to the recondensation of the resulting monomers under the intense conditions.36 Another possible reason could be the decomposition of TMAH when the temperature reaches 120 °C. As reported, TMAH decomposes at about 130 °C to generate trimethylamine and methanol,68 which may reduce or even extinguish the oxidation effect of the DES. Similarly, the reaction time also plays a key role in determining the yield of monomers. It can be observed that the maximum monomer yield was obtained at 100 °C for 2 h (Fig. 9d). Only 35.92% was obtained with a retention time of 0 min, which demonstrated that shorter reaction time cannot cleave the bonds completely. As the reaction time is extended to 4 h, the monomer yield decreases due to the condensation caused by a long reaction.36 Therefore, 100 °C and 2 h will be more suitable conditions for the catalytic oxidation of lignin in the TMAH-imidazole systems.
The effect of catalyst loading is exhibited in Fig. 9e. An inferior monomer yield (30.83%) was obtained with no catalyst loading, while it increased to 53.78%–75.96% after adding the catalyst, demonstrating the pivotal role of the catalyst in the conversion of lignin into aromatic monomers. When the C/L mass ratio reached 1:5, more than 70% of monomer yield could be achieved. In addition, an oxidative environment was necessary for achieving better lignin depolymerization performance. As shown in Fig. 9f, only 11.56% monomer yield was achieved in air, while it can increase to more than 70% when the oxygen loading reaches 1 MPa, demonstrating the significance of an oxidative environment.36,69 Therefore, a higher aromatic monomer yield can be achieved in TAMH-imidazole systems under mild reaction conditions with a small amount of catalyst, higher than the reported solvent system of other studies (Table S3†).
To elucidate the potential reaction mechanism of lignin oxidation in the TMAH-imidazole system, the structural features of the lignin after catalytic oxidation reaction were investigated by 2D-HSQC NMR. According to Fig. 9g, the typical signal peak of Aα disappeared after the catalytic reaction, which indicated that TMAH-imidazole as a catalyst can effectively break the β-O-4 linkages under the action of oxygen. The presence of obvious methoxy peaks indicated that demethoxylation may occur during catalytic oxidation. It has been reported that the demethoxylation can be achieved under the action of a manganese-containing catalyst,70 which can explain why methoxy-free phenol and benzaldehyde were the main products in the MnO2/TMAH-imidazole system. In the aromatic regions, the signal peaks of the typical lignin units were weakened or disappeared, indicating that the lignin had effectively depolymerized. The experimental results of the lignin model compound (2-phenoxy-1-phenylethanol) also demonstrate that the β-O-4 bond was broken during the catalytic oxidation process. As shown in Fig. 9i, the peak intensity of 2-phenoxy-1-phenylethanol was significantly weakened in the catalyzed sample. Meanwhile, the monomer peaks of styrene, benzaldehyde, benzyl alcohol and phenol were recorded. Therefore, the generation of aromatic monomers from lignin is mainly produced by the cleavage of the β-O-4 bond of lignin during the catalytic oxidation process.
3.5 The technical profile and recyclability of the TMAH-imidazole process
In conclusion, “lignin-first” biorefining can be achieved by a mild TMAH-imidazole process. According to the mass balance flow (Fig. 10), after the DES treatment (30%TMAH-imidazole, 50 °C-3 h), the coconut shells were fractionated into a soluble liquid fraction and an insoluble solid fraction. 91.21% cellulose and 95.14% hemicellulose were retained in the solid fraction, while 67.37% lignin was extracted due to the strong alkalinity and the strong hydrogen bonding of the solvent. Meanwhile, TMAH-imidazole extracted lignin mainly by cleavage of the lignin–carbohydrate ester bonds, resulting in the retention of most of β-O-4 linkage, which contributed highly active native lignin for efficient aromatic monomer conversion (74.54%). Taking 100 g starting biomass for example, through the DES process, 20.8 g of aromatic monomers and 40.9 g of solid components can be obtained, which recovered more than 86% of the active components. Moreover, good reusability for lignin dissolution and depolymerization was achieved. After five cycles, the lignin extraction rate could still reach 63.56%, and the aromatic monomer yield could reach 90% of that from the initial process (Fig. S10†). | ||
| Fig. 10 Mass balance flow diagram of a “lignin-first” biorefinery. | ||
4. Conclusion
COSMO-RS calculations were successfully leveraged to screen deep eutectic solvents (DESs) as an efficient and viable method. The calculations showed that TMAH-based DESs displayed good solubility for lignocellulose due to their higher excess enthalpy and strong hydrogen bonding forces. A delignification rate of 63.33%–67.37% was achieved without compromising the carbohydrate recovery by using TMAH-based DESs at lower temperatures. The cellulose and hemicellulose retention was over 90%. The cleavage of the lignin–carbohydrate ester bond was corroborated as the main route for lignin extraction, leaving a promising precursor with high β-O-4 bond content for subsequent depolymerization. In the case of lignin with a similar structure, the stronger oxidation of solvents would represent a superior catalytic oxidation performance. Therefore, excellent depolymerization performance with an aromatic monomer yield of 74.54% was obtained in the TMAH-imidazole system.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There is no conflict to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China [52276192] and the GuangDong Basic and Applied Basic Research Foundation [2023B1515020094].References
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- F. Y. Shen, C. He, Y. Wang, J. Xu, M. Huang, L. Zhao, J. Hu, D. Tian and F. Shen, Chem. Eng. J., 2024, 497, 154931 CrossRef CAS.
- T. Zhang, Science, 2020, 367, 1305–1307 CrossRef CAS PubMed.
- S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- T. Renders, S. Van Den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- C. T. Palumbo, N. X. Gu, A. C. Bleem, K. P. Sullivan, R. Katahira, L. M. Stanley, J. K. Kenny, M. A. Ingraham, K. J. Ramirez, S. J. Haugen, C. R. Amendola, S. S. Stahl and G. T. Beckham, Nat. Commun., 2024, 15, 862 CrossRef CAS.
- L. Zhao, S. Shi, M. Liu, G. Zhu, M. Wang, W. Du, J. Gao and J. Xu, Green Chem., 2018, 20, 1270–1279 RSC.
- C. He, X. Li, F. Luo, C. Mi, A. Zhan, R. Ou, J. Fan, J. H. Clark and Q. Yu, ACS Sustainable Chem. Eng., 2024, 12, 11327–11337 CrossRef CAS.
- Y. Zhu, Y. Liao, L. Lu, W. Lv, J. Liu, X. Song, J. Wu, L. Li, C. Wang, L. Ma and B. F. Sels, ACS Catal., 2023, 13, 7929–7941 CrossRef CAS.
- C. He, J. Hu, F. Shen, M. Huang, L. Zhao and J. Zou, Ind. Crops Prod., 2022, 176, 114326 CrossRef CAS.
- F. Y. Shen, S. Wu, M. Huang, L. Zhao, J. He, Y. Zhang, S. Deng, J. Hu, D. Tian and F. Shen, Green Chem., 2022, 5242–5254 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 1, 2010–2011 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- D. Tian, F. Y. Shen, J. Hu, M. Huang, L. Zhao, J. He, Q. Li, S. Zhang and F. Shen, Chem. Eng. J., 2022, 428, 131373 CrossRef CAS.
- Y. Liu, N. Deak, Z. Wang, H. Yu, L. Hameleers, E. Jurak, P. J. Deuss and K. Barta, Nat. Commun., 2021, 12, 1–15 CrossRef PubMed.
- C. He, F. Y. Shen, D. Tian, M. Huang, L. Zhao, Q. Yu and F. Shen, Int. J. Biol. Macromol., 2024, 254, 127853 CrossRef CAS.
- J. Zhao, G. Zhou, T. Fang, S. Ying and X. Liu, RSC Adv., 2022, 12, 16517–16529 RSC.
- C. Mukesh, G. Huang, H. Qin, Y. Liu and X. Ji, Biomass Bioenergy, 2024, 188, 107305 CrossRef CAS.
- M. Mohan, K. Huang, V. R. Pidatala, B. A. Simmons, S. Singh, K. L. Sale and J. M. Gladden, Green Chem., 2022, 24, 1165–1176 RSC.
- A. Casas, J. Palomar, M. V. Alonso, M. Oliet, S. Omar and F. Rodriguez, Ind. Crops Prod., 2012, 37, 155–163 CrossRef CAS.
- L. König-Mattern, A. O. Komarova, A. Ghosh, S. Linke, L. K. Rihko-Struckmann, J. Luterbacher and K. Sundmacher, Chem. Eng. J., 2023, 452, 139476 CrossRef.
- K. Yu, W. L. Ding, Y. Lu, Y. Wang, Y. Liu, G. Liu, F. Huo and H. He, J. Mol. Liq., 2022, 348, 118007 CrossRef CAS.
- V. Sharma, M. L. Tsai, C. W. Chen, P. P. Sun, A. K. Patel, R. R. Singhania, P. Nargotra and C. Di Dong, Bioresour. Technol., 2022, 360, 127631 CrossRef CAS.
- F. Gabriele, M. Chiarini, R. Germani, M. Tiecco and N. Spreti, J. Mol. Liq., 2019, 291, 111301 CrossRef CAS.
- Y. Zhang, H. Ren, H. Maarof, S. Mat Udin, Y. Liu, M. Li, H. Alias and E. Duan, J. Mol. Liq., 2023, 374, 121271 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chemie, 2017, 129, 9914–9917 CrossRef.
- M. Hu, Y. Yu, X. Li, X. Wang and Y. Liu, Green Chem., 2023, 25, 10235–10262 RSC.
- Y. R. Liu, K. Thomsen, Y. Nie, S.-J. Zhang and A. S. Meyer, Green Chem., 2016, 18, 6246–6254 RSC.
- J. Wang, Y. Guo, F. Liu, X. Zhang, W. Wang and Q. Peng, J. Mol. Liq., 2022, 349, 118139 CrossRef CAS.
- K. Huang, M. Mohan, A. George, B. A. Simmons, Y. Xu and J. M. Gladden, Green Chem., 2021, 23, 6036–6049 RSC.
- Q. Xia, Y. Liu, J. Meng, W. Cheng, W. Chen, S. Liu, Y. Liu, J. Li and H. Yu, Green Chem., 2018, 20, 2711–2721 RSC.
- A. Sluiter, B. Hames, R. O. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Biomass Anal. Technol. Team Lab. Anal. Proced., 2011, 2011, 1–14 Search PubMed.
- J. Dai, A. F. Patti, G. N. Styles, S. Nanayakkara, L. Spiccia, F. Arena, C. Italiano and K. Saito, Green Chem., 2019, 21, 2005–2014 RSC.
- A. Kumar, B. Biswas, R. Kaur, S. Rawat, B. B. Krishna, P. Kumbhar, S. Pal, S. Padmanabhan and T. Bhaskar, Bioresour. Technol., 2022, 352, 127032 CrossRef CAS PubMed.
- Q. Yu, Z. Song, X. Chen, J. Fan, J. H. Clark, Z. Wang, Y. Sun and Z. Yuan, Green Chem., 2020, 22, 6415–6423 RSC.
- Y. Lou, X. Sun, Y. Yu, S. Zeng, Y. Li, Y. Liu and H. Yu, Research, 2023, 6, 0069 CrossRef CAS PubMed.
- M. Mohan, H. Choudhary, A. George, B. A. Simmons, K. Sale and J. M. Gladden, Green Chem., 2021, 23, 6020–6035 RSC.
- M. Zhou, O. A. Fakayode, A. E. Ahmed Yagoub, Q. Ji and C. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111986 CrossRef CAS.
- S. Hong, X. J. Shen, Z. Xue, Z. Sun and T. Q. Yuan, Green Chem., 2020, 22, 7219–7232 RSC.
- O. Ershova, E. V. da Costa, A. J. S. Fernandes, M. R. Domingues, D. V. Evtuguin and H. Sixta, Cellulose, 2012, 19, 2195–2204 CrossRef CAS.
- Q. Liu, X. Zhao, D. Yu, H. Yu, Y. Zhang, Z. Xue and T. Mu, Green Chem., 2019, 21, 5291–5297 RSC.
- Q. Liu, R. Tian, Z. Lv, Y. Wu, B. Lv, X. Hao, Z. Xue and F. Peng, Carbohydr. Polym., 2023, 300, 120245 CrossRef CAS PubMed.
- R. Tian, B. Zhu, Y. Hu, Q. Liu, J. Bian, M. Li, J. Ren and F. Peng, Int. J. Biol. Macromol., 2024, 254, 127499 CrossRef CAS.
- R. Tian, B. Zhu, Q. Liu, Y. Hu, Z. Yang, J. Rao, Y. Wu, B. Lü, J. Bian and F. Peng, Bioresour. Technol., 2023, 369, 128490 CrossRef CAS PubMed.
- B. Jiang, Q. Tang, H. Yu, Q. Su, F. Y. Shen, L. Zhao, J. Hu, D. Tian and F. Shen, ACS Sustainable Chem. Eng., 2024, 12, 13960–13971 CrossRef CAS.
- Q. Su, D. He, J. Luo, X. Zhou, S. Wu, L. Zhao, F. Shen, J. Hu and D. Tian, Carbohydr. Polym., 2024, 345, 122584 CrossRef CAS.
- L. Li, M. Zhang, Y. Feng, X. Zhang and F. Xu, Carbohydr. Polym., 2024, 339, 122260 CrossRef CAS PubMed.
- A. K. Kumar, B. S. Parikh and M. Pravakar, Environ. Sci. Pollut. Res., 2016, 23, 9265–9275 CrossRef CAS.
- Y. Guo, L. Xu, F. Shen, J. Hu, M. Huang, J. He, Y. Zhang, S. Deng, Q. Li and D. Tian, Chemosphere, 2022, 286, 131798 CrossRef CAS PubMed.
- H. Li, X. Li, T. You, D. Li, H. Nawaz, X. Zhang and F. Xu, Int. J. Biol. Macromol., 2021, 193, 319–327 CrossRef CAS.
- Y. Jiang, F. Y. Shen, B. Jiang, Y. Hu, J. Hu, M. Huang, L. Zhao, H. Wu, D. Tian and F. Shen, Chem. Eng. J., 2024, 479, 147610 CrossRef CAS.
- R. Wang, D. Li, G. Zheng, Z. Cao and F. Deng, Chem. Eng. J., 2023, 455, 140916 CrossRef CAS.
- A. C. Cassoni, I. Mota, P. Costa, M. W. Vasconcelos and M. Pintado, Int. J. Biol. Macromol., 2022, 220, 406–414 CrossRef CAS.
- X. Wan, F. Shen, J. Hu, M. Huang, L. Zhao, Y. Zeng, D. Tian, G. Yang and Y. Zhang, Int. J. Biol. Macromol., 2021, 180, 51–60 CrossRef CAS PubMed.
- F. S. Mjalli, G. Murshid, S. Al-Zakwani and A. Hayyan, Fluid Phase Equilib., 2017, 448, 30–40 CrossRef CAS.
- C. E. Stauffer, J. Am. Chem. Soc., 1973, 2489–2493 Search PubMed.
- Q. Yan, X. Wu, H. Jiang, H. Wang, F. Xu, H. Li, H. Zhang and S. Yang, Coord. Chem. Rev., 2024, 502, 215622 CrossRef CAS.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
- B. Jiang, F. Y. Shen, Y. Jiang, M. Huang, L. Zhao, Y. Lei, J. Hu, D. Tian and F. Shen, Carbohydr. Polym., 2024, 323, 121452 CrossRef CAS.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- Y. Zhao, U. Shakeel, M. Saif Ur Rehman, H. Li, X. Xu and J. Xu, J. Cleaner Prod., 2020, 253, 120076 CrossRef CAS.
- J. C. Quintanilla, Encycl. Astrobiol., 2023, 52, 1377–1377 Search PubMed.
- P. Dagaut, P. Glarborg and M. U. Alzueta, Prog. Energy Combust. Sci., 2008, 34, 1–46 CrossRef CAS.
- M. Chen, W. Dai, Y. Wang, Z. Tang, H. Li, C. Li, Z. Yang and J. Wang, Fuel, 2023, 333, 126365 CrossRef CAS.
- X. Ma, J. Ma, M. Li, Y. Gu and T. Wang, Polym. Degrad. Stab., 2022, 204, 110091 CrossRef CAS.
- I. Tanczos, G. Pokol, J. Borsa, T. Tóth and H. Schmidt, J. Anal. Appl. Pyrolysis, 2003, 68–69, 173–185 CrossRef CAS.
- Y. Hu, L. Yan, X. Zhao, C. Wang, S. Li, X. Zhang, L. Ma and Q. Zhang, Green Chem., 2021, 23, 7030–7040 RSC.
- M. Ishikawa, M. Tamura, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2016, 182, 193–203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05498a |
| This journal is © The Royal Society of Chemistry 2025 |