
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Xylose-derived thionocarbamates as a synthetic handle towards a functional platform of sugar-based polymers†
James R. Runge a,
Bethan Daviesa and
Antoine Buchard*ab
a,
Bethan Daviesa and
Antoine Buchard*ab
aDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: antoine.buchard@york.ac.uk
bDepartment of Chemistry, University of York, York YO10 5DD, UK
First published on 15th July 2024
Abstract
The derivatisation of D-xylose with potassium thiocyanate is presented as a versatile synthetic handle towards functional synthetic carbohydrate polymers. The reactivity of the resulting 1,3-oxazolidine-2-thione (OZT) group with alkyl bromides is ubiquitous and, herein, has been exploited to synthesise seven bio-derived monomers with different pendant functionalities. These monomers were polymerised with dithiols to yield functional poly(ester-thioethers) with a broad spectrum of properties. From a single non-functionalised OZT-polymer, a variety of post-polymerisation modification approaches are possible to functionalise the materials with different pendant groups. These results present a novel simple methodology to tailor the properties of synthetic carbohydrate polymers to different applications.
Introduction
The ubiquitous nature of petroleum-derived plastics in modern society has prompted major concerns over their end-of-life options and the depletion of finite fossil fuel resources. Therefore, shifting towards more sustainable polymers prepared from renewable carbon feedstocks and with intrinsic degradability (e.g., through cleavable linkages) is a key step towards solving the plastic problem and achieving a circular economy.1–3 Amongst the various renewable resources used as alternatives to petrochemicals in polymer synthesis, carbohydrates stand out owing to their low cost, high natural abundance, and low toxicity. Furthermore, their diverse chemical structure allows for great functionalisation potential to tailor the properties of polymers towards a range of different applications.4–7 Monosaccharide D-xylose can for example be readily obtained from hemicellulose, a major component of lignocellulosic biomass, which has been identified as a key sustainable alternative to petrochemical feedstocks. Therefore, our group, amongst others,8–12 has been particularly interested in the use of xylose as a sustainable building block for the preparation of bio-derived polymers13–19 and functional materials.20–22Post-polymerisation modification of an existing reactive polymer is an attractive strategy to prepare materials with diverse functionalities and architectures.23 A common strategy to derivatise renewable polymers relies on post-polymerisation thiol–ene reactions, between a pendant alkene group and a thiol. For example, Greiner and co-workers have demonstrated the potential of poly(limonene carbonate), derived from citrus peels and CO2, as a platform to prepare a library of functional materials.24 However, the availability of thiols can limit the breadth of materials possible. Additionally, sugar-based polymers offer an alternative pathway towards bio-based functional materials via functionalisation of their multiple hydroxy moieties, including through the formation of ketals. However, the availability of functional ketones is limited, decreasing the number of potential structures. Developing a versatile synthetic handle towards the functionalisation, pre- or post-polymerisation, of sugar-derived polymers was therefore targeted as highly beneficial to the field of sustainable polymers. We identified organohalides as widely available and diverse compounds, which could react via nucleophilic substitution with the hydroxy groups of carbohydrates. However, the reactivity of vicinal secondary alcohols is limited, including by steric effects. Due to the greater nucleophilicity of thiols, the derivatisation of monosaccharide monomers containing sulphur was thus targeted (Fig. 1).
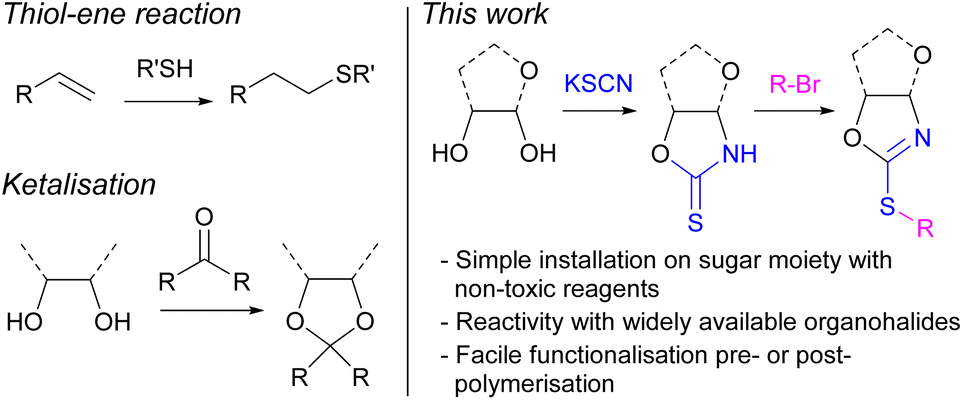 | ||
| Fig. 1 (Left) Common strategies for the functionalisation of renewable polymers; (right) thionocarbamate strategy employed here on xylose-derived monomers and polymers. | ||
The groups of Rollin and Tatibouët have pioneered methodologies to anchor 1,3-oxazolidine-2-thione (OZT) moieties onto various carbohydrate scaffolds including D-arabinose, D-ribose, and D-xylose under mild conditions and using non-toxic reagents.25,26 This interesting structural motif provides two nucleophilic sites for functionalisation reactions at either the nitrogen or sulphur atoms.27 The synthetic versatility of the cyclic thionocarbamate moiety can allow for a range of transformations with carbohydrate-anchored OZTs being studied for alkylation,28 arylation,29 vinylation,30 and palladium catalysed cross-coupling reactions.31,32
Structures bearing OZT moieties have been shown to exhibit biological activity33–35 and therefore, have been investigated as precursors for the synthesis of compounds for biological26,36,37 and agricultural applications.38 Additionally, from a synthetic perspective, OZTs find application as chiral auxiliaries in asymmetric synthesis.39,40 We hypothesised that sugar derived OZT compounds could further prove to be a promising building block for the synthesis of sustainable functional polymers and materials. There have been a few studies reporting the use of OZT containing compounds in polymer synthesis.41–44 Endo and co-workers have reported the controlled cationic ring-opening polymerisation (CROP) of an OZT derived from the amino acid L-serine using methyl trifluoromethanesulfonate (TfOMe), trifluoromethanesulfonic acid (TfOH), and boron trifluoride etherate (BF3·OEt2) to yield polythiourethanes.41 However, to the best of our knowledge there have been no reports of polymers that retain the OZT structural motif which could provide a useful synthetic toolbox for post-polymerisation functionalisation and allow for tuning of polymer properties.
Herein, we describe two α,ω-diene monomers derived from the sugar D-xylose, which incorporate an OZT synthetic handle for facile functionalisation reactions pre- or post-polymerisation. This functionality was exploited to prepare seven monomers featuring a variety of pendant functional groups. All monomers could be polymerised with dithiols via photo-initiated thiol–ene polymerisation. The presence of non-functionalised OZT moieties within the structure of some of the polymers ensured that these materials were further amenable to post-polymerisation reactions with alkyl halides. Overall, the OZT handle allowed for the introduction of functionality and modulation of properties of a renewable polymer backbone through several distinct approaches including concurrent and stepwise post-polymerisation modification or via co-polymerisation of different monomers.
Results and discussion
Synthesis of monomers
The OZT α,ω-diene monomers 1a and 2a could be prepared in a simple two-step synthesis from naturally occurring D-xylose (Scheme 1). OZT functionality can be introduced into the structure of carbohydrates through two different approaches. The more common synthetic route involves the condensation reaction of a thiocarbonyl source, e.g., thiophosgene45,46 or carbon disulfide (CS2),47 with amino sugars under basic conditions. Alternatively, a condensation reaction between thiocyanic acid, generated in situ from the reaction of potassium thiocyanate and the native sugar scaffold can be employed.25,26,36 This latter methodology is attractive as it gives access to OZT sugars using mild conditions and non-toxic reagents.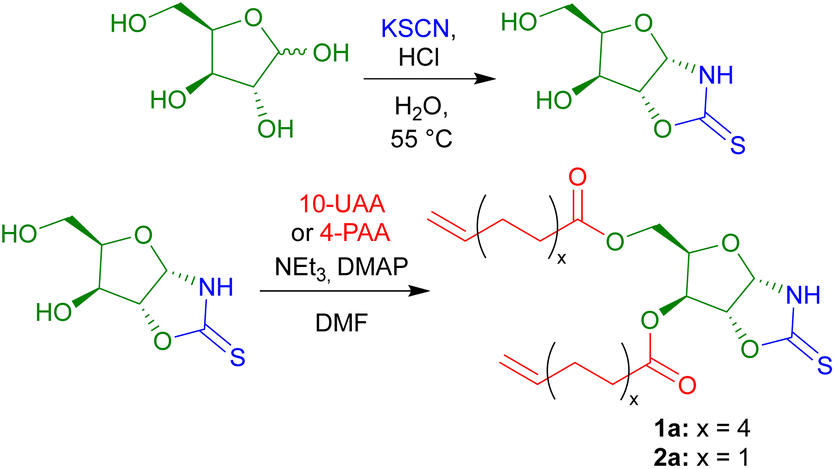 | ||
| Scheme 1 Synthesis of OZT α,ω-dienes derived from D-xylose, 1a and 2a. | ||
Following an adapted procedure from Tatibouët and co-workers,25 the OZT moiety could be directly installed onto D-xylose via reaction of the sugar with potassium thiocyanate in acidic conditions at 55 °C (Scheme 1). Purification by silica gel column chromatography yielded the OZT-xylose sugar as a white solid in a 78% yield. Characterisation by 1H and 13C{1H} NMR spectroscopy, agreed with the literature, and revealed distinctive signals at 10.72 ppm in the 1H NMR spectrum and 188.4 ppm in the 13C{1H} spectrum corresponding to the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds of the OZT functional group, respectively.
S bonds of the OZT functional group, respectively.
From this functional D-xylose core, α,ω-dienes were then prepared according to literature procedures.14,18 Unsaturated fatty acid anhydrides, 10-undecenoic acid anhydride (10-UAA) and 4-pentenoic acid anhydride (4-PAA), were synthesised by reaction of the respective acid and acetic anhydride under reflux.48 The esterification reaction of the OZT diol with either 10-UAA or 4-PAA proceeded rapidly using a combination of triethylamine and a catalytic quantity of 4-dimethylaminopyridine (DMAP) in N,N-dimethylformamide (DMF), to produce the corresponding C11 and C5 OZT α,ω-dienes within 30 minutes. Isolation by silica gel column chromatography yielded the C11 compound as a waxy solid and the C5 product as a colourless oil in 67% and 66% yields, respectively (Scheme 1).
Owing to the soft nucleophilic character of the sulphur atom, the OZT dienes were hypothesised to be capable of undergoing S-alkylation reactions with a variety of alkyl halides. To demonstrate these functionalisation capabilities, monomer 1a was subjected to a series of reactions with benzyl bromide under different conditions (Table S2†). After optimisation of the reaction conditions, near quantitative conversion of the OZT groups was observed by 1H NMR spectroscopy. When functionalised, a downfield shift of the anomeric proton signal of 1a is observed from 5.89 ppm to 6.13 ppm. Moreover, no evidence of N-alkylation under these conditions was observed by NMR analysis, with complete disappearance of the CS signal at 188.2 ppm in the 13C{1H} NMR spectrum, and appearance of a new signal at 170.3 ppm corresponding to the C–S bond, indicating the formation of the desired 2-oxazoline compound. Isolation of monomer 1b as a colourless oil was achieved by silica gel column chromatography. Next, to demonstrate the versatility of the method, we prepared a diverse range of novel α,ω-diene monomers. Reactions of 1a with various alkyl bromide compounds generated monomers 1b–1h (Scheme 2) with a diverse range of pendant functionalities, including hydrophobic (1b, 1f), hydrophilic (1g–h) and reactive groups (1c–e).
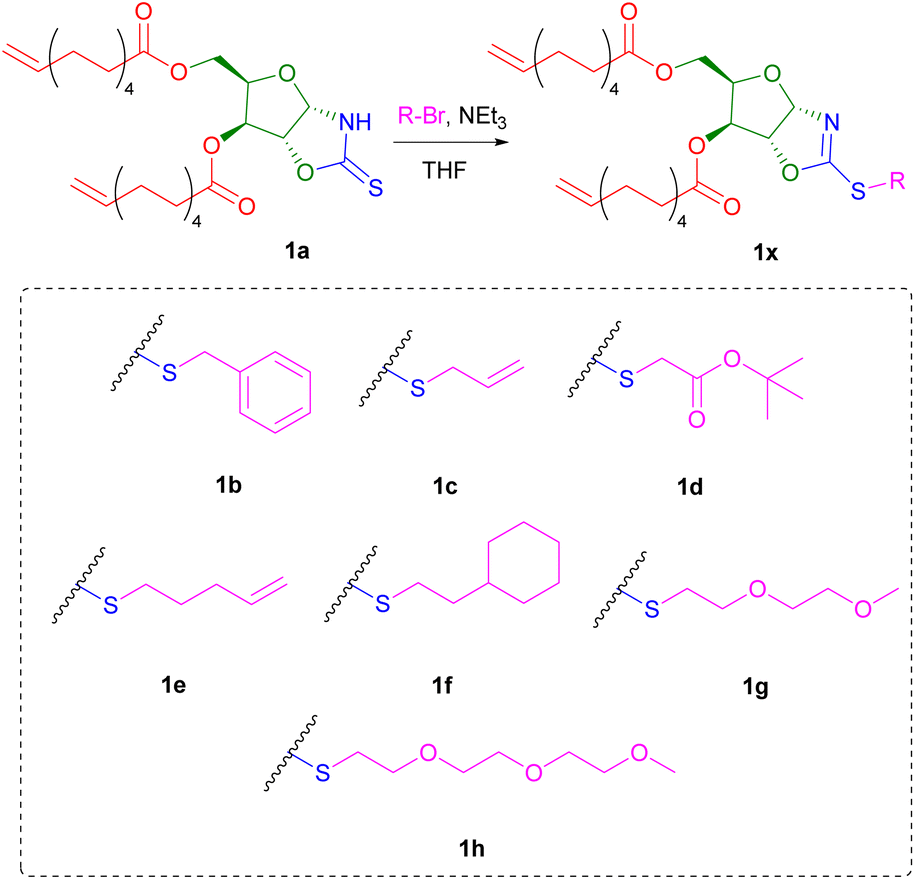 | ||
| Scheme 2 Synthesis of functionalised α,ω-dienes (1b–h) from 1a. | ||
Thiol-ene polymerisation and polymer characterisation
Previously our group has polymerised sugar-derived α,ω-diene monomers via acyclic diene metathesis (ADMET) polymerisation using Grubbs 2nd generation catalyst (G-II) to produce polyester and polyether materials.14 However, neither 1a or 2a could be polymerised by ADMET under the wide range of conditions tested, likely due to inhibition of the catalyst through coordination of the free N–H bond of the OZT group. Coordinating monomers, in particular those with unprotected amines, have been shown to have detrimental effects on the activity of Ru-based olefin metathesis catalysts.49,50 This hypothesis was validated by the fact that functionalised monomer 1b could be polymerised by ADMET (Fig. S104†).An alternative method of polymerising α,ω-dienes is via polymerisation with dithiols using thiol–ene chemistry, with recent examples reported by the groups of Becker,51 Dove,52,53 and Reineke.54 In addition, thiol–ene polymerisation avoids the use of expensive ruthenium-based catalysts and high temperatures required for ADMET. To synthesise polymers with OZT groups amenable to post-polymerisation reactions, monomers 1a and 2a were polymerised via a photo-initiated thiol–ene reaction with either 2,2′-(ethylenedioxy)diethanethiol (EDT) or 1,8-octanedithiol (ODT) in air under ambient conditions (Scheme 3; Table 1, entries 1–4). Quantitative conversion (>99%) of monomer to polymer was confirmed by 1H NMR spectroscopy from the disappearance of the olefin signals between 5.83–5.73 ppm and 5.03–4.89 ppm. Furthermore, new triplet signals at 2.57 and 2.72 ppm corresponding to methylene protons of the newly formed thioether bonds further confirmed the formation of the poly(ester-thioethers). After precipitation from cold methanol (−18 °C), the polymers were isolated as viscous yellow materials or rubbery yellow solids.
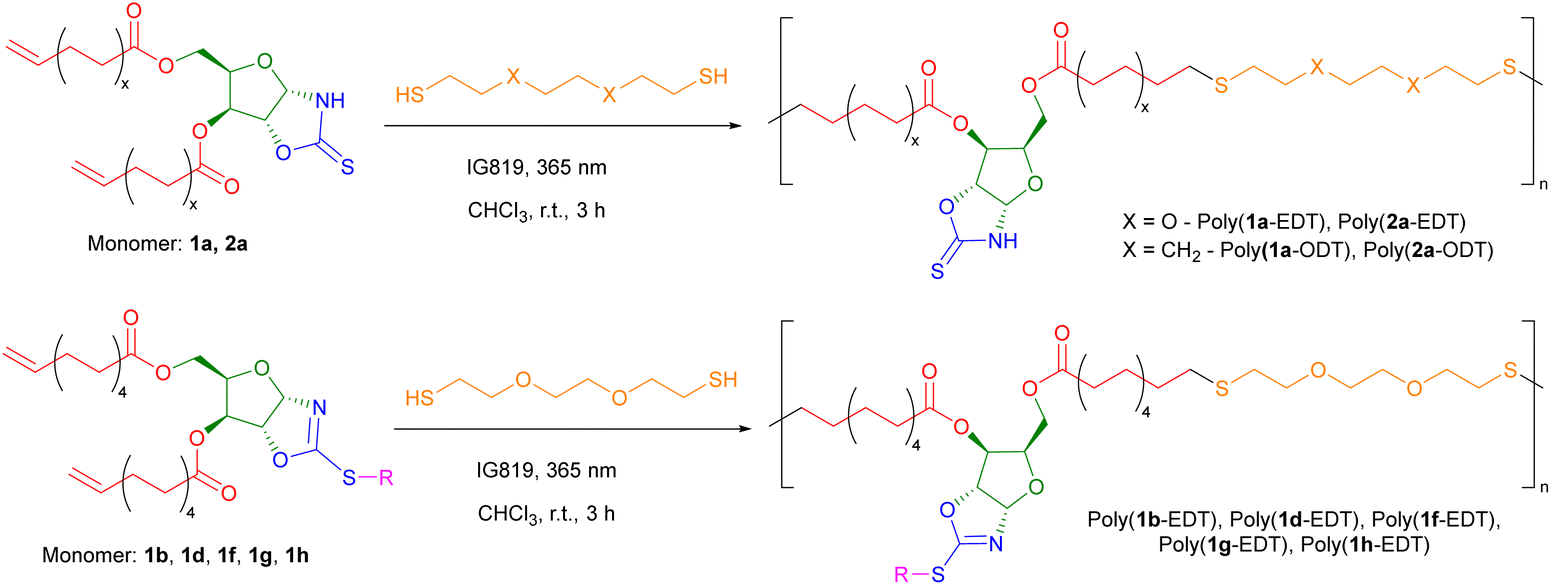 | ||
| Scheme 3 The thiol–ene polymerisation of OZT-xylose derived unsaturated α,ω-dienes with dithiols. | ||
| Entrya | Monomer | Dithiol | Polymer nomenclature | Mn, SEC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/g mol−1 b/g mol−1 |
ĐMc |
Tgd/°C |
Tmd/°C |
Td, 5%e/°C |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions unless otherwise stated: 1.00 equiv. monomer, 1.00 equiv. dithiol, 0.10 equiv. IG819, CHCl3 ([a,ω-diene] = 0.5 mol L−1), UV irradiation (λ = 365 nm, 3 h).b Calculated by SEC relative to polystyrene standards in THF eluent.c ĐM = Mw/Mn as calculated by SEC.d Determined from the second heating and cooling cycle between −60 and 150 °C in the DSC thermogram.e Determined from the 5% mass loss level by thermogravimetric analysis (TGA).f 0.50 equiv. 1b and 0.50 equiv. 1d.g Observed in the first heating and cooling cycle (Tm = 45 °C), a degree of crystallinity could be promoted by -NH interactions from the OZT functionality. | ||||||||
| 1 | 1a | EDT | Poly(1a-EDT) | 15000 |
2.89 | −22 | n.d.g | 239 |
| 2 | 1a | ODT | Poly(1a-ODT) | 11000 |
3.33 | −14 | 38 | 236 |
| 3 | 2a | EDT | Poly(2a-EDT) | 5900 | 2.05 | −7 | n.d. | 188 |
| 4 | 2a | ODT | Poly(2a-ODT) | 9500 | 3.84 | −9 | n.d. | 226 |
| 5 | 1b | EDT | Poly(1b-EDT) | 4200 | 1.94 | −41 | −13 | 228 |
| 6 | 1d | EDT | Poly(1d-EDT) | 8900 | 1.95 | −16 | n.d. | 226 |
| 7 | 1f | EDT | Poly(1f-EDT) | 7600 | 2.01 | −21 | n.d. | 251 |
| 8 | 1g | EDT | Poly(1g-EDT) | 9400 | 1.84 | −28 | n.d. | 250 |
| 9 | 1h | EDT | Poly(1h-EDT) | 6400 | 2.12 | −37 | −9 | 231 |
| 10f | 1b + 1d | EDT | Poly(1b–1d-EDT) | 6000 | 4.85 | −34 | −10 | 237 |
SEC analysis revealed number average molar masses (Mn) up to 15000 g mol−1 with high dispersities (ĐM) characteristic of step-growth polymerisation processes. Whilst this reaction offers little control over molar mass, the use of high purity monomers should allow for increased molar mass by ensuring there is little deviation from the ideal 1:1 stoichiometric ratio. Whilst efforts were made to ensure high monomer purity, it is possible that impurities resulted in a stoichiometric imbalance limiting the molar masses achieved. Polymerisation using distilled EDT did not result in the expected rise in molar mass. Additionally, increasing irradiation time appeared to have little effect on molar masses (Table S3†).
Satisfyingly, NMR analysis of the materials confirmed the OZT functionality was retained in the structure of the polymers with signals in the 13C{1H} NMR spectrum appearing at 188.9 ppm indicative of the thiocarbonyl bond. FT-IR analysis of the polymers revealed distinctive bands around 3364 cm−1, 1734 cm−1, and 1480 cm−1 indicative of the N–H, CO and C–N bonds, respectively. Thermal properties of the polymers were assessed by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Table 1). The ether-containing EDT polymers, poly(1a-EDT) and poly(2a-EDT), were found to be amorphous materials with single glass transition temperatures (Tg) at −22 °C and −7 °C, respectively.
Polymers containing aliphatic ODT were found to be either semi-crystalline, with a melting temperature (Tm) at 38 °C and low a Tg value of −14 °C (poly(1a-ODT)), or amorphous with a Tg at −9 °C (poly(2a-ODT)). Predictably, the materials based on C11 diene monomer 1a displayed lower Tg values in comparison to the C5 analogues, due to the increased chain flexibility of the longer fatty acid units. These values are lower than other bio-derived poly(ester-thioethers) reported in the literature by Reineke and co-workers.54 TGA analysis of the C11 polymers, poly(1a-EDT) and poly(1a-ODT) showed that both materials exhibited good thermal stability with Td, 5% greater than 230 °C. The C5 polymer poly(2a-EDT) displayed a lower Td, 5% of 188 °C possibly due to the lower Mn achieved.
Selected functionalised monomers (Table 1, entries 5–9) were polymerised with EDT to produce functional polymers with Mn values up to 9400 g mol−1. The effect of the different pendant groups on the thermal properties of the EDT polymers was assessed by DSC (Fig. 2A). Introduction of the flexible pendant diethylene glycol monomethyl ether (poly(1g-EDT)) chain led to an expected plasticising effect with a reduction in Tg from −22 °C to −37 °C whilst bulkier pendant tert-butyl acetate (poly(1d-EDT)) units caused an increase in Tg to −16 °C. Interestingly, the polymers possessing benzyl and diethylene glycol monomethyl ether functionality showed a degree of crystallinity with melting temperatures observed at −13 °C and −9 °C, respectively (Fig. 2B). Pendant functionality did not appear to have a significant effect on the thermal stability of the materials with all functionalised polymers displaying good thermal stability, greater than 220 °C (Fig. S97–102†). Finally, to demonstrate the capability of using this monomer platform to prepare materials variable functionality a random co-polymer was synthesised by polymerising a stoichiometric ratio of monomers 1b and 1d with EDT. The isolated sticky material was confirmed to be a single polymeric species by 1H diffusion ordered NMR spectroscopy (DOSY NMR) with all proton signals diffusing at the same rate (4.67 × 10−7 cm2 s−1) in solution (Fig. S37†). DSC analysis of this co-polymer, poly(1b–1d-EDT) revealed that the material displayed two thermal events in the second heating cycle; a Tg was observed at −34 °C whilst a Tm was found at −10 °C, likely caused by the benzyl functionalised units in the co-polymer (as for poly(1b-EDT)).
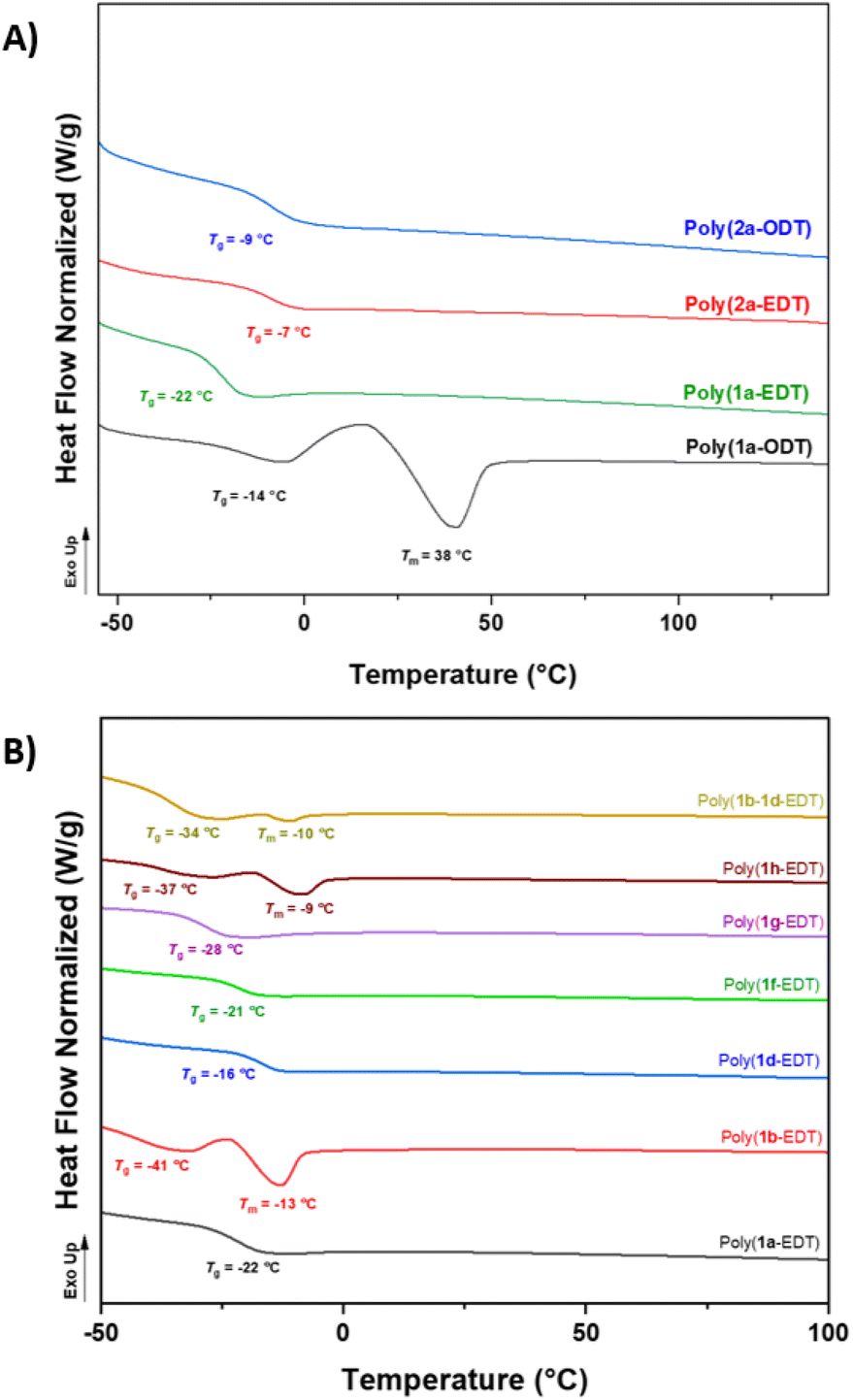 | ||
| Fig. 2 Thermal properties of OZT-derived thiol–ene polymers. (A): DSC thermograms depicting the second heating cycle between −50 and 150 °C of C11 and C5 EDT and ODT polymers. (B) DSC thermograms depicting the second heating cycle between −50 and 100 °C of functionalised thiol–ene polymers. | ||
Post-polymerisation functionalisation
Retention of the OZT functional group in the backbone of polymers of 1a and 2a (Table 1, entries 1–4) offers a synthetic handle for simple post-polymerisation functionalisation reactions. Fig. 4 represents the different post-polymerisation functionalisation approaches we undertook in this work. To investigate the post-polymerisation capabilities of the polymers, the S-benzylation of the 1a-EDT polymers was conducted using an analogous procedure to the synthesis of 1b (Table 2). Up to 91% of OZT moieties in the polymer could be benzylated after 24 hours, as determined by relative integration of the anomeric proton signals of unfunctionalised and functionalised OZT sugars. Analysis by 13C{1H} NMR spectroscopy supported successful functionalisation of the polymer with complete disappearance of the CS signal at 188.9 ppm. SEC analysis revealed the benzylated material to remain polymeric albeit with a slight increase of dispersity observed after functionalisation. FT-IR analysis of the functionalised material corroborated that benzylation was successful with complete disappearance of the broad signal at 3364 cm−1 corresponding to the N–H bond of the OZT moiety and appearance of new strong bands at 1599 cm−1 and 699 cm−1 indicative of the formation of CN and C–S bonds, respectively (Fig. 3B). Interestingly, unlike poly(1b-EDT) which showed a degree of crystallinity, the benzylated OZT polymer was found to be completely amorphous with low Tg of −37 °C (Fig. 3A). Varying the reaction conditions also allowed for different degrees of functionalisation to be achieved, 68% and 43% (Table 2, entries 2 and 3). Next, we aimed to demonstrate that we could functionalise OZT-polymers with different substituents using a concurrent post-polymerisation approach (Table 2, entries 7–9). Reaction of poly(1a-EDT) with an equimolar ratio of benzyl bromide and tert-butylbromoacetate resulted in a polymer functionalised with both pendant groups, confirmed by the presence of signals between 7.38–7.27 ppm and a large singlet at 1.47 ppm in the 1H NMR spectrum. Two new anomeric protons were also observed at 6.10 and 6.15 ppm, and the relative integration of anomeric signals revealed a 10:25:65 ratio of OZT:Bn:tBuAc units in the functionalised polymer (Fig. S103†). Varying further the ratio of alkyl halide compounds allowed for varying degrees of functionalisation to be achieved (Fig. S103†).
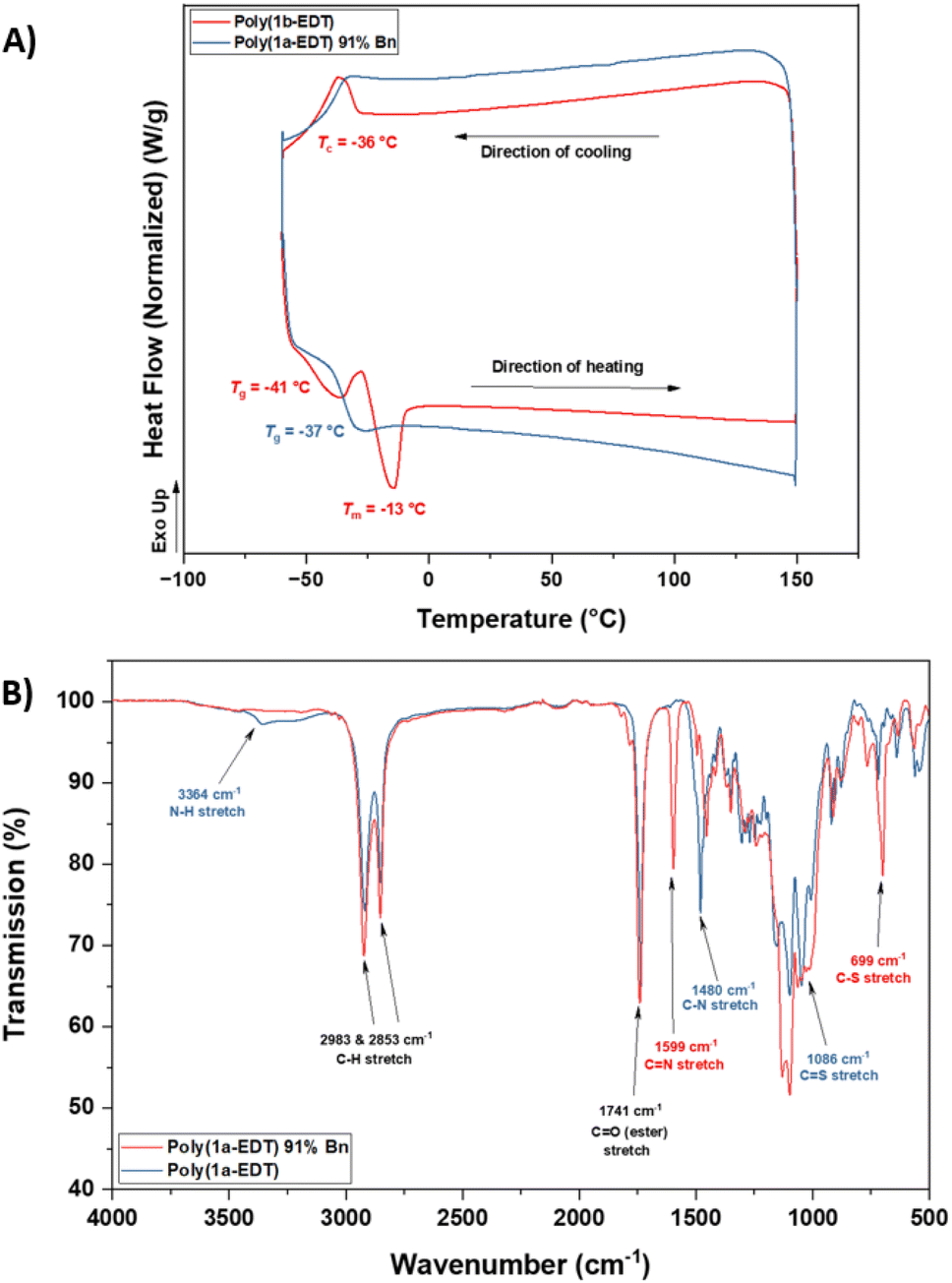 | ||
| Fig. 3 (A) Stacked DSC thermograms showing the 2nd heating and cooling cycles of poly(1b-EDT) and a 91% benzylated sample of poly(1a-EDT). (B) Stacked FT-IR spectra of poly(1a-EDT) and a 91% benzylated sample of poly(1a-EDT). | ||
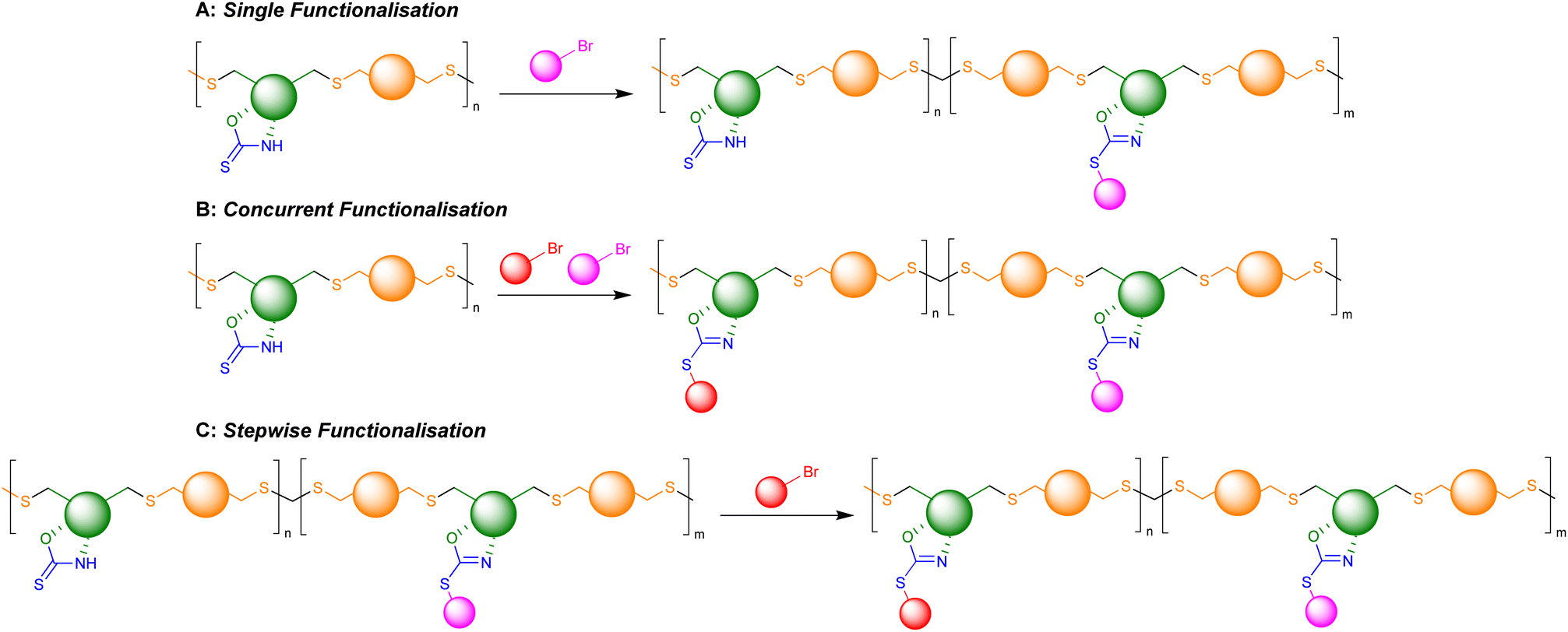 | ||
| Fig. 4 Schematic representation of possible post-polymerisation functionalisation strategies of OZT containing polymers. | ||
| Entrya | R-Br identity | R-Br/equiv. | NEt3/equiv. | [Polymer]/mol L−1 | OZT conv.b/% | Initial Mn (SEC)c/g mol−1 [ĐM]d | Mn (SEC)c/g mol−1 [ĐM]d | OZT:Bn:tBuAce |
|---|---|---|---|---|---|---|---|---|
| a General reaction conditions: polymer (1.00 equiv.), THF, 0 °C to r.t., 24 hours.b Determined by the relative integration of anomeric protons of poly(1a-EDT) (5.89 ppm) and functionalised polymer (6.10–6.15 ppm) from an aliquot of the reaction mixture.c Calculated by SEC relative to polystyrene standards in THF eluent.d ĐM = Mw/Mn as calculated by SEC.e Calculated by relative integration of anomeric protons (5.89 ppm, 6.10 ppm, 6.15 ppm).f One-pot concurrent functionalisation reaction.g Step-wise functionalisation reaction starting from a 43% benzylated polymer (from Table 2, Entry 3). | ||||||||
| 1 | BnBr | 2.00 | 4.00 | 0.50 | 91 | 10500 [2.15] |
10800 [2.40] |
— |
| 2 | BnBr | 2.00 | 4.00 | 0.10 | 68 | 13200 [3.02] |
18800 [2.87] |
— |
| 3 | BnBr | 1.00 | 2.00 | 0.50 | 43 | 13200 [3.02] |
16200 [3.12] |
— |
| 4 | BnBr | 0.50 | 1.00 | 0.50 | 42 | 13200 [3.02] |
16800 [2.87] |
— |
| 5 | BnBr | 1.00 | 2.00 | 0.10 | 35 | 15000 [2.89] |
19300 [2.91] |
— |
| 6 | BnBr | 2.00 | 1.00 | 0.10 | 34 | 15000 [2.89] |
18100 [2.93] |
— |
| 7f | BnBr + tBuBrAc | 0.50 + 0.50 | 2.00 | 0.50 | 85 | 9100 [2.87] | 7700 [3.43] | 10:25:65 |
| 8f | BnBr + tBuBrAc | 0.25 + 0.75 | 2.00 | 0.50 | 92 | 9100 [2.87] | 11900 [2.67] |
8:10:82 |
| 9f | BnBr + tBuBrAc | 0.75 + 0.25 | 2.00 | 0.50 | 89 | 9100 [2.87] | 10100 [2.82] |
11:50:40 |
| 10g | tBuBrAc | 2.00 | 4.00 | 0.50 | 100 | 16200 [3.12] |
14500 [1.95] |
0 : 43 : 57 |
Finally, we investigated whether the remaining OZT moieties in a partially functionalised polymer would be amenable to reaction with a different alkyl halide (Table 2, entry 10). Further reaction of a 43% benzylated polymer with tert-butyl bromoacetate resulted in the remaining OZT functionality being converted to the corresponding functionalised 2-oxazoline units, indicated by complete disappearance of the anomeric OZT proton signal. These results demonstrate how the OZT moiety could act as a synthetic handle to tailor polymer properties towards different applications.
Conclusions
In summary, to address the functionalisation limitations of oxygenated renewable polymers, we have synthesised a bio-derived monomer platform from D-xylose that incorporates an easily functionalisable thionocarbamate moiety. The nucleophilic nature of this synthetic handle has been exploited to prepare seven novel α,ω-dienes with varying pendant functional groups and their subsequent polymerisation with dithiols has been reported. Polymers containing some unfunctionalised OZT moieties in their backbone were also amenable to facile post-polymerisation functionalisation reactions with alkyl bromides, via a range of approaches including single, concurrent, and stepwise strategies. These results present a novel simple methodology to tailor the properties of bio-derived polymers towards different applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. JRR: all experimental work and preparation of the manuscript. BD: initial experiments which became the foundation of this work. AB: conceptualisation, manuscript writing, review and editing, supervision, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Analytical facilities were provided through the Material and Chemical Characterisation Facility (MC2) at the University of Bath. Research funding from the UK Engineering and Physical Sciences Research Council (DTP studentship to JRR), and the Royal Society (UF160021 and URF\R\221027: fellowship to A B) is also acknowledged.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- R. Xiao and M. W. Grinstaff, Prog. Polym. Sci., 2017, 74, 78–116 CrossRef CAS.
- X. Chen and R. A. Gross, Macromolecules, 1999, 32, 308–314 CrossRef CAS.
- D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Polym. Chem., 2021, 12, 5271–5278 RSC.
- A. Rizzo, G. I. Peterson, A. Bhaumik, C. Kang and T. L. Choi, Angew. Chem., Int. Ed., 2021, 60, 849–855 CrossRef CAS PubMed.
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 143, 18560–18567 CrossRef PubMed.
- Q. Chen, W. Chen and H. Zhou, J. Polym. Sci., 2023, 61, 2133–2138 CrossRef CAS.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- M. Piccini, D. J. Leak, C. J. Chuck and A. Buchard, Polym. Chem., 2020, 11, 2681–2691 RSC.
- T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed.
- T. M. McGuire, E. F. Clark and A. Buchard, Macromolecules, 2021, 54, 5094–5105 CrossRef CAS.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- M. Piccini, J. Lightfoot, B. C. Dominguez and A. Buchard, ACS Appl. Polym. Mater., 2021, 3, 5870–5881 CrossRef CAS.
- E. F. Clark, G. Kociok-Köhn, M. G. Davidson and A. Buchard, Polym. Chem., 2023, 14, 2838–2847 RSC.
- M. Oshinowo, J. R. Runge, M. Piccini, F. Marken and A. Buchard, J. Mater. Chem. A, 2022, 1, 6796–6808 RSC.
- E. L. Daniels, J. R. Runge, M. Oshinowo, H. S. Leese and A. Buchard, ACS Appl. Energy Mater., 2023, 6, 2924–2935 CrossRef CAS.
- M. Oshinowo, M. Piccini, G. Kociok-Köhn, F. Marken and A. Buchard, ACS Appl. Polym. Mater., 2024, 6, 1622–1632 CrossRef CAS PubMed.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862–11862 CrossRef CAS.
- J. Girniene, D. Gueyrard, A. Tatibouët, A. Sackus and P. Rollin, Tetrahedron Lett., 2001, 42, 2977–2980 CrossRef CAS.
- M. Domingues, J. Jaszczyk, M. I. Ismael, J. A. Figueiredo, R. Daniellou, P. Lafite, M. Schuler and A. Tatibouët, Eur. J. Org. Chem., 2020, 6109–6126 CrossRef CAS.
- A. C. Simão, J. Rousseau, S. Siliva, A. P. Rauter, A. Tatibouët and P. Rollin, in Carbohydrate Chemistry, Royal Society of Chemistry, 2009, vol. 35, pp. 127–172 Search PubMed.
- R. M. Davidson, G. D. Byrd, E. White, V. Samm, A. Margolis and B. Coxon, Magn. Reson. Chem., 1986, 24, 929–937 CrossRef CAS.
- V. Kederienė, J. Rousseau, M. Schuler, A. Šačkus and A. Tatibouët, Molecules, 2022, 27, 5597 CrossRef.
- J. Girniene, S. Tardy, A. Tatibouët, A. Sačkus and P. Rollin, Tetrahedron Lett., 2004, 45, 6443–6446 CrossRef CAS.
- S. Silva, S. Tardy, S. Routier, F. Suzenet, A. Tatibouët, A. P. Rauter and P. Rollin, Tetrahedron Lett., 2008, 49, 5583–5586 CrossRef CAS.
- S. Silva, B. Sylla, F. Suzenet, A. Tatibouët, A. P. Rauter and P. Rollin, Org. Lett., 2008, 10, 853–856 CrossRef CAS.
- Y. W. Jo, W. B. Im, J. K. Rhee, M. J. Shim, W. B. Kim and E. C. Choi, Bioorg. Med. Chem., 2004, 12, 5909–5915 CrossRef CAS.
- G. A. Youngdale, G. W. Duncan, D. E. Emmert and D. Lednicer, J. Med. Chem., 1966, 9, 155–157 CrossRef CAS.
- G. A. Johnson, E. G. Kim, S. J. Boukma, D. Lednicer and G. A. Youngdale, J. Med. Chem., 1972, 15, 327–329 CrossRef CAS.
- J. Girniene, A. Tatibouët, A. Sackus, J. Yang, G. D. Holman and P. Rollin, Carbohydr. Res., 2003, 338, 711–719 CrossRef CAS PubMed.
- S. Stairs, A. Nikmal, D.-K. Bŭar, S.-L. Zheng, J. W. Szostak and M. W. Powner, Nat. Commun., 2017, 8, 15270–15270 CrossRef CAS PubMed.
- G. Li, X. Qian, J. Cui, Q. Huang, R. Zhang and H. Guan, J. Agric. Food Chem., 2006, 54, 125–129 CrossRef CAS.
- M. T. Crimmins, B. W. King and E. A. Tabet, J. Am. Chem. Soc., 1997, 119, 7883–7884 CrossRef CAS.
- M. T. Crimmins, B. W. King, E. A. Tabet and K. Chaudhary, J. Org. Chem., 2001, 66, 894–902 CrossRef CAS PubMed.
- A. Nagai, T. Miyagawa, H. Kudo and T. Endo, Macromolecules, 2003, 36, 9335–9339 CrossRef CAS.
- A. Nagai, B. Ochiai and T. Endo, Macromolecules, 2004, 37, 7538–7542 CrossRef CAS.
- A. Nagai, B. Ochiai and T. Endo, Macromolecules, 2004, 37, 4417–4421 CrossRef CAS.
- D. Nagai, M. Sato, B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4795–4803 CrossRef CAS.
- I. Maya, Ó. López, J. G. Fernández-Bolaños, I. Robina and J. Fuentes, Tetrahedron Lett., 2001, 42, 5413–5416 CrossRef CAS.
- Ó. López, I. Maya, J. Fuentes and J. G. Fernández-Bolaños, Tetrahedron, 2004, 60, 61–72 CrossRef.
- V. M. Díaz Pérez, M. I. García Moreno, C. Ortiz Mellet, J. Fuentes, J. C. Díaz Arribas, F. J. Cañada and J. M. García Fernández, J. Org. Chem., 2000, 65, 136–143 CrossRef.
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Polym. Chem., 2017, 8, 3746–3754 RSC.
- S. N. Hancock, N. Yuntawattana, S. M. Valdez and Q. Michaudel, Polym. Chem., 2022, 13, 553–5535 RSC.
- L. Caire da Silva, G. Rojas, M. D. Schulz and K. B. Wagener, Prog. Polym. Sci., 2017, 69, 79–107 CrossRef CAS.
- Y.-H. Hsu, D. Luong, D. Asheghali, A. P. Dove and M. L. Becker, Biomacromolecules, 2022, 23, 1205–1213 CrossRef CAS PubMed.
- C. J. Stubbs, J. C. Worch, H. Prydderch, Z. Wang, R. T. Mathers, A. V. Dobrynin, M. L. Becker and A. P. Dove, J. Am. Chem. Soc., 2022, 144, 1243–1250 CrossRef CAS.
- S. R. Petersen, H. Prydderch, J. C. Worch, C. J. Stubbs, Z. Wang, J. Yu, M. C. Arno, A. V. Dobrynin, M. L. Becker and A. P. Dove, Angew. Chem., Int. Ed., 2022, 61, e202115904 CrossRef CAS PubMed.
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Polym. Chem., 2018, 9, 3272–3278 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR and FT-IR spectra for small molecules; MS data for all monomers; SEC traces, spectroscopic (NMR, FT-IR) and thermal (TGA, DSC) data for all polymers; polymer yield data, additional polymerisation data (thiol–ene, ADMET), post-polymerisation functionalisation data. See DOI: https://doi.org/10.1039/d4py00540f |
| This journal is © The Royal Society of Chemistry 2024 |