
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of the sintering parameters on the microstructural and transport properties of 60 wt% Ce0.8Gd0.2O2−δ–40 wt% FeCo2O4 composites
Liudmila
Fischer
 *ab,
Ke
Ran
cd,
Doris
Sebold
a,
Patrick
Behr
a,
Stefan
Baumann
*a,
Joachim
Mayer
cd,
Arian
Nijmeijer
b,
Henny
Bouwmeester
b,
Olivier
Guillon
ae and
Wilhelm A.
Meulenberg
ab
*ab,
Ke
Ran
cd,
Doris
Sebold
a,
Patrick
Behr
a,
Stefan
Baumann
*a,
Joachim
Mayer
cd,
Arian
Nijmeijer
b,
Henny
Bouwmeester
b,
Olivier
Guillon
ae and
Wilhelm A.
Meulenberg
ab
aInstitute of Energy and Climate Research IEK-1, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: s.baumann@fz-juelich.de
bFaculty of Science and Technology, Inorganic Membranes, University of Twente, 7500 AE, Enschede, The Netherlands
cCentral Facility for Electron Microscopy GFE, RWTH Aachen University, 52074 Aachen, Germany
dErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons ER-C, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
eJülich Aachen Research Alliance: JARA-Energy, 52425 Jülich, Germany
First published on 24th April 2024
Abstract
The dual-phase composite 60 wt% Ce0.8Gd0.2O2−δ–40 wt% FeCo2O4 (60CGO20–FCO) is shown to be an excellent oxygen transport membrane (OTM) with sufficient stability and oxygen permeance. To achieve a mechanically and chemically stable composite material an optimised microstructure should be designed. In general, the modification of the sintering profile is a classical way of achieving microstructure optimization as well as optimization of the functional properties of ceramic materials. Moreover, optimization of the sintering program would be beneficial to minimize the increase in porosity and achieve maximum densification with minimum pore and grain growth during sintering. Thus, the chosen 60CGO20–FCO composite has been subjected to different heat treatments to determine its impact on the main morphology characteristics, such as grain size, pore size, relative density, and porosity. The dwell time was varied between 0 and 100 hours at 1200 °C and seems to have an influence on the fraction of the resulting phases, revealing the formation of a high-temperature CoO phase after 100 hours. In other samples fired for 0–50 hours at 1200 °C fluorite, spinel, and perovskite phases are prevailing. To eliminate the undesired CoO phase the cooling rate has been varied. The results reveal that the 5 hour dwell time at 1200 °C is the most suitable one for the chosen composite material, resulting in a sufficiently dense membrane with well-distributed phases, and can be further utilized for scaling up OTM production.
1. Introduction
The development of ceramic dual-phase materials for oxygen transport membranes (OTM) attracts much attention in the scientific community as mixed ionic-electronic conductors (MIEC) are very promising for gas separation1–5 at elevated temperatures and have the potential for improving performance and reducing the cost of several industrial processes.6,7 MIEC materials can be used for oxygen separation from mixed gases, such as air. The oxygen separation process can include exposure to flue gases containing CO2, SOx, dust, and steam.8–10 Nowadays, dense dual- and multiphase composite materials with separate ionic and electronic conductors are among the most promising and efficient materials regarding stability for application in membrane reactors.11–16 However, these material systems are quite complex and affected by many factors, such as sintering temperature and dwell time. Thus, because of the complexity of this composite system a systematic study of the microstructure-related features is required.Rare earth doped ceria with the fluorite structure showed to be an excellent ion-conductor,17–19 in particular for application at temperatures <850 °C.20,21 The FexCo3−xO4 (0 < x < 3) spinel system is found to be most suitable and promising for the targeted application due to its stability in aggressive atmospheres and good electronic conductivity.22–24 The fluorite-spinel composite Ce0.8Gd0.2O2−δ–FeCo2O4 (CGO20–FCO) is a suitable example of such a material, providing a stable structure and sufficient oxygen permeability.25–27 The presence of both phases is key to ensure good percolation, which leads to a stable oxygen flux, and sufficient performance of the composite.14,15 This means that the concentrations of the individual phases should be above the percolating threshold, which is generally reported as 30 vol%.28,29 Under ideal conditions, all selected phases should stay separate and not interact during the heat treatment. However, recent studies reported the formation of an additional emerged orthorhombic phase of the GdFeO3-type after phase interaction.13,14,30–32 This phase contributes effectively to electronic conductivity.11,14,32
Generally, to ensure a stable and functional dual- or multi-phase material for long-term gas separation processes, the quality of the connectivity and compatibility of the two phases is crucial. Hence, a defect-free membrane with optimized phase morphology should be targeted. Under real conditions, interface between particles is imperfect and can depend on different factors, such as powder particle size, synthesis and sintering parameters. The control of the microstructure via modification of the heat treatment plays an important role in the solid-state sintering process and defines the properties of the material, such as density, strength, and transport features.18,33–35
In general, a temperature range of 1100–1500 °C is used to prepare the dual- and multiphase composites for the OTM.18,34–36 At lower temperatures, a lack of densification can be expected, and one may obtain a material with poor mechanical properties. A temperature up to 1600 °C is necessary to yield a dense functional single phase ceria membrane.37 However, the addition of the other oxides (i.e. Fe2O3) as sintering aids can lower the sintering temperature and reach sufficient densification of the dual-phase material.38 For the CGO20–FCO system, the optimal sintering temperature is 1200 °C. Above 1300 °C sintering temperature undesired effects on mechanical properties in the ceramic material, in particular, ceria and ceria-based composites were reported,33,39 which in turn might affect the transport properties of the material. Grain growth is a known phenomenon in ceria-based materials at elevated temperatures.40,41 The effect of the temperature on the CGO20–FCO composite materials is already investigated in the context of phase interaction.34,42–45
However, some questions related to the phase interaction as well as the morphology of the CGO20–FCO dual-phase composite are still open. When is the phase interaction completed in the CGO20–FCO composite, considering the complexity of this system? Is there an influence of the cooling rate on the phase composition? What is the dwell time requirement for the densification and pore removal processes at 1200 °C for the studied MIEC materials? To clarify all these aspects the correlation between the dwell time and phase interaction, final morphology, and permeation performance should be investigated.
The current study aims to present detailed characterization of the 60CGO20–FCO composites, including the quantification of all phase constituent volume fractions, grain and pore size, and density as well as the porosity, and reveal the relationships between the microstructure features with the applied heat treatment and cooling methods. Furthermore, comparatively little research has been conducted on the effect of the dwell time on the morphology features and the permeation performance of the CGO20–FCO composite, which will be presented here.
2. Experimental
2.1. Sample preparation
Ce0.8Gd0.2O2−δ (CGO20) (Cerpotech, >99%, Tiller, Norway), Fe2O3 (Merck, 99%) and Co3O4 (Merck, 99%) powders used for the experiments were synthesized by the solid-state reactive sintering method (SSRS). Fe2O3 and Co3O4 were mixed in a Fe: Co 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, resulting in spinel FeCo2O4 (FCO), which was added to commercially available Gd-doped ceria. Respective amounts of powders were weighed to create Ce0.8Gd0.2O2−δ–FeCo2O4 (CGO20–FCO) compositions with wt%-ratios of 60:40 and 80:20. The powder mixtures were ball milled in polyethylene bottles over 48 h using zirconia balls on a roller bench Roller-Mill RM1 (Zoz GmbH, Germany) at a speed of 175 rpm. After drying for 48 h in ambient air at 70 °C, powder mixtures were pressed using a uniaxial press (Paul-Otto Weber GmbH, Germany) in disc-shaped membranes in a 20-mm stainless steel die and then sintered in Muffle furnace (Nabertherm GmbH, Lilienthal, Germany) at 1200 °C for different dwell times in air with a heating rate of 5 K min−1 as listed in Table 1. At this sintering temperature, the spinel partially reduces into a high-temperature monoxide phase with a rock salt structure. To ensure complete reoxidation of the high-temperature Co/Fe-monoxide phase to a spinel phase that is stable at operating temperatures (600–1000 °C), a slow rate of 0.5 K min−1 between 900 and 800 °C was implemented in the cooling step. This rate was determined by referring to.46,47
:2 ratio, resulting in spinel FeCo2O4 (FCO), which was added to commercially available Gd-doped ceria. Respective amounts of powders were weighed to create Ce0.8Gd0.2O2−δ–FeCo2O4 (CGO20–FCO) compositions with wt%-ratios of 60:40 and 80:20. The powder mixtures were ball milled in polyethylene bottles over 48 h using zirconia balls on a roller bench Roller-Mill RM1 (Zoz GmbH, Germany) at a speed of 175 rpm. After drying for 48 h in ambient air at 70 °C, powder mixtures were pressed using a uniaxial press (Paul-Otto Weber GmbH, Germany) in disc-shaped membranes in a 20-mm stainless steel die and then sintered in Muffle furnace (Nabertherm GmbH, Lilienthal, Germany) at 1200 °C for different dwell times in air with a heating rate of 5 K min−1 as listed in Table 1. At this sintering temperature, the spinel partially reduces into a high-temperature monoxide phase with a rock salt structure. To ensure complete reoxidation of the high-temperature Co/Fe-monoxide phase to a spinel phase that is stable at operating temperatures (600–1000 °C), a slow rate of 0.5 K min−1 between 900 and 800 °C was implemented in the cooling step. This rate was determined by referring to.46,47
| Composite | 0 | 0.16 | 0.5 | 1 | 5 | 20 | 50 | 100 |
|---|---|---|---|---|---|---|---|---|
| 60:40 CGO20–FCO |
√ | √ | √ | √ | √ | √ | √ | √ |
| 80:20 CGO20–FCO |
√ | √ |
After the sintering step, all samples were ground to 1 mm thick discs in two steps, by applying SiC papers with different grits from P 800 to P 1200 (by WS FLEX 18C) by using ATM Saphir 550 machine (Germany). The surface of the membrane can contribute to the oxygen exchange process in an important, though more limited way. Modifying the surface by applying a catalytic layer is required to maximize surface area, participating in the process, and minimize all possible surface limitations. On both sides of the discs, a porous La0.58Sr0.4Co0.2Fe0.8O3−δ (LSCF) catalytic activation layer with a thickness of 5 μm was applied using a screen-printing technique. The resulting discs were then calcined in the furnace at 1100 °C for 5 hours.
The impact of the cooling method on the microstructural properties of the 60CGO20–FCO composite sintered at 1200 °C for 100 h was performed and used heat treatment parameters are listed in Table 2.
| Heat treatment and cooling method | Named further |
|---|---|
| Sintered at 1200 °C for 5 h and cooled with standard 0.5 K min−1 between 900 and 800 °C in the furnace | 5 h |
| Sintered at 1200 °C for 100 h and held additionally at 850 °C for 100 h then cooled in the furnace with 0.5 K min−1 between 900–800 °C | Slow |
| Sintered at 1200 °C for 100 h and cooled with standard 0.5 K min−1 between 900 and 800 °C in the furnace | Standard |
| Sintered at 1200 °C for 100 h and cooled freely in the air outside the furnace | Fast |
| Sintered at 1200 °C for 100 h and quenched in water | Quenching |
2.2. Characterization methods
| T, h | CGO (fluorite), Fm3m | FCO (spinel), Fd3m | Co3O4 (spinel), Fd3m | GCFCO (perovskite), Pnma | CoO Fm3m | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| F, wt% | a = b = c, Å | F wt% | a = b = c, Å | F, wt% | a = b = c, Å | F, wt% | Lattice parameter, Å | F, wt% | |||
| a | b | c | |||||||||
| CGO20 | 100.0[0] | 5.424[3] | — | — | — | — | — | — | — | — | — |
| 0 | 53.9[3] | 5.417[1] | 17.7[3] | 8.286[3] | 20.3[6] | 8.146[0] | 8.1[6] | 5.336[3] | 5.609[4] | 7.652[6] | — |
| 0.16 | 55.4[5] | 5.417[3] | 17.6[3] | 8.297[1] | 19.6[6] | 8.146[3] | 7.5[9] | 5.333 | 5.606[1] | 7.641[3] | — |
| 0.5 | 54.5[4] | 5.419[9] | 17.3[6] | 8.297[7] | 19.6[2] | 8.146[2] | 8.6[3] | 5.337[3] | 5.609[2] | 7.652[7] | — |
| 1 | 53.1[3] | 5.419[2] | 23.1[7] | 8.280[6] | 15.3[3] | 8.146[0] | 8.5[7] | 5.337[1] | 5.610[2] | 7.654[8] | — |
| 5 | 52.1[2] | 5.418[5] | 22.3[1] | 8.287[3] | 16.6[0] | 8.125[1] | 9.0[3] | 5.336[3] | 5.608[6] | 7.639[7] | — |
| 20 | 53.6[3] | 5.417[3] | 15.7[0] | 8.298[3] | 22.1[2] | 8.140[3] | 8.5[3] | 5.339[0] | 5.611[5] | 7.649[8] | — |
| 50 | 52.3[4] | 5.416[8] | 18.1[6] | 8.309[2] | 20.0[9] | 8.138[8] | 9.7[5] | 5.337[6] | 5.612[3] | 7.653[3] | — |
| 100 | 54.7[3] | 5.417[2] | 17.1[3] | 8.330[3] | 17.4[7] | 8.161[3] | 7.9[3] | 5.341[7] | 5.618[5] | 7.635[3] | 2.8[9] |
Further image analysis-based method was conducted via ImageJ software on all composite samples including ∼1000 grains utilizing so-called isodata threshold method,48 which helped to estimate the average grain size of all phases as well as area fraction of pores. However, the limited number of characterized grains/pores might induce large inaccuracy in the grain size and pore size calculations.
For transmission electron microscopy (TEM) analysis, all specimens were cut from composite pellets by focused ion beam (FIB) milling using an FEI Strata 400 system with a gallium ion beam. Further thinning and cleaning was performed using an argon ion beam in a Fischione Nanomill 1040 at beam energies of 900 eV and 500 eV. TEM and energy-filtered TEM (EFTEM) imaging was performed using an FEI Tecnai F20 at 200 kV. For high-resolution high-angle annular dark-field (HAADF) imaging and energy-dispersive X-ray spectroscopy (EDS) chemical mapping an FEI Titan G2 80–200 ChemiSTEM microscope equipped with an high brightness field emission (XFEG) gun and a probe Cs corrector was used.49
Here FAr is argon flow rate, i.e., 50 mL N min−1, XO2 and XN2 the oxygen and nitrogen concentration in the permeate gas, respectively. The membrane area available for permeation is Amem = 1.33 mm2. The factor ¼ reflects the O2/N2 ratio in the air feed assuming that the leak is not gas selective.
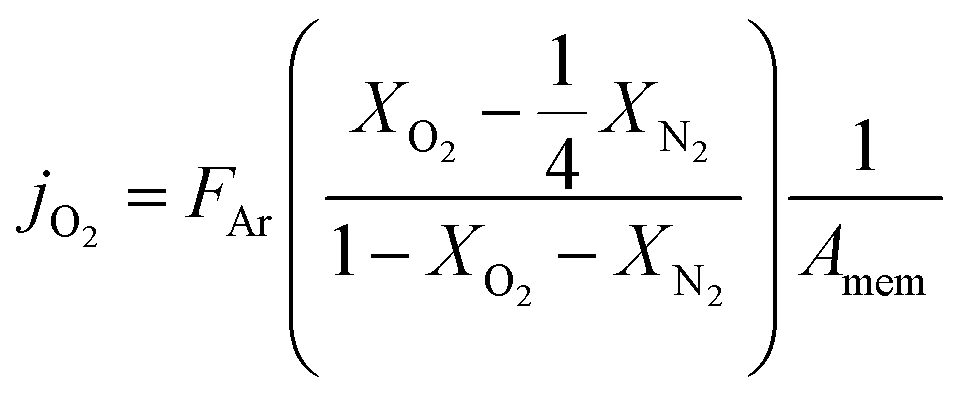
Since the oxygen partial pressure in the permeate gas is temperature dependent, the driving force for permeation is not constant during the measurement. Additionally, the sample thickness deviation after the grinding process was ±8% from the target thickness of 1 mm. Therefore, the permeance, i.e., driving force normalized permeation rate, normalized to the reference thickness L0 = 1 mm is calculated assuming Wagner's behaviour using
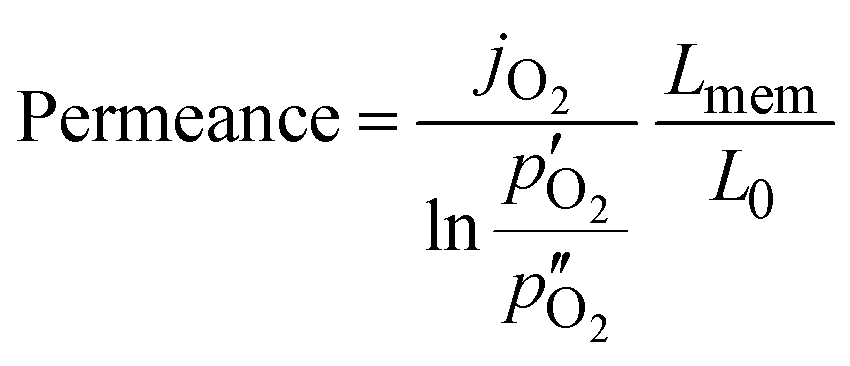
Here, 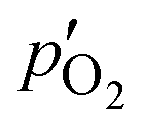 and
and 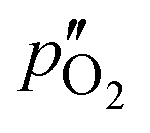 are the oxygen partial pressures in the retentate and permeate gas, respectively, and Lmem is the actual membrane thickness. The overall experimental error is assumed to be ±10%, which is well accepted in the literature.
are the oxygen partial pressures in the retentate and permeate gas, respectively, and Lmem is the actual membrane thickness. The overall experimental error is assumed to be ±10%, which is well accepted in the literature.
3. Results and discussion
Based on the realization of the complex but important microstructures of dual-phase oxygen transport membranes, the standard composite 60CGO20–FCO was subjected to sintering at 1200 °C for different dwell times and further analysed to clarify the connection between the dwell time and phase interaction, morphology as well as permeation.3.1. Microstructure analysis
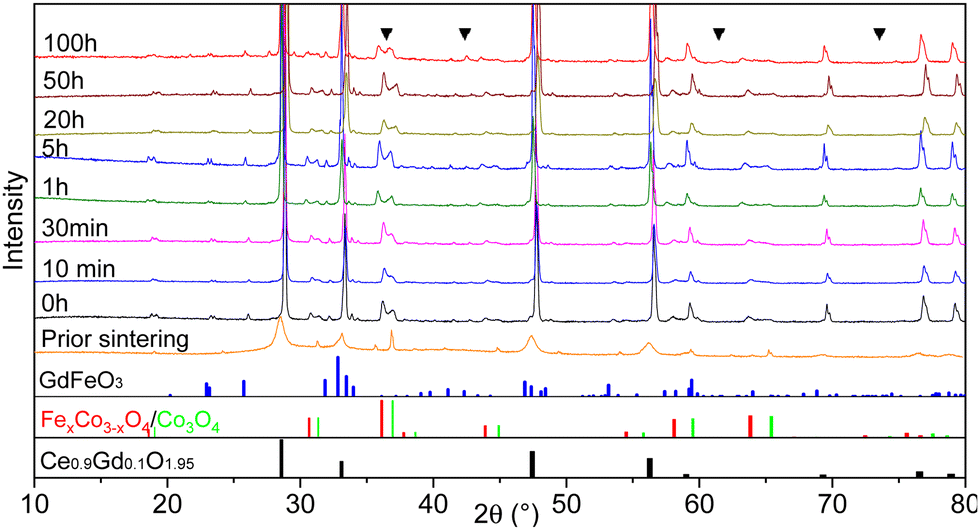 | ||
| Fig. 1 The XRD patterns of the 60CGO20–FCO composite sintered at 1200 °C with various dwell times, including an unsintered pellet. CoO rock salt phase peaks are marked as a triangle. At the bottom, reference reflections are shown for the perovskite in blue, spinel in red and green, and fluorite in black. | ||
In all sintered samples the final microstructure consists of three phases after phase interaction: a cubic fluorite structure with space group Fm3m, a donor-doped orthorhombic GdFeO3 (GCFCO) perovskite structure with space group Pnma, and Fe/Co rich phases with a cubic spinel structure with space group Fm3m. This is in agreement with45 since the formation of the Fe–Co spinel in CGO20–FCO composite starts at 900 °C and perovskite at 1050 °C. An exception is the 100 hours sintered pellet, where an additional CoO rock salt phase with space group Fm3m was observed, which is a result of incomplete re-oxidization during cooling. This has already been observed in earlier research.26,46,50 The CoO phase is difficult to distinguish in the XRD patterns since they can hardly be separated from the GCFCO and Co3O4 patterns, however, several peaks are marked are assigned to the CoO phase in Fig. 1. Moreover, with respect to the Fe–Co oxide phase diagram,47 a coexistence of the two spinel phases is detected in all samples: the nominal and inverse spinel. According to the Fe–Co phase diagram, which shows that the FeCo2O4 spinel is only stable in a narrow temperature range,51 it can be concluded that Fe-rich spinel is likely the CoFe2O4 structure in the final microstructure.
Further Rietveld refinement quantified the composition of the dual-phase composites, which are listed Table 3. The unit-cell parameters of the pure phases are known from the literature a(FeCo2O4) = 0.8244 nm, a(Fe2CoO4) = 0.8385 nm,24,51,52a(Co3O4) = 0.8142 nm52,53 and a(CGO20) = 0.54209 nm.54,55 The lattice parameter of CGO20 used in our composite before phase interaction was a = 0.54246 nm and it was found to decrease by the addition of the spinel resulting in a(CGO20) = 0.5418 ± 0.0001 nm. It can be concluded that the fluorite phase contains 10 mol% of Gd, instead 20 mol%, due to phase interaction and formation of a GCFCO emerged phase, which is in accordance with previously published work.11,56 There was no remarkable change in CGO lattice parameter along the whole investigated time range. The influence of the dwell time on the lattice parameter seems to be stronger for the inverse and normal spinel phases. The weight fraction of the fluorite remains almost unchanged, revealing a slightly lower amount for samples >1 hour dwell time, compared to <1 hour. The amount of GCFCO also remains almost constant in the range of 8.5 ± 1 wt%, regardless of the dwell time. The GdFeO3-based perovskite phase that is formed during the sintering process can be considered as a pure electron conductor according to Ramasamy, Fischer et al.,11,14 and its contribution leads to an increase in the amount of total electron conducting phase, that in turn takes place at the expense of the of ion-conducting phases. All studied samples were subjected to the analysis described in Fischer et al.,57 and the resulting volume fraction of individual phases was estimated and graphically represented in Fig. 2.
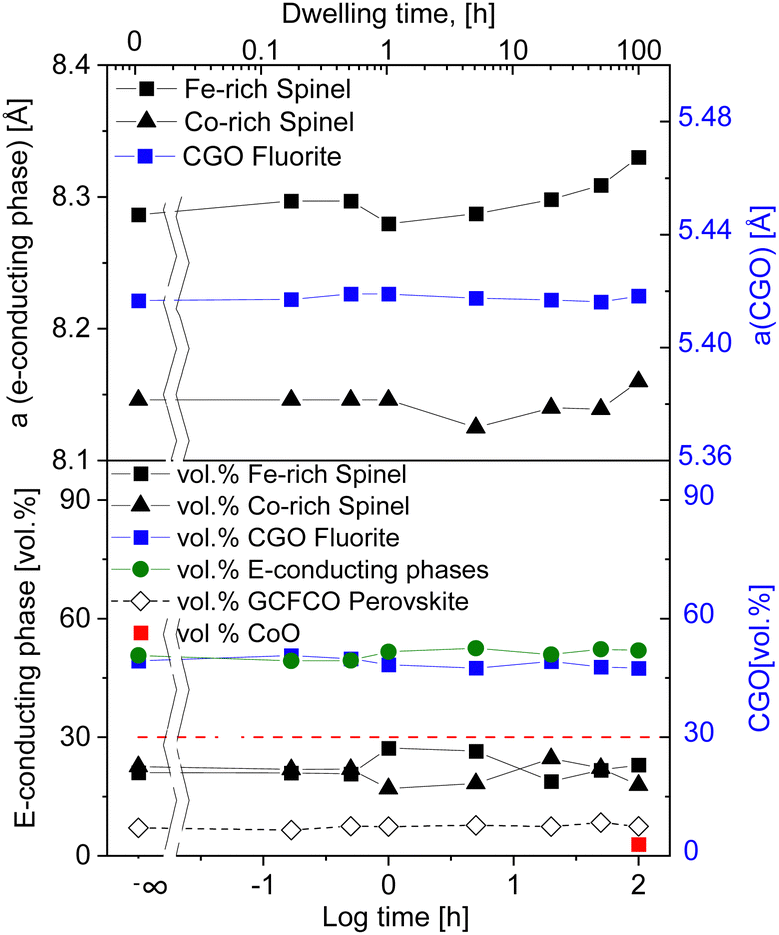 | ||
| Fig. 2 Lattice parameter and volume fraction of observed phases in 60CGO20–FCO composites sintered at 1200 °C (solid lines are a guide to the eye). | ||
The percolation threshold of 30 vol% (shown as a red dashed line in Fig. 2) has been achieved for all samples: the sum of volume fraction of all present electron conducting phases (spinels and perovskite) shown in green lays in the range of 50.5 ± 1 vol%. That explains the relatively high oxygen flux for the composites. The amount of additional CoO phase is ∼2.8 vol% in a 100 hours fired sample, which likely contributes to electronic conductivity, too.56 Due to the long dwell time the amount of CoO is increased within the composite, which in turn has not enough time for its complete reoxidation to a spinel phase.
Back scattered electron (BSE) SEM images of the surface in Fig. 3 reveal the distribution of the phases in samples fired at 1200 °C for various dwell times, including fluorite, spinel, and perovskite grains distinguished by different grey scales. It is known, that an emerging GCFCO phase appears at 1050 °C.45 In all studied samples fired at 1200 °C the perovskite phase is fully formed as indicated in light grey, dark spinel grains possess a rather uneven grain size, being coarsely distributed among white CGO grains.
 | ||
| Fig. 3 BSE-images of 60CGO20–FCO surface morphology of the samples sintered at 1200 °C with two magnifications. | ||
Generally, there are two stages of grain growth. The initial coarsening stage is between room temperature and the sintering temperature. During this stage coarsening can be responsible for grain growth, but not a conventional boundary migration. Since the grain boundaries are possibly pinned by the open pore channels in as-pressed sample. At this stage, the density of the sample is quite low, but the particle rearrangement and surface diffusion can lead to some grain growth. After reaching the sintering temperature at the typical density above 90% the second stage of grain growth begins, where grain boundary migration via a diffusion process is likely to be dominating mechanism.58 The grains grow rapidly, and pores become closed and isolated, leading also to the pore coarsening. Densification, grain, and pore growth are the three main processes that occur during sintering in the ceramic composite.45
The grain size of each phase as well as the pore size and area fraction of pores were analysed by an image analysis-based method with ImageJ software for all samples.48,59
In the not single-phase composites, the uniform distribution of the one phase in the uniformly packed matrix of the second phase is required.58 Our powder mixture consists of more than one solid prior firing process, so the grain growth is probably limited because of the second phase in the grain boundaries. Thus, in the investigated composite, the grain growth is not homogeneous and depends on the type of grains; spinel grains are slightly larger compared to the fluorite and perovskite grains.
The mean grain size of the four phases increases with increasing dwell time, as displayed in Fig. 4. Below 1 hour, the grain growth is less pronounced, with an almost constant value of 0.57 μm, followed by a gradual increase in the grain size to 0.77 μm at 50 hours. Then a drastic increase can be seen at doubling the dwell time, resulting in the largest grain size of 1.13 μm at 100 h. In general, the sintering process involves the competition of both main processes: coarsening and densification and probably depends on the dwell time at 1200 °C. It is likely that grain growth is the dominant process for the samples sintered for 50 and 100 h, while below 50 h other processes are dominant. The increased grain size in ceramic materials might lead to enhanced crack formation and poor mechanical properties.60
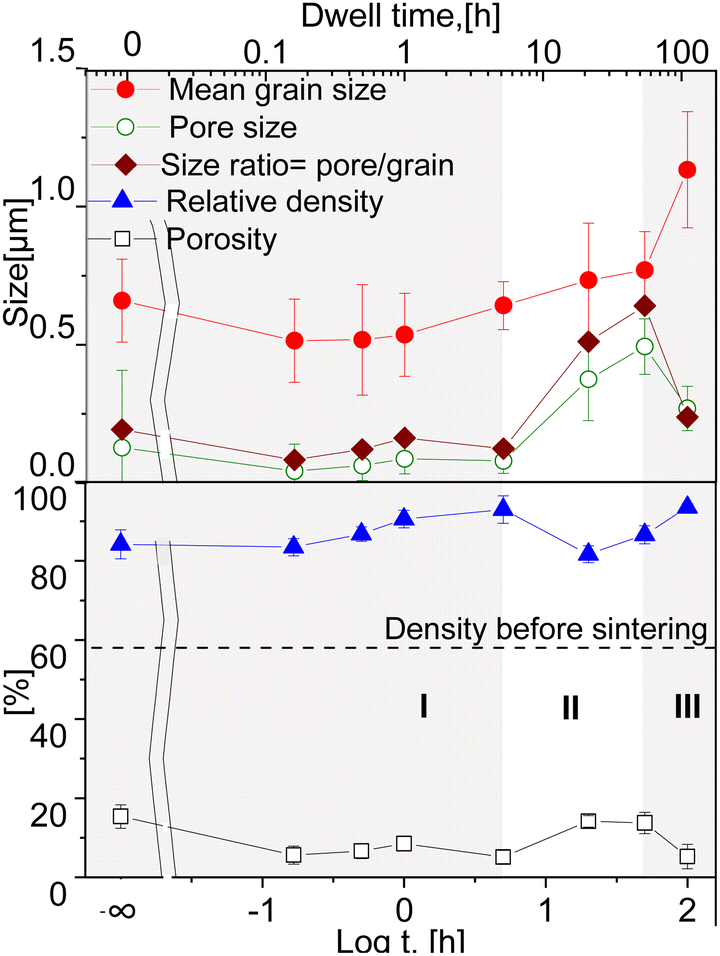 | ||
| Fig. 4 Estimated relative density and porosity over the dwell time; Average grain, pore size and the pore/grain ratio dependency on the dwell time in the 60CGO20–FCO composites sintered at 1200 °C (solid lines are a guide to the eye). | ||
An increase in density with an accompanying moderate grain growth is known for Gd-doped ceria during the first step of densification.61 Sintering at 1200 °C for dwell times between 0 and 100 hours led to a densification level between 82 and 93% of the theoretical density. It is clear that all samples undergo some densification process already at the heating phase, as the 0 h sintered sample shows a relative density >80%. In our case the samples held isothermally for less than 5 hours showed densification as the dominant process (I). The relative density of the samples gradually increases until a maximum of 93% at 5 h. Between 5 and 50 hours grain and pore growth develop concurrently with slight pore growth domination, where possibly de-densification occurs resulting in a relative density reduction (II) (similar to42). At 100 hours again an increase in densification is observed (III). This suggests that a sufficient dwell time for the 60CGO20–FCO composite at 1200 °C is 5 hours, where a fast and effective densification process takes place. Since the relationships between relative density and grain size are not linear, the densification processes and grain growth likely proceed via different diffusion routes in surface and volume, possibly due to the large difference in Ea of these diffusion mechanisms.62
The density of the 20 h and 50 h-fired samples rapidly decreased to a minimum (82% of the theoretical density) at 20 hours, where probably the de-densification phenomenon occurs. The observed loss in the density of the dual-phase composite could be attributed to a loss of weight as a result of oxygen gas production, with a reduction of Ce4+ to Ce3+ in the fluorite phase. In the spinel phase, the reduction of iron and cobalt also leads to a release of oxygen and the formation of a rock salt phase, which increases pore volume and size at elevated temperature.63
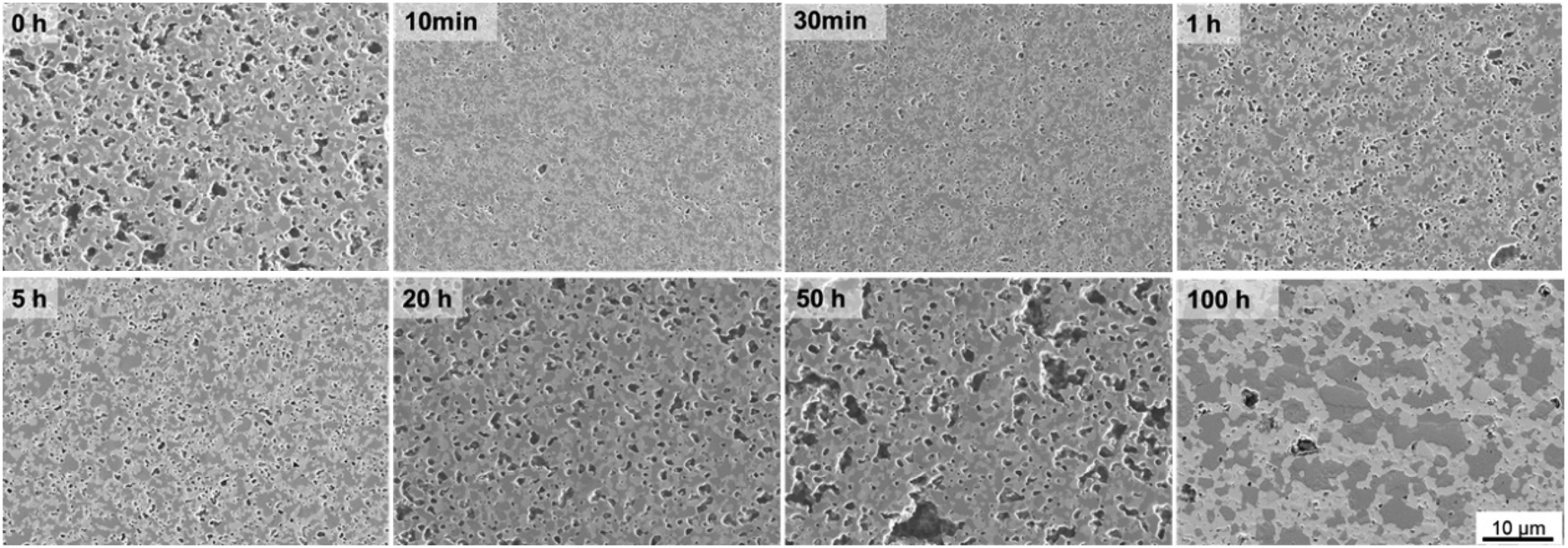 | ||
| Fig. 5 SE-images of cross-section 60CGO20–FCO composite sintered at 1200 °C for different dwell time. | ||
All samples fired below 5 hours exhibit a slightly higher level of open porosity with a homogeneous distribution of pores, corresponding to a densification ratio of 84–90%. Pores are also present in samples fired for >5 h, however, most of them are closed and large, which could be due to the clustering of the pores: the longer sample was held in the furnace, the more pores were united into larger ones, building a connected network. At dwell times >50 hours the pores are possibly unstable and hence both grain growth and pore removal can be observed, similar as was observed elsewhere.39 With a dwell time below 5 hours there is no significant influence on the pore size, it remains almost constant (0.1 μm) and afterwards increases up to 0.5 μm at 50 h dwell time, followed by shrinkage of the pore size to 0.27 μm at 100 h dwell time.
Such behaviour points out that by reaching 1200 °C the process of pore clustering starts from 5 h dwell time, with simultaneous loss in material density. Based on the analysis, three main regions corresponding to the governing process can be distinguished: densification, pore-growth, and drastic grain growth, as shown in Fig. 4.
Fluorite, spinel, and orthorhombic perovskite phases are homogeneously distributed in three samples sintered for 1, 5, and 100 hours at 1200 °C as identified by TEM (EFTEM) analysis shown in Fig. 6. According to the EFTEM elemental mapping, the Fe and Co maps show a non-uniform distribution in the spinel phase, which can be subdivided into two spinel types: a normal Co-rich and an inverse Fe-rich spinel, which is in agreement with the fact that two spinel phases coexist in the composite after the phase interaction, regardless of the dwell time. The high-temperature CoO grains are seen on the elemental map of a 100 hours fired sample. Grains are mostly situated nearby the Co-rich spinel phases, while Fe-rich spinel phases are neighbours to GCFCO-like grains.
 | ||
| Fig. 6 Energy-filtered TEM (EFTEM) analysis of 60CGO20–FCO composite sintered at 1200 °C for 1 h, 5 h and 100 h, identifying three main phases: Gd-doped ceria (red), Fe/Co-oxide (green) and the GdFeO3-based perovskite (bright magenta). | ||
Further electron diffraction analysis performed on samples sintered for 1 and 100 hours showed the presence of three main phases in both and reveals grains of an additional phase only in the 100 hours sintered sample, as shown exemplarily in Fig. 7.
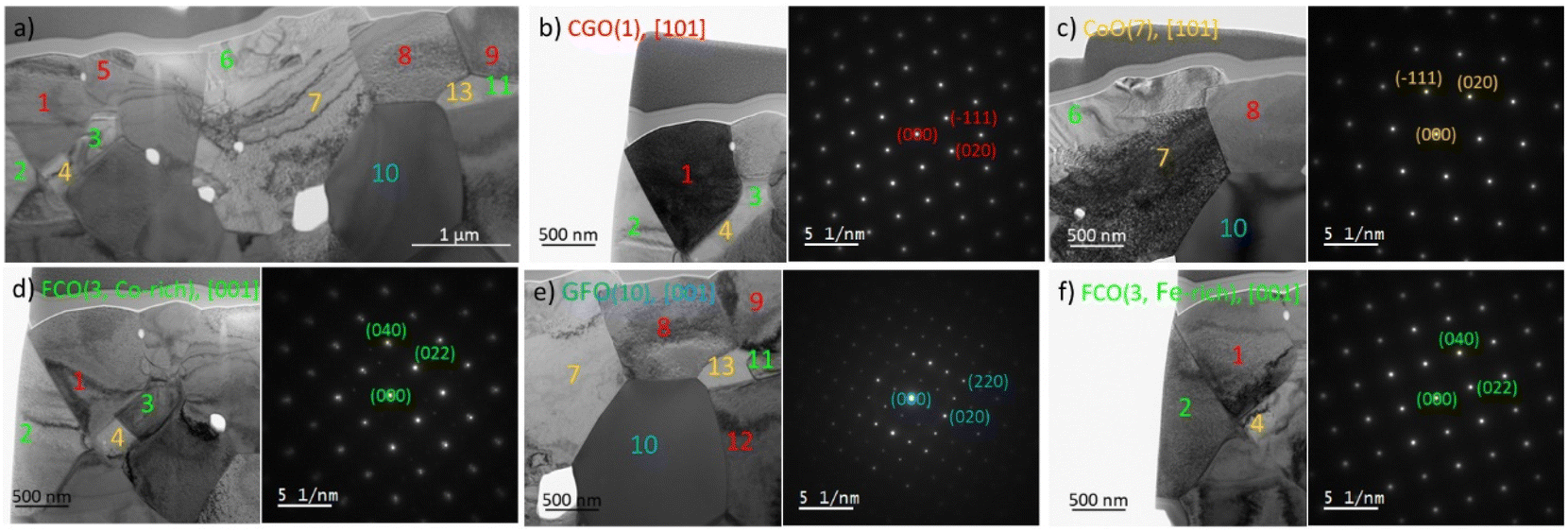 | ||
| Fig. 7 Diffraction analysis of the 60CGO20–FCO composite sintered at 1200 °C for 100 h, (a) overview of investigated sample with fluorite (red), Fe/Co spinel (green), perovskite (blue) and rock salt (orange) phases, (b)–(f) individual analysis of each grain. | ||
Together with the element mapping, the crystal structure fitting the best with our experimental results is the rock salt phase with the cubic structure. All observed CoO grains are contiguous to the spinel grains, namely cobalt-rich ones, and do not form any connected network for potential transport. CoO is the product of the Co3O4 reduction and this transformation starts already at 400 °C.64,65 At sintering temperature (1200 °C) CoO and Co3O4 are in equilibrium according to the phase diagram.47 However, below approx. 950 °C a coexistence of Co3O4 and FCO is thermodynamically favoured. Please note, that Co3O4 here refers to the crystal structure and that Fe is partly substituting Co. Since the Fe/Co-ratio in both Co3O4 and FCO varies with temperature, rearranging of the cations must occur during the cooling process, which obviously is not completed in this composite.64,65
3.2. Cooling method
Since CoO is the product of incomplete reduction of the spinel phase, some further investigation was performed to study the connection of cooling conditions with the remaining monoxide fraction. Four 100 hours sintered samples were cooled differently and analysed by XRD and Rietveld refinement. Prior to describing the results of the microstructural characterization of the 100 h sintered samples, it is important to clarify the terminology which is summarised in Table 2 in experimental section. The Slow cooling sintering program includes an additional step at 850 °C for 100 h; Fast cooling is the program, where the sample was taken out of the furnace after 100 hours sintering and cooled in ambient air, and Quenching is the sample quenched in water at room temperature after holding it for 100 hours at 1200 °C.SEM analysis in Fig. 8 shows rather large islands of the spinel phase within the microstructure and unevenly large grains of a GCFCO emerged phase in all investigated samples sintered for 100 h with different cooling cycles. In all 100 hours fired pellets the grains are closely packed and uniformly distributed. The samples are free of cracks or any other visual defects. The difference in the phase distribution is minor for all 100 h sintered samples as well as the average grain size, which is in the range of 1.2 ± 0.1 μm for all samples.
 | ||
| Fig. 8 BSE-images of surface morphology of 60CGO20–FCO subjected to different heat treatment in the furnace at (a) 1200 °C for 100 h and subsequent 850 °C for 100 h with standard cooling; (b) 1200 °C for 100 h with free cooling in air (c) 1200 °C for 100 h with quenching in water. | ||
All samples sintered for 100 hours show a low level of porosity (1–5.5%) with quite small pores (0.1–0.2 μm). There is no significant influence of the cooling method on the pore size, see Fig. 9. The sample with an additional step at 850 °C for 100 h has slightly larger pores and grains, but, however, a significantly lower level of porosity (∼1%) compared to other samples, resulting in a slightly improved relative density of 94.8%, while others have ∼92–93%. The porosity has been estimated from the SEM images via image analysis, while the relative density was estimated geometrically from the dimensions and weight of the as-sintered pellets. Moreover, the limited number of characterized images might however induce inaccuracy in the porosity calculations. The character of the pores is also slightly different. All 100 h fired samples contain both types of pores intergranular and intragranular (predominantly located in the spinel phase), while the pellet with the 850 °C dwell step contains pores located along the grain boundaries. Due to slow diffusion kinetics the removal of the pores in the grain core is almost impossible, while the reduction of the pores along the grain boundary is still possible via grain boundary diffusion at the latter sintering stage. Hence this additional step helps to eliminate pores in the grain core and along the grain boundaries, improving the quality of the final microstructure.
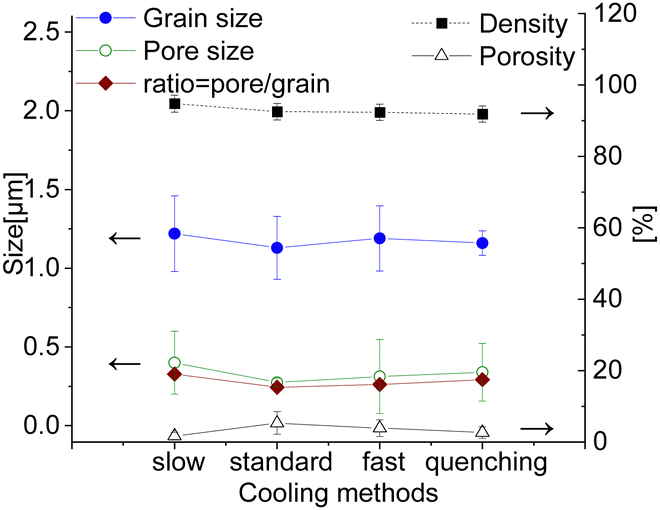 | ||
| Fig. 9 Grain size, pore size, relative density and porosity of the 100 h sintered samples cooled differently (solid lines are a guide to the eye). | ||
According to the Fe–Co–O phase diagram,46,47 the partial reduction of the spinel into a high-temperature Co/Fe monoxide phase with a rock salt structure can occur at 1200 °C sintering temperature.11,66 In the Co–O phase, the most thermodynamically stable phases are the cubic CoO and Co3O4 oxides and the transition between these two oxides is reversible.64,65,67 A slow rate of 0.5 K min−1 between 900 and 800 °C was implemented in the cooling cycle to ensure its complete reoxidation to a spinel phase that is stable at target operating temperatures (600–1000 °C). Generally, this slow cooling helps the reoxidation process and Co/Fe monoxide is no more detectable (<1 wt%), as can be seen for the sample sintered for 5 hours. The resulting XRD patterns are shown in Fig. 10. The CoO peaks were observed in the 100 hours fired samples subjected to quenching, cooling in ambient air, and the standard cooling program. Additionally, in the Slow cooling program, Co monoxide is completely oxidized and only fluorite, spinel, and perovskite phases are present, which is almost identical to the 5 hours sintered sample. Thus, this additional step in the sintering program helps to eliminate this high-temperature CoO phase and preserve normal and inverse spinel structures.
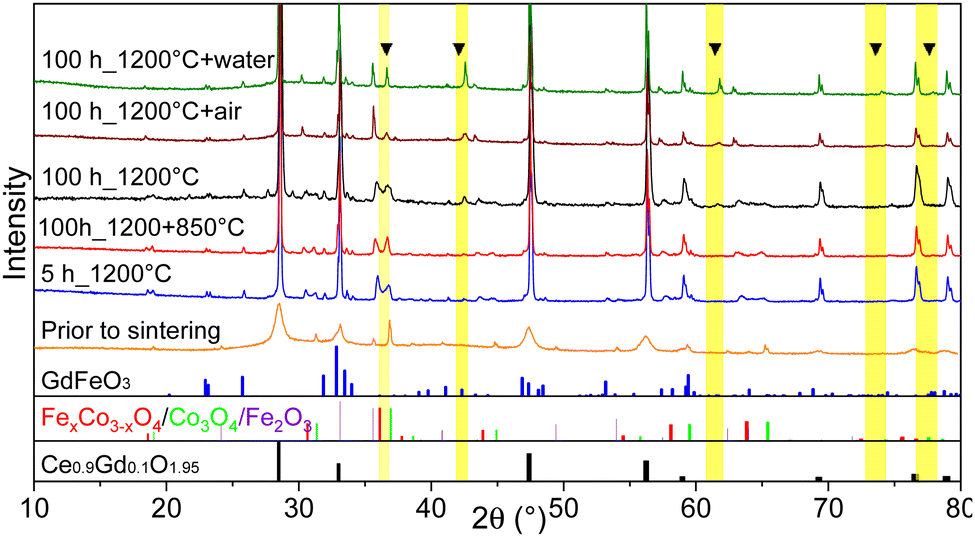 | ||
| Fig. 10 XRD patterns of 60CGO20–FCO composite subjected to different heat treatment and cooling methods. At the bottom, reference reflections are shown for the perovskite in blue, spinel in red and green, and fluorite in black. | ||
XRD and Rietveld refinement of the surface and bulk of each sample shows a small difference in the amount of fluorite phase. In the electron-conducting phases, however, the difference is more significant across the sintered pellet. While a higher amount of GCFCO can be found in the bulk, the spinel amount is higher on the surface. The Rietveld refinement confirmed the presence of the CoO in all samples fired for 100 h, except the sample cooled slowly as depicted in Fig. 11.
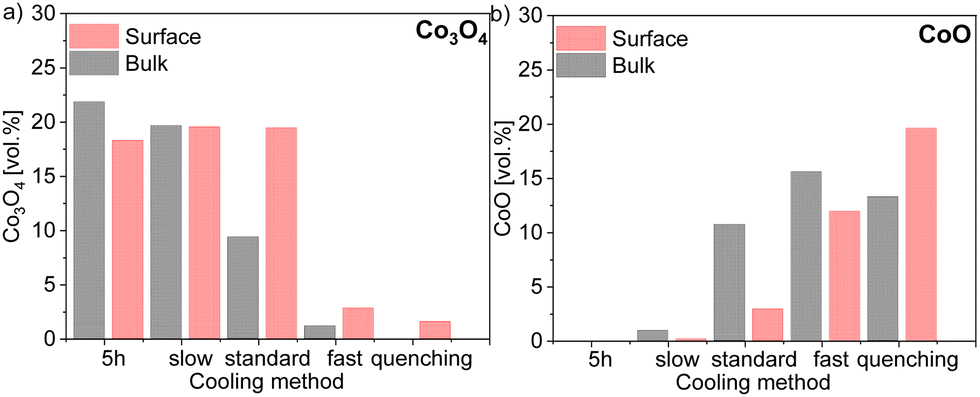 | ||
| Fig. 11 Volume fraction of 60CGO20–FCO composite subjected different heat treatment and cooling methods in (a) Co3O4 and (b) CoO amount comparison in the bulk and surface. | ||
In this pellet, only 0.2 vol% and 0.5 vol% of the monoxide was estimated on the surface and in the bulk, respectively, which is below the detection limit and thus will further be neglected. As expected, the maximum CoO fraction is found in the quenched sample and the fast cooled pellets, followed by the sample subjected to standard cooling in the furnace. The oxidation process of CoO on the surface and in the bulk encompasses different reactions and strongly depends on the diffusion paths.67 The volume fraction of CoO in the bulk is higher compared to that on the surface in pellets cooled slow, standard, and fast, as shown in Fig. 11. Due to lack of diffusion time for the CoO oxidation reaction, a certain amount of CoO remains unoxidized in the bulk.
The rock salt phase itself was probably completely oxidized into Co3O4 during the slow cooling process, due to free access to the oxygen in the air.46 In contrast, the quenched sample, where the oxidation process was terminated after 100 hours, contains a higher amount of the CoO on the surface, compared to that in the bulk. These results again support the theory that too drastic cooling gives no time for the reaction in the CoO–Co3O4 system, ending up in the high amount of 20 vol% of residual high-temperature CoO in the surface skin layer. The amount of the Co3O4 phase is strongly affected by heat treatment and the fraction of the rock salt: the amount of CoO increases, while Co3O4 decreases. Based on our observation, the rearranging of the ions happens in the cooling phase until the high-temperature phase is completely oxidized. Hence, slow cooling with an additional step is required in the sintering profile, to control the phase transformation and final morphology of the dual- or multi-phase composite. The fluorite lattice parameter remains constant independent of the cooling cycle. While the fluorite and CoO lattice parameter is almost identical for all samples in the bulk and surface, the spinel lattice parameter shows some differences, pointing out a clear trend, as shown in Fig. 12. An increase in the cooling rate leads to an increase in the lattice parameter for the iron-rich spinel and a slight increase in the cobalt-rich spinel phases, which can be attributed to the oxidation reactions upon the cooling process.68 The larger lattice parameter indicates a higher iron content in the Fe–Co spinel phase according to Vegard's law, hence by an increase in the cooling rate an increase of the iron-rich spinel fraction in the bulk is observed with a maximum in the quenched sample. This can be explained by the difference of the cations’ mobility.69 Cobalt cations are diffusing faster so they are migrating to the surface forming CoO or/and Co3O4 resulting in an increase of the Co-containing phases at the surface (Fig. 12). In consequence, the Fe-containing phases (FCO and GCFCO) are more present in the bulk and increase with a faster cooling rate.
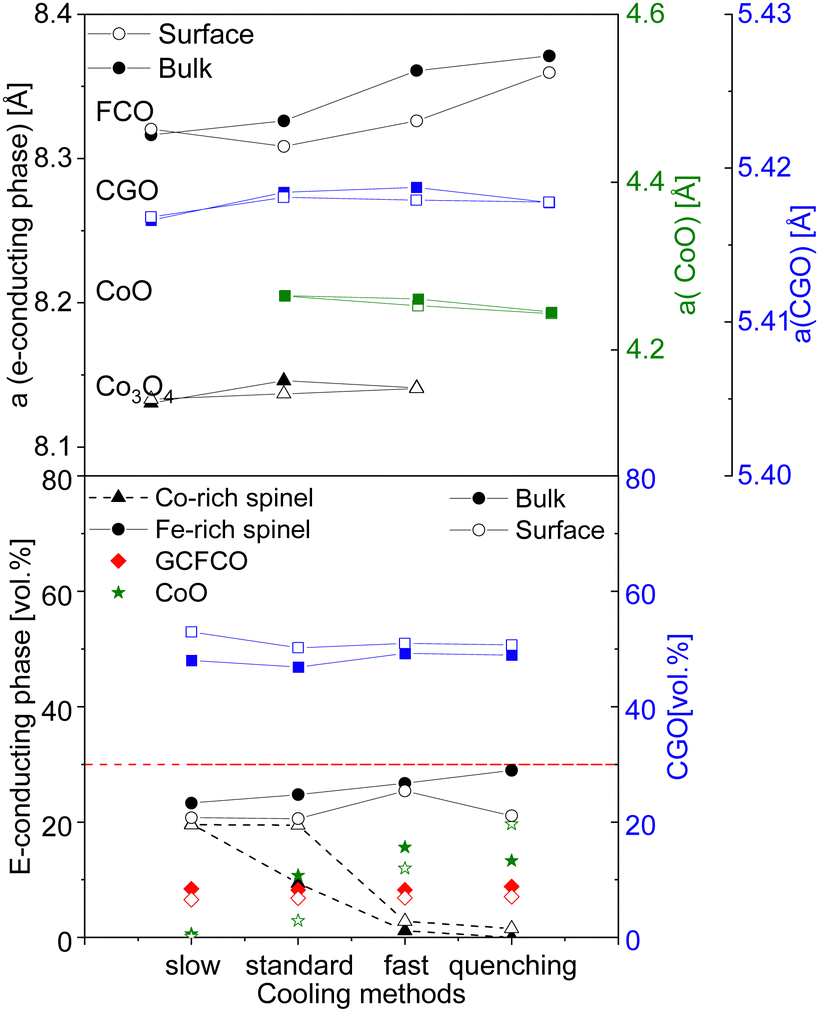 | ||
| Fig. 12 Lattice parameter difference and volume fraction of spinels and CGO in the bulk and surface (solid lines are a guide to the eye). | ||
3.3. Functional properties
The temperature-dependent oxygen permeation of the composites with nominal 60CGO20–FCO sintered at 1200 °C for different times below 1 h and above 1 h are shown along with their activation energies in Fig. 13, respectively. At short dwell time (<1 hour) all Arrhenius plots of the examined composites show a kink at 880 °C indicating a change in the rate limiting process. At 1 and 5 hours, dwell time the change in activation energy is at lower temperature, i.e., 800 °C, and much less pronounced. It completely disappears at longer dwell times ≥20 hours. Typically, in OTM this behaviour is ascribed to a change from bulk diffusion limitation at high temperature to surface exchange limitation at lower temperature. At high temperatures (>800 °C) the permeation rates for all examined composite membranes are almost identical with Ea in the range of 61–76 kJ mol−1, which indeed can be attributed to the bulk transport, i.e. the ionic conductivity of the fluorite (EaCGO20 = 72 kJ mol−154,70). The lower Ea of ∼55 kJ mol−1 for the 0 h sintered sample might be due to insufficient sintering and increased porosity impeding electronic conductivity. At low temperatures (650–800 °C) Ea is much higher 112–117 kJ mol−1 for short dwell time (<1 hour), indicating rate limitations due to surface exchange kinetics although all membranes were coated with porous LSCF activation layers on both sides.
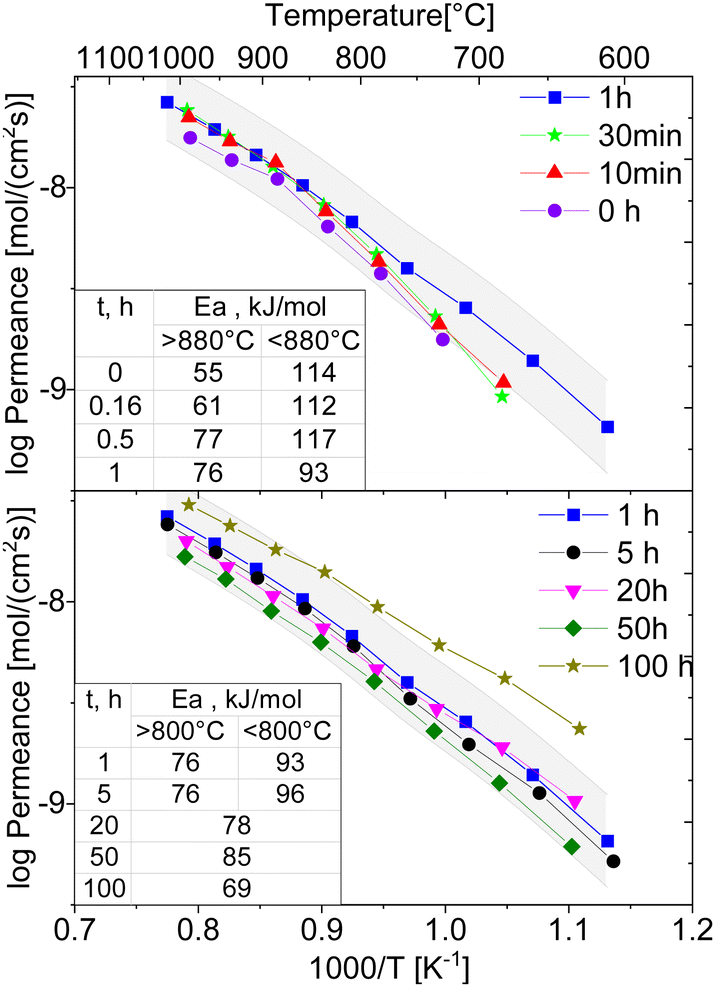 | ||
| Fig. 13 Arrhenius-type plots of oxygen flux density of the 60CGO20–FCO composite sintered at 1200 °C for 0, 1 h and 10, 30 min, 1, 5, 20, 50 and 100 hours (solid lines are a guide to the eye, grey area is ±10% error interval for the 1-hour measurement). | ||
The resulting oxygen permeances of the coated membranes are mostly within the estimated experimental error of ±10%. Along the time range between 0 and 5 h dwell time, there is a very slight increase in permeation until 1 hour followed by a slight decrease until 50 hours dwell time. But, at 100 h dwell time a sharp increase in oxygen permeation is observed with the maximum of jO2 = 0.13 mL cm−2 min at 950 °C, Fig. 14, which correlates with the grain growth shown in Fig. 4.
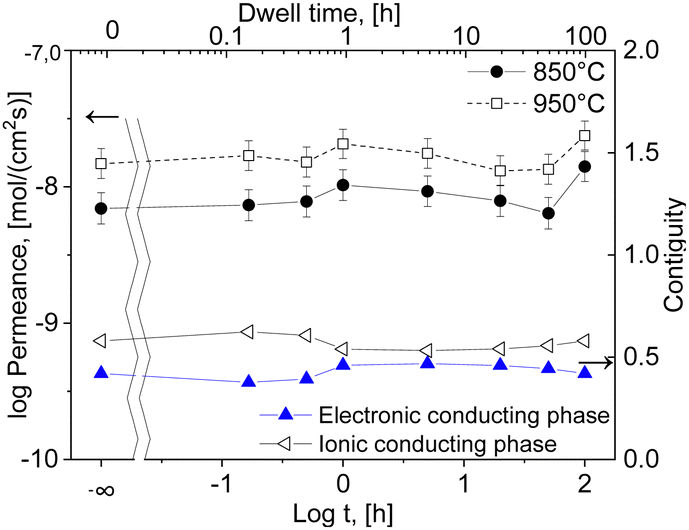 | ||
| Fig. 14 Permeation and contiguity of the phases in the 60CGO20–FCO composite sintered at 1200 °C with activation energy of the permeation depending on the dwell time (solid lines are a guide to the eye). | ||
The main challenge of the dual- and multiphase materials design is maximizing the ionic conducting volume without losing too much electronic conduction determined by the electronic conductivity of the selected phases as well as continuous percolation paths for the transport. While percolation is considering solely the volume fractions, the “contiguity” was introduced in71,72 additionally accounting for the grain sizes of the individual phases. It can be expressed as the fraction of the total interconnected surface area of one phase shared with particles of the same phase and helps to estimate percolation and conducting properties within the dual-phase OTM composite.73 Therefore, the contiguity of both the ionic conducting phase as well as the sum of electronic conducting phases was determined and shown in Fig. 14.
| Ci + Ce = 1 |
| Vi + Ve = 1 |
with
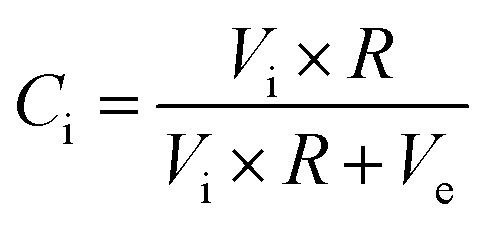
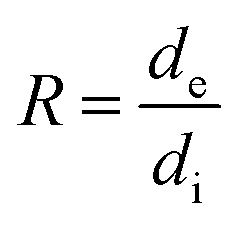 , Vi and Ve are the volume fraction of the ionic- and electron-conducting phases, respectively.
, Vi and Ve are the volume fraction of the ionic- and electron-conducting phases, respectively.
Indeed, the contiguity of electron conducting phase for the short dwell times is lower compared to higher dwell times, Fig. 14, which might be due to not fully sintered phases. However, it cannot explain the high permeance after 100 hours sintering, where the contiguity remains stable compared to 1–5 hours sintering.
The permeation of all composites at 850 °C with respect to the electron-conducting phase volume fraction is shown in Fig. 15. Among the various dwell times, the best results are found for the 100-h sintered sample of 60:40 ratio, which shows a stable dense microstructure with large grains and a low level of porosity. The large grain size is favourable in this case because less grain boundaries are involved, which typically act as barrier for the transport of charged carriers in particular ions.13
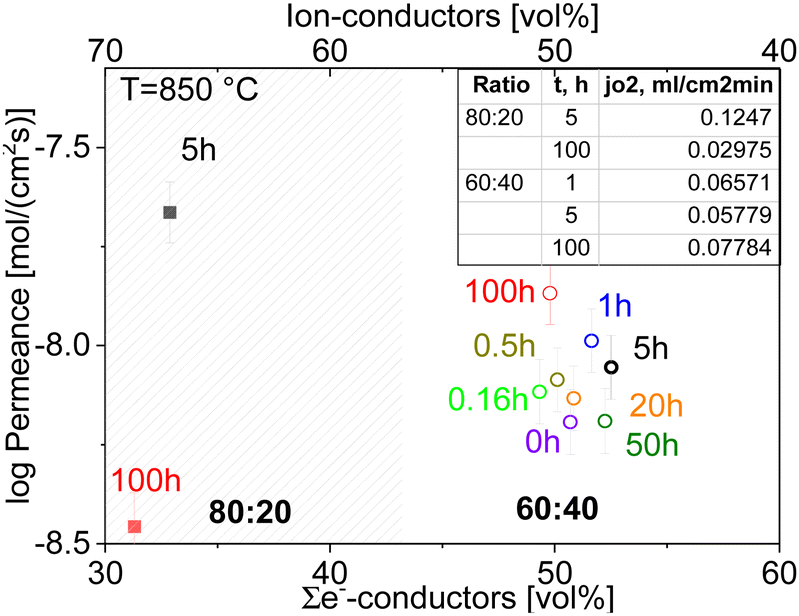 | ||
| Fig. 15 Comparison of the final permeation performance permeance of CGO20–FCO composite with ratio 60:40 and 80:20 sintered with various dwell times depending on the volume fraction of e−-conducting phases. | ||
Although the oxygen permeance of the 100 h fired sample is the best among the differently sintered multiphase 60CGO20–FCO samples, comparison of the performance with other composite variations14 shows the importance of the microstructure design. In general, the oxygen flux comparison of 60:40 with 80:20 fluorite: spinel weight ratio (Fig. 16) sintered at 1200 °C for 5 hours shows, that the reduction of the electron conducting phase volume effectively helps to improve permeation performance, reaching maximum jO2 = 0.12 and 0.22 mL cm−2 min for the 80 wt% CGO20 in composite with fine grain size at 850 °C and 1000 °C respectively.
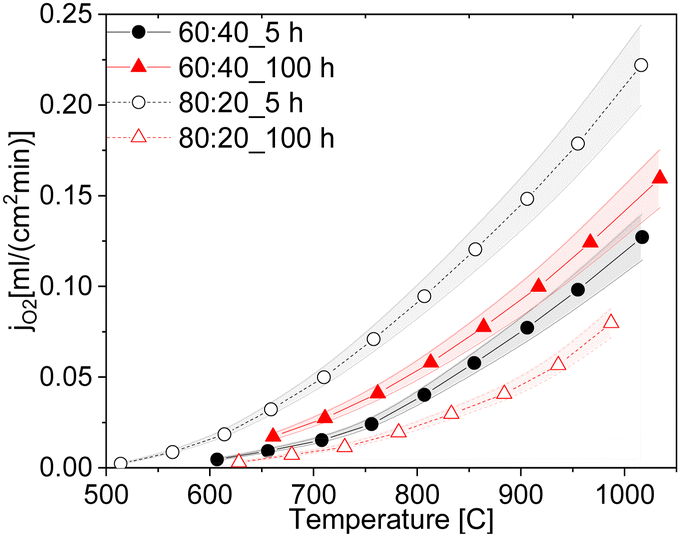 | ||
| Fig. 16 Oxygen permeation rates of the ceria-based materials with 60:40 and 80:20 ratios sintered at 5 and 100 h (solid lines are a guide to the eye). | ||
Interestingly, the 80CGO20–FCO fired for 100 hours showed a drastic drop in the oxygen flux. While in a 60:40 ratio, the amount of e-conducting phase is sufficient to provide continuous paths, regardless of the grain size, in 80:20 the large grains with a size of approx. 1.1 μm possibly lead to the insulation of the electron-conducting phase. Obviously, the coarser microstructure leads to lower electronic contiguity and overall percolation. In this case, the lower number of grain boundaries plays only a minor role.
4. Conclusions
Dual-phase dense 60 wt% Ce0.8Gd0.2O2-δ–40 wt% FeCo2O4 membranes were successfully synthesized by solid-state reactive sintering with one-step thermal processing at 1200 °C using commercially available Ce0.8Gd0.2O2−δ, Co3O4 and Fe2O3 as raw materials. The influence of the sintering parameters was systematically studied by varying the dwelling time and cooling methods.It was found that already at 0 h dwell time, the phase interaction was completed, and all phases were present: fluorite, spinel, and perovskite. All samples sintered <1 h dwell time, however, reveal insufficient densification, i.e., a high porosity and not connected matrix of the phases. That emphasizes that a longer dwell time is required to obtain a close-packed and uniformly distributed microstructure. The 100 hours sintered sample shows the best permeation performance with jO2 = 0.078 mL cm−2 min at 850 °C thanks to the improved microstructure with enlarged grains. However, the implementation of such conditions is not reasonable for future scale-up towards potential commercialization, due to very long dwell time. Although the 80:20 composite sintered for 100 h has a similar grain size, its performance is very poor. Apparently, the isolation of the minor phase (electron-conducting phase) occurs, which negatively affects the percolation and hence oxygen flux. Close to the percolation limit grain and pore size as well as porosity level are very important features in the microstructural design of composite membranes.
The influence of the sintering profile on the microstructural features such as grain size distribution, pore volume and size, as well as density were discussed. The initial mix of the oxides undergoes some modification during the sintering process and results in a multi-phase microstructure with coexistence of fluorite, perovskite, and two spinel types, except the 100 hours fired pellets, where an additional high-temperature CoO phase is observed. The grain growth as well as pore growth are dependent on the dwell time. The mean grain size increased moderately from 0.57–0.77 μm between 1 and 50 hours dwell time and reached its maximum of 1.13 μm in a 100 h fired sample. The relative density maximum of 93 and 94% were obtained for the 5 and 100 h sintered samples. The de-densification phenomenon is observed for the 20 and 50-h sintered samples, where intensive grain and pore growth occurs simultaneously.
TEM analysis indicated a few CoO grains located next to Co3O4 spinel grains in the 100 h fired sample. In this case, residual high-temperature CoO is identified due to the long dwell time, where a higher amount of CoO is formed and does not have enough time to re-oxidize during cooling completely. The control of the cooling rate can thus govern the process of the Co rearranging between the Co3O4 and CoO phases until the equilibrium is reached. Moreover, it was observed that slow cooling with an additional step for 100 hours at 850 °C helps to eliminate the CoO phase and preserve the desired fluorite, spinel, and perovskite phase mixture. This emphasizes the importance of the slow cooling for the Fe–Co spinel within our composite system.
Among all chosen conditions, the intermediate time of 5 hours at 1200 °C sintering temperature is the most suitable for the CGO20–FCO multi-phase composite showing good structure-related properties with optimised grain size and low porosity and oxygen permeation with oxygen flux of jO2 = 0.06 mL cm−2 min at 850 °C. Moreover, a cooling rate of 0.5 K min−1 between 900 and 800 °C is sufficient to re-oxidize the CoO phase to get the desired spinel structure. Future study of the stability and durability in application-oriented conditions is required in order to exploit such complex composite materials.
Author contributions
Liudmila Fischer: investigation & writing – original draft preparation; Ke Ran: investigation & writing – original draft preparation, Doris Sebold: investigation, Patrick Behr: investigation, Stefan Baumann: conceptualization, investigation, funding acquisition & writing – original draft, Joachim Mayer: funding acquisition, & writing – review & editing, Arian Nijmeijer: supervision & writing – review & editing, Henny J.M. Bouwmeester: conceptualization, Oliver Guillon resources, supervision & writing – review & editinZg; Wilhelm A. Meulenberg: resources, supervision & writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 387282673. Ke Ran acknowledges support by the Bundesministerium für Bildung und Forschung (NEUROTEC, 16ME0399, and 16ME0398K). We would like to thank Mr S. Heinz for the assistances in lab works, sample preparations and permeation measurements.Notes and references
- J. Sunarso, S. Baumann, J. M. Serra, W. A. Meulenberg, S. Liu, Y. S. Lin and J. C. Diniz da Costa, J. Membr. Sci., 2008, 320, 13 CrossRef CAS.
- R. Kiebach, S. Pirou, L. Martinez Aguilera, A. B. Haugen, A. Kaiser, P. V. Hendriksen, M. Balaguer, J. García-Fayos, J. M. Serra, F. Schulze-Küppers, M. Christie, L. Fischer, W. A. Meulenberg and S. Baumann, J. Mater. Chem. A, 2022, 10, 2152 RSC.
- G. Chen, A. Feldhoff, A. Weidenkaff, C. Li, S. Liu, X. Zhu, J. Sunarso, K. Huang, X.-Y. Wu, A. F. Ghoniem, W. Yang, J. Xue, H. Wang, Z. Shao, J. H. Duffy, K. S. Brinkman, X. Tan, Y. Zhang, H. Jiang, R. Costa, K. A. Friedrich and R. Kriegel, Adv. Funct. Mater., 2022, 32, 2105702 CrossRef CAS.
- W. Bai, J. Feng, C. Luo, P. Zhang, H. Wang, Y. Yang, Y. Zhao and H. Fan, Int. J. Hydrogen Energy, 2021, 46, 36257 CrossRef CAS.
- J. Zhao, Y. Pang, C. Su, S. Jiang and L. Ge, Energy Fuels, 2023, 37, 7042 CrossRef CAS.
- A. Leo, S. Liu and J. C. Diniz da Costa, Int. J. Greenhouse Gas Control, 2009, 3, 357 CrossRef CAS.
- S. Smart, C. X. C. Lin, L. Ding, K. Thambimuthu and J. C. Diniz da Costa, Energy Environ. Sci., 2010, 3, 268 RSC.
- M. Balaguer, J. García-Fayos, C. Solís and J. M. Serra, Chem. Mater., 2013, 25, 4986 CrossRef CAS.
- J. Garcia-Fayos, M. Balaguer, S. Baumann and J. M. Serra, J. Membr. Sci., 2018, 548, 117 CrossRef CAS.
- H. Takamura, K. Okumura, Y. Koshino, A. Kamegawa and M. Okada, J. Electroceram., 2004, 13, 613 CrossRef CAS.
- M. Ramasamy, Dual Phase Oxygen Transport Membrane for Efficient Oxyfuel Combustion, Forschungszentrum Jülich GmbH, Jülich, 2016 Search PubMed.
- W. Fang, F. Liang, Z. Cao, F. Steinbach and A. Feldhoff, Angew. Chem., Int. Ed., 2015, 54, 4847 CrossRef CAS PubMed.
- Y. Lin, S. Fang, D. Su, K. S. Brinkman and F. Chen, Nat. Commun., 2015, 6, 6824 CrossRef CAS PubMed.
- L. Fischer, K. Neuhaus, C. Schmidt, K. Ran, P. Behr, S. Baumann, J. Mayer and W. A. Meulenberg, J. Mater. Chem. A, 2022, 10, 2412 RSC.
- F. Zeng, S. Baumann, J. Malzbender, A. Nijmeijer, L. Winnubst, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Membr. Sci., 2021, 628, 119248 CrossRef CAS.
- A. Arratibel Plazaola, A. Cruellas Labella, Y. Liu, N. Badiola Porras, D. Pacheco Tanaka, M. Sint Annaland and F. Gallucci, Processes, 2019, 7, 128 CrossRef.
- M. Coduri, S. Checchia, M. Longhi, D. Ceresoli and M. Scavini, Front. Chem., 2018, 6, 526 CrossRef CAS PubMed.
- J. H. Joo, K. S. Yun, Y. Lee, J. Jung, C.-Y. Yoo and J. H. Yu, Chem. Mater., 2014, 26, 4387 CrossRef CAS.
- S. Presto, C. Artini, M. Pani, M. M. Carnasciali, S. Massardo and M. Viviani, Phys. Chem. Chem. Phys., 2018, 20, 28338 RSC.
- J. A. Kilner and M. Burriel, Annu. Rev. Mater. Res., 2014, 44, 365 CrossRef CAS.
- J. Koettgen, S. Grieshammer, P. Hein, B. O. H. Grope, M. Nakayama and M. Martin, Phys. Chem. Chem. Phys., 2018, 20, 14291 RSC.
- T. Kiefer, Entwicklung neuer Schutz- und Kontaktierungsschichten für Hochtemperatur-Brennstoffzellen, Forschungszentrum, Zentralbibliothek, Jülich 2008.
- A. J. Samson, M. Søgaard and P. Vang Hendriksen, J. Membr. Sci., 2014, 470, 178 CrossRef CAS.
- N. Bahlawane, P. H. T. Ngamou, V. Vannier, T. Kottke, J. Heberle and K. Kohse-Höinghaus, Phys. Chem. Chem. Phys., 2009, 11, 9224 RSC.
- M. Ramasamy, E. S. Persoon, S. Baumann, M. Schroeder, F. Schulze-Küppers, D. Görtz, R. Bhave, M. Bram and W. A. Meulenberg, J. Membr. Sci., 2017, 544, 278 CrossRef CAS.
- L. Fischer, K. Ran, C. Schmidt, K. Neuhaus, S. Baumann, P. Behr, J. Mayer, H. J. M. Bouwmeester, A. Nijmeijer, O. Guillon and W. A. Meulenberg, Membranes, 2023, 13 Search PubMed.
- L. Fischer, K. Ran, C. Schmidt, K. Neuhaus, S. Baumann, P. Behr, J. Mayer, H. J. Bouwmeester, A. Nijmeijer, O. Guillon and W. A. Meulenberg, Open Ceram., 2024, 17, 100519 CrossRef CAS.
- X. Zhu and W. Yang, AIChE J., 2008, 54, 665 CrossRef CAS.
- C. D. Lorenz and R. M. Ziff, J. Chem. Phys., 2001, 114, 3659 CrossRef CAS.
- M. Ge, X. Huang, H. Yan, D. Gursoy, Y. Meng, J. Zhang, S. Ghose, W. K. S. Chiu, K. S. Brinkman and Y. S. Chu, Commun. Mater., 2022, 3 Search PubMed.
- Z. Zhang, W. Zhou, Y. Chen, D. Chen, J. Chen, S. Liu, W. Jin and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 22918 CrossRef CAS PubMed.
- K. Ran, F. Zeng, L. Fischer, S. Baumann, W. A. Meulenberg, K. Neuhaus and J. Mayer, Acta Mater., 2022, 234, 118034 CrossRef CAS.
- K. Yasuda, K. Uemura and T. Shiota, J. Phys.: Conf. Ser., 2012, 339, 12006 CrossRef.
- K. Raju, S. Kim, C. J. Hyung, J. H. Yu, Y.-H. Seong, S.-H. Kim and I.-S. Han, Ceram. Int., 2019, 45, 1460 CrossRef CAS.
- J. H. Joo, G. S. Park, C.-Y. Yoo and J. H. Yu, Solid State Ionics, 2013, 253, 64 CrossRef CAS.
- Q. Li, X. Zhu, Y. He, Y. Cong and W. Yang, J. Membr. Sci., 2011, 367, 134 CrossRef CAS.
- V. V. Kharton, Solid State Ionics, 2003, 160, 247 CrossRef CAS.
- H. Luo, H. Jiang, K. Efimov, F. Liang, H. Wang and J. Caro, Ind. Eng. Chem. Res., 2011, 50, 13508 CrossRef CAS.
- J.-L. Shi, Y. Deguchi and Y. Sakabe, J. Mater. Sci., 2005, 40, 5711 CrossRef CAS.
- K.-T. Lee, D.-K. Kim, J.-H. Park and I.-J. Shon, Ceram. Int., 2009, 35, 1345 CrossRef CAS.
- T. Zhang, P. Hing, H. Huang and J. Kilner, J. Eur. Ceram. Soc., 2002, 22, 27 CrossRef CAS.
- V. Gil, J. Tartaj, C. Moure and P. Durán, J. Eur. Ceram. Soc., 2006, 26, 3161 CrossRef CAS.
- T. Zhang, P. Hing, H. Huang and J. Kilner, J. Eur. Ceram. Soc., 2001, 21, 2221 CrossRef CAS.
- E. Jud, C. B. Huwiler and L. J. Gauckler, Nippon Seramikkusu Kyokai gakujutsu ronbunshi, 2006, 114, 963 CrossRef CAS.
- F. Zeng, J. Malzbender, S. Baumann, A. Nijmeijer, L. Winnubst, M. Ziegner, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Eur. Ceram. Soc., 2021, 41, 509 CrossRef CAS.
- P. J. Murray and J. W. Linnett, J. Phys. Chem. Solids, 1976, 37, 619 CrossRef CAS.
- A. N. Hansson, S. Linderoth, M. Mogensen and M. A. Somers, J. Alloys Compd., 2005, 402, 194 CrossRef CAS.
- T. W. Ridler and S. Calvard, IEEE Trans. Syst. Man Cybern., 1978, 8, 630 Search PubMed.
- A. Kovács, R. Schierholz and K. Tillmann, J. Large Scale Res. Facil., 2016, 2 Search PubMed.
- F. Zeng, J. Malzbender, S. Baumann, W. Zhou, M. Ziegner, A. Nijmeijer, O. Guillon, R. Schwaiger and W. Albert Meulenberg, J. Am. Ceram. Soc., 2021, 104, 1814 CrossRef CAS.
- T. Ferreira, J. C. Waerenborgh, M. Mendonça, M. R. Nunes and F. M. Costa, Solid State Sci., 2003, 5, 383 CrossRef CAS.
- M. G. Brik, A. Suchocki and A. Kamińska, Inorg. Chem., 2014, 53, 5088 CrossRef CAS PubMed.
- Y. Ji, J. Kilner and M. Carolan, J. Eur. Ceram. Soc., 2004, 24, 3613 CrossRef CAS.
- V. V. Kharton, F. M. Figueiredo, L. Navarro, E. N. Naumovich, A. V. Kovalevsky, A. A. Yaremchenko, A. P. Viskup, A. Carneiro, F. M. B. Marques and J. R. Frade, J. Mater. Sci., 2001, 36, 1105 CrossRef CAS.
- C. Artini, M. Pani, M. M. Carnasciali, J. R. Plaisier and G. A. Costa, Inorg. Chem., 2016, 55, 10567 CrossRef CAS PubMed.
- F. Zeng, Mechanical reliability and oxygen permeation of Ce0.8Gd0.2O2−δ–FeCo2O4 dual-phase composite, University of Twente, Enschede, 2021 Search PubMed.
- S. J. Hong and A. V. Virkar, J. Am. Ceram. Soc., 1995, 78, 433 CrossRef CAS.
- M. N. Rahaman, Ceramic Processing and Sintering, CRC Press, 2017 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- B. Stawarczyk, M. Ozcan, L. Hallmann, A. Ender, A. Mehl and C. H. F. Hämmerlet, Clin. oral investig., 2013, 17, 269 CrossRef PubMed.
- D. Pérez-Coll, E. Sánchez-López and G. C. Mather, Solid State Ionics, 2010, 181, 1033 CrossRef.
- J. L. Shi, J. Mater. Res., 1999, 14, 1389 CrossRef CAS.
- F. Schulze-Küppers, S. Baumann, F. Tietz, H. Bouwmeester and W. A. Meulenberg, J. Eur. Ceram. Soc., 2014, 34, 3741 CrossRef.
- X. Chen, H. van Gog and M. A. van Huis, J. Mater. Chem. C, 2021, 9, 5662 RSC.
- F.-C. Kong, Y.-F. Li, C. Shang and Z.-P. Liu, J. Phys. Chem. C, 2019, 123, 17539 CrossRef CAS.
- F. Zeng, S. Baumann, J. Malzbender, A. Nijmeijer, L. Winnubst, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Membr. Sci., 2021, 628, 119248 CrossRef CAS.
- M. Żyła, G. Smoła, A. Knapik, J. Rysz, M. Sitarz and Z. Grzesik, Corros. Sci., 2016, 112, 536 CrossRef.
- A. Kaczmarska, Z. Grzesik and S. Mrowec, High Temp. Mater. Processes, 2012, 31, 371 CAS.
- L. Fuhsing, S. Tinkler and R. Diekmann, Solid State Ionics, 1993, 62, 39 CrossRef.
- V. V. Kharton, A. L. Shaula, E. N. Naumovich, N. P. Vyshatko, I. P. Marozau, A. P. Viskup and F. M. B. Marques, J. Electrochem. Soc., 2003, 150, J33 CrossRef CAS.
- J. Gurland, Brown University, Division of Engineering, United States, Office of Ordnance Research, The Measurement of Grain Contiguity in Two-phase Alloys, Division of Engineering, Brown University, 1957.
- Z. Fan, A. P. Miodownik and P. Tsakiropoulos, Mater. Sci. Technol., 1993, 9, 1094 CrossRef CAS.
- F. Zeng, J. Malzbender, S. Baumann, M. Krüger, L. Winnubst, O. Guillon and W. A. Meulenberg, J. Eur. Ceram. Soc., 2020, 40, 5646 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |