
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA paired alkaline electrolyzer for furfural oxidation and hydrogen evolution over noble metal-free NiFe/Ni and Co/MXene catalysts†
Xiaopeng
Liu‡
,
Mohammad
Albloushi‡
,
Michael
Galvin
 ,
Connor W.
Schroeder
,
Yue
Wu
* and
Wenzhen
Li
*
,
Connor W.
Schroeder
,
Yue
Wu
* and
Wenzhen
Li
*
Department of Chemical and Biological Engineering, Iowa State University, 618 Bissell Road, Ames, IA 50011, USA. E-mail: wzli@iastate.edu; yuewu@iastate.edu
First published on 11th October 2024
Abstract
Producing green hydrogen via water electrolysis using renewable energy sources holds promise for a sustainable future. However, current challenges arise from the energy-intensive oxygen evolution reaction (OER) and the potential risks associated with the mixing of H2 and O2. To address these challenges, there has been significant emphasis on replacing the OER with more thermodynamically favorable aldehyde oxidation for the production of carboxylic acids. In this work, we combined a novel two-dimensional (2D) early transition metal carbide (MXene) supported cobalt catalyst (Co/Mo2TiC2-700) for the hydrogen evolution reaction (HER) and a NiFe/Ni foam fabricated by an electrodeposition method for the furfural oxidation reaction (FOR) to design a paired flow electrolyzer. In H-type half-cell tests, the NiFe/Ni foam anode catalyst exhibited a faradaic efficiency (FE) of 47% towards 2-furoic acid (2-FA) and a conversion of 95% with 50 mM furfural at 65 mA cm−2. This FE rose to 97% at a conversion of 67% when the furfural concentration was increased to 150 mM. The optimized Co/Mo2TiC2-700 cathode catalyst exhibited outstanding HER performances of 100% FE towards H2, and low overpotentials of 244 mV and 321 mV at 100 mA cm−2 and 400 mA cm−2, respectively. A two-electrode flow cell with 2 × 2 cm2 electrodes was then assembled for simultaneous electrochemical furfural oxidation and hydrogen evolution. Remarkably, Co/Mo2TiC2-700 outperformed a commercial Pt/C electrode with the same loading of 0.5 mg-metal per cm2 by reducing the cell voltage by 150 mV at a high current density of 300 mA cm−2, while maintaining the FE-to-H2 conversion at >90%. The overall FE increased from 120% to 151% at a high current density of 200 mA cm−2, and the cell voltage dropped to 2.688 V for the electrolyzer with the Co/Mo2TiC2-700 catalyst, compared to 3.185 V when using Pt/C at the cathode.
1. Introduction
Hydrogen holds enormous promise as an energy carrier within environmentally conscientious, efficient, sustainable, and cost-effective energy systems.1 Using renewable sources, such as water or biomass, to produce hydrogen, followed by its application in fuel cells to provide power for stationary and transportation applications, leverages the environmental advantages inherent in transitioning to a hydrogen-based society.2,3 Among the various methods for hydrogen generation, water electrolysis powered by renewable energy stands as a leading and sustainable approach for green hydrogen production.1,4 However, contemporary water electrolysis systems encounter the significant challenge of the high energy costs associated with the thermodynamically and kinetically sluggish oxygen evolution reaction (OER) at the anode.5,6 Furthermore, a critical issue arises from the gas crossover phenomenon, occurring amidst disparate gas pressures, where cathodic hydrogen and anodic oxygen can potentially intermix. This could lead to the formation of explosive H2/O2 mixtures and raise safety concerns. Additionally, the generation of reactive oxygen radicals may damage the ion-exchange membranes,7,8 which, although effective and conventionally used for separating the gas products, are prohibitively expensive.9 Efforts to develop cost-effective and easily manufacturable membrane materials to address this issue remain ongoing endeavors.9,10 One strategy to mitigate the aforementioned issues is to substitute the OER with anodic reactions that offer more favorable thermodynamics and sustainability, and are economically viable by generating valuable products instead of oxygen.11 Despite noteworthy advancements in the development of paired electrolyzers tailored for hydrogen production, the current designed electrolyzers still require electrolytic voltages exceeding 1.0 V even with noble metal catalysts.12–16 Furthermore, due to the limitations on mass transport of reactants in the aqueous phase, the achievable current density typically falls below 100 mA cm−2,11–15 significantly limiting their applicability in industrial-scale settings and underscoring the urgency of further development in this area.The electrochemical oxidation of biorenewable furfural to 2-furoic acid represents an excellent alternative reaction mentioned above. In an early study conducted by Gopalan et al.17 in 1984, a comprehensive examination of the kinetics associated with furfural oxidation was carried out using ceric ammonium sulfate as the oxidizing agent. The findings suggested the involvement of a free-radical mechanism governing the furfural oxidation reaction. Recent investigations also have yielded valuable insights into electrochemical furfural oxidation. Verdeguer et al.18 optimized the catalytic oxidation of furfural to 2-furoic acid, explored a lead–platinum catalyst and identified specific conditions under which the catalytic transformation was maximized.19 Furfural electro-oxidation on gold catalysts exhibited a notable degree of activity and selectivity towards the formation of 2-furoic acid; the analysis of the reaction intermediates revealed that surface furoate species were the predominant entities in the reaction pathway. Building upon this research,20 a novel paired electrochemical conversion process was developed by a different approach, which led to the production of 2(5H)-furanone through anodic oxidation while facilitating the generation of furfuryl alcohol and/or hydrofuroin through cathodic reduction. Electrochemically, in alkaline media, aldehydes are in equilibrium with aldehyde hydrate, and the aldehyde hydrate undergoes oxidation through one electron transfer, which makes it adsorb to the catalyst's surface, and the hydroxide present in the alkaline media undergoes oxidation via one electron transfer and is adsorbed to the catalyst's surface. Finally, carboxylic acid results from H-atom abstraction from the adsorbed species onto the surface.52 In general, non-noble metal catalysts such as Ni-based catalysts are more desirable, except for the drawback that non-noble metal catalysts typically require higher anodic potentials, which may exacerbate the oxygen evolution side reaction (OER).53–55 Another noteworthy challenge encountered in the electro-oxidation process of furfural to 2-furoic acid is the Cannizzaro reaction, a homogeneous aldehyde disproportionation in high-pH solutions and concomitantly generating furfuryl alcohol alongside the desired 2-furoic acid. The Cannizzaro reaction significantly limits the maximal selectivity towards 2-furoic acid, restraining it to a mere 50%.21 Additionally, indirect electrolysis methods involving redox mediators have been studied to mitigate this issue effectively addressing the challenge of achieving a harmonized anode and cathode current alignment under potentiostatic conditions. The cathode exhibits a selectivity of 33.2% for 2-furoic acid, whereas the anode demonstrates a remarkable selectivity of 99.3%. This electrolytic process achieves a notable energy saving of approximately 12.3%. Furthermore, the stability of both the cathode and anode is maintained even after prolonged electrolysis within a flow cell configuration.21 This underscores the robustness of such an electrochemical approach and also highlights the potential for further improvements. For the hydrogen evolution reaction (HER) at the cathode, the current challenges and focus are the electrochemically driven reaction exhibiting thermal efficiency levels below the thermodynamic limits for water splitting, notably falling short of the 1.23 V threshold.22,23 The considerable overpotential of the HER in alkaline electrolytes as compared to that in acidic electrolytes is one of the prominent causes of low energy efficiency of alkaline water electrolyzers.24 Various electrocatalysts have been investigated to decrease the alkaline HER overpotential and accelerate the reaction to achieve current densities relevant at the industrial level, with the primary focus being on noble metal-based catalysts such as Pt, Pd, Ru, and Ir-based catalysts owing to their exceptional intrinsic catalytic activity.25–28 However, the scarcity and limited availability of these noble metals present a substantial obstacle to their large-scale deployment.23 Additionally, a relatively short lifespan is also a limitation that carries implications for both system efficiency and longevity. Therefore, there is an urgent need to explore electrocatalysts that are not only low-cost and highly stable, but also capable of overcoming the slow kinetics of the HER process in alkaline electrolyzers.29
2D nanomaterials with increased surface areas and number of active sites, such as transition metal sulfides, dichalcogenides, and graphitic carbon nitrides, have demonstrated enhanced performance including catalytic activity and stability as HER catalysts.30,31–33 Over the past decades, 2D transition metal carbides (MXenes) have displayed potential as effective electrocatalysts both in experimental and computational studies.34–36 Pristine MXenes have been applied in electrocatalytic hydrogen production owing to their inherent excellent electronic conductivity, abundant hydrophilic surfaces, and highly redox-active transition metal composites.37–39 Beyond their use as standalone catalysts, MXenes also function as substrates for active materials. When combined with metals, metal sulfides, and such compounds, the metal–substrate interactions (MSI) lead to orbital hybridization and enhanced charge transfer at the interface, consequently altering the adsorption behaviors of reactants and lowering the energy barriers of the reaction steps.40 MXenes serving as substrates also provide physical support for the supported materials, enabling the specific distribution of loaded metals to remain stable under electrochemical reduction conditions, which is beneficial for long-term operation and continuous performance. These processes enhance the activity beyond the active materials’ intrinsic capabilities, showing promise in designing effective non-noble metal catalysts for the HER.
In this work, we report complete noble metal-free catalysts for furfural oxidation and hydrogen evolution at the anode and cathode to develop a paired flow electrolyzer. This represents a strategic approach to reduce the production costs associated with hydrogen and 2-furoic acid. This study entailed the integration of the furfural oxidation reaction with the hydrogen evolution reaction within an electrochemical framework. The synergistic benefits of such designs resulted in the successful demonstration of a functional paired electrolyzer capable of achieving an industrially relevant current density of 200 mA cm−2 at a faradaic efficiency of 46% to 2-furoic acid at furfural conversions of 68% and 100% to hydrogen with a cell voltage of 2.688 V.
2. Experimental
2.1 Chemicals and materials
Nickel foam (1.6 mm thick, MTI Corporation), nickel(II) nitrate hexahydrate (97%, Sigma-Aldrich), ferric nitrate nonahydrate (98.7%, Fisher-Scientific), hydrochloric acid (Fisher-Scientific), furfural (99%, Sigma-Aldrich), furfuryl alcohol (98%, Sigma-Aldrich), 2-furoic acid (98%, Sigma-Aldrich), potassium hydroxide (85%, Sigma-Aldrich), sulfuric acid (Fisher-Scientific), platinum foil (0.025 mm thick, 99.9%), molybdenum (1–5 μm, 99.9%, Sigma Aldrich), aluminum (17–30 μm, 99%, Alfa Aesar), titanium (325 mesh, 99%, Alfa Aesar), graphite (7–11 μm, 99%, Alfa Aesar), hydrofluoric acid (48%, Sigma Aldrich), cobalt(II) chloride (98%, Sigma Aldrich), Nafion solution (LQ-1105 1100EW, 5 wt%, Ion Power), Nafion membrane (115, 125 μm, Ion Power), carbon paper (Freudenberg H23, Fuel Cell Store), and Pt/C (40% platinum on Vulcan XC-72R, 2–3 nm, Fuel Cell Store).2.2 Preparation of the anodic electrode
The nickel-iron on nickel foam (NiFe/NF) was prepared in the same way mentioned in our previous work.41 A piece of nickel foam was immersed in 5 M hydrochloric acid (HCl) solution. Following immersion, it underwent 20 minutes of sonication to effect the removal of any surface nickel oxide layers. Subsequently, the Ni foam was subjected to thorough rinsing sequences involving water and ethanol, and was allowed to air-dry. After these preparatory steps, the Ni foam piece was designated for deployment as the working electrode within a conventional three-electrode electrochemical cell. This electrochemical cell comprised a platinum foil serving as the counter electrode and a silver/silver chloride (Ag/AgCl) electrode functioning as the reference electrode. The electrolyte bath, employed for electrochemical processes, comprised a solution containing 3 mM nickel(II) nitrate hexahydrate and 3 mM iron(III) nitrate nonahydrate. For the purpose of electrodepositing a NiFe layer onto the surface of the Ni foam, a constant potential of −1.0 V with respect to the Ag/AgCl reference electrode was applied for a duration of 25 minutes. Concluding the electrochemical deposition, the prepared nickel foam, now adorned with the NiFe deposit, underwent a final washing procedure with water, followed by air-drying to obtain the desired state.2.3 Preparation of the cathodic electrode
The cathodic electrode was constructed by spraying the Co/Mo2TiC2 catalyst on hydrophilic carbon paper. The bulk Mo2TiAlC2 MAX precursor was first prepared by spark plasma sintering (SPS) of metal powders. Commercial powders of molybdenum (Mo), aluminum (Al), titanium (Ti), and graphite were mixed in a molar ratio of Mo/Ti/Al/C = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1:1:1.9 in a graphite die coated with boron nitride (BN). The sample was then loaded in a Fuji-211lx spark plasma sintering (SPS) system and sintered at 1450 °C under 30 MPa for 1 h. The sintered bulk Mo2TiAlC2 was pulverized in a synthetic sapphire mortar and screened through a 400-mesh sieve to obtain a fine Mo2TiAlC2 powder. Mo2TiC2 MXene was prepared by etching Mo2TiAlC2 powder with hydrofluoric acid (HF) at 55 °C for 72 h. The mixture was then washed and centrifuged with deionized water (DI) until pH reached 6 to 7. Mo2TiC2 MXene was collected by centrifugation at 9000 rpm and dried in vacuum. Co was loaded on Mo2TiC2 MXene by an incipient wetness impregnation method. Cobalt(II) chloride (CoCl2) was dissolved in DI water to form a solution of 0.125 g Co mL−1. A volume of 100 μL from this CoCl2 solution was mixed with 250 mg of Mo2TiC2 MXene powder in a polyethylene weighing boat under constant stirring. The mixture was subsequently dried in vacuum at ambient temperature, then transferred into a tube furnace and reduced under a 5% H2/N2 flow at different temperatures (500 °C, 600 °C, 700 °C). The resulting materials were designated as Co/Mo2TiC2-500, 600, and 700 catalysts, respectively. The cathodes were prepared by spray coating. The spray ink was prepared by mixing 200 mg of catalysts with 20 wt% Nafion solution and 20 mL of isopropanol with 5 min sonication. The prepared catalyst ink was sprayed on a 2 × 2 cm2 area on H23 carbon paper with a 0.1 mg cm−2 Vulcan carbon nanoparticle layer to increase the contact. The electrodes were heated at 70 °C during the spray to vaporize the isopropanol and were left on the heating plate for 10 min after the spray for complete drying. For comparative experiments, commercial 40% Pt/C was sprayed using the same method.
:1.1:1:1.9 in a graphite die coated with boron nitride (BN). The sample was then loaded in a Fuji-211lx spark plasma sintering (SPS) system and sintered at 1450 °C under 30 MPa for 1 h. The sintered bulk Mo2TiAlC2 was pulverized in a synthetic sapphire mortar and screened through a 400-mesh sieve to obtain a fine Mo2TiAlC2 powder. Mo2TiC2 MXene was prepared by etching Mo2TiAlC2 powder with hydrofluoric acid (HF) at 55 °C for 72 h. The mixture was then washed and centrifuged with deionized water (DI) until pH reached 6 to 7. Mo2TiC2 MXene was collected by centrifugation at 9000 rpm and dried in vacuum. Co was loaded on Mo2TiC2 MXene by an incipient wetness impregnation method. Cobalt(II) chloride (CoCl2) was dissolved in DI water to form a solution of 0.125 g Co mL−1. A volume of 100 μL from this CoCl2 solution was mixed with 250 mg of Mo2TiC2 MXene powder in a polyethylene weighing boat under constant stirring. The mixture was subsequently dried in vacuum at ambient temperature, then transferred into a tube furnace and reduced under a 5% H2/N2 flow at different temperatures (500 °C, 600 °C, 700 °C). The resulting materials were designated as Co/Mo2TiC2-500, 600, and 700 catalysts, respectively. The cathodes were prepared by spray coating. The spray ink was prepared by mixing 200 mg of catalysts with 20 wt% Nafion solution and 20 mL of isopropanol with 5 min sonication. The prepared catalyst ink was sprayed on a 2 × 2 cm2 area on H23 carbon paper with a 0.1 mg cm−2 Vulcan carbon nanoparticle layer to increase the contact. The electrodes were heated at 70 °C during the spray to vaporize the isopropanol and were left on the heating plate for 10 min after the spray for complete drying. For comparative experiments, commercial 40% Pt/C was sprayed using the same method.
2.4 Characterization and analytical methods
Field-emission scanning electron microscopy (SEM, FEI Quanta 250) with energy dispersive X-ray spectroscopy (EDS) was used to explore the morphology and composition change of nickel–iron on nickel foam and Co/Mo2TiC2 catalysts. The X-ray diffraction spectrometer (XRD, Siemens D500) with a Cu Kα radiation source (λ = 1.5406 Å) was used to characterize the crystalline structures of Mo2TiC2 MXene. A scanning/transmission electron microscope (STEM, 200 kV JEOL 2100) with <1.4 Å resolution was used to investigate the nanostructure of the Co/Mo2TiC2 catalyst. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping were performed using a Thermo Fisher Scientific Talos F200X S/TEM operated at 200 kV and equipped with a super-X EDS system. The H2 quantification was carried out using an SRI gas chromatograph (GC, multiple gas analyzer #5) with TCD and FID detectors. The quantification of furfural, 2-furoic acid, and furfuryl alcohol was accomplished using high-performance liquid chromatography (HPLC) with an Agilent Technologies 1260 Infinity II LC system, which was equipped with a variable wavelength detector (VWD) also from Agilent Technologies. Wavelengths of 210 nm and 260 nm were employed for the quantification of furfural and furfuryl alcohol, and furfural and 2-furoic acid, respectively. It should be noted that furoate was produced from the FOR in an alkaline electrolyte (see the FOR equation in S-4 in the ESI†), but it is 2-furoic acid that was quantified by HPLC.2.5 Electrochemical characterization
All electrochemical tests were performed using a Biologic SP-300 potentiostat with a ±2 A/±30 V booster. Electrochemical half-cell measurements were conducted in a three-electrode H-cell system, with Ag/AgCl as the reference electrode for the FOR and Hg/HgO as the reference electrode for the HER. A potentio-electrochemical impedance spectroscopy (PEIS) scan from 200 kHz to 100 mHz was first executed to ascertain the resistance existing between the working and the reference electrode. The point at which the recorded resistance intersects with the x-axis on the impedance versus resistance plot corresponded to the measured resistance value. For furfural oxidation reaction, linear sweep voltammetry (LSV) analysis was conducted to evaluate the performance of both electrodeposited Ni foam and non-deposited nickel foam in a solution containing furfural, as well as a solution without furfural. The LSV test was carried out at a scan rate of 1 mV s−1, spanning the potential range from 0 V vs. RHE to 1.8 V vs. RHE. Subsequently, electrochemical testing, specifically chronopotentiometry tests, was performed under varying constant current conditions ranging from 10 mA cm−2 to 80 mA cm−2. Furthermore, different concentrations of furfural were examined in 1 M potassium hydroxide (KOH) solution, starting from a concentration of 50 mM and incrementally increasing to 200 mM, all the while maintaining a constant current density of 65 mA cm−2. For the HER, LSV, electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV), and stepwise chronopotentiometry (CP) tests were conducted to evaluate the performance of the series of Co/Mo2TiC2 catalysts. The potentials were converted to a reversible hydrogen electrode (RHE), and all the tests except CP were automatically 85% IR corrected by the potentiostat.For electrochemical measurements in the flow cell, a 2 × 2 cm2 two-electrode flow cell was used, serving as both a current collector and a conduit for fluid flow. The flow cell was equipped with silicon gaskets to avert leakage or inundation. The inter-electrode separation was maintained using a cation exchange membrane (Nafion 115). Linear sweep voltammetry (LSV) assessments were performed employing two distinct cathodes: platinum on carbon (Pt/C) and Co/Mo2TiC2-700 catalysts. A range of constant current densities, commencing at 30 mA cm−2 and extending up to 400 mA cm−2, was applied to ascertain and the performance of the hydrogen evolution reaction was compared, when coupled with NiFe/NF for the furfural oxidation reaction.
3. Results and discussion
3.1 Catalyst morphology and microstructure
Characterization results of Mo2TiC2 and Co/Mo2TiC2 catalysts are shown in Fig. 1. X-ray diffraction (XRD) analysis was first applied to confirm the crystal structure of Mo2TiC2. Compared with the Mo2TiAlC2 precursor, the (002), (004), and (006) diffraction peaks moved to lower angles (at 2θ = 7.0°, 13.9°, and 20.9°, respectively) after the HF etching, indicating a larger c lattice parameter of Mo2TiC2 due to the removal of Al layers and the expansion along the [001] direction. The characterized peaks aligned well with previously reported Mo2TiC2 structure.42 After loading Co, the intensity of MXene peaks significantly decreased, likely due to an overall structural change, while no distinct Co peaks were observed, indicating a uniform distribution rather than large crystalline particles. The scanning electron microscopy (SEM) image of Mo2TiC2 revealed an accordion-like morphology of typical MXene materials. The two-dimensional structure of the catalyst featuring orderly arranged metal and carbon layers was clearly displayed under observation using a transmission electron microscope (TEM). A stack of several Mo2TiC2 nanosheets is shown in Fig. 1c, each comprising multiple atomic layers. The atomic layers are distinctly delineated due to the contrast difference of Mo, C, and Ti elements. The inset in Fig. 1d shows the selected area electron diffraction (SAED) pattern on the basal plane of Mo2TiC2 in the image, confirming the hexagonal symmetry structure of the Mo2TiC2 crystalline lattice. Co was loaded on the synthesized Mo2TiC2 by incipient wetness impregnation. Fig. S4† displays the EDS mappings of the Co/Mo2TiC2 catalyst, illustrating the uniform distribution of Mo and Ti with no phase segregation. Co signals were distributed throughout the surveyed area, indicating a highly dispersed distribution with no formation of large clusters or metal particles. The atomic ratio of Mo:Ti is close to 2:1, consistent with the Mo2TiC2 composition. Fig. 1e shows the lateral structure of the Co/Mo2TiC2-700 catalyst, with no discernible structures of Co existing separately observed at the nanoscale. Fig. 1f shows the ultra-thin edge region, where benefiting from high transparency and reduced background scattering at the thin layer, some distinct lattice fringes orientating differently from the planar MXene material were observed. The spacing of the lattice fringes is at 2.40–2.42 Å, corresponding to the (111) plane of CoO, suggesting the existence of oxidized Co patches on the Mo2TiC2 external surfaces. Atomic resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and EDS were employed to further investigate the structure after Co loading. Fig. 1g shows a uniform structure of Co intercalation between the Mo2TiC2 MXene layers on the Co/Mo2TiC2-700 sample, with a clear regular repeating pattern of one single atom layer of Co between each two MXene pieces observed across the sample. Furthermore, HAADF-STEM images of Co/Mo2TiC2-500 and Co/Mo2TiC2-600 in Fig. S5† show that at 500 °C, no noticeable Co intercalation was observed and the MXene exists as separated layers with a certain distance, whereas at 600 °C Co can be seen partially entering the inter-layers at the edges of the sample, forming one or several atom layers of intercalations, while Mo2TiC2 flakes farther from the surface appear to remain unaffected. This intriguing observation suggested an annealing temperature-induced enhancement of uniform metal distribution. Such phenomena have also been observed in past research,43,44 typically resulting in distribution induced metal–support interaction differences and leading to modulated performance. Therefore, testing and selecting optimal catalysts among the Co/Mo2TiC2 catalysts with the structural alterations is a worthwhile pursuit potentially enabling performance tuning.
 | ||
| Fig. 1 Mo2TiC2 MXene and Co/Mo2TiC2 cathodic catalyst. (a) XRD patterns of Mo2TiAlC2, Mo2TiC2, and Co/Mo2TiC2-700; (b) SEM image of synthesized Mo2TiC2 MXene, (c) TEM image of Mo2TiC2, (d) TEM images with the SAED pattern of Mo2TiC2 MXene, (e and f) TEM images of Co/Mo2TiC2 catalysts, (g) HAADF-STEM image of Co/Mo2TiC2-700 with EDS mapping for Mo, Ti, Co, Al, and C. | ||
The morphology and composition characteristics of the synthesized nickel–iron on nickel foam (NiFe/NF) anode, were investigated by XRD, SEM, and TEM, as shown in Fig. 2 and Fig. S8.† Comparison of the electrodes before and after the electrochemical deposition processes, as illustrated in Fig. S1,† displayed the formation of a dense uniform deposition layer widely covering the Ni foam surface. Fig. 2a presents NiFe deposition on foam over a large area. A further magnified SEM image of a small section on the framework of the nickel foam (Fig. 2b) reveals that the deposition layer exhibits a flat morphology. Some protruding particles were observed at the edges and nodal junctions of the Ni foam networks, presumably originating from the localized faster deposition rates at the uneven geometric structure of the nickel foam. Energy dispersive X-ray spectroscopy (EDS) analysis confirms the homogeneous coexistence of Fe and Ni within both the uniform deposition layer and separated particle regions (Fig. 2d and S3†). The cross-sectional area image (Fig. 2d) shows that the overall thickness of NiFe deposit is approximately 1 μm. The TEM images of the catalyst scraped from the electrode revealed the microstructure of the NiFe composite. Mostly the NiFe composite appeared as ultra-thin amorphous nanosheets, as depicted in Fig. 2e and the magnified area in Fig. 2f, corresponding to the uniform layered deposition in the SEM images. Spherical composites of approximately 500 nm size, corresponding to the protrusions described in Fig. 2c and S3b,† were also observed amidst these nanosheets, as shown in Fig. S2.† Further magnification revealed that these spheres are around 5 nm sized crystallized nanoparticles distributed within amorphous NiFe clusters (Fig. S2b†). The measured lattice fringe spacing was 2.21–2.23 Å and 1.91 Å, which deviated from that of pure nickel and iron but was close to that of the (111) and (200) planes of Ni3Fe alloy, suggesting them to be alloy nanoparticles. XRD of the NiFe/NF foam showed diffraction peaks of nickel, which further confirmed that the NiFe nanosheet has amorphous structure (Fig. S8†). The characterization collectively substantiated the co-formation of Ni and Fe and the uniform distribution of both elements within the composite structure.
 | ||
| Fig. 2 NiFe/NF anodic electrode. (a–c) SEM images of the NiFe/NF electrode; (d) SEM image of the cross-sectional area of NiFe/NF; (e and f) TEM images of NiFe nanosheets scratched off from the NiFe/NF electrode. | ||
3.2 HER electrolysis in the H-type cell
The evaluation of alkaline HER performance and selection of the best Co/Mo2TiC2 catalyst was conducted in the H-type half cell. Linear sweep voltammetry (LSV) was first conducted for the assessment of the catalytic activity of Co/Mo2TiC2-500, 600, and 700. As shown in Fig. 3a, Co/Mo2TiC2-700 gave the highest current density among the three catalysts, and exhibited a low overpotential of 244 mV @ 100 mA cm−2. At further increased current density, Co/Mo2TiC2-700 also showed excellent low overpotential of 321 mV @ 400 mA cm−2, and outperformed Pt/C (366 mV @ 400 mA cm−2) for 45 mV, highlighting its high current density advantage and potential applicability in the MEA-based flow cell electrolyzer as an efficient cathode. | ||
| Fig. 3 Electrochemical HER performance. (a) Linear sweep voltammetry (LSV) curves, (b) Tafel plots, (c) Nyquist plots, and (d) double-layer capacitance of Co/Mo2TiC2 annealed at 500, 600, and 700 °C in 1 M KOH. | ||
The Tafel curves of Co/Mo2TiC2-500, 600, and 700 are shown in Fig. 3b. The Tafel slopes are 108, 92, and 62 mV dec−1, respectively, revealing that Co/Mo2TiC2-700 exhibited the most rapid current increase with the potential, indicating the fastest reaction kinetics on this catalyst. To compare the charge transfer efficiency, the electrochemical impedance spectra (EIS) were also obtained, as shown in Fig. 3c. The solution resistance (Rs) values were approximately the same for the three catalysts, suggesting a similar electrolyte solution environment. The charge transfer resistance (Rct) of the catalysts shown in the Nyquist plots displayed Co/Mo2TiC2-500 had the largest semi-circle, while Co/Mo2TiC2-700 had the smallest Rct value, corresponding to faster charge transfer property due to the optimized metal support structure.
The electrochemically active surface area (ECSA) was also investigated by measuring the double layer capacitance (Cdl) (Fig. 3d and S6†). Cyclic voltammograms (CV) were applied at scan rates of 10, 20, 40, 60, 80, and 100 mV s−1 in the non-faradaic region (0.10–0.20 V vs. RHE). The Cdl values were calculated by plotting the charging current density differences against the scan rates. The ECSA can be obtained by dividing Cdl by the specific capacitance (Cs), which can be assumed as 40 μF cm−2. Fig. S6† and Fig. 3d show that the Cdl values of Co/Mo2TiC2-500, 600, and 700 are 7.39, 8.97, and 38.72 mF cm−2, respectively. The highest Cdl value of Co/Mo2TiC2-700 suggests the largest electrochemical active surface area among the three catalysts, demonstrating its advantage as an efficient hydrogen evolution catalyst.
Stability is also a pivotal factor for sustained and reliable operation in industrial-scale flow cells; therefore, the Co/Mo2TiC2-700 catalyst demonstrating the most superior HER catalytic performance was subjected to a stepwise chronopotentiometry (CP) stability test for 36 hours. As shown in Fig. 4, in the overall 36 h test, current densities of negative 10, 50, 100, 200, and 400 mA cm−2 were sequentially applied for 4 h periods. The potential remained stable at each current density and quickly returned to the corresponding potential value at every current decreasing step, indicating no degradation following these tests. The sustained performance under extended testing conditions underscored the robustness and durability of the catalyst, marking it as a viable candidate for long-term applications.
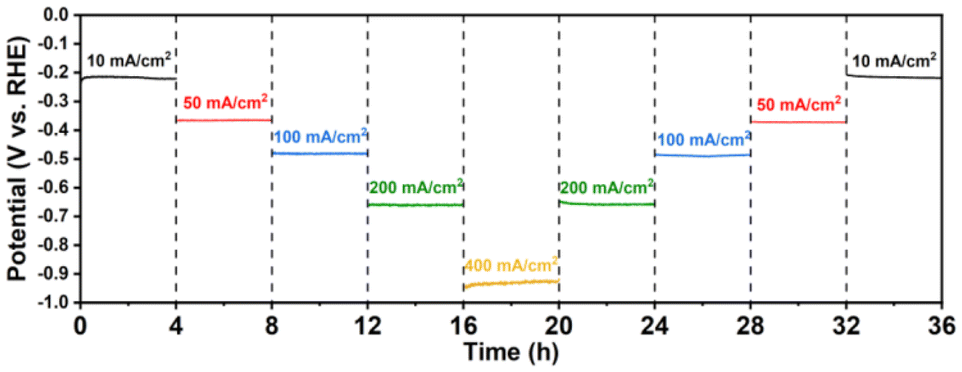 | ||
| Fig. 4 Stability test. Stepwise chronopotentiometry profiles of Co/Mo2TiC2-700 in 1 M KOH without IR correction. | ||
3.3 FOR electrolysis in the H-type cell
Previous works were heavily focused on noble metal catalysts for heterogeneous catalytic oxidation (Pt, Au, and Pd) that require aerobic conditions,45–48 or electrocatalytic oxidation.49 The NiFe-based catalysts have been identified and documented as effective catalysts for the oxygen evolution reaction (OER).50,51 Given its high intrinsic suitability, low costs of NiFe, and favorable thermodynamics for aldehyde oxidation, non-noble bimetal NiFe could be a possible catalyst for furfural oxidation. However, the faradaic efficiencies towards 2-furoic acid (furfural oxidation product) and oxygen (OER product) at high overpotentials have been rarely studied. Furthermore, the effects of the homogeneous Cannizzaro reaction of furfural at anodic potential, and its effect on the FOR need to be better understood. Aiming to acquire deeper understanding of the FOR over NiFe catalysts, an LSV experiment was conducted, comparing its catalytic activity with that of pure nickel foam (NF) under two distinct conditions: with/without the presence of furfural, as depicted in Fig. 5c. The fundamental challenge in the present study is illustrated, as the oxygen evolution reaction (OER) represents a potential competing reaction with the furfural oxidation reaction (FOR). This challenge is notably heightened by the fact that the thermodynamic potential of the OER is measured at 1.229 V vs. RHE. The LSV results distinctly demonstrated that the FOR process occurred at potentials exceeding the thermodynamic potential of the OER, with an apparent on-site potential registering at approximately 1.38 V vs. RHE. This test also gives an insight into how the concentration of furfural can affect the onset potential of the FOR. For instance, the onset potential at the furfural concentration of 50 mM was 1.41 V vs. RHE, while the FOR onset potential at the furfural concentration of 200 mM slightly dropped to 1.39 V vs. RHE. | ||
| Fig. 5 FOR electrolysis in H-cell. (a) Faradaic efficiency of 2-furoic acid, and concentrations of 2-furoic acid, furfuryl alcohol, and unreacted furfural after 1-hour electrolysis of furfural at different current densities. (b) Faradaic efficiency of 2-furoic acid, and concentrations of 2-furoic acid, furfuryl alcohol, and unreacted furfural after 1-hour electrolysis of furfural at different starting concentrations of furfural. (c) Linear sweep voltammetry of NF and NiFe/NF when furfural is present and absent. (d) Potential profile of 65 mA cm−2 for different furfural initial concentrations. (e) Potential profiles of different currents at 50 mM furfural in 1 M KOH. | ||
To further test the catalyst selectivity and conversion from furfural to 2-furoic acid, different constant currents were applied on the NiFe/NF anode, as shown in Fig. 5a. The various current densities employed in this study exhibited a discernible reduction in faradaic efficiency as their magnitudes were elevated. Nonetheless, a noteworthy observation was the concomitant increase in the conversion of furfural, reaching an approximate near-quantitative conversion rate of 100%. This observation highlights the remarkable selectivity towards the targeted product of 2-furoic acid on NiFe catalysts. Fig. 5e shows that the FOR performance was stable throughout the one-hour electrolysis even at a higher current density when about 90% of the furfural was converted, and the FE dropped. Upon near-complete consumption of furfural, a conspicuous phenomenon emerged in the form of gas bubbles in the solution, becoming increasingly evident with time. This observation served as a clear indicator of the transition from the FOR to the side reaction: the oxygen evolution reaction (OER) within the electrochemical process. It is reasonable to infer that the dwindling availability of furfural as a reactant contributes to the ascendance of the OER, thereby elucidating the concomitant decline in the overall faradaic efficiency (FE). For instance, at a current density of 80 mA cm−2, the recorded FE stood at 40%, signifying that 40% of the generated charge was directly used for the FOR, while the remaining 60% was for the OER. It should be noted that this FE reduction should not be misconstrued as a decline in the catalyst's efficacy towards the FOR. Instead, it primarily reflects the near exhaustion of the furfural reactant. Furthermore, to acquire deep insight into this phenomenon, a series of experiments were conducted employing varying concentrations of furfural at a fixed current density of 65 mA cm−2, aiming to assess the feasibility of sustaining high-efficiency levels over a prolonged 1-hour duration, as delineated in Fig. 5b. The conducted test has illuminated that FE of 80% or higher can be sustained within the system when ample supply of reactant is available. However, it is crucial to consider that excessively high aldehyde concentration triggers the risk of occurrence of the Cannizzaro reaction, which is the disproportionate transformation of two molecules of a non-enolizable aldehyde (furfural) into one primary alcohol (furfuryl alcohol) molecule and one carboxylic acid (2-formic acid) molecule. This reaction is regarded as undesired in the context of the present electro-oxidation process. To mitigate the undesired side reactions and minimize the formation of byproducts, the faradaic reaction pathway converting furfural directly into 2-furoic acid is more desirable. Nevertheless, it was observed that, at higher furfural concentrations of 150 mM and 200 mM, the side product furfuryl alcohol was still generated to some extent. Moreover, the potential profile depicted in Fig. 5d reveals a reduction in potential corresponding to an increase in furfural concentration. Enhanced performance was observed at 100 mM furfural, representing an improvement over the 50 mM concentration. However, concentrations of 150 mM and 200 mM furfural did not yield a significant enhancement in performance. Notably, these higher concentrations concomitantly promoted the Cannizzaro reaction during the electrolysis, resulting in the production of furfuryl alcohol. Subsequently, it was determined that the optimal concentration for the electrochemical FOR in the flow cell is 100 mM of furfural in 1 M potassium hydroxide (KOH) electrolyte. This concentration reaches a balance between minimizing the production of furfuryl alcohol and concurrently upholding high FE. It is worth noting that these experiments were conducted within our H-type cell, a setup with a limited reactant capacity of up to 15 mL, and all faradaic efficiency shown in Fig. 5 accounted only for 2-furoic acid produced electrochemically (via faradaic process).
3.4 FOR–HER paired electrolysis in the MEA flow cell
An MEA-based flow cell was used for experimental investigation of FOR–HER paired electrolysis, as depicted in Fig. 6. Its basic feature was the insertion of a cation exchange membrane (CEM) in a zero-gap setting situated between the cathode and the anode. In this configuration, the inter-electrode resistance can be substantially reduced. The resistance within this configuration averaged around 0.925 Ω. Internal resistance was automatically compensated at 85% for LSV to reduce the overall cell voltage, particularly when the anodic reaction exhibited increased on-site potential owing to the use of non-noble NiFe catalyst, as evidenced in Fig. 5c. Within this electrochemical environment, furfural underwent electrocatalytic transformation facilitated by the NiFe catalyst with a nickel foam as the substrate at the anode, while water reduction was driven by either the Pt/C or Co/Mo2TiC2-700 catalysts with a carbon paper as the cathodic substrate. The primary objective was to experimentally validate the feasibility of paired electrolysis with complete noble-metal-free catalysts, and the performance of the two cathodic catalysts (Pt/C and Co/Mo2TiC2-700) when employed with the same anodic catalyst was compared. | ||
| Fig. 6 MEA-based flow cell. Diagram of the two-electrode MEA-based flow cell for the FOR paired with HER tests. | ||
A 12-hour electrolysis test at 50 mA cm−2 with a Pt/C electrode was conducted to assess the endurance of NiFe/NF over long time operation. The electrolyzer was supplied with freshly prepared 100 mM furfural in 1 M KOH at the 6th hour of the test. Throughout the test duration, a consistent trend emerged wherein the FE progressively decreased with the conversion increasing, as shown in Fig. 7. Notably, the anode consistently maintained a reasonable FE to 2-furoic acid, with any observed drop attributed to an insufficient quantity of reactant in the electrolyte, as evidenced by the concurrent increase in conversion over time. Additionally, the test demonstrated a stable cell voltage throughout most of the testing period, with a sudden drop occurring during a reaction pause for electrolyte replacement, as previously mentioned. Before the electrolysis for paired HER and FOR, the LSV of cobalt-based and platinum-based cathode catalysts for the HER and NiFe/NF for the FOR was first conducted in the flow cell. The LSV results, as shown in Fig. 8a, clearly demonstrated a noteworthy decrease in the cell voltage when Co/Mo2TiC2-700 was used at high current density operation, for example, decreasing from an initial 4.5 V to close to 2.6 V at a current density of 400 mA cm−2, in contrast to the usage of Pt/C as the cathodic catalyst, while the metal loading in each of the cathodic catalysts was kept the same (0.5 mg cm−2 for each: Pt or Co).
 | ||
| Fig. 7 A durability test for 12 hour electrolysis of furfural oxidation to 2-furoic acid on NiFe/NF paired with the hydrogen evolution reaction on Pt/C. Faradaic efficiency and conversion are referred to the FOR. | ||
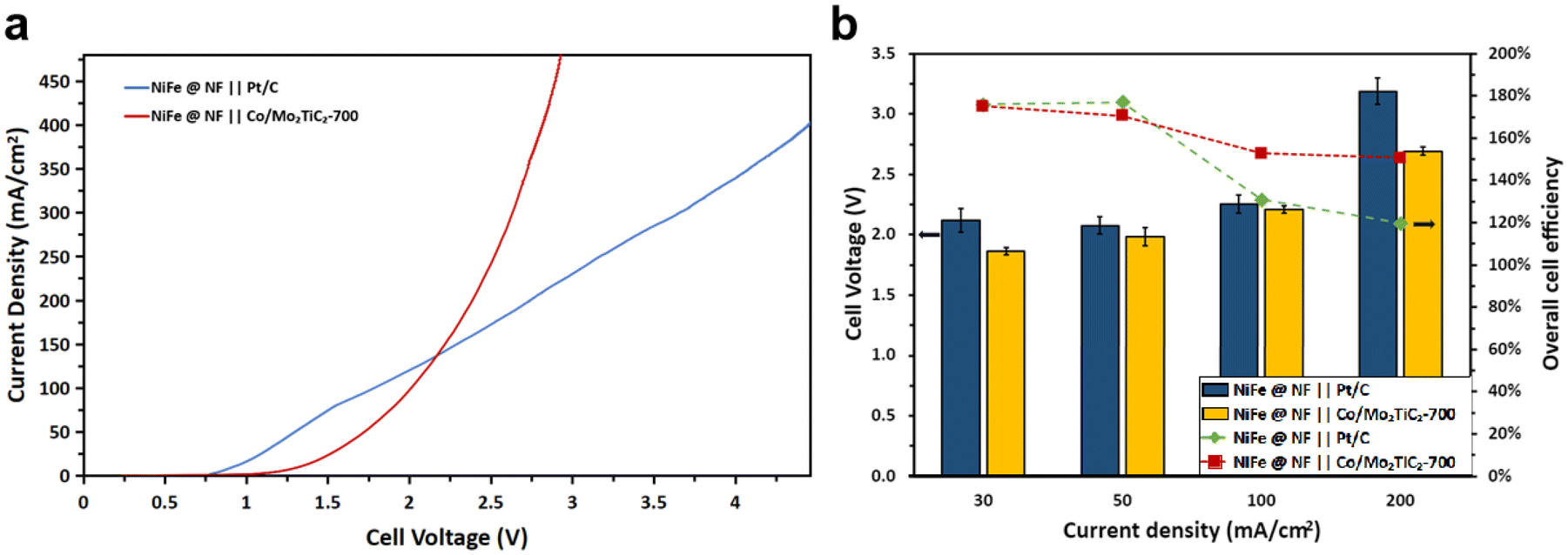 | ||
| Fig. 8 Electrolyzer performance when comparing the cobalt-based catalyst with the platinum-based catalyst at the cathode for the HER when paired with NiFe/NF for the FOR. (a) Linear sweep voltammetry of cobalt-based catalyst and platinum-based catalyst at the cathode for the HER when paired with NiFe/NF for the FOR. (b) The flow cell performance (cell voltage and faradaic efficiency) over 3 hours of electrolysis to compare the cobalt-based and platinum-based catalysts. | ||
Bulk electrolysis experiments were finally conducted using the FOR–HER paired electrolyzer by applying constant current densities ranging from 30 mA cm−2 to 200 mA cm−2, each maintained for three hours. Flow cell performances of distinct cathodes were systematically investigated. As illustrated in Fig. 8b, it was consistently observed that when employing Co/Mo2TiC2-700 as the cathode material, the cell voltage exhibited lower values in comparison with Pt/C. Furthermore, a noteworthy correlation was established between lower cell voltages and an increased likelihood of the anodic electrode displaying selectivity towards the formation of furoic acid (FOR) rather than participating in the OER, a phenomenon notably evident at current densities of 100 mA cm−2 and 200 mA cm−2. As previously analyzed, the faradaic efficiency considers only the 2-furoic acid produced electrochemically. Furthermore, a comprehensive examination of furfural consumption, 2-furoic acid production, and side product furfuryl alcohol generation was conducted over the course of the three-hour electrolysis. These assessments were conducted with a focus on the cathodic electrodes utilized in the electrochemical process. Intriguingly, the electrosynthesis of 2-furoic acid exhibited equivalence when employing Co/Mo2TiC2-700, and in certain instances even exceeded that observed when Pt/C was used as the cathode (Fig. 9a and b). Notably, at 200 mA cm−2 approximately 46% faradaic efficiency of furfural was obtained when Co/Mo2TiC2-700 served as the cathode, while on Pt/C an FE of only 21% was achieved under identical conditions. The decreased FE was attributed to the elevated voltages within the electrolyzer generated when employing Pt/C, potentially impacting anode performance and promoting a shift toward the OER. Consequently, the occurrence of the Cannizzaro reaction increased, resulting in greater furfural consumption, leading to the production of both 2-furoic acid and furfuryl alcohol.
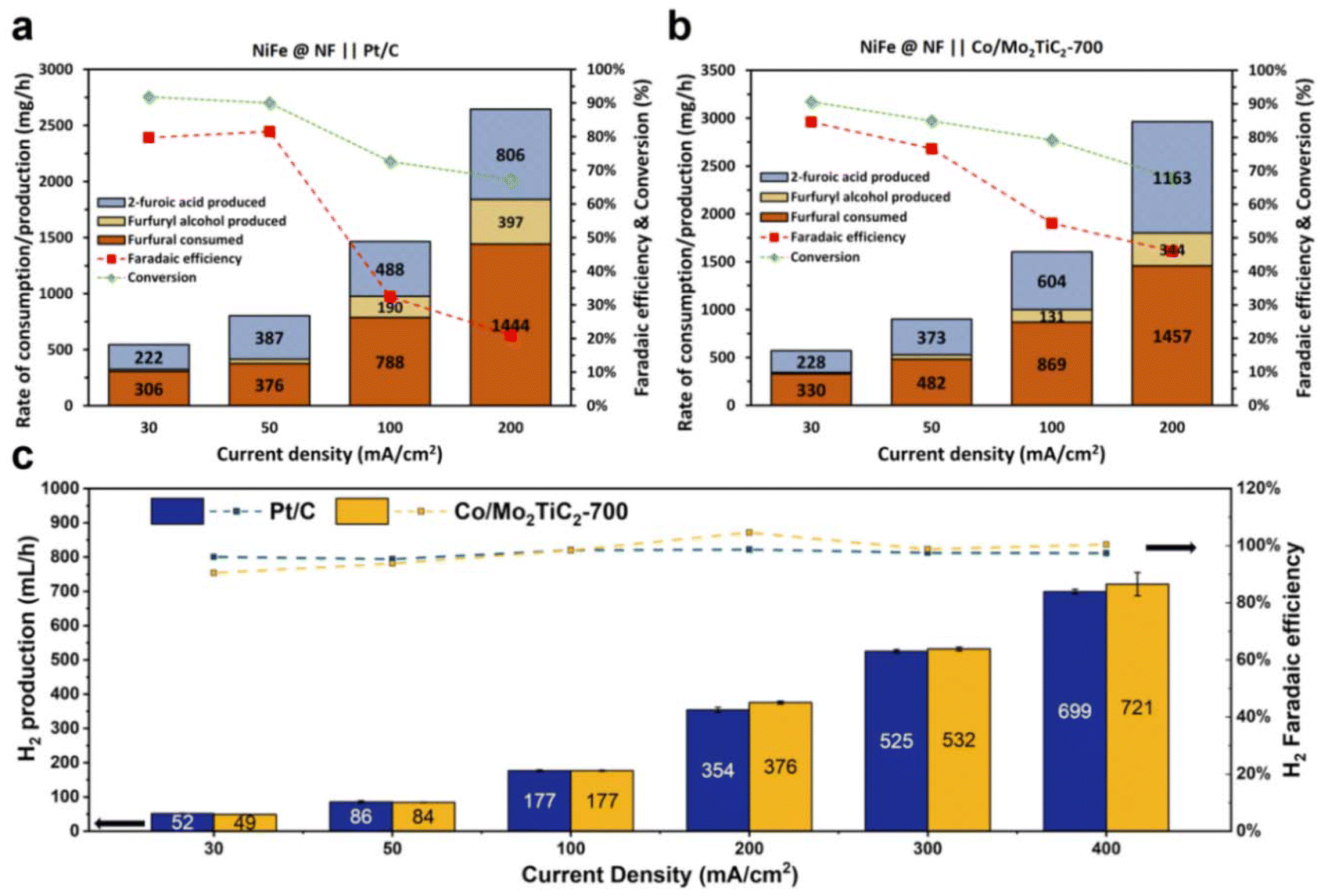 | ||
| Fig. 9 Alkaline electrolyzer performance for the cathode and anode. (a) Anode performance when the cathode contains a platinum-based catalyst. (b) Anode performance when the cathode contains a cobalt-based catalyst. (c) Hydrogen production and faradaic efficiency when comparing a cobalt-based catalyst with a platinum-based catalyst. | ||
Co/Mo2TiC2-700 at the cathode exhibited notable performance comparable to the state-of-the-art Pt/C for the HER, as depicted in Fig. 9c. Both catalysts gave over 90% H2 faradaic efficiency across all current ranges. Particularly, in the range of 100 to 400 mA cm−2, the Co/Mo2TiC2-700 exhibited higher than 98% H2 efficiency, and the quantity of produced H2 and faradaic efficiency surpassed that of Pt/C. At 400 mA cm−2, Co/Mo2TiC2-700 had a higher cell voltage than Pt/C, and this disparity in catalyst performance can be mitigated by the cost differential between the cobalt-based catalyst and platinum. Though the low cell voltage advantage of Co/Mo2TiC2-700 at 400 mA cm−2 is not as pronounced as the lower range, i.e., at 200 mA cm−2 (Fig. 8), considering the lower cost of non-precious materials compared to expensive platinum, the overall reduction in the industrial production cost is still considerably significant. The tests indicate that the Co/Mo2TiC2-700 catalyst screened for H-cell according to the best intrinsic performance is indeed also highly efficient in the flow electrolyzer, and facilitated the co-production of H2 when paired with furfural oxidation.
We further analyzed the distribution of overpotentials originating from anode and cathode reaction kinetics and internal resistance (ohmic loss), as shown in Tables S3 and S4.† It is interesting to find that the discrepancies between measured and calculated cell voltages are in a reasonable range of 0.22–0.55 V. The discrepancies can be attributed to (1) the omission of anolyte and catholyte overpotentials in the analytical framework, and (2) the direct use of the anodic/cathodic potentials obtained from the half-cell tests. It is worth mentioning that this reported HER/FOR paired electrolyzer has demonstrated fairly high current density (200 mA cm−2) operation as compared with previous work (Table S5†). However, based on the overpotential distribution analysis, there are some future directions to reduce the cell voltage. For example, despite the maximum resistance being only 1.48 ohm, there exists a notable ohmic overpotential at high current densities. This overpotential underscores the significance of internal resistance even at seemingly low resistive values. The amelioration of internal resistance can be achieved by employing nickel foam with further reduced thickness or a thinner cation exchange membrane. Such modifications can further decrease the internal resistance, subsequently leading to cell voltage reduction. Lowering the cell voltage is pivotal in augmenting the furfural conversion rate, as previously observed that higher conversion rates occur at decreased cell voltages. This optimization not only enhanced conversion efficiency, but also curtails the expense associated with product–reactant separation. Maximizing the transformation of furfural to 2-furoic acid diminishes the presence of unreacted furfural, thereby potentially minimizing separation costs.
4. Conclusions
In summary, a versatile and flexible paired alkaline electrolyzer based on a compact membrane electrode assembly (MEA) configuration was developed for co-production of hydrogen and valuable carboxylic acid from water and biorenewable aldehydes. Specifically, the continuous and stable production of 2-furoic acid and H2 at 200 mA cm−2 was achieved by coupling the FOR on a NiFe/NF anode with the HER on a Co/Mo2TiC2-700 cathode. This paired electrolysis exhibited a combined faradaic efficiency (FE) of 150%, corresponding to a furfural conversion of 67.9% and cathodic FE towards the HER of 98.6%, at a cell voltage of ∼2.7 V. Moreover, the compact design of the MEA-based electrolyzer enabled its operation at a high current density of 200 mA cm−2 (800 mA). This study underscored the significance of employing a zero-gap MEA configuration in advanced electrochemical system design, specifically for the electrochemical upgrading of biomass-derived feedstocks. Furthermore, the approach of this work facilitated the co-production of H2 and valuable carboxylic acids using complete non-noble metal electrocatalysts, paving a cost-effective avenue toward the sustainable and distributed manufacturing of high-value chemicals in the chemical industry in the future.Author contributions
Conceptualization: W. L. and Y. W.; methodology: X. L., M. A., W. L. and Y. W.; investigation: X. L., M. A., M. G., C. W. S., Y. W., and W. L.; resources: X. L., M. A., M. G., C. W. S., Y. W., and W. L.; writing – original draft: X. L. and M. A.; writing – review & editing: W. L. and Y. W.; funding acquisition: W. L., and Y. W.; and supervision: W. L., and Y. W.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported this paper.Acknowledgements
This research was supported by NSF CBET 1947435, NSF EPSCoR 2316481, and USDA 2021-67021-34650. In particular, M. Albloushi would like to thank Dr Warren E. Straszheim for his help with SEM characterization. M. Galvin would like to thank Mickie and Gary Griswold for their philanthropic efforts to sponsor Michael's research through the Griswold internship program. M. Albloushi and X. Liu would like to thank Dr Yifu Chen, Dr Hengzhou Liu, Tianlei Li, and Sujin Kang for their insightful comments and suggestions. W. Li acknowledges his Herbert L. Stiles Faculty Fellowship and the ISU Presidential Interdisciplinary Research Initiative (PIRI) grant.References
- J. Turner, G. Sverdrup, M. K. Mann, P. C. Maness, B. Kroposki, M. Ghirardi, R. J. Evans and D. Blake, Int. J. Energy Res., 2008, 32, 379–407 CrossRef CAS.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS PubMed.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- H. Liu and W. Li, Curr. Opin. Electrochem., 2021, 30, 100795 CrossRef CAS.
- G. F. Chen, Y. Luo, L. X. Ding and H. Wang, ACS Catal., 2018, 8, 526–530 CrossRef CAS.
- H. Liu, N. Agrawal, A. Ganguly, Y. Chen, J. Lee, J. Yu, W. Huang, M. Mba Wright, M. J. Janik and W. Li, Energy Environ. Sci., 2022, 15, 4175–4189 RSC.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed.
- R. Gopalan and E. Kannamma, Indian J. Chem. A, 1984, 23, 518–519 Search PubMed.
- P. Verdeguer, N. Merat, L. Rigal and A. Gaset, J. Chem. Technol. Biotechnol., 1994, 61, 97–102 CrossRef CAS.
- A. M. Román, N. Agrawal, J. C. Hasse, M. J. Janik, J. W. Medlin and A. Holewinski, J. Catal., 2020, 391, 327–335 CrossRef.
- Y. Cao, J. Knijff, A. Delparish, M. F. N. d'Angelo and T. Noël, ChemSusChem, 2021, 14, 590–594 CrossRef CAS PubMed.
- X. Li, L. Cong, H. Lin, F. Liu, F. Han and N. Lin, Sci. China: Chem., 2022, 65, 2576–2587 CrossRef CAS.
- S. K. Dash, S. Chakraborty and D. Elangovan, Energies, 2023, 16, 1141 CrossRef CAS.
- N. Zaman, T. Noor and N. Iqbal, RSC Adv., 2021, 11, 21904–21925 RSC.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu and B. J. Ge, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- M. Luo, T. Wu, S. Xu, R. Wang and F. Huang, Chem. Commun., 2022, 58, 6204–6207 RSC.
- C. Meng, T. Ling, T. Y. Ma, H. Wang, Z. Hu, Y. Zhou, J. Mao, X. W. Du, M. Jaroniec and S. Qiao, Adv. Mater., 2017, 29, 1604607 CrossRef PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- D. S. A. Pratama, A. Haryanto and C. W. Lee, Front. Chem., 2023, 11, 1141361 CrossRef CAS PubMed.
- Z. Kang, M. A. Khan, Y. Gong, R. Javed, Y. Xu, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9, 6089–6108 RSC.
- P. Prabhu, V. Jose and J. M. Lee, Matter, 2020, 2, 526–553 CrossRef.
- S. J. Rowley Neale, D. A. Brownson, G. C. Smith, D. A. Sawtell, P. J. Kelly and C. E. Banks, Nanoscale, 2015, 7, 18152–18168 RSC.
- H. Wang, X. Xiao, S. Liu, C.-L. Chiang, X. Kuai, C.-K. Peng, Y.-C. Lin, X. Meng, J. Zhao, J. Choi, Y. G. Lin, J. M. Lee and L. Gao, J. Am. Chem. Soc., 2019, 141, 18578–18584 CrossRef CAS PubMed.
- G. Gao, A. P. O'Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang, Q. Chen and J. Wang, Adv. Sci., 2016, 3, 1600180 CrossRef PubMed.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2017, 1, 173–180 CrossRef.
- S. Li, P. Tuo, J. Xie, X. Zhang, J. Xu, J. Bao, B. Pan and Y. J. Xie, Nano Energy, 2018, 47, 512–518 CrossRef CAS.
- W. Yuan, L. Cheng, Y. An, H. Wu, N. Yao, X. Fan and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 8976–8982 CrossRef CAS.
- X. Wang, J. Ding, W. Song, X. Yang, T. Zhang, Z. Huang, H. Wang, X. Han and W. Hu, Adv. Energy Mater., 2023, 13, 2300148 CrossRef CAS.
- M. Albloushi, Master thesis, Iowa State University, 2022 Search PubMed.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- L. Dai, Y. Shen, J. Z. Chen, L. Zhou, X. Wu, Z. Li, J. Wang, W. Huang, J. T. Miller, Q. Wang, A. Cao and Y. Wu, Small, 2022, 18, 2105226 CrossRef CAS PubMed.
- Z. Li, Z. Qi, S. Wang, T. Ma, L. Zhou, Z. Wu, X. Luan, F. Y. Lin, M. Chen, J. T. Miller, H. Xin, W. Huang and Y. Wu, Nano Lett., 2019, 19, 5102–5108 CrossRef CAS PubMed.
- P. Parpot, A. P. Bettencourt, G. Chamoulaud, K. B. Kokoh and E. M. Belgsir, Electrochim. Acta, 2004, 49, 397–403 CrossRef CAS.
- M. Manzoli, F. Menegazzo, M. Signoretto, G. Cruciani and F. Pinna, J. Catal., 2015, 330, 465–473 CrossRef CAS.
- B. Zhao, M. Chen, Q. Guo and Y. Fu, Electrochim. Acta, 2014, 135, 139–146 CrossRef CAS.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J. D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- Y. Qiu, L. Xin and W. Li, Langmuir, 2014, 30, 7893–7901 CrossRef CAS PubMed.
- T. Suga, N. Shida and M. Atobe, Electrochem. Commun., 2021, 124, 106944 CrossRef CAS.
- W. Chen, J. Shi, C. Xie, W. Zhou, L. Xu, Y. Li, Y. Wu, B. Wu, Y. C. Huang, B. Zhou, M. Yang, J. Liu, C. L. Dong, T. Wang, Y. Zou and S. Wang, Natl. Sci. Rev., 2023, 10, nwad099 CrossRef CAS PubMed.
- W. Chen, Y. Wang, B. Wu, J. Shi, Y. Li, L. Xu, C. Xie, W. Zhou, Y. C. Huang, T. Wang, S. Du, M. Song, D. Wang, C. Chen, J. Zheng, J. Liu, C. L. Dong, Y. Zou, J. Chen and S. Wang, Adv. Mater., 2022, 34, 2105320 CrossRef CAS PubMed.
- W. Chen, J. Shi, Y. Wu, Y. Jiang, Y.-C. Huang, W. Zhou, J. Liu, C.-L. Dong, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316449 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04447a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |