
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design principles for (efficient) excited-state absorption-based blue-to-UV upconversion phosphors with Pr3+†
Tom
Förster
a,
Josefine
Reifenberger
a,
Tugce
Moumin
a,
Justus
Helmbold
a,
Željka
Antić
 bc,
Miroslav D.
Dramićanin
bc and
Markus
Suta
*a
bc,
Miroslav D.
Dramićanin
bc and
Markus
Suta
*a
aInorganic Photoactive Materials, Institute of Inorganic and Structural Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: markus.suta@hhu.de
bNational Institute of Research and Development for Electrochemistry and Condensed Matter, INCEMC, Timisoara, Romania. E-mail: zeljkaa@gmail.com; dramican@vinca.rs
cCentre of Excellence for Photoconversion, Vinča Institute of Nuclear Sciences, National Institute of the Republic of Serbia, University of Belgrade, P. O. Box 522, 11351 Belgrade, Serbia
First published on 2nd June 2025
Abstract
UV light generation is generally not very efficient, expensive, or may even require toxic elements such as mercury. In contrast, blue light (λ = 450 nm) is cheaply available from semiconductor LEDs and its use in phosphor-converted LEDs is technologically mature and could be envisioned as an intense, sustainable light source in an upconversion scheme. The electronic energy level landscape of the 4f2 ion Pr3+ does allow such a blue-to-UV upconversion (UC) by resonantly exciting the 3PJ (J = 0, 1, 2) levels with blue light, followed by absorption of a second blue photon, thus populating the 4f15d1 configuration states located in the UV range. While the second absorption step is expected to be efficient based on selection rules, no clear guidelines on how to optimize the expected upconversion efficiency for Pr3+ by appropriate choice of a surrounding host are known up to now. Within this work, selected halidoelpasolites, oxyfluorides, garnets, silicates and borates are activated with Pr3+ to understand the relation between ESA-based UC efficiency, the energy and configurational offset of the 4f15d1 states as well as the excited-state dynamics. For that purpose, quantum yield measurements, as well as steady-state, time-resolved and temperature-dependent luminescence spectroscopy with different excitation sources and powers are combined. It turns out that several parameters must be carefully mutually matched within a host compound for efficient ESA-based blue-to-UV UC with Pr3+. Not only does the decay time of the intermediate 3P0 level have to be particularly long in an excited-state absorption upconversion scheme, but also the non-radiative crossover from the excited 4f15d1 states needs to be limited. All these conditions are particularly well fulfilled in the Pr3+-activated chloridoelpasolite Cs2NaYCl6:Pr3+, which shows the highest upconversion quantum yield (ΦUC = 0.11%, P = 0.59 W cm−2) among all investigated compounds within this work and even surpasses the efficiency of well-known upconverters in this field such as Lu3Al5O12:Pr3+ (LuAG:Pr3+) or β-Y2Si2O7:Pr3+ (YPS:Pr3+). The relatively high efficiency of this compound compared to the other standards is a consequence of its low cut-off phonon energy and rigid, densely packed structure with large mutual distances between the rare-earth ions.
1 Introduction
The generation of ultraviolet (UV) light is essential for the fields of photocatalysis,1,2 antimicrobial treatments,3–5 cancer therapy6 and persistent optical tags.7 Historically, UV light is directly generated from sealed mercury (253.7 nm), xenon (190–1100 nm), or deuterium (190–370 nm) gas-based light sources. The current industrial standard for UVC light sources is the electric discharge of mercury, which has many disadvantages such as short lifetimes, large heat generation, and safety hazards. In addition to these, excimer lasers are used as high-energy UV light sources.8 These are also characterised by high voltages and safety hazards due to handling with poisonous gases (halogens).9 Due to their more complex set-ups, mercury lamps and excimer lasers are very limited in size.Another modern possibility would be the use of UV light emitting diodes (UV-LEDs), which however show a strong drop in their external quantum efficiencies for emission wavelengths below 365 nm and potent doped semiconductors are not always readily available.10–13
An alternative approach is the generation of UV light through blue-to-UV upconversion (UC).7,14 Despite a theoretical maximum quantum yield of 50% (ref. 15) due to the absorption of two photons, there is the cost-effective and technologically established possibility of applying a powdered phosphor to a blue light LED chip analogous to a phosphor-converted LED (pc-LED)16 to generate UV light.
By absorbing two or more long-wavelength photons, both organic and inorganic compounds can convert these into a UV light photon in an anti-Stokes process.17 For organic compounds, usually triplet–triplet annihilation is the working mechanism, giving rise to decently efficient blue-to-UV upconversion (UC efficiency ηUC ∼20%, UC quantum yield ΦUC ≤ 5.7%).18–20 In inorganic materials, mostly lanthanide-activated host compounds have emerged as UV upconversion systems, especially Yb3+/Tm3+ co-activated materials for a NIR-to-UV upconversion process.21–23 However, this is a 5-photon process, which results in an overall low efficiency for the generation of UV light (ΦUC(250–375 nm) ≈10−5 for LiYF4: 0.4 mol% Tm3+, 16.5 mol% Yb3+ single crystal at 10 W cm−2 (ref. 24)).
In the last few years, Pr3+-activated ([Xe]4f2) materials have become increasingly interesting for blue-to-UV upconversion.25–29 The electronic structure of Pr3+ allows upconversion to the states of the excited [Xe]4f15d1 configuration upon irradiation with intense blue light. Two basic mechanisms are particularly relevant for upconversion, which are majorly dependent on the activator concentration. For low concentrations, two sequential one-photon absorption processes give rise to collective single-ion upconversion based on excited-state absorption (ESA). This mechanism is typically rather inefficient.17 At higher activator fractions, it is possible that two neighboring ions can be excited and upconversion can occur by an energy transfer process.30 This process is referred to as energy transfer upconversion (ETU) and typically more efficient than single ion-based ESA. For Pr3+, intense blue excitation into the 3PJ levels (J = 0, 1, 2) results in a fast decaying (∼10 ns) broadband UV emission due to an electric-dipole allowed 4f15d1 → 4f2 transition.25,31–33 This type of electric-dipole allowed transitions is widely known from other lanthanides, such as Ce3+ (ref. 34–36) and divalent lanthanides like Eu2+ (ref. 37 and 38), Yb2+ (ref. 39 and 40) or Sm2+ (ref. 41 and 42). Due to the involvement of the 5d orbitals and the resulting crystal field splitting, the energy of the excited 4f15d1 states can be tuned by the chemical composition, Pr–ligand distance, and local site symmetry of the Pr3+ ions.43,44 Precise tuning of the energy position of the 4f15d1 configuration states is mandatory: if the energy of the 4f15d1 states is too high (ΔE(3H4, 4f15d1) > 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 445 cm−1), two blue photons will not lead to a resonant upconversion process. If the energy of the 4f15d1 states is, however, too low, the excess energy of the two blue photons may provoke a non-radiative crossover or even thermal ionization into the conduction band ultimately quenching the 4f15d1 → 4f2-based emission (see Fig. 1).45–47
445 cm−1), two blue photons will not lead to a resonant upconversion process. If the energy of the 4f15d1 states is, however, too low, the excess energy of the two blue photons may provoke a non-radiative crossover or even thermal ionization into the conduction band ultimately quenching the 4f15d1 → 4f2-based emission (see Fig. 1).45–47
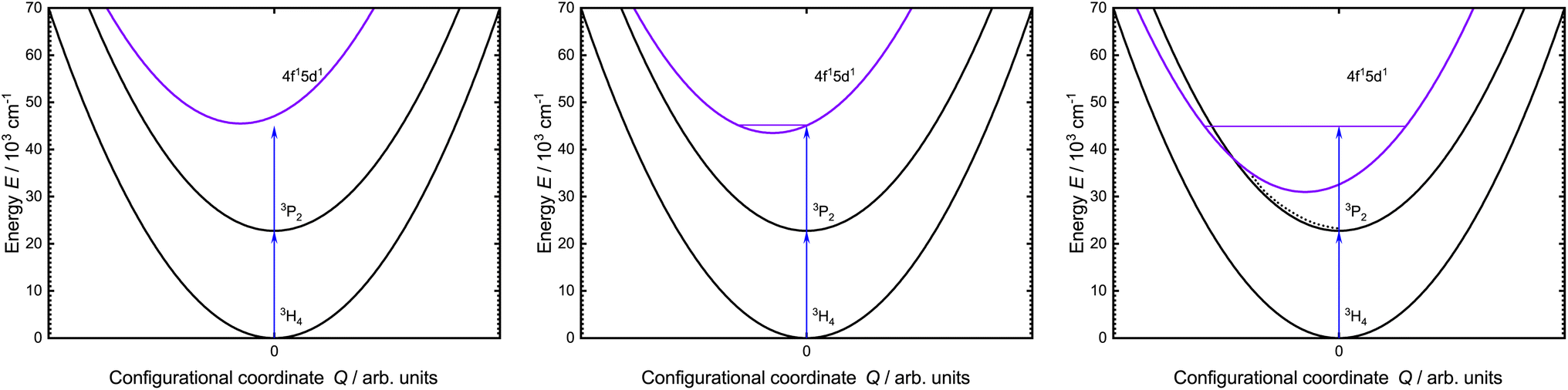 | ||
| Fig. 1 Simplified configurational coordinate diagram of Pr3+ showing the involved states of the blue-to-UV upconversion process. (Left) 4f15d1 states are not accessible via blue-to-UV upconversion. (Middle) 4f15d1 states are in the right range for blue-to-UV upconversion. (Right) Blue-to-UV upconversion results in an excitation above the crossover of the 4f15d1 states, which results in non-radiative relaxation into the lower-lying 4f2 levels (dotted line). | ||
A very well-established upconversion phosphor is β-NaYF4:Er3+,Yb3+, in which the Yb3+ efficiently absorb NIR light resulting in upconverted green and red emission by the Er3+ ions.48–52 The Yb3+/Er3+ NIR-to-Vis UC follows the cooperative ETU mechanism53 and is quite efficient, as upconversion quantum yields up to 10% can be achieved in β-NaYF4: 2% Er3+, 18% Yb3+ by now.54–56 This is the result of decades of research into optimising this cooperative ETU pair.57,58 However, for Pr3+, UC quantum yields are much lower (ΦUC < 1%),59 as there are no guidelines for designing Pr3+-based upconversion phosphors yet, although many examples are already known in the literature.29,47,60–65 Unfortunately, the well-known host compounds for upconversion, α-/β-NaYF4 and LiYF4 are not suitable for the blue-to-UV upconversion of Pr3+. In the case of α-NaYF4, the 4f15d1 states are energetically above the 1S0 (4f2) level, resulting in quantum cascade luminescence.66 For β-NaYF4 and LiYF4, on the other hand, the excitation of the 4f15d1 states is outside the energy range (>42700 cm−1).67–69
A special feature of the electronic energy level landscape of Pr3+ is that both ESA and ETU are in principle possible as UC mechanisms (Fig. 2). ETU can be controlled via the activator concentration in a given host compound. Since ETU is more efficient than ESA, a high concentration of Pr3+ may appear as an obvious choice. However, both the excited 3P0 and 1D2 levels of Pr3+ also show a high tendency of cross-relaxation at elevated activator concentrations, which increases their non-radiative decay rates.70–72 This results in an additional difficulty for ETU-based blue-to-UV UC with Pr3+. Consequently, the ESA process should first be optimised at low concentrations of Pr3+ for a better understanding of the control of the upconversion process in general.
 | ||
| Fig. 2 Possible blue-to-UV upconversion mechanisms of Pr3+ after first excitation with blue light populating the 3P2 level. Solid arrows represent absorption, while dotted arrows show non-radiative processes. (a) Excited-state absorption involving 3P0 as intermediate state. (b) Excited-state absorption involving 1D2 as intermediate state. (c) Energy transfer upconversion involving 3P0 as intermediate state. (d) Energy transfer upconversion involving 1D2 as intermediate state. | ||
A long-lived intermediate state is required for efficient ESA. The decay rate of the 3P0 level is thus very crucial for Pr3+, which is mainly controlled by multiphonon relaxation (MPR) at low concentrations.73 According to Hund's rule, the spin multiplicity of the lowest 4f15d1 state should be dominantly triplets given the still limited degree of spin–orbit coupling. Consequently, ESA from the intermediate 3P0 level is expected to be more effective than from the 1D2 level.60,74,75 There have been recent reports about a potentially important role of the lower energetic 1D2 level in the blue-to-UV upconversion with Pr3+ in the case of Sr3(BO3)2:Pr3+ (ref. 61). Within this work, however, we will explicitly focus on the ESA-based upconversion mechanism and the importance of the 3P0 level, while a direct inclusion of the 1D2 level will be part of future studies.
The efficiency of ESA-based blue-to-UV upconversion in Pr3+-activated phosphors is critically affected by the decay rate of the 3P0 state and the crossover energy of the 4f15d1 states. To prove this, we systematically elucidated different microcrystalline phosphors activated with 0.5 mol% Pr3+, namely YAl3(BO3)4 (YAB),28 Na3Y(BO3)2 (NYB),76 β-Y2Si2O7 (YPS),75,77 X2-Y2SiO5 (YSO),78–80 Lu3Al5O12 (LuAG),81,82 Y3Al5O12 (YAG),82 Y7O6F9 (V-YOF)27 and Cs2NaYCl6.83 These host compounds differ in their cut-off phonon energies, but also have small sites and matching energies of the 4f15d1 configuration states. The host compounds are activated with a low concentration of Pr3+ to prevent cross-relaxation and ETU. It is important to note that the optimum Pr3+ concentration also depends on the explicit structure of the surrounding host compounds, which has an immediate impact on the achievable (internal) quantum yield.
2 Results and discussion
2.1 Structural analysis
The X-ray powder diffraction (XRPD) patterns of the synthesized microcrystalline powders are depicted in Fig. S1,† showing no additional diffraction peaks and therefore no detectable impurities for most of the powders. Small traces of α-cristobalite (SiO2, ICSD depository no.: 77452 (ref. 84)) are detected in the XRPD pattern of YPS due to the employed excess of TEOS in the synthesising process. Furthermore, other Bragg reflections can be observed in LuAG, which can be assigned to the used aluminum sample holder. Based on Rietveld refinement, the obtained lattice parameters were slightly larger than in the pure compounds (Tables S1–S8†). This observation can be attributed to the higher ionic radius of Pr3+ compared to Y3+ and Lu3+.85
2.2 Photophysical properties
All the Pr3+-activated samples show luminescence in the visible range under blue light excitation into the 3P2 level (Fig. 3). There are up to nine sets of emission bands in the spectra, displaying several transitions from the 3P1, 3P0 and 1D2 levels into the energetically lower 3HJ (J = 4, 5, 6) and 3FJ′ (J′ = 2, 3, 4) levels. The Pr3+ 4f2–4f2-based emission in V-YOF is comparably broadened, which can be attributed to the presence of four crystallographically independent Y3+ sites in the structure.32 The most evident difference among the luminescence spectra of the various presented Pr3+-activated compounds is the intensity of the 1D2 → 3H4 transition: in hosts with a high cut-off phonon energy, e.g. borates and silicates (exact values of cut-off phonon energies are compiled in Table 1, as derived from the IR spectra shown in the ESI†), the 1D2 → 3H4 transition is the most intense transition in the spectrum, while in hosts with comparably low cut-off phonon energies, it is almost absent in favor of 3P0-based luminescence. That striking difference is in line with the energy gap law of multiphonon transitions, which states that the non-radiative decay rate is exponentially damped with increasing number of required phonons to bridge the energy gap between two energetically neighboring energy levels. The energy gap ΔE between the 3P0 and 1D2 levels of Pr3+ is about 3700 cm−1.90 Therefore, in host compounds with high cut-off phonon energies such as borates (see Table 1 and Fig. S8–S15†), the 3P0 level quickly decays non-radiatively to the lower energetic 1D2 level, while in hosts with a comparably low cut-off phonon energy such as a chloride, non-radiative decay is slow compared to radiative decay resulting in a longer decay time of the 3P0 level (see Fig. 4 and Table 1). Another possibility for non-radiative relaxation from the 3P0 to the 1D2 level are intervalence charge transfer (IVCT) states between Pr3+ and transition metal ions with a d0 valence electron configuration such as Ti4+, V5+, Zr4+ or Nb5+.91–93 In the host compounds considered within this work, however, IVCT states are not observable and a potential impact can be disregarded.
 | ||
| Fig. 3 Normalized photoluminescence emission spectra of the synthesized samples under 450 nm excitation into the 3P2 level at 298 K. | ||
| τ(3P0) (77 K)/μs | τ(1D2) (298 K)/μs | ℏω eff/cm−1 | |
|---|---|---|---|
| YAB:Pr3+ | 0.85 ± 0.03 | 15.52 ± 0.04 | 1400 |
| NYB:Pr3+ | 0.86 ± 0.04 | 13.99 ± 0.03 | 1395 |
| YPS:Pr3+ | 1.03 ± 0.01 | 156.19 ± 1.09 | 1113 |
| YSO:Pr3+ | 1.96 ± 0.01 | 71.35 ± 1.32 | 990 |
| LuAG:Pr3+ | 11.75 ± 0.39 | 150.84 ± 0.92 | 850 |
| YAG:Pr3+ | 12.01 ± 0.66 | 153.48 ± 1.43 | 840 |
| V-YOF:Pr3+ | 13.48 ± 0.03 | 66.35 ± 1.21 | 520 |
| Cs2NaYCl6:Pr3+ | 172.67 ± 0.29 | 1107.96 ± 7.68 | 287 |
 | ||
| Fig. 4 Photoluminescence decay curves of the synthesized samples of the 3P0 level under 450.9 nm excitation (3P2 ← 3H4) at 77 K monitoring the 3P0 → 3H4-based emission around 488 nm. Due to strongly differing decay times, decay curves are shown for different delay times. Values of the measured decay times are given in Table 1. | ||
The decay curves of the 1D2-based luminescence under direct excitation were also measured (see Fig. S5† and Table 1). In line with these observations, also the 1D2 level follows this general trend, although its decay time is generally longer than that of the 3P0 level based on its energetically more isolated nature (ΔE = 7000 cm−1 to the lower 1G4 level90).
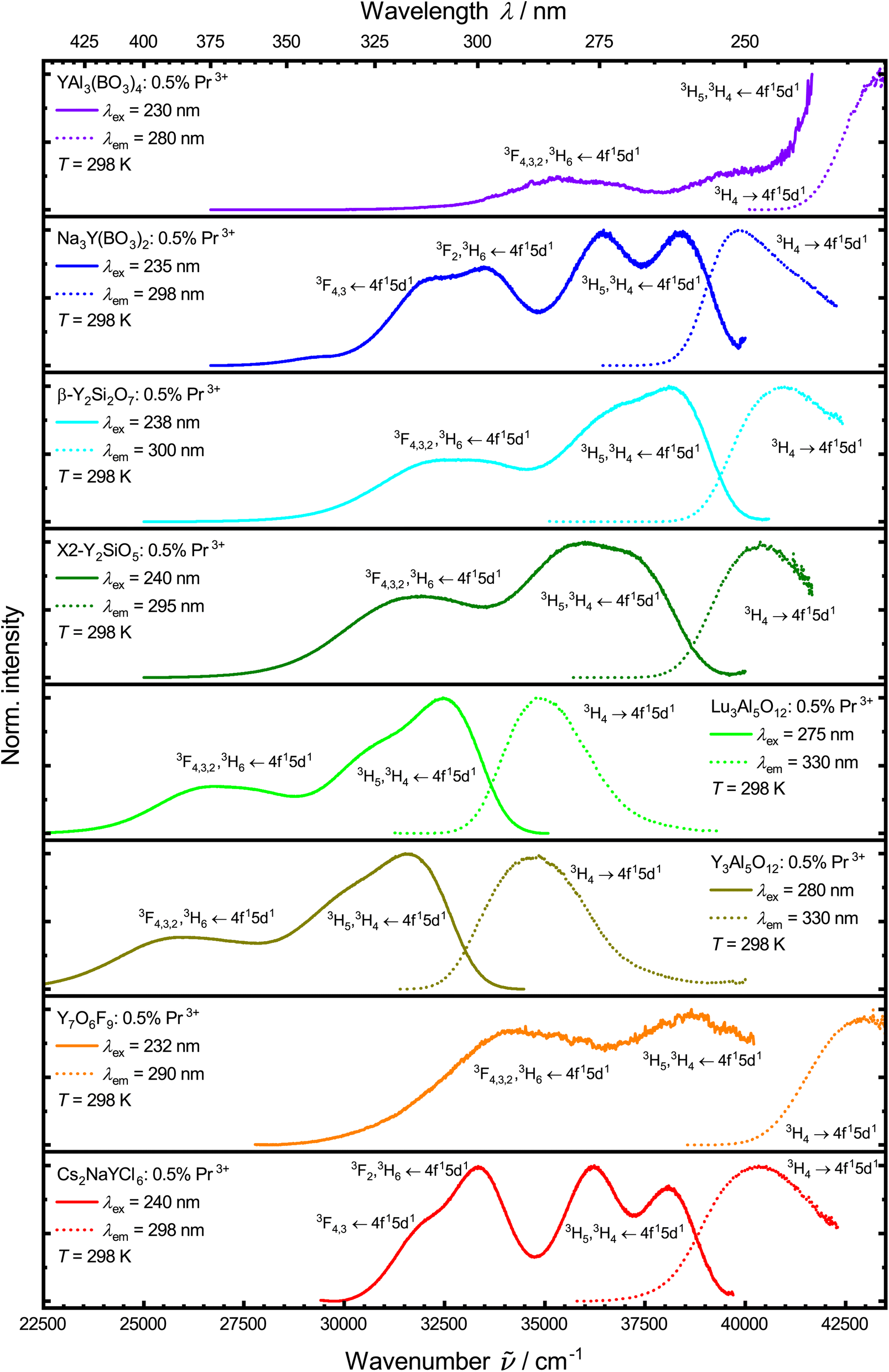 | ||
| Fig. 5 Normalized photoluminescence emission (solid lines) and excitation spectra (dotted lines) of the synthesized samples at 298 K in UV range showing emission and excitation of 4f15d1 states. Spectra were measured against wavelengths and converted to wavenumbers using a Jacobian transformation.99 | ||
The variation of the 4f15d1-related decay times among the various host compounds has several reasons. On the one hand, the decay time depends on the thermal activation barrier for non-radiative crossover to the lower energy 4f2 (3PJ, 1D2) states.46,47,100,101 Photoluminescence excitation spectra monitoring the respective 3P0- or 1D2-based emission can give additional insights here as a broad excitation band in the UV range indicates indirect excitation via the 4f15d1 states by non-radiative crossover. The other influences are the wavelength of the 4f15d1 → 4f2-based emission and the refractive index of the host compound at that given wavelength, which affects the available photonic density of states and thus, the radiative decay probability itself.102,103
From the 4f15d1 ↔ 4f2-based broadband emission and excitation spectra of Pr3+ in the UV range at 77 K (Fig. S6†), Stokes shifts can be estimated that give additional insights into the degree of non-radiative relaxation by thermal crossover. They are compiled in Table 2. For that, emission spectra recorded in wavelengths were converted to wavenumbers using a Jacobian transformation.99 Emission and excitation spectra were fitted using Gaussian fits, calculating the Stokes shift between the lowest energy (excitation) and the highest energy (emission) maximum.
The Stokes shift of the Pr3+-activated YAB was not determined due to limitations in the experimental setup. From the depicted spectra at 298 K (Fig. 5), it can be inferred, however, that the 4f15d1 → 4f2-based emission in YAB:Pr3+ has a small Stokes shift. Pr3+ shows the lowest Stokes shift in NYB (1390 cm−1), which is in good agreement with Pr3+ in other borates, like YBO3 (1800 cm−1).104 The determined Stokes shift in Cs2NaYCl6 (1520 cm−1) is in good agreement with previous results (1028 cm−1 (ref. 105) at 10 K). The Stokes shifts of the respective emission of Pr3+ in YAG (2550 cm−1) and LuAG (1970 cm−1) are also in good agreement with previous reports.46,95,106 The lower Stokes shift in LuAG compared to YAG results from the smaller rare earth site the Pr3+ ions occupy.104 Previously reported values of the Stokes shift of Pr3+ in YSO are in a similar range (≈2420 cm−1 (ref. 106); ≈ 2852 cm−1 (ref. 74), calculated from emission and excitation maxima) to the one determined here (2530 cm−1). For Pr3+ in YPS, our determined Stokes shift (2010 cm−1) fits well to those estimated by other groups (≈2265 cm−1 (ref. 75), calculated from emission and excitation maxima).
Due to limitations in the experimental setup, a Stokes shift of Pr3+-activated V-YOF could not be determined from spectra at 77 K. Therefore, the Stokes shift was taken from spectra at 298 K for the sake of comparison. Pr3+-activated V-YOF shows the highest Stokes shift of the investigated materials (3150 cm−1). This value is in agreement with previously published results (≈3499 cm−1 (ref. 27), calculated from emission and excitation maxima). As there is weak, but still observable UV emission from Pr3+ in V-YOF, non-radiative relaxation from 4f15d1 → 4f2 and 4f15d1 radiative decay starts to compete. This competition is particularly severe at room temperature with a connected Stokes shift of around 3000 cm−1.107,108
Due to the involvement of the spatially more extended 5d orbitals in the excited 4f15d1 configuration, a shift in the configuration coordinate diagram is to be expected. This leads to a crossover with lower lying 4f2 levels, via which the 4f15d1 state can be non-radiatively depopulated upon thermal activation. It is already known that the position of the crossover barrier correlates with the Stokes shift.45,46 The crossover barrier is also relevant for the thermal quenching of the 4f15d1 → 4f2-based broadband emission and can be probed by the temperature-dependent decay times of the 4f15d1 states (Fig. 6).
 | ||
| Fig. 6 Luminescence decay times of 4f15d1 configuration states of Pr3+ in the synthesized host compounds as a function of temperature with a fit to a Mott-Seitz law (eqn (1) or (3)). Decay times were derived from the exponential fit using eqn (S1).† | ||
The temperature dependence follows a Mott-Seitz law109 (eqn (1)) and allows the estimation of the crossover barrier and thermal quenching temperature T50 (eqn (2)) defined as the temperature at which the decay time has decreased to 50% of its original value at sufficiently low temperatures (77 K). The quenching temperatures for the different regarded Pr3+-activated compounds are compiled in Table 3.
 | (1) |
 | (2) |
| T 50/K | |
|---|---|
| YAB:Pr3+ | — |
| NYB:Pr3+ | ∼634 |
| YPS:Pr3+ | ∼467 |
| YSO:Pr3+ | ∼350 |
| LuAG:Pr3+ | ∼552 |
| YAG:Pr3+ | ∼318 |
| V-YOF:Pr3+ | <318 |
| Cs2NaYCl6:Pr3+ | ∼845 |
For YPS:Pr3+, LuAG:Pr3+ and Cs2NaYCl6:Pr3+ an increase of 4f15d1-related decay time can be observed with increasing temperature before quenching. This behaviour is also known for the electric-dipole allowed 4f65d1 → 4f7 transition of Eu2+ due to mixing of spin-forbidden components into the spin-allowed component.110–112 This could also happen for Pr3+ as the 4f15d1 states can have spin triplet and singlet character.113,114 Consequently, the regular Mott-Seitz law can no longer be fully applied for this compounds. Therefore, the Mott-Seitz law is extended by a Boltzmann expression for τ0 (ref. 115) in eqn (1), which takes into account a thermal coupling between the singlet and triplet states of the excited 4f15d1 configuration of Pr3+. The added Boltzmann expression takes into account an effective energy gap between coupled singlet and triplet states ΔEST, the decay rate of the singlet state kr,S and the degeneracy g1 for the lower excited state |1〉 and the degeneracy g2 for the higher excited state |2〉 (g = 3 for triplet and g = 1 for singlet states), respectively,
 | (3) |
The lowest quenching temperature was determined for YAG:Pr3+ at ∼318 K, which is in good agreement with previous results.46,82,106 Quenching of the 4f15d1 → 4f2-based luminescence of Pr3+ in YAG starts to become noticeable even at 150 K. This fundamental issue will also lower the overall expected upconversion quantum yield of this luminescent compound as the 4f15d1 state of Pr3+ in YAG shows a strong tendency for thermally activated non-radiative decay even below room temperature. Quenching at room temperature is also a problem for YSO:Pr3+ with T50 ∼ 350 K for the 4f15d1 → 4f2-based broadband emission. This value is in line with previous work.100,106 The other regarded Pr3+-activated samples were characterized by significantly higher quenching temperatures of the 4f15d1 → 4f2-based luminescence.
Quenching temperatures of the samples are in good agreement with the determined Stokes shifts as a high Stokes shift generally scales with a low quenching temperature. Based on that argument, a very low T50 can be assumed for V-YOF:Pr3+, probably even lower than YAG:Pr3+.
 | ||
| Fig. 7 Comparison of UV emission under direct excitation (colored lines) and high power blue light excitation (black lines) for the different samples. Emission under blue light was only corrected for PMT sensitivity. Because of measurement artefacts, spectra are only shown up to 340 nm. | ||
The number of photons n involved in the upconversion process can be estimated from the slope of a double logarithmic plot of the integrated upconversion intensity I against the incident pump power P since116
| I ∝ Pn | (4) |
Fig. 8 depicts power dependence for the investigated Pr3+-activated compounds within this work. The slope implies a two-photon process for most of the samples, in agreement with expectations. The low upconversion intensity of Pr3+ in YAB does not allow the determination of a reliable number of involved photons in this upconversion process as the intensity of the upconverted 4f15d1-based luminescence is barely distinguishable from background noise and scattering light (Fig. S16†). The same observation can be made for NYB: 0.5 mol% Pr3+ at lower pump power. Previous research obtained a slope near two using Gd3+ as a sensitizer,28 so a two-photon process for YAB can also be assumed. Due to additionally active non-radiative processes in many of the investigated Pr3+-activated compounds at room temperature, the resulting linear fits give slopes lower than the value of 2.116 At higher incident pump power, YSO and Cs2NaYCl6 show saturation effects, which indicate that the intermediate level shows a small decay compared to the pump rate (see the ESI†).
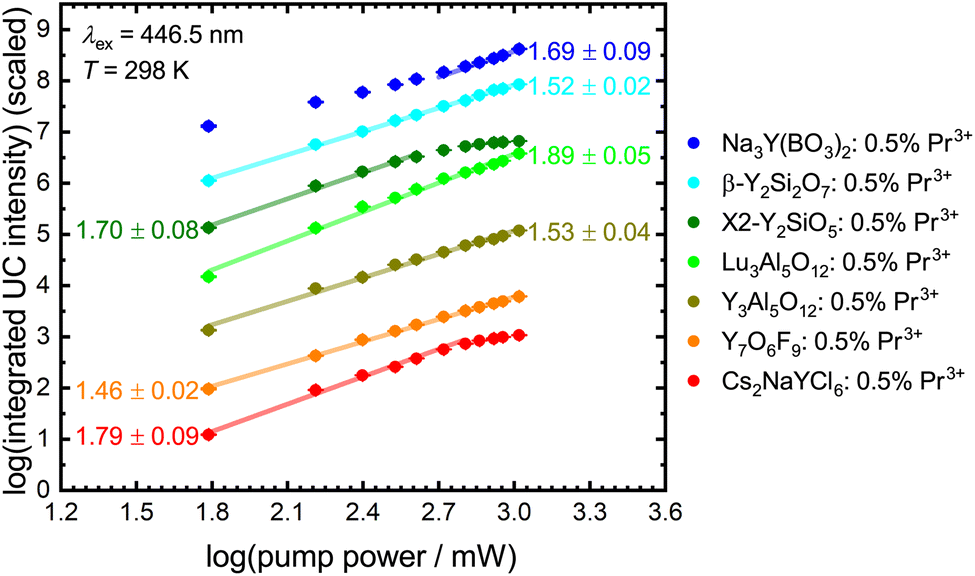 | ||
| Fig. 8 Double logarithmic plot of integrated intensity of the upconverted 4f15d1-based emission of Pr3+ against blue laser (λex = 446.5 nm) pump power at 298 K with linear regression. UC intensity is scaled to allow a better comparison. Slopes are given in the diagram. | ||
Due to the low Pr3+ concentration, we assume ESA as a primary UC mechanism. However, to fully differentiate between ESA and ETU as a UC mechanism, kinetic rate equations according to Pollnau et al.116 and Sun et al.78 were used to describe the behaviour in the limits of low and high pump rates compared to the decay rate of the intermediate level. A complete derivation of the power-dependent excited-state kinetics is detailed in the ESI.†
It becomes evident that the UC mechanism of Pr3+-activated compounds cannot be determined via the intensity of the 4f15d1 configuration states, as this has the same dependence on the irradiated power for both ESA and ETU (Table 4). However, the intensities of the 3P0 and the 1D2 level show a change in the power dependence with the onset of saturation. As depicted in Fig. 8, only Pr3+-activated YSO and Cs2NaYCl6 show saturation effects with our simple experimental setup. For YSO activated with 0.5 mol% Pr3+, ETU is assumed to be the dominant UC mechanism based on estimates,78 which can be explained by the presence of two different Y sites with a mutual distance of only 3.51 Å in the host lattice.117 Therefore, a review of the mechanism for the promising host Cs2NaYCl6 is recommended. Compared to the other compounds mentioned, Cs2NaYCl6 with an elpasolite-type structure is characterized by largely separated rare earth sites with a closest distance of about 7.59 Å.118 Since this compound fulfills many of the presented requirements for efficient ESA-based upconversion, we investigated its power-dependent blue-to-UV upconversion also at lower temperatures (77 K) to avoid temperature-induced decay processes of the intermediate 3P0 level based on its uncommonly low cut-off phonon energy of 287 cm−1 (Table 1 and Fig. S15†). Due to the lack of emission from the 1D2 level in Pr3+-activated Cs2NaYCl6 (Fig. 3), only the dependence on the emission of the 3P0 level and the 4f15d1 states is considered.
| ESA | ETU | |||
|---|---|---|---|---|
| Low pump rate | High pump rate | Low pump rate | High pump rate | |
| 1D2 | ∝I | ∝I−1 | ∝I | ∝Const. |
| 3P0 | ∝I | ∝Const. | ∝I |

|
| 4f15d1 | ∝I2 | ∝I | ∝I2 | ∝I |
Under excitation with low incident power of the blue light source, the aforementioned slopes in the range of 2 for the UC luminescence and 1 for the luminescence of the intermediate 3P0 level are observed for Pr3+-activated Cs2NaYCl6 (Fig. 9, middle). After saturation, a drop in the slope is observed, with the power dependence of the intensity of the 4f15d1-derived states decreasing to n = 1.65 ± 0.15 and the dependence of the intensity of 3P0 decreasing to n = 0.77 ± 0.07. As the slope of the 4f15d1 upconverted emission intensity has not yet reached the anticipated value of n = 1 for an ESA mechanism, it can be assumed that saturation is not yet complete and therefore the slope of the 3P0 intensity will continue to decrease.
 | ||
| Fig. 9 Double logarithmic plot of integrated upconversion and 3P0 intensity against laser pump power with linear regression for Cs2NaYCl6: 0.25 mol% Pr3+ (top), Cs2NaYCl6: 0.5 mol% Pr3+ (middle) and Cs2NaYCl6: 1 mol% Pr3+ (bottom) at 77 K. Intensity is scaled for comparison purposes. Slopes are given in the diagram. (Right) Integrated 3P0 intensity around the saturation point. | ||
Since no precise statement can be made about the UC mechanism in Cs2NaYCl6: 0.5 mol% Pr3+ given the power limitations of our experimental setup, the measurements were repeated with a concentration of 0.25 mol% Pr3+ and 1 mol% Pr3+ (see the ESI† for XRPD and Rietveld refinement). A small change in the Pr3+ concentration in Cs2NaYCl6 does not lead to a decrease in the decay time of the 3P0 level indicating that cross-relaxation can be neglected at these low concentration levels in the chloridoelpasolite (see Fig. S24†). The slopes for the upconverted UV luminescence and the luminescence of the intermediate 3P0 level can be also reproduced in the other Pr3+-activated compounds in principle. However, a different behaviour is observed for the intensity of the 3P0 level after the commencement of saturation effects. After a drop in the slope at the saturation point, a renewed increase in intensity with power is observed shortly afterwards (see Fig. 9 right). According to Pollnau et al.,116 this behaviour is observable when ESA and ETU exist simultaneously, and was also reported for Re4+ in Cs2ZrCl6.119,120 This behaviour is observed in all Pr3+-activated chloridoelpasolites, but is very slight in the sample with 0.25 mol% and 0.5 mol% Pr3+. Consequently, it can be assumed that ETU plays a non-negligible role in Cs2NaYCl6: 1 mol% Pr3+. Similar conclusions have been anticipated for YSO:Pr3+.78
The obtained upconversion quantum yields with the corresponding standard deviation for the Pr3+-activated compounds are given in Table 5. The VPL-450 used here has a power density of 0.59 W cm−2. This power density is therefore lower than the power density of modern LED chips, which can be up to 7.2 W cm−2.121 With the used integrating sphere setup, UC luminescence was only observed for Pr3+-activated YAG, LuAG and Cs2NaYCl6, with YAG:Pr3+ showing the lowest quantum yield of the three regarded compounds in the range of 0.009%. Based on these findings, it is anticipated that the quantum yields of the remaining compounds are lower than the one of YAG:Pr3+. The higher UC quantum yield of Cs2NaYCl6:Pr3+ compared to YAG:Pr3+ and LuAG:Pr3+ is also indicated in the intensity of the upconverted 4f15d1 → 4f2-based emission spectra upon blue-light excitation recorded under the same conditions (Fig. 10). Furthermore, it is necessary to mention that Cs2NaYCl6:Pr3+ could not be measured in the ampoule and thus the hygroscopic properties could not be suppressed that would limit long-term applications. An even higher upconversion quantum yield is thus conceivable for Cs2NaYCl6:Pr3+.
| Upconversion quantum yield ΦUC/% | |
|---|---|
| YAB:Pr3+ | <0.009 |
| NYB:Pr3+ | <0.009 |
| YPS:Pr3+ | <0.009 |
| YSO:Pr3+ | <0.009 |
| LuAG:Pr3+ | 0.025 ± 0.006 |
| YAG:Pr3+ | 0.009 ± 0.001 |
| V-YOF:Pr3+ | <0.009 |
| Cs2NaYCl6:Pr3+ | 0.105 ± 0.019 |
 | ||
| Fig. 10 Comparison of the intensities of the UC luminescence of Pr3+ in LuAG, YAG and Cs2NaYCl6 measured under the same conditions (under air at 298 K) and with the same measurement parameters (1 nm emission bandwidth, 0.1 nm step size, 0.3 s dwell time). | ||
The UC quantum yields for the Pr3+-activated compounds are much lower than the ones known for the Yb3+/Er3+ UC couple, which can be up to ΦUC = 2% (P = 0.6 W cm−2) in β-NaYF4.54,56
A better comparison of the UC quantum yields of Pr3+ and Yb3+/Er3+ than the value at the same power density is provided by the additional consideration of the photon flux qp, which depends on the power density P and the excitation wavelength λ of the used excitation source,
 | (5) |
The corrected UC quantum yield thus results from the ratio of the photon flux, which equals the ratio of the excitation wavelengths at a given power density.
 | (6) |
Taking this into account, the UC quantum yield of Yb3+/Er3+ in comparison to the one of Pr3+ is ΦUC,corr = 0.92%, which is higher than the one of Pr3+-activated Cs2NaYCl6 with ΦUC = 0.11%. This is consistent with the different UC mechanisms, as UC from Yb3+/Er3+ is known to dominantly occur via ETU, which shows a higher efficiency.17
Although a UC quantum yield for Pr3+ could only be determined in three host compounds, the trend shows an increasing quantum yield with decreasing cut-off phonon energy and increasing quenching temperature T50 (Fig. 11). The influence of the quenching temperature T50 of the 4f15d1-based emission becomes particularly evident when considering YAG and LuAG, as LuAG:Pr3+ has a UC quantum yield more than twice as large as YAG:Pr3+ despite similar phonon energies.
 | ||
| Fig. 11 3D bar chart of the upconversion quantum yield in relation to the cut-off phonon energy of the host compound and the quenching temperature T50 of the 4f15d1-based emission. | ||
For the already well-studied Pr3+-activated YSO, no UC quantum yield could be determined with our setup. Previous work indicates a UC efficiency of 0.0019% (P = 1.65 mW cm−2)122 for YSO:Pr3+,Li+. As the UC efficiency for YSO:Pr3+,Li+ was determined via biodosimetry, no exact comparison can be made here. It is also noteworthy that the quantum yield of Pr3+-activated YSO seems to be lower despite a more efficient ETU mechanism for concentrations higher than 0.02 mol% of Pr3+.78 A UC quantum yield could also not be determined for Pr3+-activated YPS, which shows a higher UC efficiency than YSO:Pr3+ according to other studies.75 While the 4f15d1-based emission in YSO shows slight quenching at room temperature, no decrease in the 4f15d1 decay time is observed in YPS (Fig. 6). Consequently, the absence of UC luminescence in the quantum yield cannot be due to thermal quenching, which is also consistent with the power-dependent measurements (Fig. 7), but must be related to the high decay rate of the 3P0 level (Fig. 4 and Table 1).
A similarly low ESA-based UC efficiency of Pr3+ in the analysed silicates is also shown in YAB and NYB. For both Pr3+-activated compounds, no quantum yields can be measured with our setup, which is also due to the high decay rate of the 3P0 level (Fig. 4 and Table 1). Quenching of the 4f15d1-based emission at room temperature can also be ruled out for both host compounds (Table 2 and Fig. 6). Thus, the Pr3+-activated borates are capable UV-emitting phosphors, but poor ESA-based blue-to-UV upconverters.
3 Conclusions
In this work, the different influences of host compounds on the excited-state absorption-based blue-to-UV upconversion of Pr3+ are presented. A high decay time of the intermediate state is required for this mechanism. Using time-resolved measurements, it is shown that the decay rate of the intermediate 3P0 level increases with increasing cut-off phonon energy of the host compound. This is consistent with the energy gap law and the resulting multiphonon relaxation into the energetically lower 1D2 level.Furthermore, non-radiative decay of the excited 4f15d1 states must be limited in order for them to exhibit a high quantum yield. This pathway is minimized if the thermal activation barrier to the crossover point with the low-energy 4f2 levels is high and thus only a small shift in the configurational coordinate is present. Rigid host compounds are suited choices to fulfill these requirements, as is the case in many borates, silicates and phosphates. Alternatively, this can also be achieved if Pr3+ substitutes smaller cation sites with yet high coordination numbers. Here, for example, a substitution of Lu3+, In3+ or Sc3+ would be suitable.
Those criteria fit especially well with the studied Pr3+-activated Cs2NaYCl6. With a small cut-off phonon energy ℏωeff of 287 cm−1 and a quenching temperature T50 of 845 K, an upconversion quantum yield of ΦUC = 0.11% for a power density of P = 0.59 W cm−2 is achieved. In direct comparison with the already known LuAG:Pr3+ and YPS:Pr3+, Cs2NaYCl6:Pr3+ thus shows the highest blue-to-UV efficiency for Pr3+-activated inorganic compounds reported to date.
The results of this work provide new guidelines for designing blue-to-UV phosphors with Pr3+ using ESA as a mechanism. The low quantum yield indicates that there are still limitations in the technical applications of Pr3+-activated solids for blue-to-UV ESA-based upconversion. Creating guidelines for designing Pr3+-activated solids that affect the ETU mechanism offers the possibility of further optimisation given the several orders of magnitude higher efficiency.
Data availability
Source data generated in this study, which are presented in the main text and ESI,† are provided as Source Data files via the Zenodo repository under accession code https://doi.org/10.5281/zenodo.15589798. Source data are also available from the corresponding author upon request.Author contributions
T. Förster performed the measurements, planned the experiments, supervised data acquisition and curated the data. He performed quantum yield measurements and wrote the major part of the manuscript. J. Reifenberger, T. Moumin, and J. Helmbold synthesized relevant compounds, and acquired spectroscopic data. Ž. Antić, M. D. Dramićanin, and M. Suta conceptualized the project, acquired funding and reviewed the manuscript. T. Förster and M. Suta calculated the excited-state dynamics. M. Suta gave suggestions about the respective compounds, details in the synthesis and supervised the whole project. He reviewed and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. gratefully acknowledges a materials cost allowance of the Fonds der Chemischen Industrie e.V. and a scholarship of the ”Junges Kolleg” of the North-Rhine Westphalian Academy of Sciences and Arts. Moreover, M. S. is grateful for generous funding by an Exploration Grant of the Boehringer Ingelheim Foundation (BIS). M. D. D. and Ž. A. would like to acknowledge funding by Romania's National Recovery and Resilience Plan (NRRP) (Grant no. C9-I8-28/FC 760107/2023). The authors are thankful for a Project-Related Personal Exchange of the German Academic Exchange Service (Grant no. 57657161).References
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- G. Goti, K. Manal, J. Sivaguru and L. Dell'Amico, Nat. Chem., 2024, 16, 684–692 CrossRef CAS.
- Y.-M. Yang, Z.-Y. Li, J.-Y. Zhang, Y. Lu, S.-Q. Guo, Q. Zhao, X. Wang, Z.-J. Yong, H. Li, J.-P. Ma, Y. Kuroiwa, C. Moriyoshi, L.-L. Hu, L.-Y. Zhang, L.-R. Zheng and H.-T. Sun, Light: Sci. Appl., 2018, 7, 88 CrossRef PubMed.
- U. Kumar, C. R. Fox, E. Kolanthai, C. J. Neal, K. Kedarinath, Y. Fu, E. Marcelo, B. Babu, G. D. Parks and S. Seal, ACS Appl. Mater. Interfaces, 2022, 14, 40659–40673 CrossRef CAS.
- L. G. Leanse, S. Marasini, C. dos Anjos and T. Dai, Antibiotics, 2023, 12, 1437 CrossRef CAS PubMed.
- J. Itoh and Y. Itoh, Front. Biosci.-Scholar, 2022, 14, 27 CrossRef CAS.
- X. Wang, Y. Chen, F. Liu and Z. Pan, Nat. Commun., 2020, 11, 2040 CrossRef CAS.
- J. Shao, X. Liang, Y. Lin, Q. Shen, J. Ren and J. Han, Opt Laser. Technol., 2024, 176, 110974 CrossRef.
- D. Basting and U. Stamm, Z. Phys. Chem., 2001, 215, 1575 CAS.
- M. Kneissl, T.-Y. Seong, J. Han and H. Amano, Nat. Photonics, 2019, 13, 233–244 CrossRef CAS.
- C. J. Zollner, S. P. DenBaars, J. S. Speck and S. Nakamura, Semicond. Sci. Technol., 2021, 36, 123001 CrossRef CAS.
- R. Xu, Q. Kang, Y. Zhang, X. Zhang and Z. Zhang, Micromachines, 2023, 14, 844 CrossRef.
- Y. Yao, H. Li, M. Wang, P. Li, M. Lam, M. Iza, J. S. Speck, S. P. DenBaars and S. Nakamura, Opt. Express, 2023, 31, 28649 CrossRef CAS PubMed.
- X. Zhao, F. Liu, T. Shi, H. Wu, L. Zhang, J. Zhang, X. jun Wang and Y. Liu, Adv. Photonics Res., 2022, 3, 2200106 CrossRef CAS.
- C. M. S. Jones, A. Gakamsky and J. Marques-Hueso, Sci. Technol. Adv. Mater., 2021, 22, 810–848 CrossRef.
- G. B. Nair, H. Swart and S. Dhoble, Prog. Mater. Sci., 2020, 109, 100622 CrossRef CAS.
- F. Auzel, Chem. Rev., 2003, 104, 139–174 CrossRef PubMed.
- T. J. B. Zähringer, J. A. Moghtader, M.-S. Bertrams, B. Roy, M. Uji, N. Yanai and C. Kerzig, Angew. Chem., Int. Ed., 2023, 62, e202215340 CrossRef.
- M. Uji, N. Harada, N. Kimizuka, M. Saigo, K. Miyata, K. Onda and N. Yanai, J. Mater. Chem. C, 2022, 10, 4558–4562 RSC.
- T. Kashino, R. Haruki, M. Uji, N. Harada, M. Hosoyamada, N. Yanai and N. Kimizuka, ACS Appl. Mater. Interfaces, 2022, 14, 22771–22780 CrossRef CAS PubMed.
- H.-L. Wei, W. Zheng, X. Zhang, H. Suo, B. Chen, Y. Wang and F. Wang, Adv. Opt. Mater., 2022, 11, 2201716 CrossRef.
- Y. Shang, T. Chen, T. Ma, S. Hao, W. Lv, D. Jia and C. Yang, J. Rare Earths, 2022, 40, 687–695 CrossRef CAS.
- F. Li, L. Tu, Y. Zhang, D. Huang, X. Liu, X. Zhang, J. Du, R. Fan, C. Yang, K. W. Kraämer, J. Marques-Hueso and G. Chen, Nat. Photonics, 2024, 18, 440–449 CrossRef CAS.
- B. Purohit, E. Jeanneau, Y. Guyot, D. Amans, B. Mahler, M.-F. Joubert, C. Dujardin, G. Ledoux and S. Mishra, ACS Appl. Nano Mater., 2023, 6, 2310–2326 CrossRef CAS.
- L. Rößmann, F. Schröder and T. Jüstel, J. Lumin., 2023, 263, 120033 Search PubMed.
- G. Flizikowski, V. Zanuto, A. Novatski, L. Nunes, L. Malacarne, M. Baesso and N. Astrath, J. Lumin., 2018, 202, 27–31 CrossRef CAS.
- Y. Sun, Y. Wang, C. Hu, X. Zhou, J. Hao, W. Li and H. Li, Materials, 2022, 15, 7680 Search PubMed.
- D. Yu, H. Li, D. Zhang, Q. Zhang, A. Meijerink and M. Suta, Light:Sci. Appl., 2021, 10, 236 CrossRef CAS.
- N. Rebrova, P. Zdeb, K. Lemański, B. Macalik, O. Bezkrovnyi and P. J. Dereń, Inorg. Chem., 2024, 63, 3028–3036 CrossRef CAS PubMed.
- Z. Wang and A. Meijerink, J. Phys. Chem. C, 2018, 122, 26298–26306 Search PubMed.
- S. Wang, C. Hua, L. Wang, C. Wang, L. Liu, J. Ren and J. Zhang, Adv. Opt. Mater., 2023, 12, 2302086 CrossRef.
- M. Li, Q. Shi, K. V. Ivanovskikh, J. Qiao, L. Wang, H. Guo, P. Huang, S. A. Kiselev and V. A. Pustovarov, J. Lumin., 2023, 263, 120134 CrossRef CAS.
- I. Romet, E. Tichy-Racs, K. Lengyel, E. Feldbach, L. Kovacs, G. Corradi, K. Chernenko, M. Kirm, D. Spassky and V. Nagirnyi, J. Lumin., 2024, 265, 120216 Search PubMed.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077–2084 CrossRef CAS.
- S. Hu, C. Gu, S. Lu, Y. Hong, Q. Wu, M. Fu, P. Xiong and Y. Wang, Dyes Pigm., 2023, 215, 111216 CrossRef CAS.
- J. Qiao, S. Jia, S. He, J. Liu and Y. Ma, J. Mater. Sci.:Mater. Electron., 2023, 34, 1127 CrossRef CAS.
- F. Ruegenberg, A. García-Fuente, M. Seibald, D. Baumann, S. Peschke, W. Urland, A. Meijerink, H. Huppertz and M. Suta, Adv. Opt. Mater., 2021, 9, 2101643 Search PubMed.
- K. M. Rießbeck, D. S. Wimmer, M. Seibald, D. Baumann, K. Wurst, G. Heymann and H. Huppertz, Dalton Trans., 2023, 52, 4900–4910 RSC.
- K. Mizoi, Y. Fujimoto, D. Nakauchi, M. Koshimizu, T. Yanagida and K. Asai, J. Lumin., 2022, 245, 118797 Search PubMed.
- J. Zhang, H. Song, Y. Zhang, X. Wu, B. Li and J. Yu, J. Lumin., 2022, 242, 118611 CrossRef CAS.
- B. Bendel and M. Suta, J. Mater. Chem. C, 2022, 10, 13805–13814 RSC.
- C. van Aarle, K. W. Krämer and P. Dorenbos, J. Lumin., 2024, 266, 120329 CrossRef CAS.
- L. van Pieterson, M. F. Reid, R. T. Wegh, S. Soverna and A. Meijerink, Phys. Rev. B:Condens. Matter Mater. Phys., 2002, 65, 045113 Search PubMed.
- L. van Pieterson, M. F. Reid, G. W. Burdick and A. Meijerink, Phys. Rev. B:Condens. Matter Mater. Phys., 2002, 65, 045114 Search PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer Berlin Heidelberg, 1994 Search PubMed.
- K. V. Ivanovskikh, J. M. Ogiegło, A. Zych, C. R. Ronda and A. Meijerink, ECS J. Solid State Sci. Technol., 2012, 2, R3148–R3152 CrossRef.
- Y. Du, Z. Jin, Z. Li, T. Sun, H. Meng, X. Jiang, Y. Wang, D. Peng, J. Li, A. Wang, H. Zou, F. Rao, F. Wang and X. Chen, Adv. Opt. Mater., 2024, 12, 2400971 CrossRef CAS.
- G. B. Nair, S. Tamboli, S. J. Dhoble and H. C. Swart, ACS Appl. Nano Mater., 2023, 6, 15255–15265 CrossRef CAS.
- S. Zhou, K. Deng, X. Wei, G. Jiang, C. Duan, Y. Chen and M. Yin, Opt. Commun., 2013, 291, 138–142 CrossRef CAS.
- C. Mi, J. Wu, Y. Yang, B. Han and J. Wei, Sci. Rep., 2016, 6, 22545 CrossRef CAS PubMed.
- P. S. Solanki, S. Balabhadra, M. F. Reid and J.-P. R. Wells, J. Appl. Phys., 2023, 133, 035104 CrossRef CAS.
- I. P. Machado, J. de Wit, A. J. van Bunningen, C. C. Pedroso, L. C. Rodrigues, H. F. Brito and A. Meijerink, J. Alloys Compd., 2023, 942, 169083 CrossRef CAS.
- R. B. Anderson, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. Lett., 2013, 5, 36–42 CrossRef.
- M. Kaiser, C. Würth, M. Kraft, I. Hyppänen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS PubMed.
- C. Würth, B. Grauel, M. Pons, F. Frenzel, P. Rissiek, K. Rücker, M. Haase and U. Resch-Genger, Nano Res., 2022, 15, 9639–9646 CrossRef.
- H. Wu, Z. Hao, L. Zhang, X. Zhang, Y. Xiao, G.-H. Pan, H. Wu, Y. Luo, H. Zhao and J. Zhang, J. Phys. Chem. C, 2018, 122, 9611–9618 CrossRef CAS.
- C. Koepke, K. Wisniewski, M. Żelechower and E. Czerska, J. Lumin., 2018, 204, 278–283 CrossRef CAS.
- F. Schröder, P. Pues, D. Enseling and T. Jüstel, Luminescence, 2023, 38, 702–708 CrossRef PubMed.
- Y. Kitagawa, H. Nakamura and K. Shinozaki, J. Mater. Chem. C, 2024, 12, 18865–18876 RSC.
- P. Zdeb, N. Rebrova and P. J. Dereń, J. Phys. Chem. Lett., 2024, 15, 9356–9360 CrossRef CAS.
- N. Rebrova, P. Zdeb and P. J. Dereń, J. Phys. Chem. C, 2024, 128, 9090–9098 CrossRef CAS.
- K. Lemański, O. Bezkrovna, N. Rebrova, R. Lisiecki, P. Zdeb and P. J. Dereń, Molecules, 2024, 29, 2084 CrossRef PubMed.
- O. Bezkrovna, R. Lisiecki, B. Macalik and P. J. Dereń, Materials, 2024, 17, 1771 CrossRef CAS PubMed.
- N. Rebrova, A. Grippa, P. Zdeb and P. J. Dereń, Scr. Mater., 2025, 255, 116395 CrossRef CAS.
- J. Sommerdijk, A. Bril and A. de Jager, J. Lumin., 1974, 8, 341–343 CrossRef CAS.
- W. Piper, J. DeLuca and F. Ham, J. Lumin., 1974, 8, 344–348 CrossRef CAS.
- D. Wang, S. Huang, F. You, S. Qi, Y. Fu, G. Zhang, J. Xu and Y. Huang, J. Lumin., 2007, 122–123, 450–452 Search PubMed.
- B. Herden, J. Nordmann, R. Komban, M. Haase and T. Jüstel, Opt. Mater., 2013, 35, 2062–2067 CrossRef CAS.
- V. Naresh and B. S. Ham, J. Alloys Compd., 2016, 664, 321–330 CrossRef CAS.
- R. Naccache, F. Vetrone, A. Speghini, M. Bettinelli and J. A. Capobianco, J. Phys. Chem. C, 2008, 112, 7750–7756 CrossRef CAS.
- J. Collins, M. Geen, M. Bettinelli and B. Di Bartolo, J. Lumin., 2012, 132, 2626–2633 CrossRef CAS.
- C. De Mello Donegá, A. Meijerink and G. Blasse, J. Phys. Chem. Solids, 1995, 56, 673–685 CrossRef.
- E. L. Cates, A. P. Wilkinson and J.-H. Kim, J. Lumin., 2015, 160, 202–209 CrossRef CAS.
- E. L. Cates and F. Li, RSC Adv., 2016, 6, 22791–22796 RSC.
- Q. Shi, J. Cui, F. You, S. Huang, H. Peng and Y. Huang, J. Lumin., 2019, 213, 489–493 CrossRef CAS.
- X. Xu, Z. Xiao, Y. Wang, Y. Yan, J. Shen, Y. Nie, W. You, D. Wu, L. Han and F. Lai, Opt. Mater., 2022, 134, 113191 CrossRef CAS.
- C. L. Sun, J. F. Li, C. H. Hu, H. M. Jiang and Z. K. Jiang, Eur. Phys. J. D, 2006, 39, 303–306 CrossRef CAS.
- E. L. Cates, M. Cho and J.-H. Kim, Environ. Sci. Technol., 2011, 45, 3680–3686 CrossRef CAS.
- F. Lai, X. Xu, J. Shen, Y. Wang, Y. Yan, Y. Nie, W. You, D. Wu, L. Han and Z. Xiao, Silicon, 2022, 15, 1913–1923 CrossRef.
- P. Pues, M. Laube, S. Fischer, F. Schröder, S. Schwung, D. Rytz, T. Fiehler, U. Wittrock and T. Jüstel, J. Lumin., 2021, 234, 117987 CrossRef CAS.
- F. Schröder and T. Jüstel, Opt. Mater.:X, 2021, 12, 100117 Search PubMed.
- M. Laroche, M. Bettinelli, S. Girard and R. Moncorgé, Chem. Phys. Lett., 1999, 311, 167–172 CrossRef CAS.
- W. W. Schmahl, I. P. Swainson, M. T. Dove and A. Graeme-Barber, Z. Kristallogr., 1992, 201, 125–145 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Lazarowska, S. Mahlik, M. Krosnicki, M. Grinberg and M. Malinowski, J. Phys.:Condens. Matter, 2012, 24, 115502 CrossRef CAS PubMed.
- M. Y. A. Yagoub, H. C. Swart, P. Bergman and E. Coetsee, AIP Adv., 2016, 6, 025204 CrossRef.
- M. Runowski, P. Woźny, I. R. Martín, V. Lavín and S. Lis, J. Lumin., 2019, 214, 116571 CrossRef CAS.
- F. B. Xiong, X. Luo, W. B. Yang, Y. Yang, H. F. Lin, X. G. Meng, E. Ma and W. Z. Zhu, J. Mater. Sci.:Mater. Electron., 2022, 33, 2619–2630 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef CAS.
- P. Boutinaud, R. Mahiou, E. Cavalli and M. Bettinelli, Chem. Phys. Lett., 2006, 418, 185–188 CrossRef CAS.
- P. Boutinaud, E. Cavalli and M. Bettinelli, J. Phys.: Condens. Matter, 2007, 19, 386230 CrossRef.
- Z. Barandiarán, M. Bettinelli and L. Seijo, J. Phys. Chem. Lett., 2017, 8, 3095–3100 CrossRef PubMed.
- F. You, S. Huang, C. Meng, D. Wang, J. Xu, Y. Huang and G. Zhang, J. Lumin., 2007, 122–123, 58–61 CrossRef CAS.
- A. Srivastava, J. Lumin., 2016, 169, 445–449 CrossRef CAS.
- M. Nikl, H. Ogino, A. Yoshikawa, E. Mihokova, J. Pejchal, A. Beitlerova, A. Novoselov and T. Fukuda, Chem. Phys. Lett., 2005, 410, 218–221 CrossRef CAS.
- K. Ivanovskikh, A. Meijerink, F. Piccinelli, A. Speghini, E. Zinin, C. Ronda and M. Bettinelli, J. Lumin., 2010, 130, 893–901 CrossRef CAS.
- S. Mahlik, M. Malinowski and M. Grinberg, Opt. Mater., 2011, 34, 164–168 CrossRef CAS.
- J. Mooney and P. Kambhampati, J. Phys. Chem. Lett., 2013, 4, 3316–3318 CrossRef CAS.
- J. Pejchal, M. Nikl, E. Mihokova, A. Novoselov, A. Yoshikawa and R. Williams, J. Lumin., 2009, 129, 1857–1861 CrossRef CAS.
- K. Fiaczyk, S. Omagari, A. Meijerink and E. Zych, J. Lumin., 2018, 198, 163–170 CrossRef CAS.
- A. Zych, M. de Lange, C. de Mello Donegá and A. Meijerink, J. Appl. Phys., 2012, 112, 013536 CrossRef.
- P. Dorenbos, IEEE Trans. Nucl. Sci., 2010, 57, 1162–1167 CAS.
- G. Blasse, J. van Vliet, J. Verwey, R. Hoogendam and M. Wiegel, J. Phys. Chem. Solids, 1989, 50, 583–585 CrossRef CAS.
- P. A. Tanner, C. S. K. Mak, M. D. Faucher, W. M. Kwok, D. L. Phillips and V. Mikhailik, Phys. Rev. B:Condens. Matter Mater. Phys., 2003, 67, 115102 CrossRef.
- J. Pejchal, M. Nikl, E. Mihóková, J. A. Mareš, A. Yoshikawa, H. Ogino, K. M. Schillemat, A. Krasnikov, A. Vedda, K. Nejezchleb and V. Múčka, J. Phys. D: Appl. Phys., 2009, 42, 055117 CrossRef.
- A. de Vries and G. Blasse, Mater. Res. Bull., 1986, 21, 683–694 CrossRef CAS.
- A. Srivastava, M. Jennings and J. Collins, Opt. Mater., 2012, 34, 1347–1352 CrossRef CAS.
- B. Canny and D. Curie, in Non-Radiative Relaxation of Solids: Different Pathways to the Ground State, ed. B. Di Bartolo, Springer US, 1991, vol. 249, pp. 1–28 Search PubMed.
- A. Meijerink and G. Blasse, J. Lumin., 1990, 47, 1–5 CrossRef CAS.
- C. Duan, A. Meijerink, R. Reeves and M. Reid, J. Alloys Compd., 2006, 408–412, 784–787 CrossRef CAS.
- S. Zhang, Y. Huang and H. J. Seo, Opt. Mater., 2010, 32, 1545–1548 CrossRef CAS.
- S. Huang, F. You, C. Meng, D. Wang, Y. Tao and G. Zhang, J. Lumin., 2007, 122–123, 25–27 CrossRef CAS.
- M. Krośnicki, A. Kędziorski, L. Seijo and Z. Barandiarán, J. Phys. Chem. A, 2013, 118, 358–368 CrossRef.
- M. Suta, Opt. Mater.:X, 2022, 16, 100195 CAS.
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B:Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS.
- B. Maksimov, V. Ilyukhin, Y. A. Kharitonov and N. Belov, Kristallografiya, 1970, 15, 926–932 CAS.
- D. R. Gamelin and H. U. Güdel, J. Phys. Chem. B, 2000, 104, 10222–10234 CrossRef CAS.
- D. R. Gamelin and H. U. Güdel, J. Am. Chem. Soc., 1998, 120, 12143–12144 CrossRef CAS.
- D. R. Gamelin and H. U. Güdel, Inorg. Chem., 1999, 38, 5154–5164 CrossRef CAS.
- L. Y. Kuritzky, A. C. Espenlaub, B. P. Yonkee, C. D. Pynn, S. P. DenBaars, S. Nakamura, C. Weisbuch and J. S. Speck, Opt. Express, 2017, 25, 30696 Search PubMed.
- S. L. Cates, E. L. Cates, M. Cho and J.-H. Kim, Environ. Sci. Technol., 2014, 140205070115003 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01862e |
| This journal is © The Royal Society of Chemistry 2025 |