
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAffinity peptide ligands: new tools for chasing non-canonical N-phosphoproteome†
He
Wang‡
 ab,
Xiaoyu
Zhang‡
a,
Dongdong
Wang
a,
Qianqian
Jiang
ab,
Yue
Sun
a,
Baofeng
Zhao
a,
Zhen
Liang
a,
Guangyan
Qing
*a,
Bo
Jiang
*a,
Lihua
Zhang
a and
Yukui
Zhang
a
ab,
Xiaoyu
Zhang‡
a,
Dongdong
Wang
a,
Qianqian
Jiang
ab,
Yue
Sun
a,
Baofeng
Zhao
a,
Zhen
Liang
a,
Guangyan
Qing
*a,
Bo
Jiang
*a,
Lihua
Zhang
a and
Yukui
Zhang
a
aState Key Laboratory of Medical Proteomics, National Chromatographic R&A Center, CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: qinggy@dicp.ac.cn; jiangbo@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th April 2025
Abstract
The enrichment of protein N-phosphorylation encounters substantial challenges due to the inherent instability of the N–P bond, severely impeding the manifestation of its biological functions. Traditional enrichment methods often rely on antibodies, organic solvents and metal ion interactions, which are limited by lack of universality, potential degradation of sample integrity, or reduced selectivity for N-phosphorylation. To overcome these challenges, we innovatively capitalized phage display technology to identify affinity peptides that specifically bind to the N–PO3 group. By functionalizing magnetic nanoparticles with the affinity peptide, we developed a novel, organic solvent- and metal-free enrichment strategy that enhanced both the selectivity and efficiency for all three types of N-phosphopeptide capture under neutral conditions, ensuring superior preservation of sample integrity and allowing more accurate proteomic analysis. This strategy has demonstrated robust enrichment capabilities for both prokaryotic and eukaryotic samples. In HeLa cells, 1995 novel N-phosphorylation sites were identified, representing a substantial increase of 2- to 5-fold in detection depth over previous approaches and significantly expanding the scale of the N-phosphoproteome database. Additionally, it was discovered that N-phosphorylation modification was highly concentrated in the nucleus. By integrating the nuclear isolation technique, 1296 N-phosphorylation sites were identified for the first time, offering new leads for uncovering the functions of N-phosphorylation in nuclear proteins. Finally, in conjunction with the quantitative proteomics method, the dynamic changes in N-phosphorylation modification during the progression of Alzheimer's disease were investigated, providing fresh perspectives on the research of AD pathogenesis. Overall, this work not only presents a new approach for efficient enrichment of N-phosphopeptides but also advances the functional study of N-phosphorylated proteins in physiological and pathological processes.
Introduction
Reversible protein phosphorylation, one of the most extensively studied post-translational modifications (PTMs), is intertwined with nearly all life processes. Phosphorylation acts as a critically important regulatory switch that exerts a determinant influence on the implementation of a wide array of fundamental biological functions.1–3 Nine phosphorylated amino acid residues are currently known: serine, threonine, tyrosine, histidine, lysine, arginine, aspartic acid, glutamic acid, and cysteine. These residues exhibit distinct isoelectric points and reactivities, endowing phosphorylation with diverse properties.4 For example, O-phosphorylation, taking place on the hydroxyl groups of the side chains of serine (pSer), threonine (pThr), and tyrosine (pTyr), has been the focus of extensive research owing to its remarkable stability. These O-phosphorylated proteins have emerged as crucial drug targets, spurred by the remarkable success of several protein tyrosine kinase inhibitors in cancer treatment.5At the same time, N-phosphorylation, which occurs on the side-chain amino groups of histidine (pHis), lysine (pLys), and arginine (pArg), has drawn increasing attention.6 As advances in biotechnology continue, the functions of N-phosphorylation are being gradually revealed. For instance, pHis was recognized as an intermediate within the two-component regulatory system in prokaryotes.7–9 Two mammalian phosphohistidine kinases, namely NME1 and NME2, and three phosphohistidine phosphatases, PHPT1, LHPP, and PGAM5, were successfully verified.10–13 NME2 and PHPT1 were demonstrated to regulate the immune system via the phosphorylation and dephosphorylation of ion-channel proteins.14–16 Additionally, in Bacillus subtilis, McsB and YwIE were confirmed to function as arginine kinase and phosphatase, respectively. These two proteins played crucial roles in regulating the stress response and protein degradation processes.17–19 Despite these advances, however, the function of protein N-phosphorylation remains insufficient.
Compared to O-phosphorylation, this gap in research is primarily due to the inherent instability of N-phosphorylation. The most notable chemical characteristic of N-phosphorylation is the high-energy phosphoramide bond (N–P), which leads to its lability in acidic environments and lower thermodynamic stability. For instance, pHis is rapidly hydrolyzed into phosphoric acid and histidine with a half-life of less than 30 s in 1 M HCl.20 Conventional enrichment methods, such as immobilized metal ion chromatography (IMAC), metal oxide affinity chromatography (MOAC), and strong cation exchange chromatography (SCX), demand acidic conditions. As a result, they are unsuitable for N-phosphorylation enrichment. Although polyclonal and even monoclonal pHis antibodies have been developed to capture pHis peptides, they exhibit cross reactivity and are ineffective in enriching pArg and pLys peptides.21–25 An affinity ligand capable of recognizing phosphate is considered an alternative approach.26,27 Fe3+-IMAC can be used for pHis enrichment under mild acidic conditions.28 Meanwhile, bis(zinc(II)-dipicolylamine) has been utilized as an affinity ligand for N-phosphopeptide enrichment under neutral conditions.29–31 Nevertheless, the use of organic solvent in enrichment buffer and the interaction between high-abundance negatively charged peptides and metal ions decrease the sample integrity and enrichment selectivity. Thus, there is an urgent necessity to develop novel affinity ligands for the selective enrichment of N-phosphopeptides bypassing the above-mentioned unfavorable factors.
Instead of metal ion ligands leading to contamination of other molecules due to coordination and electrostatic interactions, peptide ligands have shown more specific target-binding capabilities.32,33 Phage display technology provides an efficient platform to screen for peptide ligands with high affinity for the N–PO3 group. However, screening for a high-affinity ligand targeting such a small, compact structure poses significant challenges. Through rational target design and optimization of the screening process, we successfully identified peptides with exceptional specificity and affinity for the N–PO3 group, overcoming the inherent difficulties of selecting ligands for small, structurally constrained targets. Furthermore, the screening was carried out under mild, neutral conditions, preserving the stability of the N–P bond. A negative screening strategy was employed to further enhance specificity, resulting in a substantial improvement in selectivity. Subsequently, the peptide was immobilized on Sepharose magnetic beads, which functioned as a potent tool for the facile enrichment of N-phosphopeptides in neutral buffer solutions. This enrichment approach circumvented the detrimental effects of organic solvents on the structure of N-phosphopeptides and the surface properties of materials. As a result, it ensured highly efficient enrichment and more accurate analysis in proteomics.
The newly developed enrichment strategy enabled comprehensive coverage analysis of the N-phosphoproteome across a wide spectrum ranging from prokaryotes to mammals with high efficiency and specificity. In conclusion, this study introduces an organic solvent- and metal-free N-phosphopeptide enrichment method based on affinity peptides for landscape mapping of non-canonical N-phosphorylation, substantially advancing our understanding of N-phosphorylation in both physiological and pathological contexts, with particular implications for neurodegenerative diseases. Our research made substantial contributions to the development of atypical N-phosphorylated proteomics, significantly propelling its forward progress.
Results and discussion
Screening and characterization of affinity peptide ligands
Phage display stands out as a powerful tool for the direct discovery of high-affinity peptide ligands targeting a wide range of substrates.34,35 It offers a completely blind and high-throughput strategy for selecting ligands from vast and diverse peptide libraries via top-down affinity selection.36 Through iterative biopanning, it enables the screening of optimal peptide ligands without prior knowledge of their molecular characteristics, presenting an opportunity for ligand discovery in N-phosphorylation studies.As shown in Fig. 1A and B, we employed M13 phage dodecapeptide libraries to conduct a comprehensive biopanning procedure, incorporating BSA reverse screening and four rounds of affinity screening, to identify affinity peptides. In our experiment, BSA served the purpose of minimizing nonspecific binding of phage clones and N-phosphopeptide pHFF (Fig. S1A†) was employed for subsequent screening. pHFF could form hydrophobic assemblies that were immobilized on the well plate, exposing the hydrophilic N–PO3 group for specific interactions during screening. Notably, to eliminate peptides with off-target binding to imidazole or histidine residues, we implemented a negative screening step with a non-phosphorylated HFF peptide before proceeding to each affinity screening round. This HFF counter-selection effectively removed phage clones that bound to the backbone structure (HFF) rather than the critical N–PO3 group, thereby dramatically enhancing the specificity of the selected peptides for phospho-specific interactions. The biopanning conditions were progressively intensified in the last two rounds to ensure stringent selection. The increased enrichment efficiency observed after each round of affinity screening indicated the successful selection of high-affinity phages (Table S1†). Subsequently, we randomly selected 15 phage clones from the final round for sequencing analysis.
 | ||
| Fig. 1 Selection and characterization of the affinity peptide for N-phosphorylation. (A) Target design for phage display screening. The N-phosphopeptide pHFF (Fig. S1A†) was used for affinity screening, with HFF employed for negative screening. The peptides consist of two components: a hydrophilic segment containing either histidine or phosphohistidine, followed by a hydrophobic assembly designed to immobilize the peptide onto the well plate. This setup exposes the target group, ensuring its accessibility for efficient screening. (B) Schematic of the phage display process employed to obtain the affinity peptide. (C) The sequences and relative occurrence frequencies of the four identified peptides. (D) Assessment of affinity between identified peptides and the N-phosphopeptide via phage ELISA. (E) ITC raw data and the corresponding fitting curves of P-1 upon the titration of the phosphorylated peptide (TSpHYSIMAR) in HEPES (20 mM, pH = 7.2) at 25 °C. (F) Molecular docking of P-1 and the 3-pHis standard peptide (TSpHYSIMAR). The potential binding model (left) between the 3-pHis standard peptide (shown as sticks) and P-1 (shown as blue lines and surface) was demonstrated using PyMOL after molecular docking with AutoDock. The right panel shows the extracted binding amino acids between pHis and P-1 from the left panel. Binding interactions are displayed as green dashed lines. | ||
The sequences of the four identified peptides, along with their relative frequencies of occurrence, are presented in Fig. 1C. Notably, after conducting BLAST analysis on NCBI, no homologous sequences were matched, indicating that these four peptides were all reported for the first time. Moreover, during the analysis, we observed overlaps between these sequences and certain phosphatase segments, further suggesting their potential recognition for phosphopeptides. Next, we evaluated affinity of identified peptides to pHFF using a phage enzyme-linked immunosorbent assay (ELISA). As depicted in Fig. 1D, P-1 exhibited significantly stronger affinity. All peptides displayed markedly higher binding affinities than the blank control, confirming the effectiveness of the biopanning procedure. Taken together, P-1 emerged as a promising candidate for N-phosphopeptides enrichment. We synthesized TSpHYSIMAR (Fig. S1B–D†) to validate the interaction. We determined the interaction by isothermal titration calorimetry37 and the result confirmed the high binding affinity between P-1 and TSpHYSIMAR with the dissociation constant (Kd) being 8.5 μM, according to the fitting curve of exothermic data (Fig. 1E). Furthermore, molecular docking simulations provided detailed insights into the interaction. P-1 and the N-phosphopeptide formed a closely-knit complex, with multiple hydrogen bonds established between the various amino acids of the N-phosphopeptide and P-1 (Fig. 1F and S2A†), which indicated strong binding affinity. In contrast, we also conducted molecular docking between P-1 and the non-phosphorylated peptide. Interestingly, there was no evident interaction between them (Fig. S2B†), further validating the specificity of the affinity peptide.
Development of an affinity peptide-based enrichment method for N-phosphoproteome
To attain a specific enrichment of N-phosphorylated targets from complex mixtures, we employed carboxyl-functionalized agarose magnetic beads (with a diameter of 10 μm) to conjugate P-1 onto their surface. Surface-functionalized magnetic beads exhibit remarkable surface activity and excellent non-specific adsorption resistance, which enables their extensive application in the separation field. Additionally, their magnetic properties facilitate quick and easy separation, significantly simplifying the sample preparation process. As shown in Fig. 2A, we successfully functionalized magnetic beads through the covalent coupling of the N-terminal amino groups of P-1 with the carboxyl groups on the bead surface, and the resulting product was denoted as MB@P1. Based on the thermogravimetric analysis (TGA) results presented in Fig. 2B, the content of P-1 was determined to be approximately 4.38 wt%. | ||
Fig. 2 Development and evaluation of the affinity peptide-based N-phosphopeptide enrichment strategy. (A) The synthesis process of P-1 functioned magnetic beads (MB@P1). (B) TGA curves and SEM images of MB@P1. (C) The relative quantitative elemental analysis of MB@P1 by XPS. (D) The magnetic hysteresis loop of magnetic beads with or without P-1. (E) Schematic of enrichment workflow for N-phosphopeptides using MB@P1. (F) MALDI-TOF/TOF spectra of TSpHYSIMAR from β-casein digests at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (m/m) before and (G) after enrichment using MB@P1. The blue rhombus, square and red asterisk represented ion peaks of the original, oxidized and N-phosphorylated peptide, respectively. :10 (m/m) before and (G) after enrichment using MB@P1. The blue rhombus, square and red asterisk represented ion peaks of the original, oxidized and N-phosphorylated peptide, respectively. | ||
Scanning electron microscopy (SEM) revealed a spherical morphology with a diameter of approximately 10 μm. Moreover, the surface exhibited a mesh-like cross-linked structure typical of agarose. The elemental composition of the beads was characterized by X-ray photoelectron spectroscopy (XPS) as well (Fig. 2C, Table S2†). The relative quantitative elemental analysis clearly demonstrated a remarkable increase in the nitrogen (N) element content after the P-1 modification. In the N 1s and C 1s spectra of MB@P1, the presence of the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond could be attributed to the amide bond between beads and P-1. Additionally, both the hysteresis loop analysis and the magnetic adsorption experiment demonstrated the successful preparation of MB@P1 and a rapid magnetic response (Fig. 2D and S3†). Indeed, these results offered robust evidence of the substantial alterations induced by the P-1 modification process, thus validating the successful preparation of the material.
O bond could be attributed to the amide bond between beads and P-1. Additionally, both the hysteresis loop analysis and the magnetic adsorption experiment demonstrated the successful preparation of MB@P1 and a rapid magnetic response (Fig. 2D and S3†). Indeed, these results offered robust evidence of the substantial alterations induced by the P-1 modification process, thus validating the successful preparation of the material.
The schematic of the enrichment workflow using MB@P1 is presented in Fig. 2E. Taking advantage of the fact that the affinity peptide screening was conducted in neutral buffer solutions, MB@P1 was capable of enriching N-phosphopeptides under the same conditions (20 mM HEPES, pH = 7.2) while maintaining high affinity. This enrichment mode effectively mitigated the impact of organic solvents on the structure of N-phosphopeptides and the surface properties of materials. Consequently, it contributed significantly to enhancing the enrichment selectivity and the identification coverage. After eliminating interfering peptides, the enriched N-phosphopeptides were eluted with ammonia solution. This was because alkaline conditions disrupted the strength of the hydrogen bonds between MB@P1 and N-phosphopeptides.
The enrichment capability was initially evaluated by capturing TSpHYSIMAR from β-casein digests (with a mass ratio of 1:10, Fig. 2F and G). Following the enrichment process, the majority of interfering peptides were eliminated, leaving N-phosphopeptides. This result was highly significant as it clearly demonstrated the selectivity of the method. By specifically targeting and retaining N-phosphopeptides while effectively removing other interfering components, it laid a solid foundation for in-depth analysis of N-phosphopeptides. Subsequently, to assess the sensitivity and reproducibility of our method, the target N-phosphopeptide was spiked into the HeLa cell digests at a proportion of 1/1000 or 1/10000 (m/m). After the enrichment procedure, the N-phosphopeptides were identified in both experiments, exhibiting identical MS/MS spectra and retention time (Fig. S4A and B†). This consistency in spectra and retention time indicated that the enrichment process was highly reliable. Even at extremely low spiking ratios, the successful identification of N-phosphopeptides highlighted the excellent sensitivity of our method, which could broaden the scope of applications, particularly in samples with inherently low abundances of N-phosphopeptides.
In addition, similar signal intensities were identified in three independent replicates as well (Fig. S4C†), demonstrating the high reproducibility of the strategy. This high reproducibility also provided strong support for the use of our enrichment strategy in large-scale N-phosphoproteome profiling studies. Motivated by these highly promising results, we carried out a comprehensive N-phosphoproteome analysis of both prokaryotes and eukaryotes. This comprehensive analysis not only facilitated a deeper understanding of the fundamental biological processes related to N-phosphorylation, but also had the potential to uncover regulatory mechanisms that were of great significance to diverse biological and medical research fields.
N-Phosphoproteome analysis of E. coli
The widespread presence and complexity of bacterial phosphorylation signals endow bacteria with a powerful regulatory ability over all aspects of their life processes. Among the diverse phosphorylation signaling mechanisms in bacteria, the bacterial two-component system (TCS) and the phosphotransferase system (PTS) were widely regarded as classic signaling mechanisms.7,38 Although significant progress was made, the specific functions of N-phosphorylated proteins in signaling pathways and the mechanisms governing this modification remained largely elusive. Moreover, other potential functions of N-phosphorylated proteins still awaited exploration. Therefore, we employed MB@P1 to enrich N-phosphopeptides from E. coli lysate, aiming to facilitate the functional study of bacterial N-phosphorylation.Overall, after removing the pLys/pArg peptides located at the C-terminus, a total of 308 N-phosphorylation sites corresponding to 232 proteins were identified. Some of these proteins were involved in TCSs and PTSs, which demonstrated high reliability of the method (Fig. 3A). Among these N-phosphorylation sites, 262 sites (localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5) were identified, including 98 pHis, 141 pLys and 23 pArg sites (Fig. S5A, Dataset S1†). In addition to known sites (pgm-pH147 and ppsA-pH421), a number of new sites were also identified by our strategy. Compared with the results obtained using SiO2@DpaZn, the number of identified sites increased significantly from 31 to 262 (Fig. 3B). This was attributed to the crucial advantages of using neutral buffers in the enrichment processes. The results demonstrated the advantages of the strategy in improving the identification coverage of N-phosphorylation sites.
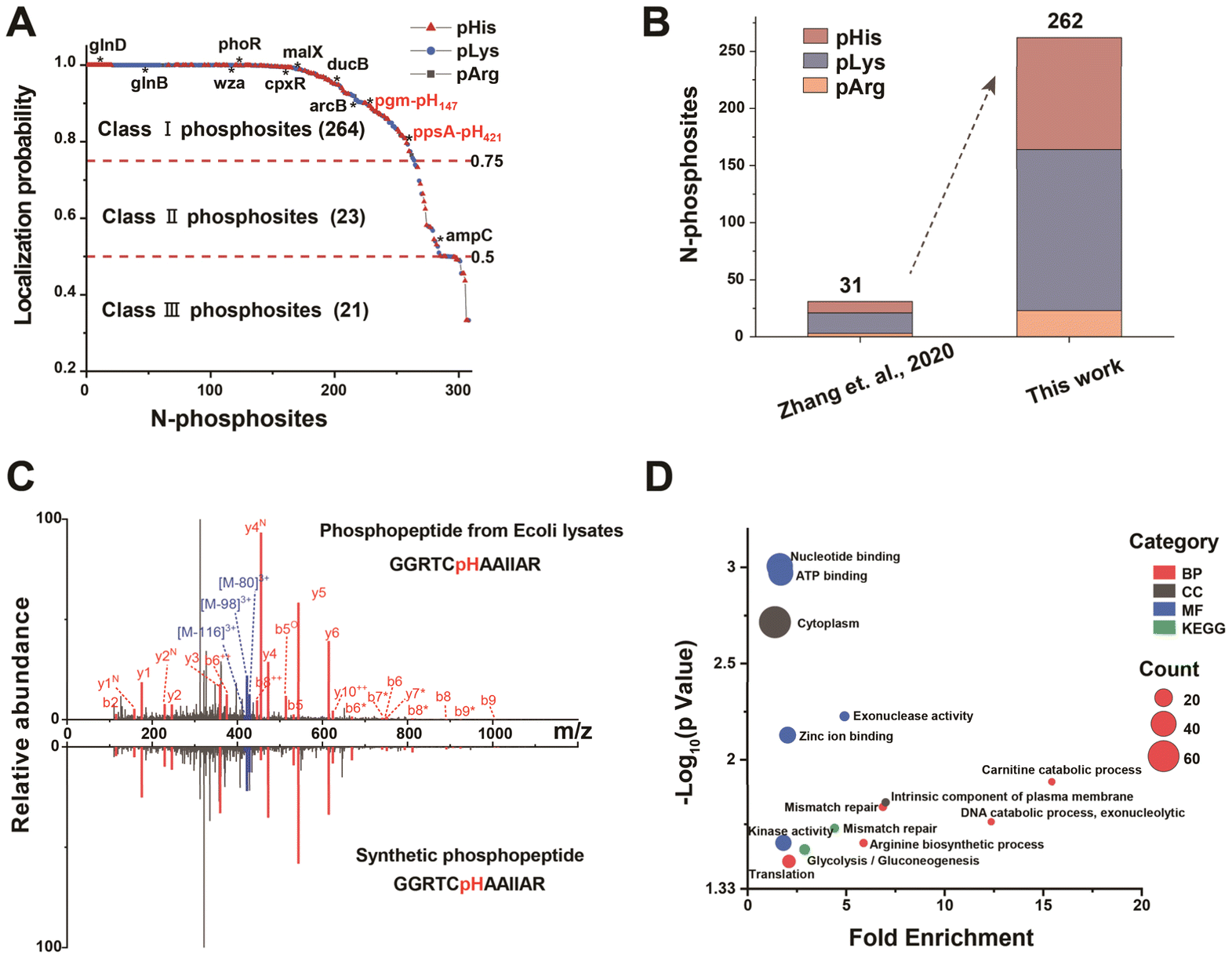 | ||
| Fig. 3 N-Phosphoproteome profiling in E. coli by the MB@P1 based enrichment strategy. (A) Distribution of localization probability scores for identified N-phosphorylation sites in E. coli. Class I, II, and III were cut off by scores ≥ 0.75, between 0.5 and 0.75, <0.5, respectively. The N-phosphoproteins involved in TCSs and PTSs were indicated by asterisks. (B) Comparison of identified N-phosphorylation sites in E. coli between our work and other methods (Zhang et al., 2020; the data of Zhang et al. were derived from database searching again using the same method applied in this work). (C) MS/MS spectra of the endogenous pHis peptide (GGRTCH(+79.98)AAIIAR) from ppsA in E. coli and that of the same synthetic phosphopeptide. The superscript symbols represent dephosphorylated (asterisks), deamination (N), and dehydrated (O) ions. (D) The main GO enrichment terms (biological process, cellular component, molecular function and KEGG pathways) of identified N-phosphoproteins in E. coli. | ||
To confirm the authenticity of the identified N-phosphorylation sites, N-phosphopeptides were synthesized for MS identification. The MS/MS spectrum of the synthetic peptide was compared with that obtained from in vivo peptides. For example, phosphoenolpyruvate synthase (ppsA) was reported as an enzyme responsible for catalyzing the phosphorylation of pyruvate to phosphoenolpyruvate, in which it was experimentally identified previously that there was N-phosphorylation at the 421 position of H.21 The MS/MS spectrum of the peptide from in vivo (GGRTCH(+79.98)AAIIAR) with a mass shift of +79.98 Da at the histidine residue was the same as that of the synthetic peptide with a phosphate group on histidine (Fig. 3C). Moreover, triple neutral loss peaks of phosphate were found. On the basis of highly similar fragmentation, the nearly same retention time of the in vivo peptide and the synthetic peptide further demonstrated the authenticity of the identified site (Fig. S5B†).
The subsequent gene ontology (GO) enrichment analysis showed that N-phosphoproteins were intimately linked to regulation of gene expression and energy metabolism (Fig. 3D), which was also supported by protein functions such as nucleic acid metabolism, metabolite interconversion and transport (Fig. S5C†). The research significantly expanded the database of the E. coli N-phosphoproteome, thereby verifying the widespread presence of N-phosphorylation in prokaryotes. This expansion not only provided a more comprehensive resource for understanding the phosphorylation events in E. coli but also offered valuable insights into the potential regulatory mechanisms associated with N-phosphorylation across different prokaryotic species.
N-Phosphoproteome profiling in eukaryotes
A body of research has indicated that N-phosphorylation exerts a regulatory function in tumor invasion and neuroinflammation within mammalian organisms.39 Deep coverage of mammalian N-phosphorylation, regarded as a fundamental prerequisite, is crucial for elucidating its potential function.In the case of HeLa cells, the number of N-phosphorylation sites identified by our method increased to 2876 (Fig. S6†) and the number of sites with localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5 was 2307. These included 829 pHis, 904 pLys and 574 pArg, corresponding to 2038 peptides and 1482 proteins, respectively (Fig. 4A, Dataset S1). We discovered that the strategy exhibited outstanding enrichment specificity for N-phosphorylation. Compared to non-phosphoproteins, phosphoproteins yielded higher scores, counts of peptides and MS/MS spectra (Fig. S7†). In addition, the number of N-phosphorylation sites identified by this method exceeded that of existing methods by 2- to 5-fold (Fig. S8A†).29,40,41 Moreover, more key functional proteins, including protein kinases, phosphatases, and transmembrane signal receptors, were detected (Fig. S8B†).
 | ||
| Fig. 4 MB@P1 based enrichment strategy enabled N-phosphoproteome profiling in HeLa cells. (A) The number of N-phosphorylation sites (localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5) and corresponding peptides and proteins enriched with MB@P1 in HeLa cells. (B) The overlap of identified N-phosphorylation sites between our work and published lists or databases (the data of Zhang et al. were derived from database searching again using the same method applied in this work). (C) Related biological processes for proteins with different types of N-phosphorylation sites (pHis, pLys or pArg). (D) Sequential characteristics of N-phosphorylation sites. The conserved motifs were detected with rmotifx. (E) The correlation scatter plots of LFQ intensity for N-phosphorylation sites in three LC-MS/MS replicates and biological replicates, to assess the quantitative reproducibility of the MB@P1 based enrichment strategy. | ||
This finding demonstrated the stronger enrichment capability of the affinity peptide and advantages of enrichment in neutral buffer solutions. However, even when the same human cell line was analyzed, there was a low overlap of sites among different enrichment methods (Fig. 4B and S8C†). This could be attributed to the high dynamic nature and complexity of N-phosphorylation,4,7 high false positives for identification results due to low abundance of N-phosphorylation,42 spectral mismatch and cross-talk of O-phosphorylation,43 and the various interactions in the enrichment methods. On the other hand, the work contributed 1995 new sites (Fig. 4B), significantly expanding the existing N-phosphorylation databases.44,45 Moreover, it was worth noting that the existing data processing approach involved simultaneously searching all raw files. Due to the need to balance the false discovery rate (FDR), this method resulted in a decrease in the number of N-phosphorylation sites. When we carried out separate searches of the raw files without considering O-phosphorylation as a variable modification, the number of identified sites increased to 5649. However, this situation had the potential to increase false positives. In any case, these sites needed to be verified through multiple means.
GO analysis of the 1482 identified N-phosphoproteins revealed a significant enrichment of biological processes including nucleic acid processing, cell activity regulation, organization, protein transport and phosphorylation (Fig. S9†). This discovery suggested that N-phosphorylation exerted vital functions in a diverse range of cellular processes, thus underscoring its extensive presence across mammalian species. Interestingly, different kinds of phosphorylation sites seemed to exhibit a distinct inclination in their functions. As shown in Fig. 4C, the proteins with pHis and pLys predominantly participated in the crucial process of DNA transcription regulation. On the other hand, proteins that contained only pLys were found to be closely related to chromatin remodeling and DNA repair. Furthermore, proteins with pArg had various functions in mRNA processing and the cell cycle. Overall, this differential distribution of functions among proteins with different N-phosphorylation sites underscores the intricate and precise nature of cellular regulation at the molecular level.
Furthermore, it was noted that a number of important kinase families and phosphatases underwent N-phosphorylation modification and were visualized in the interaction network with several known substrates (Fig. S10 and S11†). This result implied that well-characterized O-phosphorylation kinases or phosphatases could potentially undergo N-phosphorylation modification. Moreover, they might possess the enzymatic capabilities to N-phosphorylate or N-dephosphorylate their respective substrates. Motif analysis was subsequently carried out to assess the characteristics of the N-phosphoproteins. ±10 residue sequence windows were generated from all N-phosphorylation sites against the HeLa proteome background. The data demonstrated enrichment of serine, proline, and leucine residues in the vicinity of the N-phosphorylation sites (Fig. 4D and S12†). This suggested possible cross-talk between O-phosphorylation and N-phosphorylation. It was reported that the P–N bond could translocate to the P–COO bond and further to the P–O bond when kinases have a catalysis function.46,47 We speculated that the formation of the intrinsically unstable N–P bond might be the intermediate process of the stable O–P bond according to the possible translocation, thus leading to the significant enrichment of serine and glutamic acid residues in the proximity of N-phosphorylation sites. The results further confirm the possibility of N-phosphorylation taking place in O-phosphorylated kinases.
Finally, the remarkable quantitative reproducibility of our method was validated using the high Pearson correlation coefficients (PCCs) of label-free quantification (LFQ) intensities for the identified sites in triplicate (both biological and LC-MS replicates). These PCCs exceeded 0.81 (Fig. 4E), which effectively verified the feasibility of the quantitative analysis of the MB@P1-based enrichment strategy. Overall, the above results clearly demonstrated the outstanding enrichment performance of MB@P1, indicating the success of the screening strategy for the affinity peptide. This novel enrichment method has made a significant breakthrough in enhancing our understanding of the function of N-phosphorylation.
Comprehensive analysis of the N-phosphorylation landscape within the nucleus
Eukaryotic cells are highly compartmentalized. Protein functions are closely intertwined with their subcellular localization, owing to the distinct chemical microenvironments present in different subcellular compartments.48 In view of the localization of N-phosphorylation proteins in whole cells according to the above results (Fig. S13†), N-phosphorylation might be significantly enriched in the nucleus and played a critical role. The nucleus, being the control center of the cell, regulates various essential cellular processes. The significant enrichment of N-phosphorylation proteins in this region implied that they could be involved in crucial functions such as gene regulation, DNA replication, and repair. To verify our hypothesis, immunofluorescence was performed using an affinity peptide labeled with fluorescein isothiocyanate (FITC) (Fig. 5A). As shown in Fig. 5B and S14,† a substantial spatial overlap was observed between the fluorescent peptide and the commercial nuclear dye. This result indicated that a large amount of nuclear proteins tend to undergo extensive N-phosphorylation. To conduct a comprehensive exploration of the nuclear N-phosphoproteome, the nuclei were meticulously isolated using a separation kit. Subsequently, with the assistance of MB@P1, N-phosphorpeptides were efficiently enriched before being subjected to LC-MS/MS analysis. For the first time, a total of 1296 N-phosphorylation sites (localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5) including 455 pHis, 450 pLys and 391 pArg were identified from nuclei lysates (Fig. 5C, Dataset S1). Compared with the whole cell results, there was a significant increase in the proportion of pArg sites. This phenomenon could be attributed to the relatively high abundance of arginine residues in nuclear proteins. Furthermore, motif analysis revealed an enrichment of leucine residues around the N-phosphorylation sites, which was distinct from the results obtained from whole cells (Fig. S15†). These findings demonstrated the spatial specificity of N-phosphorylation. | ||
| Fig. 5 Subcellular N-phosphoproteome mapping by the MB@P1 based enrichment strategy. (A) The extensive occurrence of N-phosphorylation in the cell nucleus was demonstrated by immunofluorescence and LC-MS/MS analysis after enrichment. (B) The fluorescence images of HeLa cells incubated with P-1-FITC and commercial nuclear dye Hoechst 33342 and the intensity profile of images. Scale bar: 10 μm. (C) The number of N-phosphorylation sites (localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5) and corresponding peptides and proteins enriched with MB@P1 in nuclei obtained by the kit. (D) The main GO enrichment terms of identified N-phosphoproteins in the nucleus. | ||
GO analysis revealed that the identified proteins were distributed in various sub-compartments within the nucleus, where they performed binding functions. These proteins were mainly associated with processes such as DNA repair, transcriptional regulation, and nucleocytoplasmic transport (Fig. 5D). By focusing on the proteins related to DNA repair, which are vital for maintaining genomic integrity, stability and the normal functions of the entire cell, some important transcription factors were detected. These transcription factors featured N-phosphorylation sites that were seldom reported before (Fig. S16†). For example, the breast cancer type 2 susceptibility protein (BRCA2) played a crucial role in the DNA damage response and DNA repair through its function in homologous recombination, which was mainly regulated by phosphorylation.49 Multiple O-phosphorylation sites of BRCA2 were identified,50,51 yet research on N-phosphorylation sites of BRCA2 was extremely scarce. Our findings could offer some novel perspectives on the regulatory mechanism of the DNA repair process.
Based on the above results, our strategy enabled the large-scale identification of the N-phosphoproteome in the cell nucleus with high selectivity while circumventing the interference of other high-abundance proteins effectively, which imparts to it the potential of N-phosphoproteome plotting in other subcellular compartments.
Analysis of dynamic changes in the N-phosphoproteome of brain tissue in Alzheimer's disease mice
During the pathogenesis of Alzheimer's disease (AD), neurofibrillary tangles caused by hyperphosphorylation initially emerge in the parahippocampal gyrus at an early stage and subsequently spread to neocortical regions, resulting in neuronal damage and cognitive decline.52 Therefore, mapping the phosphoproteome of the cerebral cortex is of great significance for elucidating the pathology of AD. However, the N-phosphoproteome associated with AD has been scarcely reported. After successfully demonstrating the ubiquity of N-phosphorylation in mammals, we then employed the LFQ method with MB@P1 to define the differences in the N-phosphoproteome of the cerebral cortex and other brain regions between wild-type (WT) and PS19 (AD) mice. The tissues of the cerebral cortex and other brain regions were collected from 6-month-old WT and AD mice (n = 3) and then subjected to enrichment.As a result, a total of 1254 N-phosphorylation sites (with a localization probability of ≥0.75, a score of ≥40, and a score difference of ≥5, including 418 pHis, 441 pLys, and 395 pArg) were identified. These sites were assembled into 1051 peptides and 781 proteins (Fig. 6A, Dataset S2). Excitingly, 9 rarely reported N-phosphorylation sites of tau (from Homo sapiens, 1N4R isoform) were discovered. Notably, five of these sites exhibited an increase of more than 3-fold in the brain tissue of Alzheimer's disease (AD) mice, particularly in the cerebral cortex (Fig. 6B and C). These results verified the reliability of the MS results and provided noteworthy N-phosphorylation sites that may potentially affect tau aggregation. The phosphorylation sites pR182 and pR201 are located within the proline-rich domain (PRD) of tau, while pK292 and pK366 are in the microtubule-binding domain (MBD), which drives an increase in negative charge in the PRD and a decrease in positive charge in the MBD, consistent with the features critical for AD intervention.53 The phosphorylation site pH378 is located in the C-terminal region on tau, that may affect the stability and resistance to proteolysis by changing inter- or intra-molecular interactions.54,55 As shown in the MS/MS spectra of the 5 quantified tau sites in the cerebral cortex (Fig. 6D and S17†), the corresponding N-phosphopeptides were well fragmented into y ions and b ions and underwent triple neutral loss, facilitating the high-confidence identification of the N-phosphorylation sites.
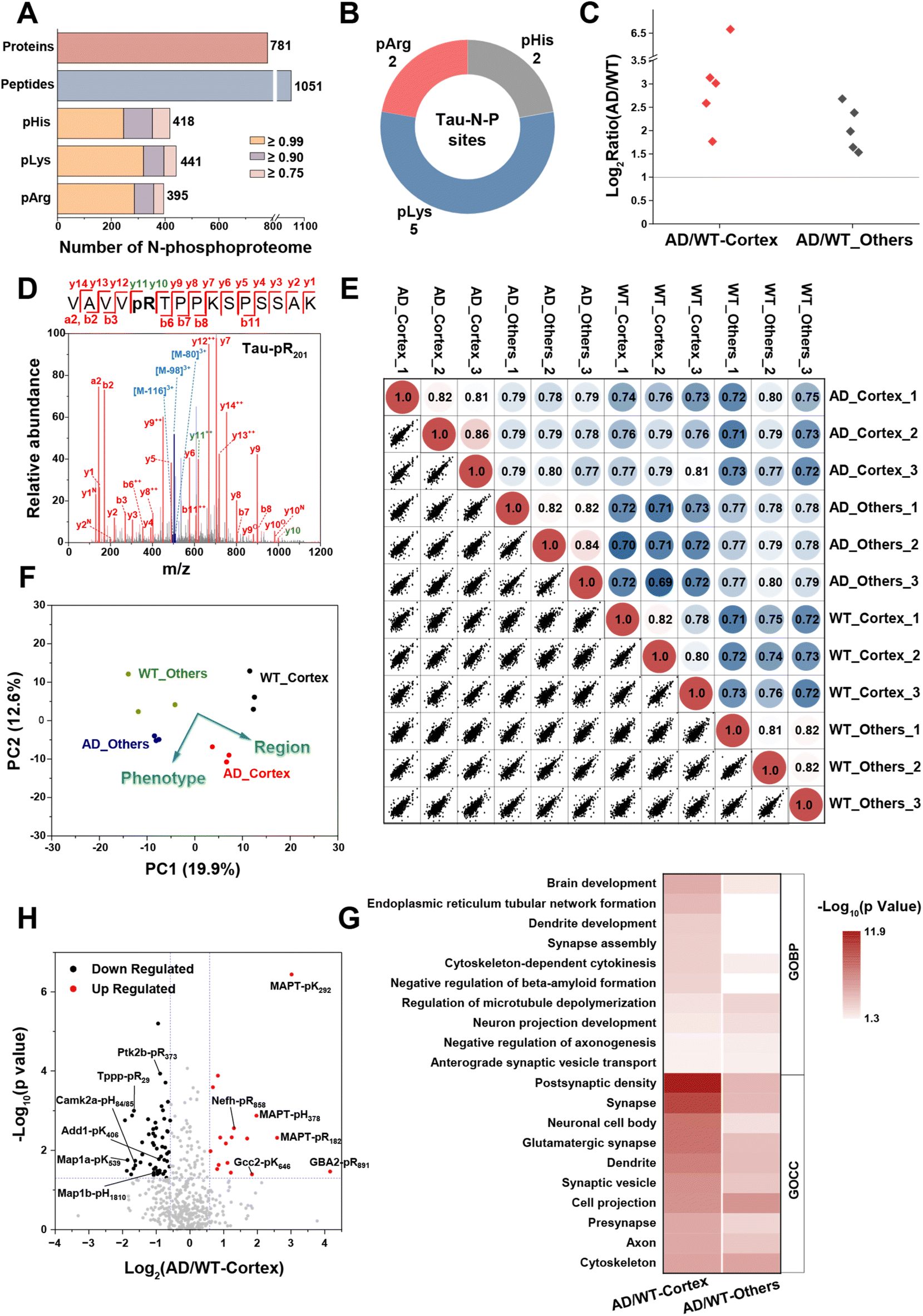 | ||
| Fig. 6 Quantitative analysis of N-phosphoproteome in an AD mouse brain via the MB@P1 based enrichment strategy. (A) The number of N-phosphorylation sites (localization probability ≥ 0.75, score ≥ 40, and score difference ≥ 5) and corresponding peptides and proteins enriched with MB@P1 in the brain of 6-month-old WT and AD mice. (B) The identified N-phosphorylation sites of the tau protein. (C) Fold changes of quantified N-phosphorylation sites for tau in the cerebral cortex or other brain regions between AD and WT mice. (D) MS/MS spectrum for pArg in position 201 of tau. (E) The correlation plots matrix from LFQ intensities of N-phosphorylation sites in three biological replicates. The color code indicates the value of the PCC. (F) PCA analysis of N-phosphorylation sites from all samples. (G) The main GO enrichment terms of proteins with differentially expressed N-phosphorylation sites in the cerebral cortex and other brain regions. (H) The volcano plot of identified N-phosphorylation sites from the cerebral cortex in AD mice compared with WT mice. | ||
More importantly, the robust reproducibility of our enrichment and quantification method was demonstrated using the PCCs of quantified sites across three biological replicates, all of which were approximately 0.80. In addition, there were slight differences observed between diverse phenotypes and brain regions (Fig. 6E). The principal component analysis (PCA) of the quantified sites further visualized these differences (Fig. 6F). Notably, the N-phosphorylation sites from the cerebral cortex and other brain regions were separated along component 1, and those from Alzheimer's disease (AD) and wild-type (WT) mice were clearly separated along component 2. This suggests that the expression levels of N-phosphorylation undergo extensive remodeling during the progression of AD and different brain regions exert varying effects on this process.
Based on the preliminary evaluation of the N-phosphoproteome, statistical analysis of the quantified sites in AD mice compared with WT mice was carried out subsequently. In total, 74 and 50 significant sites exhibited differential expression with a change of more than 1.5-fold (p-value < 0.05) in the cerebral cortex and other brain regions, respectively (Fig. S18†). From a proteomic viewpoint, GO enrichment analysis revealed that the corresponding N-phosphoproteins in the cerebral cortex were intimately involved in AD-related processes, such as brain and dendrite development, endoplasmic reticulum (ER)-mediated transport, regulation of beta-amyloid formation, synaptic activities and so forth. Meanwhile, those in other brain regions were more closely related to cytoskeleton and microtubule depolymerization (Fig. 6G). This indicated the crucial role that the cerebral cortex plays in the progression of AD.
By focusing on in-depth analysis of the differential sites in the cerebral cortex shown in Fig. 6H, we discovered that these sites were intricately associated with AD pathology. They interacted with AD-related processes by modulating signal transduction in multiple aspects, including metabolism, inflammation, and microtubule-related functions. The most prominently upregulated site was pArg at position 891 of β-glucocerebrosidase 2 (GBA2). GBA2 is an enzyme responsible for catalyzing the degradation of glucosylceramide. It has been identified as a compensatory factor for GBA1 in lysosomal storage disorders.56 Notably, the N-phosphorylation of GBA2 has not been previously reported, and it may potentially play a role in regulating the degradation of impaired proteins resulting from lysosomal dysfunction. Moreover, the N-phosphorylation levels of cytoskeletal proteins, microfilaments, and microtubule/actin-binding proteins exhibited diverse changes. In our findings, MAPT, Nefh, Gcc2, and Septin2 demonstrated hyper N-phosphorylation, whereas MAP1a/b, Add1/2, Tppp, Ina, Mlc1, and Septin5/7 exhibited hypo N-phosphorylation during AD. These alterations in phosphorylation facilitated microtubule depolymerization and the formation of neurofibrillary tangles, ultimately resulting in neuronal damage.
In addition, the N-phosphorylation levels of key protein kinases Camk2a and Ptk2b, which are involved in the calcium-signaling pathway, were downregulated. Camk2a contributes to the abnormal phosphorylation of tau when activated by Ca2+ or calmodulin and autophosphorylated due to a large influx of calcium.57 The downregulation of pHis at positions 84/85 of Camk2a may represent a regulatory mechanism in response to calcium homeostasis, preventing over-activation. This downregulation is also likely to implicate the hyperphosphorylation of tau and the deposition of Aβ, which are prevalent in the AD brain. Regarding the Ca2+-dependent tyrosine kinase Ptk2b, a susceptibility gene for AD, it has been reported to directly interact with and phosphorylate tau. Simultaneously, it contributes to tau pathology through neuroinflammation in microglia.58,59 The downregulated pArg at position 373 of Ptk2b can provide insights into how inflammatory factors activate kinase pathways.
In summary, the N-phosphoproteome mapping of the cerebral cortex in AD mice revealed the extensive and profound involvement of N-phosphorylation in the progression of AD and contributed some valuable perspectives for exploring the molecular mechanisms underlying AD.
Conclusions
In conclusion, phage display technology grounded in meticulous target screening was employed to identify affinity peptides capable of specifically binding to the N–PO3 group. We then developed an affinity peptide-based strategy for the highly efficient enrichment of all forms of N-phosphopeptides without the use of organic solvents and metal ions. This platform was applicable to a wide variety of biological systems, ranging from E. coli and HeLa cells to cell nuclei and brain tissues. The strategy allowed us to study the global landscape of the N-phosphoproteome in a neutral buffer environment, which effectively avoided the potential adverse effects of organic solvents on peptide properties. Our proteomic analysis results not only confirmed the reliability of the enrichment method, but also significantly expanded the existing N-phosphorylation databases by identifying 1995 new N-phosphorylation sites specifically. Furthermore, large-scale studies on cell nuclear N-phosphorylation revealed that N-phosphorylation exhibited subcellular heterogeneity. Finally, our research demonstrated that N-phosphorylation was highly expressed in AD. It was intricately implicated in the disease progression through regulating the activities of proteins associated with microtubules, metabolism, and inflammation. This discovery offered novel perspectives on the regulatory factors underpinning the pathogenesis of AD, thereby providing new leads for follow-up research and potential therapeutic approaches.Taken together, this research introduced a novel strategy for the efficient enrichment of N-phosphopeptides and further promoted the functional investigation of N-phosphorylated proteins in physiological and pathological processes. The strategy's high efficiency, selectivity, and robustness enable it as a versatile tool for N-phosphoproteome studies in various scenarios. Dynamic mapping of N-phosphorylation networks within subcellular niches can be foreseen by integrating with proximity labelling methods. Furthermore, the phage display platform holds potential for developing affinity ligands targeting other PTMs, facilitating exploration of cross-talk between PTM networks. Although comprehensive analysis for N-phosphoproteome has been achieved in this work, higher site coverage and specificity can be expected by screening more specific ligands for the N–PO3 group with multiple targets (such as pKFF and pRFF). Besides, high false positives arising from chemical instability of N-phosphorylation and spectral similarities with O-phosphorylation need further mitigation as well, which requires exploration of N-phosphorylation-specific diagnostic ions or spectral matching criteria to enhance identification accuracy.
Data availability
The data supporting the findings of this study are available within the article and in the ESI.† The mass spectrometric data generated in this study have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository60 and are publicly available with the dataset identifier PXD058797.Author contributions
L. Z. administrated and directed the project. B. J. and G. Q. conceived and designed the research. H. W. performed the proteomic experiments and manuscript writing. X. Z. carried out the peptide screening, material preparation and manuscript writing. D. W., Q. J. and Y. S. analyzed the data. B. Z., Z. L. and Y. Z. contributed to the project conceptualization. All authors discussed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2022YFC3401204, 2024YFC3405400, and 2022YFC3400800), the National Natural Science Foundation of China (22393931; 22174138; 22474136; 22074140), the DICP Innovation Funding (DICP-I202243, I202316 and I202229), and the United Foundation of Dalian Institute of Chemical Physics, Chinese Academy of Sciences and the Second Hospital of Dalian Medical University (DMU-2&DICP UN202405 and DMU-2&DICP UN202301).Notes and references
- Q. Liu, J. Zhang, C. Guo, M. Wang, C. Wang, Y. Yan, L. Sun, D. Wang, L. Zhang, H. Yu, L. Hou, C. Wu, Y. Zhu, G. Jiang, H. Zhu, Y. Zhou, S. Fang, T. Zhang, L. Hu, J. Li, Y. Liu, H. Zhang, B. Zhang, L. Ding, A. I. Robles, H. Rodriguez, D. Gao, H. Ji, H. Zhou and P. Zhang, Cell, 2024, 187, 184–203 CrossRef CAS PubMed.
- Y. Lu, J. Xu, Y. Li, R. Wang, C. Dai, B. Zhang, X. Zhang, L. Xu, Y. Tao, M. Han, R. Guo, Q. Wu, L. Wu, Z. Meng, M. Tan and J. Li, Sci. Transl. Med., 2024, 16, eade8647 CrossRef CAS PubMed.
- B. Bai, X. Wang, Y. Li, P.-C. Chen, K. Yu, K. K. Dey, J. M. Yarbro, X. Han, B. M. Lutz, S. Rao, Y. Jiao, J. M. Sifford, J. Han, M. Wang, H. Tan, T. I. Shaw, J.-H. Cho, S. Zhou, H. Wang, M. Niu, A. Mancieri, K. A. Messler, X. Sun, Z. Wu, V. Pagala, A. A. High, W. Bi, H. Zhang, H. Chi, V. Haroutunian, B. Zhang, T. G. Beach, G. Yu and J. Peng, Neuron, 2020, 105, 975–991 CrossRef CAS PubMed.
- T. Hunter, Mol. Cell, 2022, 82, 2190–2200 CrossRef CAS PubMed.
- A. Frando, V. Boradia, M. Gritsenko, C. Beltejar, L. Day, D. R. Sherman, S. Ma, J. M. Jacobs and C. Grundner, Nat. Microbiol., 2023, 8, 548–561 CrossRef CAS PubMed.
- N. M. Leijten, A. J. R. Heck and S. Lemeer, Nat. Methods, 2022, 19, 827–828 CrossRef CAS PubMed.
- A. Frando and C. Grundner, mSystems, 2024, 9, e00289–e00224 CrossRef PubMed.
- A. F. Alvarez and D. Georgellis, Biochem. Soc. Trans., 2022, 50, 1859–1873 CrossRef CAS PubMed.
- S. R. Fuhs and T. Hunter, Curr. Opin. Cell Biol., 2017, 45, 8–16 CrossRef CAS PubMed.
- R. Kalagiri and T. Hunter, Biochem. J., 2021, 478, 3575–3596 CrossRef CAS PubMed.
- X. Cai, S. Srivastava, S. Surindran, Z. Li and E. Y. Skolnik, Mol. Biol. Cell, 2014, 25, 1244–1250 CrossRef PubMed.
- S. K. Hindupur, M. Colombi, S. R. Fuhs, M. S. Matter, Y. Guri, K. Adam, M. Cornu, S. Piscuoglio, C. K. Y. Ng, C. Betz, D. Liko, L. Quagliata, S. Moes, P. Jenoe, L. M. Terracciano, M. H. Heim, T. Hunter and M. N. Hall, Nature, 2018, 555, 678–682 CrossRef CAS PubMed.
- P. V. Attwood and T. Wieland, Naunyn-Schmiedeberg's Arch. Pharmacol., 2015, 388, 153–160 CrossRef CAS PubMed.
- S. Srivastava, Z. Li, K. Ko, P. Choudhury, M. Albaqumi, A. K. Johnson, Y. Yan, J. M. Backer, D. Unutmaz, W. A. Coetzee and E. Y. Skolnik, Mol. Cell, 2006, 24, 665–675 CrossRef CAS PubMed.
- S. Srivastava, S. Panda, Z. Li, S. R. Fuhs, T. Hunter, D. J. Thiele, S. R. Hubbard and E. Y. Skolnik, Elife, 2016, 5, e16093 CrossRef PubMed.
- S. Panda, S. Srivastava, Z. Li, M. Vaeth, S. R. Fuhs, T. Hunter and E. Y. Skolnik, Mol. Cell, 2016, 63, 457–469 CrossRef CAS PubMed.
- D. B. Trentini, M. J. Suskiewicz, A. Heuck, R. Kurzbauer, L. Deszcz, K. Mechtler and T. Clausen, Nature, 2016, 539, 48–53 CrossRef CAS PubMed.
- A. Schmidt, D. B. Trentini, S. Spiess, J. Fuhrmann, G. Ammerer, K. Mechtler and T. Clausen, Mol. Cell. Proteomics, 2014, 13, 537–550 CrossRef CAS PubMed.
- M. J. Suskiewicz and T. Clausen, Cell Chem. Biol., 2016, 23, 888–890 CrossRef CAS PubMed.
- X. Chang, Y. Zhu, Y. Chen and L. Li, Comput. Biol. Med., 2024, 170, 108079 CrossRef CAS PubMed.
- R. C. Oslund, J. M. Kee, A. D. Couvillon, V. N. Bhatia, D. H. Perlman and T. W. Muir, J. Am. Chem. Soc., 2014, 136, 12899–12911 CrossRef CAS PubMed.
- J. M. Kee, R. C. Oslund, A. D. Couvillon and T. W. Muir, Org. Lett., 2015, 17, 187–189 CrossRef CAS PubMed.
- M. Lilley, B. Mambwe, M. J. Thompson, R. F. W. Jackson and R. Muimo, Chem. Commun., 2015, 51, 7305–7308 RSC.
- J. M. Kee, R. C. Oslund, D. H. Perlman and T. W. Muir, Nat. Chem. Biol., 2013, 9, 416–421 CrossRef CAS PubMed.
- S. R. Fuhs, J. Meisenhelder, A. Aslanian, L. Ma, A. Zagorska, M. Stankova, A. Binnie, F. Al-Obeidi, J. Mauger, G. Lemke, J. R. Yates, III and T. Hunter, Cell, 2015, 162, 198–210 CrossRef CAS PubMed.
- E. T. Yuan, Y. Ino, M. Kawaguchi, Y. Kimura, H. Hirano, E. Kinoshita-Kikuta, E. Kinoshita and T. Koike, Electrophoresis, 2017, 38, 2447–2455 CrossRef CAS PubMed.
- G. Hardman, EMBO J., 2019, 38, e100847 CrossRef CAS PubMed.
- C. M. Potel, M.-H. Lin, A. J. R. Heck and S. Lemeer, Nat. Methods, 2018, 15, 187–190 CrossRef CAS PubMed.
- Y. Hu, B. Jiang, Y. Weng, Z. Sui, B. Zhao, Y. Chen, L. Liu, Q. Wu, Z. Liang, L. Zhang and Y. Zhang, Nat. Commun., 2020, 11, 6226 CrossRef CAS PubMed.
- Y. Hu and B. Jiang, Talanta, 2022, 247, 123580 CrossRef CAS PubMed.
- Y. Hu, B. Jiang, J. Liu, H. Wang, Z. Sui, B. Zhao, Z. Liang, L. Zhang and Y. Zhang, Talanta, 2022, 243, 123384 CrossRef CAS PubMed.
- Z. Shi, X. Zhang, X. Yang, X. Zhang, F. Ma, H. Gan, J. Chen, D. Wang, W. Sun, J. Wang, C. Wang, L. Lyu, K. Yang, L. Deng and G. Qing, Adv. Mater., 2023, 35, 2302560 CrossRef CAS PubMed.
- A. Spiliotopoulos, S. K. Maurer, M. T. Tsoumpeli, J. A. F. Bonfante, J. P. Owen, K. C. Gough and I. Dreveny, Methods Mol. Biol., 2023, 2591, 189–218 CrossRef CAS PubMed.
- G. P. Smith, Angew. Chem., Int. Ed., 2019, 58, 14428–14437 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, H. Gao and G. Qing, Theranostics, 2022, 12, 2041–2062 CrossRef CAS PubMed.
- X. Zhang, B. Zhong, Y. Sun, D. Liu, X. Zhang, D. Wang, C. Wang, H. Gao, M. Zhong, H. Qin, Y. Chen, Z. Yang, Y. Li, H. Wei, X. Yang, Y. Zhang, B. Jiang, L. Zhang and G. Qing, Chem. Sci., 2025, 16, 2634–2647 RSC.
- H. Keren-Shaul, A. Spinrad, A. Weiner, O. Matcovitch-Natan, R. Dvir-Szternfeld, T. K. Ulland, E. David, K. Baruch, D. Lara-Astaiso, B. Toth, S. Itzkovitz, M. Colonna, M. Schwartz and I. Amit, Cell, 2017, 169, 1276–1290 CrossRef CAS PubMed.
- M. H. Saier Jr, J. Mol. Microbiol. Biotechnol., 2015, 25, 73–78 Search PubMed.
- K. Pang, W. Wang, J. X. Qin, Z. D. Shi, L. Hao, Y. Y. Ma, H. Xu, Z. X. Wu, D. Pan, Z. S. Chen and C. H. Han, MedComm, 2022, 3, e175 CrossRef CAS PubMed.
- K. Adam, S. Fuhs, J. Meisenhelder, A. Aslanian, J. Diedrich, J. Moresco, J. La Clair, J. R. Yates and T. Hunter, bioRxiv, 2019, preprint, DOI:10.1101/691352, https://www.biorxiv.org/content/10.1101/691352v1.
- G. Hardman, S. Perkins, P. J. Brownridge, C. J. Clarke, D. P. Byrne, A. E. Campbell, A. Kalyuzhnyy, A. Myall, P. A. Eyers, A. R. Jones and C. E. Eyers, EMBO J., 2019, 38, e100847 CrossRef CAS PubMed.
- R. Kalagiri, R. L. Stanfield, J. Meisenhelder, J. J. La Clair, S. R. Fuhs, I. A. Wilson and T. Hunter, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010644118 CrossRef CAS PubMed.
- T. Hunter, Mol. Cell, 2007, 28, 730–738 CrossRef CAS PubMed.
- J. Zhao, L. Zou, Y. Li, X. Liu, C. Zeng, C. Xu, B. Jiang, X. Guo and X. Song, J. Proteomics, 2021, 243, 104262 CrossRef CAS PubMed.
- J. Zhao, M. Zhuang, J. Liu, M. Zhang, C. Zeng, B. Jiang, J. Wu and X. Song, BMC Bioinf., 2022, 23, 399 CrossRef CAS PubMed.
- K. I. Varughese, I. Tsigelny and H. Zhao, J. Bacteriol., 2006, 188, 4970–4977 CrossRef CAS PubMed.
- S. D. Lahiri, G. Zhang, D. Dunaway-Mariano and K. N. Allen, Science, 2003, 299, 2067–2071 CrossRef CAS PubMed.
- E. Lundberg and G. H. H. Borner, Nat. Rev. Mol. Cell Biol., 2019, 20, 285–302 CrossRef CAS PubMed.
- R. Roy, J. Chun and S. N. Powell, Nat. Rev. Cancer, 2012, 12, 68–78 CrossRef CAS PubMed.
- Å. Ehlén, C. Martin, S. Miron, M. Julien, F.-X. Theillet, V. Ropars, G. Sessa, R. Beaurepere, V. Boucherit, P. Duchambon, A. El Marjou, S. Zinn-Justin and A. Carreira, Nat. Commun., 2020, 11, 1819 CrossRef PubMed.
- M. Julien, R. Ghouil, A. Petitalot, S. M. Caputo, A. Carreira and S. Zinn-Justin, Biomolecules, 2021, 11, 1060 CrossRef CAS PubMed.
- M. A. Busche and B. T. Hyman, Nat. Neurosci., 2020, 23, 1183–1193 CrossRef CAS PubMed.
- H. Wesseling, W. Mair, M. Kumar, C. N. Schlaffner, S. Tang, P. Beerepoot, B. Fatou, A. J. Guise, L. Cheng, S. Takeda, J. Muntel, M. S. Rotunno, S. Dujardin, P. Davies, K. S. Kosik, B. L. Miller, S. Berretta, J. C. Hedreen, L. T. Grinberg, W. W. Seeley, B. T. Hyman, H. Steen and J. A. Steen, Cell, 2020, 183, 1699–1713 CrossRef CAS PubMed.
- P. Flores-RodrÃ-guez, M. A. Ontiveros-Torres, M. a. C. Cárdenas-Aguayo, J. P. Luna-Arias, M. A. Meraz-RÃ-os, A. Viramontes-Pintos, C. R. Harrington, C. M. Wischik, R. l. Mena, B. Florán-Garduño and J. Luna-Muñoz, Front. Neurosci., 2015, 9, 33 Search PubMed.
- C. M. Nadel, S. Pokhrel, K. Wucherer, A. Oehler, A. C. Thwin, K. Basu, M. D. Callahan, D. R. Southworth, D. A. Mordes, C. S. Craik and J. E. Gestwicki, Nat. Commun., 2024, 15, 7972 CrossRef CAS PubMed.
- A. Massimo, S. Maura, L. Nicoletta, M. Giulia, M. Valentina, C. Elena, P. Alessandro, B. Rosaria and S. Sandro, Neurochem. Res., 2015, 41, 210–220 CrossRef PubMed.
- M. Ge, J. Zhang, S. Chen, Y. Huang, W. Chen, L. He and Y. Zhang, Neuropsychiatr. Dis. Treat., 2022, 18, 487–498 CrossRef CAS PubMed.
- C. Li, J. Götz and P. H. Reddy, J. Alzheimer's Dis., 2018, 64, 205–221 CrossRef CAS PubMed.
- Y. Guo, C.-K. Sun, L. Tang and M.-S. Tan, Curr. Alzheimer Res., 2023, 20, 692–704 CrossRef CAS PubMed.
- J. Ma, T. Chen, S. Wu, C. Yang, M. Bai, K. Shu, K. Li, G. Zhang, Z. Jin, F. He, H. Hermjakob and Y. Zhu, Nucleic Acids Res., 2019, 47, D1211–D1217 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01557j |
| ‡ H. W. and X. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |