
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfinement-supported aurophilic interaction†‡
Youssoupha
Diack
a,
Sonia
Mallet-Ladeira
b,
Denis
Lesage
c,
João V. S.
Guerra
 d,
Didier
Bourissou
*a and
György
Szalóki
*a
d,
Didier
Bourissou
*a and
György
Szalóki
*a
aCNRS/Université de Toulouse, Laboratoire Hétérochimie Fondamentale et Appliquée (UMR 5069), 118 Route de Narbonne, 31062, Toulouse cedex 9, France. E-mail: didier.bourissou@univ-tlse3.fr; gyorgy.szaloki@univ-tlse3.fr
bInstitut de Chimie de Toulouse (UAR 2599), 118 Route de Narbonne, 31062 Toulouse cedex 9, France
cSorbonne Université, Institut Parisien de Chimie Moléculaire (UMR 8232), 4 Place Jussieu, 75252, Paris cedex 5, France
dBrazilian Center for Research in Energy and Materials, Brazilian Biosciences National Laboratory, Rua Giuseppe Máximo Scolfaro, 10000, Bosque das Palmeiras, Campinas, SP 13083-100, Brazil
First published on 28th April 2025
Abstract
An anionic cyclotricatechylene cage (H) is shown (NMR, mass spectrometry and X-ray diffraction) to encapsulate cationic Au(I) complexes. As a result, an unprecedented bimetallic Au(I) complex (GG@H) has been characterized, in which confinement supports aurophilic interaction, even without bridging ligands.
Aurophilic interactions occupy a forefront position in gold chemistry and attract considerable interest.1 Such unusual attractive interactions between closed-shell gold(I) centres are of fundamental interest. They also noticeably influence the physicochemical properties of gold complexes and have thus important consequences on their applications in catalysis, sensing and nanoelectronics.2
Intramolecular (‘supported’ or ‘semi-supported’) aurophilic interactions occur in complexes where the Au⋯Au contact is sustained by bridging bi- or multidentate ligands (Fig. 1A and B). Here, the aurophilic interaction is present both in the solid state and in solution. Many examples of such intramolecular Au⋯Au interactions have been described in the literature.3 In contrast, the formation of unsupported complexes (Fig. 1C) lacks this entropic driving force and is mainly governed by secondary intermolecular interactions (H-bonding, π-stacking). Of note, the relatively weak aurophilic interaction (∼20–40 kJ mol−1) alone is usually not sufficient to stabilize these species. As a result, intermolecular Au⋯Au interactions are much scarcer than their intramolecular counterparts,1 and only a few unsupported aurophilic interactions have been substantiated through short Au⋯Au distances (2.9–3.4 Å) in the solid state.4 Cationic gold(I) bis-phosphine complexes represent an even more challenging case where the aurophilic interaction is largely outrun by the lack of favorable secondary interactions as well as the presence of unfavorable coulombic and steric repulsions. Consequently, no aurophilic interaction has been observed in such complexes so far, either in the solid state, or in solution.
 | ||
| Fig. 1 Aurophilic interactions in fully-supported (A), semi-supported (B), unsupported (C) and confinement-supported (D) gold(I) phosphine complexes. | ||
In this context, we envisioned that confinement within the cavity of an anionic supramolecular cage could be a powerful strategy to bring together two cationic complexes, by counterbalancing the previously mentioned unfavorable interactions (Fig. 1D). The anionic tetrahedral cage developed by Raymond et al. has shown great potential in confining small cationic monometallic gold(I)/gold(III) complexes and modifying their reactivity.5 However, according to our calculations, the cavity of this cage (VH ∼ 340 Å3) is too small to encapsulate larger monometallic or bimetallic complexes.6,7 To this end, the construction of larger anionic cages is highly sought after, not only for the mere synthetic challenge but also to enable the encapsulation of larger guest molecules. However, not many anionic cages have been documented in the literature, much less than cationic and neutral cages. Anionic cyclotricatechylene (C) clusters of [C4M6]n− general formula (M = Mn, VO, Cu) have attracted our attention. They have been known since the 2010s, but their instability prevented their application in supramolecular chemistry.8 In 2018, improved stability was reported for an analogous cage with M = SiR.9 One year later, Kabe and co-workers developed a robust methodology, based on dynamic covalent chemistry, to synthesize a family of such [C4(SiR)6]6− cages.10 Except for proof-of-concept studies with pyridiniums, the host–guest chemistry of these cages has not been investigated further, either with organic cations, or with transition metal complexes. This anionic cage thus offers an ideal starting point for our study and we report here an unprecedented supramolecular approach to support aurophilic interaction between two linear gold(I) bis-phosphine complexes. According to our strategy, the cationic complexes are encapsulated within an anionic supramolecular cage, driven by favorable CH–π and electrostatic interactions. For the first time, these species are characterized both in solution and in the solid state, without the need for a supporting ligand.
To start with, the cavity size of the cage [C4(SiR)6]6− (R = Ph) was calculated. Taking into account our recent evaluation of cavity characterization softwares,6 we used the rolling probe method and the pyKVFinder software.11 Accordingly, the [C4(SiPh)6]6− cage was found to possess a significantly larger cavity (VH = 558 Å3) than Raymond's cage.7 Next, suitable guests were identified, referring to the packing coefficient (PC = VG/VH × 100 (%), with VG = guest volume). Based on Rebek's guideline, the optimal PC is about 55 ± 9%.12 Therefore, strong encapsulation is expected for guests in the size-range of 307 ± 50 Å3. The volume of a series of cationic gold(I) bis-phosphine complexes was calculated using Vega ZZ (Fig. 2).13 Based on the corresponding PC values, encapsulation of complexes 1 (PC = 49%) and 2 (PC = 52%) is expected, while larger complexes 3 (PC = 85%) and 4 (PC = 90%) should not enter the cavity. More importantly, Au(PMe3)2+ (G) was identified as a guest with an optimal size (VG = 176 Å3) to achieve double encapsulation. Indeed, the PC value of the corresponding bimetallic complex (GG) falls well within the “Rebek's range” (2 × VG = 352 Å3, PC = 63%).
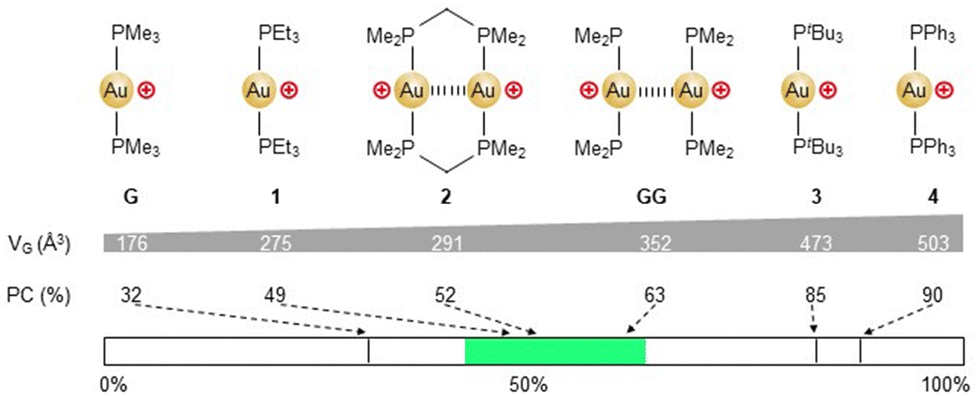 | ||
| Fig. 2 Gold(I) bis-phosphine complexes considered in this study. The “Rebek's range” is depicted in green on the bottom scale. | ||
After considering these metric parameters, the [C4(SiPh)6]6− cage and gold complexes (G and 1–4) were synthesized following known or slightly modified literature protocols.7,13 At this stage, we noticed that the reported [C4(SiPh)6]6− cage and the complexes G, 1–4 were only sparingly soluble in solvents other than DMSO or DMF. Therefore, we envisioned to synthesize a new [C4(SiR)6]6− cage that would be soluble in less polar solvents such as MeOH, MeCN, acetone or THF. Thus, we expected that encapsulation would be promoted in these solvents due to the insolubility of the gold complexes (G, 1–4). Accordingly, a new n-octyl Si-substituted cage H (R = nC8H17) was prepared (Scheme 1). Thanks to a simplified workup, it was obtained in 87% yield on multigram scale. Satisfactorily, it turned out to be significantly more soluble in less polar solvents such as MeOH (Fig. S26 and 27, ESI‡).14
 | ||
| Scheme 1 Synthesis of H. | ||
First, H was characterized by multinuclear NMR. 1H-DOSY NMR showed the presence of a single species in solution (Fig. S19, ESI‡). Its diffusion coefficient (D = 3.2 × 10−10 m2 s−1) corresponds to a structure with a Stokes radius of 12.6 Å (calculated by the Stokes–Einstein equation). Furthermore, ESI-FTICR analysis revealed the presence of multi-anionic species of general formula [C4(SinC8H17)6NaaDMSOb]c− (a = b = c = 3, m/z = 863.97547; a = c = 3, b = 4, m/z = 889.98052; a = b = 4, c = 2, m/z = 1346.46276) (Fig. S55–S58, ESI‡). Finally, single crystals of H were obtained by slow evaporation of a DMSO solution and XRD analysis unambiguously confirmed the structure (Fig. 3). It resembles those of the [C4M6]n− cages previously characterized crystallographically.8–10 Here, a Na+ ion sits within the cavity, while the five remaining Na+ counter ions are located at the openings. Three of these ions are coordinated to a single catecholate moiety, while the other two are bound to two catecholate moieties of different cages, resulting in a Na-bridged dimeric structure in the solid state (Fig. S71, ESI‡). According to these crystallographic data, the size of the cage is estimated to be d ∼ 25 Å, in good agreement with that determined by diffusion experiments in solution.
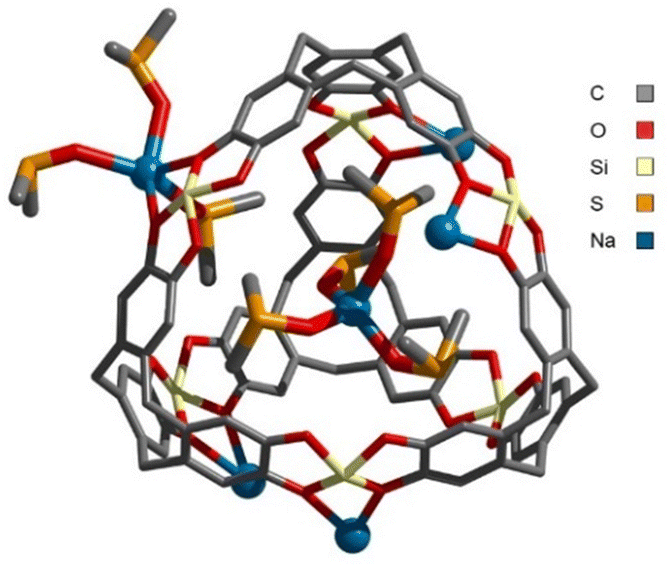 | ||
| Fig. 3 X-ray structure of H. For the sake of clarity, hydrogen atoms, the n-octyl chains at Si, some of the coordinating DMSO molecules and the second cage are omitted. | ||
Next, we interrogated the ability of H to encapsulate the dinuclear complex (2) in which the Me2PCH2PMe2 (dmpm) ligand fully supports aurophilic interaction. 1H NMR analysis of a 1/1 mixture of H and 2 revealed a significant upfield shift of the methylene and methyl protons (Δδ = −1.59 and −1.64 ppm, respectively). This shielding effect is characteristic of inclusion complexes involving supramolecular cages (Fig. S28, ESI‡).14 Moreover, encapsulation was also supported by 1H DOSY analysis, where H and 2 showed an identical diffusion coefficient (D = 2.9 × 10−10 m2 s−1, Fig. S31, ESI‡). Moreover, HRMS analysis revealed different multianionic species, corresponding to the 2/H adduct (Fig. S59–S62, ESI‡). Finally, the structure of the 2@H inclusion complex was unambiguously confirmed by XRD analysis (Fig. S72, ESI‡).7 In addition to the endohedral guest (2@H), two exohedral guests (2ex/H) were found outside the cage, ensuring overall charge neutrality. Of note, confinement induces small deformations of the endohedral guest, as apparent from the difference in PAuAuP dihedral angles (2@H: 165.41(9)° vs.2ex/H: 180.00(12)°/179.98(10)°) and Au⋯Au distances (2@H: 2.9823(8) Å vs.2ex/H: 3.0247(12)/3.0305(10) Å).
Next, the host–guest chemistry of H and G was thoroughly studied. 1H NMR titration experiments revealed 3 distinct regimes depending on the amount of G used (Fig. 4a and Fig. S32, ESI‡). Between 0 and 1 equiv., a significant upfield shift of the methyl protons (Δδ = −1.85 ppm) suggested an important shielding and therefore a close contact between the PMe3 moieties and the aromatic groups of the cyclotricatechylene. A 1H DOSY experiment carried out on a 1/1 mixture of H and G revealed the same diffusion coefficient (D = 3.2 × 10−10 m2 s−1) for both components (Fig. S35, ESI‡). In addition, 1H NOESY experiment showed through space correlations between the methyl protons of G and the aromatic protons of H (Fig. S40, ESI‡). These data strongly support the formation of a G@H inclusion complex. Between 1 and 2 equiv. of G per H, the gradual formation of a new species was observed, which also showed an important shift of the Me 1H NMR signal (Δδ = −1.61 ppm). According to 1H DOSY NMR analysis, the diffusion coefficient of this species (D = 3.2 × 10−10 m2 s−1) is identical to those of H and G@H (Fig. S37, ESI‡). Similarly to G@H, correlations between the H and G protons were observed by 1H NOESY NMR (Fig. S41, ESI‡) and this species was assigned to the GG@H inclusion complex. Finally, addition of more than 2 equiv. of G per H resulted in the appearance of an additional broad 1H NMR signal at around δ = 1.58 ppm. Based on the minimal upfield shift of this signal (Δδ = −0.03 ppm) and literature precedents,15 it is attributed to the exohedrally coordinated guest (Gex/H). It is important to note that in each case, the integral ratios of the H and G1H NMR signals were in good agreement with the amounts of G introduced per H (Fig. S34, 36 and 38, ESI‡). The successive formation of G@H, GG@H and Gex/H was further corroborated by 31P NMR analysis that also showed 3 regimes (Fig. 4b and Fig. S33†).
 | ||
| Fig. 4 1H (a) and 31P NMR (b) spectra of G, H and G/H mixtures of different ratios. | ||
Besides NMR spectroscopy, different multianionic species with G/H = 1 and G/H = 2 stoichiometries were unambiguously identified by ESI-FTICR mass spectrometry, where species of [C4(SinC8H17)6NaaDMSObGd]c− general formula (a = b = 2, c = 3, d = 1, m/z = 946.66092; a = b = 1, c = 3, d = 2, m/z = 1029.34517; a = b = 3, c = 2, d = 1, m/z = 1470.48920; a = b = c = d = 2, m/z = 1594.52971) were observed (Fig. S63–S67, ESI‡).
In addition, single crystals could be obtained from a MeOH solution of a 10/1 G/H mixture. The resulting XRD data are of very good quality (Ra1 = 0.0423), higher than all the previously reported XRD structures for [C4M6]n− cages. Two G complexes sit within the cavity, confirming the formation of GG@H (Fig. 5). Four Na+ ions are located at the openings, coordinated to the catecholate moieties, achieving charge neutrality. Remarkably, the intermolecular Au⋯Au distance is short (3.0532(2) Å) and falls in the midpoint of the aurophilic range (2.5–3.5 Å).1 For comparison, the Au⋯Au distance in the previously mentioned, fully supported, non-confined ((dmpm)Au)2 complex (2ex/H) is 3.0247(12)/3.0305(10) Å. Such an interaction has not been obwhich make themserved in unsupported gold(I) bis-phosphine complexes. Even in the smallest member of this family, i.e. (PMe3)2Au+ (G), the PMe3 ligands prevent the approach of the two gold(I) centres.16 In order to minimize steric repulsions, the two linear complexes are oriented perpendicularly in GG@H (PAuAuP dihedral angle 88.09°, Fig. S73, ESI‡). Importantly, all PMe3 ligands have one methyl group pointing towards a catecholate moiety with short CH–π distances (between 2.6 and 2.9 Å). These numerous CH–π interactions parallel the important upfield shift observed by 1H NMR spectroscopy for the encapsulated complex GG@H (Δδ = −1.61 ppm). Note that the XRD refinement showed 3 different orientations of the bimetallic complex (with 96, 3 and 1% occupancies). They all display ca perpendicular arrangement of the two G motifs and similar CH–π interactions. Interestingly, the volume of the staggered GG guest is slightly smaller than two times the volume of G (VGG = 333 Å3vs. 2 × VG = 352 Å3). As a result, the packing coefficient calculated from the XRD structure is 59% (vs. 63% predicted for the encapsulation of 2 × G), very close to the ideal value proposed by Rebek (55 ± 9%).12 The slightly tighter conformation of GG may play a role in the stabilization of the inclusion complex GG@H.
 | ||
| Fig. 5 X-ray crystal structure of GG@H. For the sake of clarity, hydrogen atoms of H, the n-octyl chains at Si and three Na(MeOH)3+ are omitted. | ||
Finally, the encapsulation of 1, 3 and 4 was also investigated by 1H, 31P and 1H-DOSY NMR spectroscopy (Fig. S42–S54, ESI‡). As expected, the smaller guest 1 (PC = 49%) was readily and quantitatively encapsulated in the cage to give the inclusion complex 1@H, while the larger complexes 3 (PC = 85%) and 4 (PC = 90%) did not enter the cavity.7
In conclusion, encapsulation and confinement in a supramolecular cage has been shown to enable and support intermolecular aurophilic interactions even without a bridging ligand. A new anionic cyclotricatechylene cage [C4(SinOct)6]6− with improved solubility has been synthesized and exploited. An inclusion adduct with two Au(PMe3)2+ complexes within the cavity has been authenticated by NMR spectroscopy, mass spectrometry and X-ray crystallography. It displays short Au⋯Au contact and comparison with the inclusion adduct of the supported complex [Au(Me2PCH2PMe2)+]22 provides the first hints of the distortion confinement that may be induced on aurophilic complexes. Combining cavity and guest size assessment and referring to Rebek's guideline is found to be a relevant and useful predictive tool for encapsulation studies. Diphosphine-bridged dinuclear gold(I) complexes such as 2 possess rich optical and redox properties, which make them powerful photocatalysts.17 It is intriguing and worth exploring how confinement in supramolecular cages affects the behaviour of such systems and if confinement enables extension of the field to unsupported dinuclear gold(I) complexes.
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, the Agence Nationale de la Recherche (CONFICAT ANR-23-CE07-0018), the French Ministry of Higher Education and Research (PhD fellowship to YD) and the IR INFRANALYTICS FR2054 (FT-ICR analyses) is kindly acknowledged. This work was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001 [Grant Number 88887.928702/2023-00] (to J. V. S. G.). We are grateful for the access to the High-Performance Computing Cluster (HPCC Marvin) and the scientific infrastructure provided by the Brazilian Biosciences National Laboratory (LNBio) at the Center for Research in Energy and Materials (CNPEM), a non-profit organization under the supervision of the Brazilian Ministry for Science, Technology, and Innovations (MCTI). The NMR service of the Institut de Chimie de Toulouse – UAR 2599 (Marc Vedrenne) is acknowledged for assistance with the DOSY experiments. Thanks are also due to Saloua Chelli-Lakhdar (LHFA) and Salomé Guilbert for the preparation of some starting materials (cyclotricatechylene) and some exploratory experiments. We are also grateful to Gregori Ujaque Perez for our insightful discussions.
Data availability
The data supporting this article have been included as part of the ESI.‡ Supplementary crystallographic data for H (2427801), GG@H (2427802) and 2@H (2427803) can be obtained from the Cambridge Crystallographic Data Centre (CCDC) and can be accessed via the following URL: https://www.ccdc.cam.ac.uk/structures. The processed structural data files for the cages ([C4(SiPh)6]6−, Raymond's cage and H) and gold complexes (G, GG and 1–4) along with the calculated cavity sizes are available on Zenodo via the following URL: https://zenodo.org/records/14795286.Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- H. Schmidbaur and B. A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- (a) X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111 CrossRef CAS; (b) T. P. Seifert, V. R. Naina, T. J. Feuerstein, N. D. Knöfel and P. W. Roesky, Nanoscale, 2020, 12, 20065 RSC; (c) W. Wang, C.-L. Ji, K. Liu, C.-G. Zhao, W. Li and J. Xie, Chem. Soc. Rev., 2021, 50, 1874 RSC.
- (a) C. D. McTiernan, M. Morin, T. McCallum, J. C. Scaiano and L. Barriault, Catal. Sci. Technol., 2016, 6, 201 RSC; (b) T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044 RSC.
- (a) K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472, 371 CrossRef CAS; (b) D. M. P. Mingos, J. Yau, S. Menzer and D. J. Williams, J. Chem. Soc., Dalton Trans., 1995, 319 RSC; (c) Z. Tang, A. P. Litvinchuk, H.-G. Lee and A. M. Guloy, Inorg. Chem., 1998, 37, 4752 CrossRef CAS PubMed; (d) P. Römbke, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 2001, 2482 RSC; (e) J. Qu, W. Gu and X. Liu, J. Coord. Chem., 2008, 61, 618 CrossRef CAS.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447 CrossRef CAS.
- J. V. S. Guerra, L. F. G. Alves, D. Bourissou, P. S. Lopes-de-Oliveira and G. Szalóki, J. Chem. Inf. Model., 2023, 63, 3772 CrossRef CAS.
- See the ESI,‡ for more information.
- (a) B. F. Abrahams, N. J. FitzGerald and R. Robson, Angew. Chem., Int. Ed., 2010, 49, 2896 CrossRef CAS; (b) B. F. Abrahams, B. A. Boughton, N. J. FitzGerald, J. L. Holmes and R. Robson, Chem. Commun., 2011, 47, 7404 RSC.
- J. L. Holmes, B. F. Abrahams, A. Ahveningen, B. A. Boughton, T. A. Hudson, R. Robson and D. Thinagaran, Chem. Commun., 2018, 54, 11877 RSC.
- Y. Kawakami, T. Ogishima, T. Kawara, S. Yamauchi, K. Okamoto, S. Nikaido, D. Souma, R.-H. Jin and Y. Kabe, Chem. Commun., 2019, 55, 6066 RSC.
- J. V. S. Guerra, H. V. Ribeiro-Filho, G. E. Jara, L. O. Bortot, J. G. C. Pereira and P. S. Lopes-de-Oliveira, BMC Bioinf., 2021, 22, 1 CrossRef.
- S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016 CrossRef CAS.
- A. Pedretti, A. Mazzolari, S. Gervasoni, L. Fumagali and G. Vistoli, Bioinformatics, 2020, 37, 1174 CrossRef.
- M. Yamashina, Y. Tanaka, R. Lavendomme, T. K. Ronson, M. Pittlekow and J. R. Nitschke, Nature, 2019, 574, 511 CrossRef CAS PubMed.
- M. D. Pluth, B. E. F. Tiedemann, H. van Halbeek, R. Nunlist and K. N. Raymond, Inorg. Chem., 2008, 47, 1411 CrossRef CAS PubMed.
- The Au(PMe3)2+ complexes previously characterized crystallographically show Au⋯Au distances exceeding 5.8 Å, see: (a) K. Angermaier, E. Zeller and H. Schmidbaur, J. Organomet. Chem., 1994, 472, 371 CrossRef CAS; (b) U. E. I. Horvath and H. G. Raubenheimer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m567 CrossRef CAS; (c) W. M. Khairul, M. A. Fox, N. N. Zaitseva, M. Gaudio, D. S. Yufit, B. W. Skelton, A. H. White, J. A. K. Howard, M. I. Bruce and P. J. Low, Dalton Trans., 2009, 610 RSC.
- M. Zidan, S. Rohe, T. McCallum and L. Barriault, Catal. Sci. Technol., 2018, 8, 6019 RSC.
Footnotes |
| † This article is dedicated to Prof. Hubert Schmidbaur. |
| ‡ Electronic supplementary information (ESI) available: Characterizations and experimental details. CCDC 2427801–2427803. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01540e |
| This journal is © The Royal Society of Chemistry 2025 |